

Abstract
Scientists at the University of Kentucky are unravelling the details of DNA damage repair in the melanocyte, with an eye towards finding druggable targets for melanoma prevention. Jarret et al. report in this issue three new assays that can yield mechanistic information about nucleotide excision repair (NER) stimulated by cAMP-dependent signaling downstream of the melanocortin-1 receptor (MC1R).
Molecularly-targeted prevention
Protecting the lives of individuals and increasing the productivity of society by stopping disease before it causes harm is of the greatest aspirations of medicine; and as the molecular etiologies of cancer are discovered, there will be opportunities to apply targeted molecular strategies to its prevention, much as they have been applied to therapeutics. Consider the development of targeted therapy for melanoma utilizing BRAF inhibitors (Puzanov and Flaherty, 2010). This accomplishment resulted directly from the elucidation of the molecular mechanism of melanocyte transformation (V600E mutation of BRAF) and subsequent identification of a drug that could specifically antagonize the mutated protein. To develop targeted prevention agents, it will be necessary to elucidate targetable molecular pathways that predispose a cell to transformation and then to identify drugs that can successfully and safely target these pathways. Melanoma is a good candidate disease for the development of targeted prevention agents because several melanoma predisposition genes have been identified. One of the most common and well characterized of these is MC1R (Abdel-Malek et al., 2014), the target of investigation in an article from this issue by Jarrett et al (Jarrett et al., 2015).
MC1R and DNA damage repair
Epidemiological studies have found a strong correlation between the carriage of loss-of-function (LOF) mutations in MC1R (which encodes a 7-pass transmembrane G-protein-coupled receptor) and both the red hair phenotype and melanoma risk (Pasquali et al., 2015). In cell culture, treatment of melanocytes that express the wild-type MC1R (Figure 1), with the agonist alpha-melanocyte stimulating hormone (α-MSH), elicits a variety of responses, including synthesis of eumelanin, reduction of UV-induced oxidative stress, stimulation of adenylyl cyclase and cAMP-dependent signaling, and enhancement of DNA repair via base-excision repair and nucleotide-excision repair (NER) mechanisms (Abdel-Malek et al., 2014). NER repairs UV-photoproducts such as cyclobutane pyrimidine dimers (CPDs) and 6′-4′-pyrimdine-pyrimidone photoproducts (6-4-PPs). If not repaired properly, CPD formation can result in C>T transitions, which are a signature mutation in melanomas (discussed in (Jarrett et al., 2014)). Thus, impaired NER in the skin cells of individuals with LOF MC1R mutations likely contributes to their vulnerability to melanoma, and it is therefore a rational target for prevention. Until recently, the mechanistic details (i.e. demonstrable, quantifiable and targetable molecular events) linking MC1R activation to NER have been hazy. Then, it was reported last year that pre-treatment of melanocytes with α-MSH augmented their DNA damage response by increasing phosphorylation of DNA-damage sensing proteins ataxia telangiectasia and Rad3-related protein (ATR, at serine 428) and ataxia telangiectasia mutated (ATM, at serine 1981) and enhancing formation of phosphorylated γH2AX at nuclear sites of DNA repair (Swope et al., 2014). And now, in this issue, comes the report of an MC1R-dependent cascade of molecular events involving phosphorylation of ATR at a second serine, which directs the recruitment of the key NER protein xeroderma pigmentosum complementation group A (XPA) to sites of DNA damage (Jarret et al., JID 2015).
Figure 1. MC1R signaling controls repair of UV-induced DNA damage and suppression of mutations via elevation of intracellular levels of cyclic AMP.
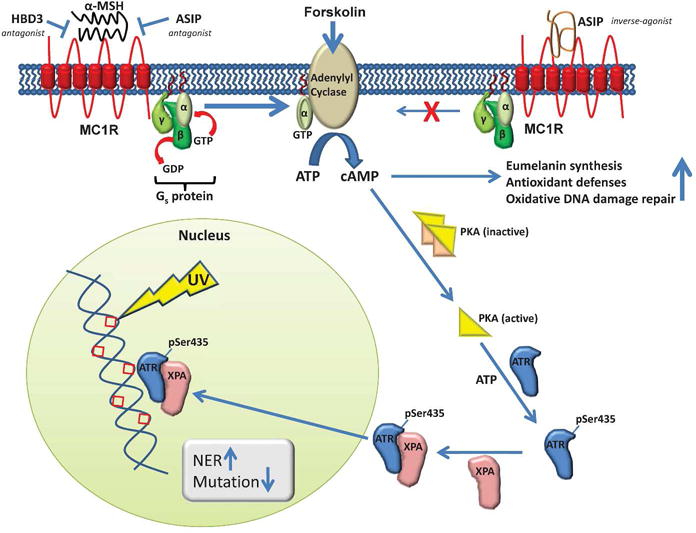
MC1R is a G-protein-coupled 7-pass transmembrane receptor expressed on the cell surface of melanocytes. Upon binding the agonist alpha-melanocyte-stimulating hormone (α-MSH), conformational changes in MC1R elicit the activation of the heterotrimeric G-protein Gs by exchange of bound GDP for GTP. The α-subunit of the G-protein (bound to GTP) can then activate adenylyl cyclase, which in turn catalyzes the cyclization of ATP to give cyclic AMP (cAMP). ASIP and HBD3 interfere with the binding of α-MSH to MC1R (top left). There is also some baseline activity of unstimulated MC1R, which is negatively affected by inverse-agonist activity of ASIP (top right). cAMP interacts with the inactive heterotetrameric form of PKA, freeing the catalytically active monomeric catalyze the phosphorylation of ATR on serine 435 to give ATR(pSer435). The physical interaction between ATR(pSer435) and XPA drives the recruitment of XPA to sites of nuclear photodamage, thereby facilitating repair of the DNA and decreased rates of mutation.
How cAMP and PKA affect recruitment of critical DNA damage machinery to sites of photodamage
The group headed by John D’Orazio at the University of Kentucky have contributed to our understanding of how MC1R affects NER in two reports ((Jarrett et al., 2014) and in this issue). The 2014 publication demonstrates that MC1R-mediated activation of cAMP-dependent protein kinase (PKA) leads to phosphorylation of ATR at a previously unrecognized phosphorylation site on serine 435 (S435, Figure 1). They show that phosphorylation of ATR S435 results in an ATR-mediated recruitment of the NER protein xeroderma pigmentosum complementation group A (XPA) to sites of nuclear photodamage. In this way XPA accelerates the clearance of UV photolesions, including CPDs and 6-4-PPs, and reduces the frequency of UV-induced mutations. Finally, they demonstrate clinical relevance by showing that all of these MC1R-mediated activities are diminished in cells bearing LOF mutations in MC1R and that the activities can be restored by treatment of melanocytes with forskolin, a small-molecule activator of adenylyl cyclase that can bypass the MC1R deficiency (Figure 1). Thus, the authors have elucidated a detailed mechanism by which MC1R is able to reduce UV-induced DNA damage and the resulting mutations, and they have identified a pathway that should be targeted for melanoma prevention in individuals who have are deficient in this repair capacity due to mutations in MC1R.
New assays will aid in the identification and development of new melanoma prevention agents
In their paper published here (Jarrett et al., JID this issue), Jarrett et al., describe three new assays that they use to define the kinetics and dose-response correlations for MC1R-mediated ATR phosphorylation and DNA damage repair. These assays enable the authors to carry out detailed evaluations of the activities of MC1R agonists α-MSH and adrenocorticotripic hormone (ACTH) and MC1R antagonists agouti signaling protein (ASIP) and human beta-defensin 3 (HBD3). The first assay is a high-throughput, fluorescence-based assay that quantifies phosphorylation of biotinylated 14-mer peptides that correspond to the PKA recognition sequence containing S435 on ATR. Using this assay they characterize the kinetics of ATR S435 phosphorylation by the recombinant catalytic subunit of PKA. Then they use nuclear lysates from melanocytes to determine the effects of MC1R antagonists ASIP and HBD3 on the EC50s of α-MSH and forskolin. They find that both antagonists increase the EC50 of α-MSH but that they have no effect on the potency of forskolin, consistent with the schemes illustrated in Figure 1.
The second assay is adapted by the authors from the oligonucleotide retrieval assay (ORA), originally published by Shen et al. (Shen et al., 2014). ORA can be used to quantify the capacity of cells or cell lysates to repair UV-photodamaged DNA. Repair is detected by PCR-based amplification of biotinylated oligos, which cannot take place unless the damaged bases are removed. Jarrett et al. use the assay to show that in human melanocyte lysates, α-MSH pre-treatment enhances the repair of photodamaged oligos, while concomitant treatment with ASIP or HBD3 and α-MSH abolishes this effect. They also show that ASIP decreases baseline repair, which is consistent with other reports of the inverse-agonist activity of ASIP for the MC1R ((Hida et al., 2009) and Figure 1).
The third assay is a method to quantify the ATR-pS435-dependent recruitment of XPA to photodamaged DNA in nuclear extracts. Nuclear lysates of treated melanocytes (containing unknown levels of ATR-pS435 and XPA) are incubated with the same biotinylated UV-damaged oligonucleotides described above. Once the ATR-pS435 and XPA in the lysates have bound to the UV-damaged oligonucleotides, the oligos are retrieved using streptavidin-coated beads, and bound ATR-pS435 and XPA are quantified by Western blot. Using this assay the authors found low baseline levels of XPA and ATR-pS435 in unstimulated cells, and 3–4 fold increases following pre-incubation with α-MSH or forskolin.
Using these three assays, researchers in the field will now be able to systematically evaluate MC1R-dependent repair of UV-damaged DNA, as measured by the quantity of 1) ATR-pS435, 2) repair of UV-damaged oligonucleotides and 3) XPA and ATR-pS435 recruited to damaged DNA. This tool-kit paves the way for testing other candidate prevention agents with the capacity to enhance impaired MC1R functions. Agents that could be evaluated using these methods include α-MSH analogs (Abdel-Malek et al., 2009), thymidine dinucleotides (pTpT) designed as mimics of UV-induced DNA damage (Goukassian et al., 1999), and other pharmacologic agents that might increase levels of cAMP in the epidermis (D’Orazio 2006).
Several investigative groups have reported (Beaumont et al., 2007; Kadekaro et al., 2010) that different combinations of MC1R mutations result in varying degrees of impairment of cAMP generation in α-MSH-treated melanocytes. The new assays reported here will allow the examination in finer detail, of the molecular responses to α-MSH in melanocytes with homo-or heterozygous LOF mutations in MC1R. These new methodologies will also facilitate the investigation of molecular underpinnings of the intriguing new findings from meta-analyses of MC1R epidemiological studies that suggest that melanoma risk is highest in MC1R mutation carriers with darker skin (Type III or higher), and without red hair or freckles (Pasquali et al., 2015). Determining whether these phenomena are caused by altered MC1R function, interactions between mutated and wild-type MC1Rs, or, alternatively, by some behavioral phenomenon (e.g. less-effective sun-protective behaviors in individuals who do not sunburn easily), will be vital to both treatment and risk assessment of patients with MC1R mutations who do not have the canonical red-hair phenotype. These patients may merely need to be reminded that a tan is not healthy for them and they need to wear a hat, or that they might benefit specifically from a pharmacological agent designed to address the deficiency in NER in their melanocytes. The studies and assays discussed here will help in the design optimal protocols for melanoma prevention in these patients.
Questions and future directions
As with all important advances in science, this report raises as many new issues and questions as it answers. First, these assays will need to be adapted for use in human tissue samples in order to realize their full potential. It is still uncertain whether the effects described in these cell-based systems will prove to be relevant in vivo. If it is true that one of the major underlying causes of increased risk for melanoma in MC1R mutation carriers is diminished NER secondary to decreased ATR-pS435, then how and why has this variation been preserved evolutionarily? Does the increase in vitamin D synthesis in fair skinned individuals trump the skin cancer risk because skin cancer occurs later in life, after reproduction has been accomplished? Is it possible that in vivo there are redundant mechanisms in place that can overcome the loss of function of MC1R, perhaps by altering the activities of α-MSH, ACTH, HBD3 or ASIP? Is it possible that because of the role of HBD3 in innate immunity and inflammation in the skin, that inflammatory conditions (e.g. sunburns, chronic wounds) or infection could increase HBD3 levels and simultaneously reduce NER in melanocytes? And finally, it is intriguing that repair of UV-induced DNA damage in melanocytes is regulated hormonally. What biological advantage is conferred by maintaining intermediate levels of repair that can be up- and down-regulated by hormonal factors produced locally and centrally? Is it possible that DNA repair is playing a role in the balance between cell survival, senescence, and immortalization of cancerous melanocytes? Could reduction of repair mechanisms through antagonism of MC1R function make melanocytes or melanoma cells more vulnerable to immunologic attack (e.g. in vitiligo or immunotherapy for melanoma) or sensitize these cells to traditional DNA damaging chemotherapeutics? These questions and ideas are certainly not the only ones that could be raised, but they will hopefully increase readers’ appreciation of the clinical relevance of this work as well as the implications that extend from the fields of pigmentation and melanoma to inflammation, immunology, and, even, infectious disease.
Highlight.
Discovery of the molecular mechanisms of DNA damage repair downstream of MC1R will enable the design of targeted prevention agents and risk assessment for melanoma.
Abbreviations
- 6-4-PP
6′-4′-pyrimdine-pyrimidone
- ATM
ataxia telangiectasia mutated
- ATR
ataxia telangiectasia and Rad3-related protein
- CPD
cyclobutane pyrimidine dimer
- LOF
loss-of-function
- MC1R
melanocortin-1 receptor
- NER
nucleotide excision repair
- XPA
xeroderma pigmentosum complementation group A
References
- Abdel-Malek ZA, Ruwe A, Kavanagh-Starner R, et al. alpha-MSH tripeptide analogs activate the melanocortin 1 receptor and reduce UV-induced DNA damage in human melanocytes. Pigment Cell Melanoma Res. 2009;22:635–44. doi: 10.1111/j.1755-148X.2009.00598.x. [DOI] [PubMed] [Google Scholar]
- Abdel-Malek ZA, Swope VB, Starner RJ, et al. Melanocortins and the melanocortin 1 receptor, moving translationally towards melanoma prevention. Arch Biochem Biophys. 2014;563:4–12. doi: 10.1016/j.abb.2014.07.002. [DOI] [PubMed] [Google Scholar]
- Beaumont KA, Shekar SN, Newton RA, et al. Receptor function, dominant negative activity and phenotype correlations for MC1R variant alleles. Hum Mol Genet. 2007;16:2249–60. doi: 10.1093/hmg/ddm177. [DOI] [PubMed] [Google Scholar]
- Hida T, Wakamatsu K, Sviderskaya EV, et al. Agouti protein, mahogunin, and attractin in pheomelanogenesis and melanoblast-like alteration of melanocytes: a cAMP-independent pathway. Pigment Cell Melanoma Res. 2009;22:623–34. doi: 10.1111/j.1755-148X.2009.00582.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarrett SG, Wolf Horrell EM, Boulanger MC, et al. Defining the contribution of MC1R physiological ligands to ATR phosphorylation at Ser435, a predictor of DNA repair in melanocytes. J Invest Dermatol. 2015;XXX:XX–XXX. doi: 10.1038/jid.2015.280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarrett SG, Wolf Horrell EM, Christian PA, et al. PKA-mediated phosphorylation of ATR promotes recruitment of XPA to UV-induced DNA damage. Mol Cell. 2014;54:999–1011. doi: 10.1016/j.molcel.2014.05.030. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Kadekaro AL, Leachman S, Kavanagh RJ, et al. Melanocortin 1 receptor genotype: an important determinant of the damage response of melanocytes to ultraviolet radiation. FASEB J. 2010;24:3850–60. doi: 10.1096/fj.10-158485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasquali E, Garcia-Borron JC, Fargnoli MC, et al. MC1R variants increased the risk of sporadic cutaneous melanoma in darker-pigmented Caucasians: a pooled-analysis from the M-SKIP project. Int J Cancer. 2015;136:618–31. doi: 10.1002/ijc.29018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puzanov I, Flaherty KT. Targeted molecular therapy in melanoma. Semin Cutan Med Surg. 2010;29:196–201. doi: 10.1016/j.sder.2010.06.005. [DOI] [PubMed] [Google Scholar]
- Shen JC, Fox EJ, Ahn EH, et al. A rapid assay for measuring nucleotide excision repair by oligonucleotide retrieval. Sci Rep. 2014;4:4894. doi: 10.1038/srep04894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swope V, Alexander C, Starner R, et al. Significance of the melanocortin 1 receptor in the DNA damage response of human melanocytes to ultraviolet radiation. Pigment Cell Melanoma Res. 2014;27:601–10. doi: 10.1111/pcmr.12252. [DOI] [PubMed] [Google Scholar]