

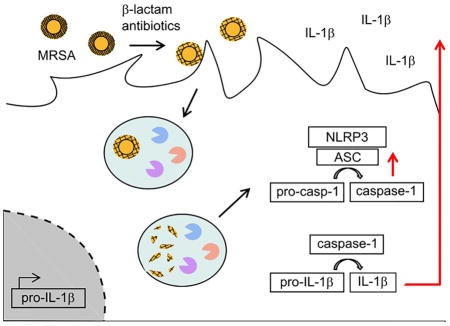
Summary
Methicillin-resistant S. aureus (MRSA) is a leading health problem. Compared to methicillin-sensitive S. aureus, MRSA infections are associated with greater morbidity and mortality but the mechanisms underlying MRSA pathogenicity are unclear. Here we show that the protein conferring β-lactam antibiotic resistance, penicillin-binding protein 2A (encoded by the mecA gene), directly contributes to pathogenicity during MRSA infection. MecA induction leads to a reduction in peptidoglycan cross-linking that allows for enhanced degradation and detection by phagocytes, resulting in robust IL-1β production. Peptidoglycan isolated from β-lactam-challenged MRSA strongly induces the NLRP3 inflammasome in macrophages but these effects are lost upon peptidoglycan solubilization. Mutant MRSA bacteria with naturally-occurring short peptidoglycan cross-links induce high IL-1β levels in vitro, and cause increased pathology in vivo. β-lactam treatment of MRSA skin infection exacerbates immunopathology, which is IL-1-dependent. Thus, antibiotic-induced expression of mecA during MRSA skin infection contributes to immunopathology by altering peptidoglycan structure.
Graphical Abstract
Introduction
Methicillin-resistant Staphylococcus aureus (MRSA) is the most common cause of skin and soft tissue infections in the US, but is also a frequent cause of severe invasive diseases (Hersh et al., 2008, Taylor, 2013). In the past decade, community-associated MRSA (CA-MRSA) strains USA300 and USA400 have spread endemically in the US (Kennedy et al., 2008, Oliveira et al., 2001). Compared to infections with methicillin-sensitive S. aureus (MSSA), MRSA infections cause greater morbidity and mortality (Kopp et al., 2004, Antonanzas et al., 2015, Ganga et al., 2009). The underlying reason is not entirely clear, but some studies suggest that as-yet-unidentified pathogenic factors contribute to the poor outcome (Watkins et al., 2012).
The genetic determinant that renders MRSA resistant to nearly all β-lactam antibiotics is the acquired gene mecA (Beck et al., 1986). It codes for a penicillin-binding protein (PBP), PBP2A, which has a lower affinity for β-lactam antibiotics compared to the native PBPs that are the targets of this class of antibiotics (Chambers et al., 1985). PBPs are essential for the generation of the large cell wall polymer peptidoglycan (PGN). In addition to extending the glycan strands, they connect individual glycan strands in a process termed transpeptidation (Sauvage et al., 2008). The domain of PBPs catalyzing this transpeptidation reaction is inhibited by β-lactam antibiotics. Hence, in the presence of β-lactam antibiotics endogenous PBPs are inactivated (Tomasz, 1979). Not constitutively expressed, mecA is strongly up-regulated upon challenge of MRSA with β-lactam antibiotics, and all transpeptidation events are then carried out by PBP2A (de Jonge and Tomasz, 1993). Therefore, MRSA survives at concentrations of β-lactam antibiotics usually administered to patients.
Even though MRSA survives in the presence of β-lactam antibiotics, its PGN is structurally different from PGN made by native PBPs. β-lactam challenge of MRSA leads to a reduction of muropeptide cross-linking (de Jonge and Tomasz, 1993). PBP2A is able to connect muropeptides to form dimers, but is unable to accept dimers as substrates for further cross-linking (de Jonge et al., 1992). Hence, if PBP2A is the only functional PBP enzyme left due to antibiotic-inactivation of native PBPs, the degree of PGN cross-linking is low.
S. aureus modifies its PGN in a way that makes it resistant to intracellular degradation by immune cells (Bera et al., 2005). We have previously shown that PGN from a S. aureus mutant with reduced resistance to intracellular PGN degradation stimulates greater production of inflammatory cytokines by macrophages than native PGN (Wolf et al., 2011, Shimada et al., 2010). This increased inflammatory response is especially evident in the activation of the NLRP3 inflammasome and increased production of IL-1β. Degradation-sensitive bacteria are more efficiently killed by host immune cells in vivo, but cause increased IL-1β-induced immunopathology in a mouse skin infection model (Shimada et al., 2010).
Because challenge of MRSA with β-lactam antibiotics causes reduced muropeptide cross-linking of PGN in the cell wall, we hypothesized that these bacteria might induce an altered inflammatory response. Such an altered innate inflammatory response could lead to increased immunopathology and might contribute to the noted difference in morbitity between MRSA and MSSA infections.
Results
PGN made by PBP2A is poorly cross-linked and strongly induces IL-1β
To investigate whether antibiotic resistance is positively correlated with inflammatory responses, we first examined the effects of β-lactam antibiotics on PGN structure. Consistent with published findings (de Jonge and Tomasz, 1993), HPLC analysis showed that PGN from MRSA (USA300 JE2 strain) is highly cross-linked when grown in the absence of antibiotics (Fig. 1A), but is poorly cross-linked when the bacteria are challenged with the β-lactam antibiotic cefoxitin, to which the bacteria are fully resistant (Fig. 1B).
Figure 1. PGN made by PBP2A has reduced muropeptide cross-links and induces robust IL-1β secretion by macrophages.

(A–B) PGN was purified from MRSA, grown overnight with or without cefoxitin. HPLC profiles of muropeptides from the PGN are shown. Fragments with reduced cross-links have a short elution time. (C–D) PGN from untreated or cefoxitin-treated MRSA was used to stimulate (C) murine BMDM, or (D) human monocyte-derived macrophages (80 μg/ml PGN) (rep. data of 3 exp. for (C)). (E) PGN from MRSA treated with cefoxitin (Cef), ampicillin (Amp), or nafcillin (Naf) was used to stimulate murine BMDM (rep. data of 4 exp.). (F) mecA mRNA from MRSA grown for 4 h with or without cefoxitin was measured by qRT-PCR and standardized against expression of 16S rRNA (rep. data of 2 exp.). (G) IL-1β release by murine BMDM incubated with live MRSA pretreated with cefoxitin for 4 h (rep. data of 3 exp.). (H) IL-1β stimulatory activity of clinical MRSA strains grown in the presence or absence of cefoxitin (rep. data of 2 exp.). See also Fig. S1.
To test whether β-lactam treatment of MRSA leads to increased cytokine production due to structural changes in PGN, we purified PGN from cefoxitin-challenged and unchallenged MRSA, and used it to stimulate primary mouse bone marrow-derived macrophages (BMDM). PGN from MRSA treated with β-lactam antibiotics induced higher IL-1β, IL-6, TNF-α, and IL-1α levels compared to PGN from untreated MRSA (Fig. 1C, S1A–C), though among these cytokines, the β-lactam effect was most pronounced on IL-1β. Human monocyte-derived macrophages showed a similar increase in IL-1β secretion after stimulation with PGN from bacteria exposed to cefoxitin (Fig. 1D). This effect was not specific to cefoxitin, but was also seen when BMDM were stimulated with PGN from ampicillin-challenged or nafcillin-challenged MRSA (Fig. 1E). MRSA grown in the presence of different concentrations of cefoxitin strongly upregulated mecA (Fig. 1F), and these whole bacteria induced higher IL-1β secretion from macrophages compared to untreated MRSA (Fig. 1G). Similar to the data presented for cefoxitin, MRSA challenged with ampicillin produced PGN with reduced cross-links and induced increased cytokine levels as mecA is induced (Fig. S1D–F). In the presence of β-lactams, PBP2A expression is induced, and as the dose of antibiotic increases, which progressively inhibits native PBPs, transpeptidation reactions are performed more prominently by PBP2A. Increasing use of PBP2A would lead to a decrease in PGN cross-linking up to the point when transpeptidation is performed exclusively by PBP2A. In addition to the USA 300 JE2 strain, other MRSA clinical strains triggered increased IL-1β release after challenge with cefoxitin (Fig. 1H). Besides MRSA, the human opportunistic pathogen S. epidermidis also synthesizes PBP2A to resist killing by β-lactam antibiotics. A mecA-positive S. epidermidis strain or PGN isolated from it also induced higher IL-1β levels after cefoxitin treatment (Fig. S1G–I). Taken together, mecA expression in MRSA leads to structural changes in PGN composition, which correlates with increased IL-1β secretion by macrophages.
PGN made by PBP2A in the presence of β-lactam antibiotics is a potent activator of the inflammasome
PGN-induced secretion of IL-1β by macrophages requires stimulation of IL-1β mRNA production, synthesis of pro-IL-1β protein, and activation of the NLRP3 inflammasome to process and release mature IL-1β (Agostini et al., 2004, Garlanda et al., 2013). We stimulated mouse BMDM with PGN from bacteria treated or untreated with cefoxitin and observed that induction of IL-1β mRNA was not different (Fig. 2A). This suggests that PGN made by PBP2A (from cefoxitin-challenged MRSA) is a more potent activator of the inflammasome. To isolate inflammasome activation from IL-1β mRNA induction, we primed macrophages with lipopolysaccharide (LPS) to strongly induce IL-1β mRNA and then measured the ability of PGN from antibiotic-treated MRSA to trigger release of IL-1β in a short time period (6 h). PGN from untreated MRSA hardly induced IL-1β secretion in this time frame whereas PGN from antibiotic-treated MRSA strongly induced IL-1β release (Fig. 2B) and caspase-1 activation (Fig. 2C). In addition, we confirmed release of cleaved IL-1β in the culture supernatants by immunoblotting (Fig. S2A). In accordance with IL-1β mRNA levels, protein levels of pro-IL-1β were not different in cell lysates from LPS-primed PGN-stimulated BMDM (Fig. S2B). We similarly observed that antibiotic-treated, heat-killed (HK) MRSA activated caspase-1 cleavage more effectively than untreated HK MRSA in LPS-primed macrophages (Fig. S2C). The increase in IL-1β secretion in response to PBP2A-made PGN is completely dependent on the NLRP3 inflammasome and caspase 1 (Fig. 2D). Activation of the inflammasome by strong stimuli such as ATP or nigericin is commonly accompanied by pyroptosis. However, even though we observed higher IL-1β levels in response to PGN from cefoxitin-challenged MRSA compared to unchallenged MRSA, we did not observe increased cell death (Fig. S2D). This is in line with our previous study showing that PGN from S. aureus does not induce pyroptosis in BMDM (Shimada et al., 2010). Together, the data suggest that the increased IL-1β secretion in response to PBP2A-made PGN and antibiotic-treated MRSA is due to stronger activation of the inflammasome.
Figure 2. PGN made by PBP2A requires intracellular degradation to activate the inflammasome.

PGN from MRSA grown overnight in the absence or presence of cefoxitin was used in macrophage stimulation assays. (A) IL-1β mRNA expression (relative to β-actin mRNA) by BMDM challenged with PGN (40 μg/ml) for 6 h (n=3). (B) IL-1β release by LPS-primed BMDM incubated with PGN (40 μg/ml) (rep. data of 3 exp.). (C) Detection of cleaved caspase-1 in supernatants of LPS-primed PGN-stimulated BMDM (rep. data of 4 exp.). (D) IL-1β release by WT, Nlrp3−/−, and Casp1−/− BMDM after overnight stimulation with PGN (80 μg/ml) (rep. data of 4 exp.). (E) Effect of lysostaphin degraded PGN (40 μg/ml) from cefoxitin-challenged or unchallenged MRSA on IL-1β release by BMDM (rep. data of 3 exp.). (F–H) IL-1β release by BMDM treated with cytochalasin D (F), bafilomycin A1 (G), or E64d (H) and stimulated with PGN (40 μg/ml) (rep. data of 3 exp.). (I) Degradation of PGN by purified lysosomal extract after 24 h (n=4). (J) Effect of cefoxitin on MRSA survival within BMDM. The assay was repeated with ampicillin-treated MRSA (Fig. S2G). (K) Growth curve of MRSA initially cultured with or without cefoxitin, and subsequently grown in antibiotic-free media. See also Fig. S2.
To determine whether inflammasome activation is induced by PGN and not by some unknown co-purifying factor that is produced by antibiotic-exposed MRSA, we digested MRSA PGN with lysostaphin, an enzyme that specifically cleaves the cross-linking pentaglycine bridges in PGN from Staphylococci. The enzyme completely solubilized the particulate material and abrogated its ability to stimulate IL-1β secretion by macrophages (Fig. 2E), suggesting that the IL-1β production is not due to a factor other than PGN. We have previously observed that inflammasome activation in response to PGN requires its degradation within phagosomes (Shimada et al., 2010). Inhibition of phagocytosis with cytochalasin D (Fig. 2F), inhibition of acidification of the endolysosomal/phagolysosomal compartment with bafilomycin A1 (Fig. 2G), and inhibition of proteolytic enzymes with the protease inhibitor E64d (Fig. 2H) markedly decreased IL-1β secretion upon macrophage stimulation with PBP2A-made PGN. The PGN made by PBP2A and the PGN made by native PBPs were internalized equivalently by macrophages, ruling out differences in contact and phagocytosis efficiency as factors in the differential IL-1β response (Fig. S2E–F). We hypothesized that the poor cross-linking of PGN made by cefoxitin-treated bacteria makes PGN more susceptible to degradation by macrophages. To test this, we purified lysosomes from BMDM and incubated the lysosomal extract with PGN from either untreated or β-lactam-treated MRSA. As predicted, PGN from cefoxitin-challenged MRSA was more efficiently degraded by lysosomal enzymes compared to PGN from unchallenged MRSA (Fig. 2I). In line with this finding, MRSA treated with cefoxitin or ampicillin showed reduced intracellular survival in macrophages (Fig. 2J, S2G). This was not due to a general growth disadvantage of antibiotic-challenged MRSA since they regrew normally in antibiotic-free medium after antibiotic challenge (Fig. 2K). Together the data suggest that the structural change in PGN caused by PBP2A in the presence of β-lactam antibiotics causes it to become a more potent activator of the inflammasome.
Reduced PGN cross-linking is sufficient to enhance PGN-induced IL-1β production
To test the hypothesis that reducing the degree of cross-linking in PGN leads to greater inflammatory responses, we digested PGN from unchallenged MRSA with various concentrations of lysostaphin. At low lysostaphin concentrations (at which PGN cross-links are not yet fully degraded and the material is still particulate) the PGN made by endogenous PBPs induced IL-1β release to the same extend as PGN made after PBP2A induction with antibiotics (Fig. 3A). As the lysostaphin concentration was increased and the PGN was solubilized, it lost its ability to stimulate IL-1β production. Thus, reducing the cross-linking of particulate PGN is sufficient to cause it to stimulate more IL-1β production.
Figure 3. Alteration in PGN cross-links affects IL-1β release from macrophages.

(A) PGN derived from untreated or cefoxitin-treated MRSA was digested with lysostaphin for 2 h, and then used to stimulate LPS-primed BMDM. (B–C) Effect of low cefoxitin concentrations on (B) MSSA-induced or (C) PGN-induced IL-1β release by BMDM. (D–E) IL-1β release by BMDM stimulated with (D) live JE2 WT, pbpD-Tn mutant (reduced PGN cross-links), or clpP-Tn mutant (excessive PGN cross-links) bacteria, or (E) PGN (80 μg/ml) isolated from these bacterial strains. (F–G) WT mice were infected on the right flank with WT JE2 (107 CFUs), and on the left flank with JE2 pbpD-Tn mutant (107 CFUs). (F) Skin lesions measured on day 1, and (G) on consecutive days. Also shown are two images of the lesions (n=2 with 19 total animals). Wilcoxon rank-sum test was used; * p<0.05, ** p<0.01. (A–E) (rep. data of 2–5 exp.). See also Fig. S3
MSSA exposed to sub-lethal doses of β-lactam antibiotics also leads to poorly cross-linked PGN presumably by reducing the efficiency of antibiotic-sensitive PBPs (de Jonge et al., 1992). If so, this too should increase inflammasome activation. Indeed, when a mecA-transposon (Tn) mutant of the MRSA JE2 strain was challenged with sub-lethal doses of cefoxitin (Fig. 3B) or ampicillin (Fig. S3A) and then used to infect macrophages, the antibiotic treatments led to increased IL-1β secretion. PGN purified from the low-dose cefoxitin-treated mecA-Tn mutant strongly induced IL-1β secretion from macrophages, while PGN from the untreated mecA-Tn mutant did not (Fig. 3C).
We also used a genetic approach to specifically manipulate PGN cross-linking. It was recently reported that MRSA mutants for PBP4 and the protease ClpP show an abnormal low or high degree of PGN cross-linking respectively (Baek et al., 2014, Memmi et al., 2008). When isogenic strains of these bacteria (Fig. 3D), or PGN isolated from them (Fig. 3E) were used to infect macrophages, the pbpD-Tn mutant proved to be more potent and the clpP-Tn mutant proved to be less active in inducing IL-1β. This is consistent with our hypothesis that the degree of PGN cross-linking directly determines the amount of IL-1β that is secreted and that this correlates with the degradability of PGN by lysosomal enzymes (Fig. S3B–C). In vivo, mice infected with pbpD-Tn mutant had bigger skin lesions compared to WT MRSA (Fig. 3F–G), which was not due to a higher bacterial burden of the pbpD-Tn mutant compared to WT MRSA (Fig. S3D). Overall these data show that reducing PGN cross-linking is sufficient to cause enhanced inflammatory responses in vitro and in vivo.
β-lactam-challenged MRSA causes more severe IL-1-dependent skin immunopathology in vivo
To determine if β-lactam treatment of MRSA in vivo induces greater skin immunopathology, we next injected WT mice subcutaneously (s.c.) with PGN from cefoxitin-challenged and unchallenged MRSA. Higher IL-1β levels and neutrophil recruitment were detected in the skin of mice that had received PGN from antibiotic-treated MRSA (Fig. 4A, S4A). Next, we injected HK MRSA, treated or untreated with cefoxitin, into the skin of mice. HK bacteria were used to avoid effects on skin pathology that might arise from differential expression of toxins in response to antibiotic challenge (Ohlsen et al., 1998). HK antibiotic-challenged MRSA induced significantly higher levels of IL-1β, abscess weight, and abscess volume compared to unchallenged HK MRSA (Fig. 4B–C, S4B). Finally, we infected mice s.c. with live MRSA, and treated the mice starting one day after and twice daily with either PBS or nafcillin. Antibiotic treatment of the MRSA infection significantly increased lesion size in comparison to no treatment (Fig. 4D, S4C). In support of our hypothesis that increased inflammation rather than bacterial virulence contributes to immunopathology, we found reduced numbers of bacteria in lesions from antibiotic-treated animals (Fig. 4E). Furthermore, we found increased numbers of neutrophils in lesions from MRSA-infected mice treated with nafcillin compared to PBS-treated mice (Fig. 4F). This is consistent with the interpretation that although the bacteria are resistant to the antibiotic, they are killed more effectively by phagocytes in the presence of the antibiotic. The immunopathology induced by β-lactams appeared to be dependent on IL-1 signaling, as the differences in abscess weight and volume induced by antibiotic-treated HK MRSA compared to untreated HK MRSA were abrogated in IL1R1−/− mice (Fig. 4G–H). Overall, these data suggest that the well-studied antibiotic resistance factor PBP2A is also an as-yet unrecognized pathogenic factor during MRSA infections. β-lactam antibiotics are therefore not only ineffective against MRSA infections, but actually contribute to immunopathology by inducing mecA.
Figure 4. MRSA and PGN induce greater IL-1β levels and immunopathology in vivo after β-lactam treatment.

(A) Skin IL-1β levels in WT mice injected s.c. with PGN derived from untreated or cefoxitin-treated MRSA. (B) Skin IL-1β levels and (C) abscess weight on day 3 from WT mice injected s.c. with HK, untreated or cefoxitin-pretreated MRSA (5×108 CFUs). (D–F) WT mice infected s.c. with MRSA (3×107 CFUs) were treated starting on day 1 and twice a day s.c. with PBS or 5 mg/ml nafcillin at the site of infection. (D) Average lesion sizes, (E) bacterial burden, and (F) neutrophils at the site of infection on day 3 (n=2). (G–H) HK, untreated or cefoxitin-pretreated MRSA (108 CFUs) were injected s.c. into WT or IL-1R1−/− mice. (G) Abscess weight and (H) abscess volume after 3 days. Mann Whitney U-test was used. * p<0.05. See also Fig. S4.
Discussion
Many studies have compared the outcome of MSSA and MRSA infections. A meta-analysis of these studies determined that MRSA infections are associated with worse outcomes compared to MSSA even after adjusting for comorbidities and severity of initial diseases (Cosgrove et al., 2003). Several underlying reasons are offered for the finding, including reduced efficacy of many anti-MRSA antibiotics (Deresinski, 2007), delay in appropriate treatment because of failure to recognize MRSA infection (Kim et al., 2004), and the possibility that MRSA is more pathogenic compared to MSSA. Though the association of MRSA with increased pathogenicity has been suggested by several clinical studies, pertinent pathogenic factors have not been identified (Barrios Lopez et al., 2013, Watkins et al., 2012). In this study we demonstrated that PBP2A, the factor that makes MRSA resistant to β-lactam treatment, contributes to the pathogenicity of MRSA infection by changing PGN structure and augmenting immunopathology, although this effect is not observed until mecA is induced by β-lactam antibiotics.
This finding provides some insight on the difference in outcome between MRSA and MSSA infections. Based on reports, between 30–80% of individuals infected with MRSA were inappropriately treated, often with β-lactam antibiotics (Paul et al., 2010, Kim et al., 2004, Rodriguez-Bano et al., 2009). Studies confirmed that initial inappropriate antibiotic treatment is an independent predictive factor for mortality during MRSA bacteremia (Gasch et al., 2013). Although lack of awareness of MRSA likely contributed to some of these cases, technical limitations in rapid diagnosis of MRSA and the pressure to not over-prescribe vancomycin, the gold standard for MRSA treatment, will likely lead to more prescription of β-lactam antibiotics (e.g. ceftriaxone). Our study, upon additional clinical corroboration, may therefore suggest caution in the use of β-lactams which may lead to worse outcome compared to inaction. Aside from MRSA, treatment of MSSA with sub-lethal doses of β–lactam antibiotics could also lead to a reduction of PGN cross-links, and this would be predicted to induce increased inflammation. However, the use of low doses of β-lactam antibiotics is likely less clinically relevant.
Our data suggest that reduction of muropeptide cross-linking increases MRSA susceptibility to intracellular degradation followed by killing by macrophages. We have previously shown that S. aureus PGN structure and PGN degradability by immune cells impact cytokine production by macrophages and dendritic cells, and that IL-1β responses are particularly strongly affected (Shimada et al., 2010, Wolf et al., 2011). IL-1β is thought to be a critical cytokine for effective control of S. aureus skin infections (Miller et al., 2006), being particularly important for recruitment of neutrophils (Cho et al., 2012). However, increased neutrophil recruitment and activation can potentially cause immunopathology as observed in our study, so the process must be balanced carefully (Fournier and Parkos, 2012, Yang et al., 2013).
One recent publication reported that nafcillin treatment in vivo “sensitizes” MRSA to killing by the innate immune system, although the mechanism of this response was not clear (Sakoulas et al., 2014). This is in line with our in vitro data showing increased susceptibility of β-lactam-challenged MRSA to macrophage-mediated killing, as well as reduced CFU numbers in vivo upon nafcillin treatment. Interestingly a murine and two human studies recently showed that the combination of β-lactam antibiotics with vancomycin or daptomycin is more effective in MRSA infections compared to vancomycin or daptomycin treatment alone, leading to enhanced bacterial clearance (Dilworth et al., 2014, Yang et al., 2010, Dhand et al., 2011). Our work provides a possible mechanistic explanation for the improved bacterial clearance observed in these studies.
Since many antibiotics target PGN assembly by S. aureus and other bacteria, it is important to understand how the development of bacterial resistance affects the overall immune response to these pathogens. Here we provide evidence that the resistance machinery of MRSA, through mecA induction, results in increased inflammation in response to the pathogen.
Experimental Procedures
Animals
C57BL/6 mice were purchased from Charles River and were between 7 and 12 weeks of age during experiments. IL-1R1−/− mice (The Jackson Laboratory) were bred at Cedars-Sinai Medical Center.
Bacterial strains
Unless otherwise indicated, the MRSA USA300 strain JE2 was used for all experiments. JE2 transposon insertion mutants of mecA, pbpD, and clpP from The Nebraska Transposon Mutant Library were obtained from the NARSA repository. The USA400 strain MW2 and two clinical strains isolated at Cedars-Sinai Medical Center (MRSA strain CST10 and a mecA-positive S. epidermidis strain, Fig. S1G) were also studied.
Cell culture
BMDM were generated as previously described (Wolf et al., 2011). BMDM were cultured in 1640 RPMI without glucose (Life Technologies), 10% FCS (Sigma), 1% penicillin/streptomycin, 1% L-glutamine, and 5 mM glucose (United States Biological). Nlrp3−/− (Kuida et al., 1995) and Casp1−/− (Mariathasan et al., 2006) BMDM were kindly provided by Dr. Moshe Arditi.
Macrophage killing assay
The assay was performed as previously described (Muller et al., 2014). In brief, macrophages plated in 24 well plates were infected with MRSA (MOI 10) grown in antibiotics overnight. After 45 min, antibiotic-free cell culture medium was replaced by medium containing 100 μg/ml gentamicin to kill the remaining extracellular bacteria. Macrophages were lysed at indicated time points and bacterial CFUs were determined.
Skin infection
Mice were shaved the day before injection. When PGN was injected, 200 μg PGN was administered in a total volume of 100 μl PBS. 108 or 5×108 HK MRSA were injected in 100 μl PBS. For injection of live MRSA, bacteria were prepared as described in supplemental experimental procedures, and 3×107 CFUs were injected s.c. in 100 μl PBS. If not indicated otherwise skin lesions were excised, homogenized, and plated to determine CFUs on day 3. Afterwards the homogenate was centrifuged for 5 min and the supernatant was used for cytokine measurement. The remaining pellet was further processed in some experiments for FACS analysis.
Ethic statement
This study was performed under strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals. The protocol was approved by the institutional animal use and care committee of the Cedars-Sinai Medical Center (IACUC 3402).
Statistical analysis
When a representative experiment is shown, data are presented as mean ± SD of replicates (depicted as: rep. data of X exp.). Otherwise, data are presented as mean ± SE, or individual data points are shown in graphs. Statistical analysis was performed using non-parametric Mann-Whitney U Test or Wilcoxon Signed-Rank Test. p-values less than 0.5 were considered significant.
Supplementary Material
Acknowledgments
We thank Dr. Colleen Moody for help with HPLC analysis, Dr. Jargalsaikhan Dagvadorj for providing Nlpr3−/− and Casp1−/− BMDM, and Christian Leal, Christopher Reyes, and Dr. David Taylor for outstanding technical assistance. We thank Dr. Moshe Arditi for providing IL-1R1−/− mice. This study was supported by NIH grant R21AI097741 to GYL and DMU and R01GM085796 to DMU. IDI is supported by NIH grant DK098310.
Footnotes
All authors declare that they have no competing interests.
Author contributions
G.Y.L. and D.M.U. formulated the original hypothesis. G.Y.L., D.M.U., and S.M. designed the study and wrote the manuscript. S.M. and I.D.I. performed the experiments and analyzed the data. A.J.W. provided advice and technical support. B.L.B. provided technical support. All authors provided comments on the manuscript and data before submission.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- AGOSTINI L, MARTINON F, BURNS K, MCDERMOTT MF, HAWKINS PN, TSCHOPP J. NALP3 forms an IL-1beta-processing inflammasome with increased activity in Muckle-Wells autoinflammatory disorder. Immunity. 2004;20:319–25. doi: 10.1016/s1074-7613(04)00046-9. [DOI] [PubMed] [Google Scholar]
- ANTONANZAS F, LOZANO C, TORRES C. Economic features of antibiotic resistance: the case of methicillin-resistant Staphylococcus aureus. Pharmacoeconomics. 2015;33:285–325. doi: 10.1007/s40273-014-0242-y. [DOI] [PubMed] [Google Scholar]
- BAEK KT, GRUNDLING A, MOGENSEN RG, THOGERSEN L, PETERSEN A, PAULANDER W, FREES D. beta-Lactam resistance in methicillin-resistant Staphylococcus aureus USA300 is increased by inactivation of the ClpXP protease. Antimicrob Agents Chemother. 2014;58:4593–603. doi: 10.1128/AAC.02802-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BARRIOS LOPEZ M, GOMEZ GONZALEZ C, ORELLANA MA, CHAVES F, ROJO P. Staphylococcus aureus abscesses: methicillin-resistance or Panton-Valentine leukocidin presence? Arch Dis Child. 2013;98:608–10. doi: 10.1136/archdischild-2012-302695. [DOI] [PubMed] [Google Scholar]
- BECK WD, BERGER-BACHI B, KAYSER FH. Additional DNA in methicillin-resistant Staphylococcus aureus and molecular cloning of mec-specific DNA. J Bacteriol. 1986;165:373–8. doi: 10.1128/jb.165.2.373-378.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BERA A, HERBERT S, JAKOB A, VOLLMER W, GOTZ F. Why are pathogenic staphylococci so lysozyme resistant? The peptidoglycan O-acetyltransferase OatA is the major determinant for lysozyme resistance of Staphylococcus aureus. Mol Microbiol. 2005;55:778–87. doi: 10.1111/j.1365-2958.2004.04446.x. [DOI] [PubMed] [Google Scholar]
- CHAMBERS HF, HARTMAN BJ, TOMASZ A. Increased amounts of a novel penicillin-binding protein in a strain of methicillin-resistant Staphylococcus aureus exposed to nafcillin. J Clin Invest. 1985;76:325–31. doi: 10.1172/JCI111965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CHO JS, GUO Y, RAMOS RI, HEBRONI F, PLAISIER SB, XUAN C, GRANICK JL, MATSUSHIMA H, TAKASHIMA A, IWAKURA Y, CHEUNG AL, CHENG G, LEE DJ, SIMON SI, MILLER LS. Neutrophil-derived IL-1beta is sufficient for abscess formation in immunity against Staphylococcus aureus in mice. PLoS Pathog. 2012;8:e1003047. doi: 10.1371/journal.ppat.1003047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- COSGROVE SE, SAKOULAS G, PERENCEVICH EN, SCHWABER MJ, KARCHMER AW, CARMELI Y. Comparison of mortality associated with methicillin-resistant and methicillin-susceptible Staphylococcus aureus bacteremia: a meta-analysis. Clin Infect Dis. 2003;36:53–9. doi: 10.1086/345476. [DOI] [PubMed] [Google Scholar]
- DE JONGE BL, CHANG YS, GAGE D, TOMASZ A. Peptidoglycan composition of a highly methicillin-resistant Staphylococcus aureus strain. The role of penicillin binding protein 2A. J Biol Chem. 1992;267:11248–54. [PubMed] [Google Scholar]
- DE JONGE BLM, TOMASZ A. Abnormal Peptidoglycan Produced in a Methicillin-Resistant Strain of Staphylococcus aureus Grown in the Presence of Methicillin: Functional Role for Penicillin-Binding Protein 2A in Cell Wall Synthesis. Antimicrob Agents Chemother. 1993;37:342–346. doi: 10.1128/aac.37.2.342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DERESINSKI S. Counterpoint: Vancomycin and Staphylococcus aureus--an antibiotic enters obsolescence. Clin Infect Dis. 2007;44:1543–8. doi: 10.1086/518452. [DOI] [PubMed] [Google Scholar]
- DHAND A, BAYER AS, POGLIANO J, YANG SJ, BOLARIS M, NIZET V, WANG G, SAKOULAS G. Use of antistaphylococcal beta-lactams to increase daptomycin activity in eradicating persistent bacteremia due to methicillin-resistant Staphylococcus aureus: role of enhanced daptomycin binding. Clin Infect Dis. 2011;53:158–63. doi: 10.1093/cid/cir340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DILWORTH TJ, IBRAHIM O, HALL P, SLIWINSKI J, WALRAVEN C, MERCIER RC. beta-Lactams enhance vancomycin activity against methicillin-resistant Staphylococcus aureus bacteremia compared to vancomycin alone. Antimicrob Agents Chemother. 2014;58:102–9. doi: 10.1128/AAC.01204-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FOURNIER BM, PARKOS CA. The role of neutrophils during intestinal inflammation. Mucosal Immunol. 2012;5:354–66. doi: 10.1038/mi.2012.24. [DOI] [PubMed] [Google Scholar]
- GANGA R, RIEDERER K, SHARMA M, FAKIH MG, JOHNSON LB, SHEMES S, KHATIB R. Role of SCCmec type in outcome of Staphylococcus aureus bacteremia in a single medical center. J Clin Microbiol. 2009;47:590–5. doi: 10.1128/JCM.00397-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GARLANDA C, DINARELLO CA, MANTOVANI A. The interleukin-1 family: back to the future. Immunity. 2013;39:1003–18. doi: 10.1016/j.immuni.2013.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GASCH O, CAMOEZ M, DOMINGUEZ MA, PADILLA B, PINTADO V, ALMIRANTE B, LEPE JA, LAGARDE M, RUIZ DE GOPEGUI E, MARTINEZ JA, MONTEJO M, TORRE-CISNEROS J, ARNAIZ A, GOENAGA MA, BENITO N, RODRIGUEZ-BANO J, PUJOL M, GROUPS RGS. Predictive factors for early mortality among patients with methicillin-resistant Staphylococcus aureus bacteraemia. J Antimicrob Chemother. 2013;68:1423–30. doi: 10.1093/jac/dkt016. [DOI] [PubMed] [Google Scholar]
- HERSH AL, CHAMBERS HF, MASELLI JH, GONZALES R. National trends in ambulatory visits and antibiotic prescribing for skin and soft-tissue infections. Arch Intern Med. 2008;168:1585–91. doi: 10.1001/archinte.168.14.1585. [DOI] [PubMed] [Google Scholar]
- KENNEDY AD, OTTO M, BRAUGHTON KR, WHITNEY AR, CHEN L, MATHEMA B, MEDIAVILLA JR, BYRNE KA, PARKINS LD, TENOVER FC, KREISWIRTH BN, MUSSER JM, DELEO FR. Epidemic community-associated methicillin-resistant Staphylococcus aureus: recent clonal expansion and diversification. Proc Natl Acad Sci USA. 2008;105:1327–32. doi: 10.1073/pnas.0710217105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KIM SH, PARK WB, LEE KD, KANG CI, BANG JW, KIM HB, KIM EC, OH MD, CHOE KW. Outcome of inappropriate initial antimicrobial treatment in patients with methicillin-resistant Staphylococcus aureus bacteraemia. J Antimicrob Chemother. 2004;54:489–97. doi: 10.1093/jac/dkh366. [DOI] [PubMed] [Google Scholar]
- KOPP BJ, NIX DE, ARMSTRONG EP. Clinical and economic analysis of methicillin-susceptible and -resistant Staphylococcus aureus infections. Ann Pharmacother. 2004;38:1377–82. doi: 10.1345/aph.1E028. [DOI] [PubMed] [Google Scholar]
- KUIDA K, LIPPKE JA, KU G, HARDING MW, LIVINGSTON DJ, SU MS, FLAVELL RA. Altered cytokine export and apoptosis in mice deficient in interleukin-1 beta converting enzyme. Science. 1995;267:2000–3. doi: 10.1126/science.7535475. [DOI] [PubMed] [Google Scholar]
- MARIATHASAN S, WEISS DS, NEWTON K, MCBRIDE J, O’ROURKE K, ROOSE-GIRMA M, LEE WP, WEINRAUCH Y, MONACK DM, DIXIT VM. Cryopyrin activates the inflammasome in response to toxins and ATP. Nature. 2006;440:228–32. doi: 10.1038/nature04515. [DOI] [PubMed] [Google Scholar]
- MEMMI G, FILIPE SR, PINHO MG, FU Z, CHEUNG A. Staphylococcus aureus PBP4 is essential for beta-lactam resistance in community-acquired methicillin-resistant strains. Antimicrob Agents Chemother. 2008;52:3955–66. doi: 10.1128/AAC.00049-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MILLER LS, O’CONNELL RM, GUTIERREZ MA, PIETRAS EM, SHAHANGIAN A, GROSS CE, THIRUMALA A, CHEUNG AL, CHENG G, MODLIN RL. MyD88 mediates neutrophil recruitment initiated by IL-1R but not TLR2 activation in immunity against Staphylococcus aureus. Immunity. 2006;24:79–91. doi: 10.1016/j.immuni.2005.11.011. [DOI] [PubMed] [Google Scholar]
- MULLER S, FAULHABER A, SIEBER C, PFEIFER D, HOCHBERG T, GANSZ M, DESHMUKH SD, DAUTH S, BRIX K, SAFTIG P, PETERS C, HENNEKE P, REINHECKEL T. The endolysosomal cysteine cathepsins L and K are involved in macrophage-mediated clearance of Staphylococcus aureus and the concomitant cytokine induction. FASEB J. 2014;28:162–75. doi: 10.1096/fj.13-232272. [DOI] [PubMed] [Google Scholar]
- OHLSEN K, ZIEBUHR W, KOLLER KP, HELL W, WICHELHAUS TA, HACKER J. Effects of subinhibitory concentrations of antibiotics on alpha-toxin (hla) gene expression of methicillin-sensitive and methicillin-resistant Staphylococcus aureus isolates. Antimicrob Agents Chemother. 1998;42:2817–23. doi: 10.1128/aac.42.11.2817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- OLIVEIRA DC, TOMASZ A, DE LENCASTRE H. The evolution of pandemic clones of methicillin-resistant Staphylococcus aureus: identification of two ancestral genetic backgrounds and the associated mec elements. Microb Drug Resist. 2001;7:349–61. doi: 10.1089/10766290152773365. [DOI] [PubMed] [Google Scholar]
- PAUL M, KARIV G, GOLDBERG E, RASKIN M, SHAKED H, HAZZAN R, SAMRA Z, PAGHIS D, BISHARA J, LEIBOVICI L. Importance of appropriate empirical antibiotic therapy for methicillin-resistant Staphylococcus aureus bacteraemia. J Antimicrob Chemother. 2010;65:2658–65. doi: 10.1093/jac/dkq373. [DOI] [PubMed] [Google Scholar]
- RODRIGUEZ-BANO J, MILLAN AB, DOMINGUEZ MA, BORRAZ C, GONZALEZ MP, ALMIRANTE B, CERCENADO E, PADILLA B, PUJOL M GEIH/GEMARA/REIPI. Impact of inappropriate empirical therapy for sepsis due to health care-associated methicillin-resistant Staphylococcus aureus. J Infect. 2009;58:131–7. doi: 10.1016/j.jinf.2008.11.003. [DOI] [PubMed] [Google Scholar]
- SAKOULAS G, OKUMURA CY, THIENPHRAPA W, OLSON J, NONEJUIE P, DAM Q, DHAND A, POGLIANO J, YEAMAN MR, HENSLER ME, BAYER AS, NIZET V. Nafcillin enhances innate immune-mediated killing of methicillin-resistant Staphylococcus aureus. J Mol Med. 2014;92:139–49. doi: 10.1007/s00109-013-1100-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SAUVAGE E, KERFF F, TERRAK M, AYALA JA, CHARLIER P. The penicillin-binding proteins: structure and role in peptidoglycan biosynthesis. FEMS Microbiol Rev. 2008;32:234–58. doi: 10.1111/j.1574-6976.2008.00105.x. [DOI] [PubMed] [Google Scholar]
- SHIMADA T, PARK BG, WOLF AJ, BRIKOS C, GOODRIDGE HS, BECKER CA, REYES CN, MIAO EA, ADEREM A, GOTZ F, LIU GY, UNDERHILL DM. Staphylococcus aureus evades lysozyme-based peptidoglycan digestion that links phagocytosis, inflammasome activation, and IL-1beta secretion. Cell Host Microbe. 2010;7:38–49. doi: 10.1016/j.chom.2009.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TAYLOR AR. Methicillin-resistant Staphylococcus aureus infections. Prim Care. 2013;40:637–54. doi: 10.1016/j.pop.2013.06.002. [DOI] [PubMed] [Google Scholar]
- TOMASZ A. The mechanism of the irreversible antimicrobial effects of penicillins: how the beta-lactam antibiotics kill and lyse bacteria. Annu Rev Microbiol. 1979;33:113–37. doi: 10.1146/annurev.mi.33.100179.000553. [DOI] [PubMed] [Google Scholar]
- WATKINS RR, DAVID MZ, SALATA RA. Current concepts on the virulence mechanisms of meticillin-resistant Staphylococcus aureus. J Med Microbiol. 2012;61:1179–93. doi: 10.1099/jmm.0.043513-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WOLF AJ, ARRUDA A, REYES CN, KAPLAN AT, SHIMADA T, SHIMADA K, ARDITI M, LIU G, UNDERHILL DM. Phagosomal degradation increases TLR access to bacterial ligands and enhances macrophage sensitivity to bacteria. J Immunol. 2011;187:6002–10. doi: 10.4049/jimmunol.1100232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- YANG Q, GHOSE P, ISMAIL N. Neutrophils mediate immunopathology and negatively regulate protective immune responses during fatal bacterial infection-induced toxic shock. Infect Immun. 2013;81:1751–63. doi: 10.1128/IAI.01409-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- YANG SJ, XIONG YQ, BOYLE-VAVRA S, DAUM R, JONES T, BAYER AS. Daptomycin-oxacillin combinations in treatment of experimental endocarditis caused by daptomycin-nonsusceptible strains of methicillin-resistant Staphylococcus aureus with evolving oxacillin susceptibility (the “seesaw effect”) Antimicrob Agents Chemother. 2010;54:3161–9. doi: 10.1128/AAC.00487-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.