

Summary
Understanding how antibiotics impact bacterial metabolism may provide insight into their mechanisms of action and could lead to enhanced therapeutic methodologies. Here, we profiled the metabolome of Escherichia coli after treatment with three different classes of bactericidal antibiotics (beta-lactams, aminoglycosides, quinolones). These treatments induced a similar set of metabolic changes after 30 minutes that then diverged into more distinct profiles at later timepoints. The most striking changes corresponded to elevated concentrations of central carbon metabolites, active breakdown of the nucleotide pool, reduced lipid levels, and evidence of an elevated redox state. We examined potential end-target consequences of these metabolic perturbations and found that antibiotic-treated cells exhibited cytotoxic changes indicative of oxidative stress, including higher levels of protein carbonylation, malondialdehyde adducts, nucleotide oxidation, and double-strand DNA breaks. This work shows that bactericidal antibiotics induce a complex set of metabolic changes that are correlated with the buildup of toxic metabolic by-products.
Keywords: antibiotics, E. coli, metabolomics, oxidative stress, reactive oxygen species, lipid peroxidation, protein carbonylation DNA damage, double-strand breaks
Graphical Abstract

Introduction
During the last several decades, much of the research effort on newly identified antibiotics has focused on profiling their direct targets. These approaches have largely been successful, but resulted in a simplified view that antibiotic-induced cell death is exclusively a consequence of target-specific inhibition. The increased availability of high-throughput technologies has enabled the analysis of system-wide bacterial responses to toxic stress. This work has revealed that antibiotic-mediated cell death is a complex, multi-faceted process that cannot be fully accounted for by the direct interactions of antibiotics with their cellular targets (Dwyer et al., 2014; 2012; 2015; Zhao et al., 2015). The current antibiotic crisis has made understanding these cellular responses essential for improving existing therapies and identifying novel therapeutic approaches (Dwyer et al., 2012; 2014; 2015; Wright, 2012; Zhao et al., 2015).
While the majority of systems biology efforts on antibiotics have focused on understanding transcriptional regulatory networks, a number of studies have revealed that changes to cellular metabolic states may play a role in modulating antibiotic susceptibility (Cohen et al., 2013; Gomez and McKinney, 2004; Van Acker et al., 2014). Alterations in cellular carbon flux, for example, have been demonstrated to influence antibiotic susceptibility in multiple bacterial species (Brynildsen et al., 2013; Chittezham Thomas et al., 2013) most notably in Mycobacterium tuberculosis (Baek et al., 2011). Additionally, perturbations to the TCA cycle have been found to reduce antibiotic sensitivity in vitro and TCA cycle defects have been identified in numerous clinical isolates (Chittezham Thomas et al., 2013; Rosato et al., 2014). Metabolic perturbations have been hypothesized to induce a protective state in bacteria by reducing overall cellular growth (Baek et al., 2011), inhibiting antibiotic uptake (Allison et al., 2011), and/or by directly reducing the production of cytotoxic metabolic by-products (Dwyer et al., 2014).
Characterizing antibiotic-induced metabolic changes, and understanding how these alterations impact bacterial cell viability, could be vital to current efforts directed towards enhancing our antibiotic arsenal. To identify global changes to bacterial metabolism following antibiotic treatment, we profiled metabolic alterations in E. coli resulting from treatment with three different bactericidal antibiotics: ampicillin (a β-lactam), kanamycin (an aminoglycoside), and norfloxacin (a quinolone). We found that all three antibiotics induce a similar, initial metabolic response that then becomes more distinctively individualized for each antibiotic at later timepoints. Further, we found that the antibiotic-induced metabolic alterations are associated with oxidative damage to critical cellular components as well as the activation of antioxidant responses. Our results suggest that bactericidal antibiotics induce a complex set of metabolic changes in bacteria, downstream of their direct target interaction, that correlate with the production of reactive oxygen species (ROS) that can damage key cellular components.
Results
Antibiotics induce metabolic alterations in bacteria
We profiled the E. coli metabolome to explore global metabolic alterations induced by bactericidal antibiotics – ampicillin (Amp), kanamycin (Kan), and norfloxacin (Nor) – after 30, 60, and 90 minutes of treatment compared to the initial untreated state (UNT0). Antibiotic concentrations were selected to minimize cell death and lysis at the 30-minute timepoint and to achieve substantial lethality, without lysis, at later timepoints (Figures S1-2) (Kohanski et al., 2007). These conditions can provide a comparison of the initial metabolic response prior to death to that found during the death process. An ultrahigh performance liquid/gas chromatography/electrospray ionization tandem mass spectrometry (LC/MS/MS and GC/MS/MS) platform (Evans et al., 2009) was used to determine the relative concentration of detectable intracellular metabolites.
A total of 195 metabolites were robustly identified (present in at least three out of the five replicates in all tested conditions), spanning 49 sub-pathways and eight super-pathways. A complete set of bar charts can be found in Supplemental Data S1, and Supplemental Data S2 contains a spreadsheet of normalized metabolite measurements and pathway associations. Figure 1 shows the fold change (with respect to UNT0) in relative concentration for the detected metabolites across all treatment conditions, grouped into the six most biologically relevant super-pathways. We observed both increases and decreases in relative concentrations, suggesting that antibiotic treatments have broad, complex effects on metabolism and do not simply quench all metabolic activity.
Figure 1. Bactericidal antibiotics induce broad metabolic perturbations in bacteria.

Bar plots depicting fold change in the relative concentration of individual metabolites (mean value, n=5), with respect to t=0 (the UNT0 control), for E. coli treated with Amp (3 μg/mL), Kan (7.5 μg/mL), or Nor (150 ng/mL) 30, 60, and 90 minutes post antibiotic treatment. Blue indicates decreased concentration and red indicates increased concentration. Fold change values were log-transformed and plotted on a y-axis of −5 to 5.
A number of common metabolic changes were observed for the three antibiotic treatments across the profiled timepoints. Namely, the relative concentrations of nucleotides and lipids were generally seen to decrease upon treatment with antibiotics, whereas the relative concentrations of carbohydrate, energy, and cofactor & vitamin metabolites were generally found to increase. Antibiotic-specific trends were more evident for the amino acid metabolites, with Nor-treated cells showing a larger number of decreased metabolites compared to Kan- or Amp-treated cells at 30 minutes post-treatment, and Amp-treated cells showing increased amino acid metabolites at 60 and 90 minutes post-treatment (Figure 1). A reduction in concentration of most lipid and nucleotide metabolites was evident at 30 minutes for all three of the treatment conditions and persisted for the remainder of the timepoints. In particular, we found that medium chain fatty acids as well as most metabolites categorized as lysolipids were reduced under all treatment conditions (Figure 1).
Antibiotic-induced cell death is thought to be a progressive process (Dwyer et al., 2012; 2014; Kohanski et al., 2010b; 2008), a phenomenon that can be observed in our bar chart visualization (Figure 1). To further elucidate the time-dependent impact of each tested antibiotic, we separately performed hierarchical clustering on the relative metabolite concentration data from the three different treatment timepoints (Figure 2). At the 30-minute timepoint, prior to cell death, the metabolic profiles of the treated samples appeared similar to one another and collectively different from the UNT0 sample. The clustering algorithm was unable to distinguish between the Kan and Amp biological replicates at this timepoint, suggesting that these antibiotics in particular elicited a similar metabolic response. The Nor replicates formed a separate cluster due to a set of uniquely decreased metabolites (largely populated by amino acid metabolites); however, Nor still showed increases and decreases in metabolite concentrations (with respect to UNT0) that were similar to those for Kan and Amp. At the 30-minute timepoint, all of the treatment types were significantly enriched (p<0.05, supplemental data set) for carbohydrates among the increased metabolites and for nucleotides among the decreased metabolites.
Figure 2. Bactericidal antibiotics induce a common set of initial metabolic alterations.

Hierarchical clustering of the metabolic profiling data at 30, 60, and 90 minutes after treatment with Amp (3 μg/mL), Kan, (7.5 μg/mL) or Nor (150 ng/mL). UNT0 (black bars), Kan (green bars), Amp (red bars), and Nor (blue bars). Relative concentration measurements were scaled along each row (i.e., for each metabolite) using a z-score prior to clustering, and red and blue indicate increased and decreased concentrations, respectively
At the later timepoints (60 and 90 minutes), the clustering analysis showed divergence into profiles specific to each treatment type – suggesting that after longer exposure to antibiotics the induction of cellular death leads to differing metabolic outcomes that may be more tightly linked to the target-specific effects. Notably, treatment with Nor led to a general decrease in metabolite concentrations and treatment with Amp led to a general increase in metabolite concentrations – observations that became more pronounced as treatment time and cell death progressed. The differential metabolic changes at 60 and 90 minutes may result from divergent extent of bacterial death and death kinetics as seen in Figure S1. For example, Amp induced 10- to 100-fold less cellular toxicity than Nor or Kan at 60 and 90 minutes (Figure S1). At those same time points, metabolite concentrations in Amp-treated cells became dramatically elevated, accounting for the majority of the differential clustering (Figure 2). It is possible that reducing bacterial death provides additional time for metabolic changes to progress.
Overall, hierarchical clustering of the metabolomics data showed that despite functionally unique modes of action, the distinct antibiotic classes tested elicited substantially similar alterations to the metabolite levels from the central metabolic pathways queried. Similar to previous findings that identified a common stress response upon treatment with antibiotics (Dwyer et al., 2012; 2014; 2015; 2007; Kohanski et al., 2007; Zhao et al., 2015), we saw commonality in the metabolic response to antibiotics in the first 30 minutes of antibiotic exposure. This, along with the separation into antibiotic-specific metabolic profiles at later timepoints, suggests that prior to cell death the target-specific effects of antibiotic treatment are less metabolically evident. We corroborated these findings using principal component analysis (Figure S3); as with hierarchical clustering, commonality was observed for the 30-minute timepoint followed by separation into antibiotic-specific clusters, and all of the treated samples clustered away from UNT0.
To further explore the commonality observed at the 30-minute timepoint, we examined in more detail the relative concentration data for several metabolites (Figure 3). With respect to central carbon metabolism, the most marked changes seen upon antibiotic treatment were within the TCA cycle (Figure 3A). For all three antibiotic classes, citrate and succinate were among metabolites with the largest increases in concentration. Further, our results show elevated levels of nicotinamide adenine dinucleotide (NAD+) as well as NAD precursors and intermediates (Figure 3B). Other notable cofactor/vitamin metabolism alterations indicated in our data include increased levels of coenzyme A (Figure 3C). Together, these findings imply that bactericidal antibiotics lead to changes in TCA cycle activity and oxidative phosphorylation. These results are consistent with recent metabolomic studies in M. tuberculosis that linked antibiotic treatment to changes in central energy metabolism and the TCA cycle (Dwyer et al., 2012; 2014; 2015; Nandakumar et al., 2014; Wright, 2012; Zhao et al., 2015). In addition, recent publications suggest that bactericidal efficacy is linked to bacterial respiration – specifically, bactericidal antibiotics were found to accelerate cellular respiration and this induction was critical for antibiotic lethality (Cohen et al., 2013; Dwyer et al., 2014; Gomez and McKinney, 2004; Lobritz et al., 2015; Van Acker et al., 2014).
Figure 3. Bactericidal antibiotics induce a common set of metabolic perturbations.

E. coli were treated with Amp (3 μg/mL), Kan (7.5 μg/mL), or Nor (150 ng/mL) for 30 minutes before samples were taken for metabolic profiling. Shown are fold changes in relative metabolic concentrations in comparison to UNT0. Data shown reflect mean ± SEM of n = 5. Statistical significance is shown (*: p ≤ 0.05; **: p ≤ 0.01; ***: p ≤ 0.001) using the untreated control for determination. A complete data set for all metabolites presented as box plots can be found in Supplemental Data S1.
In addition to central carbon metabolism and oxidative phosphorylation, we detected marked changes to nucleotide metabolism. We observed a striking decrease in nucleoside, nucleotide, and purine/pyrimidine base levels in response to all antibiotic treatments (Figure 3D), suggestive of a diminishing pool of nucleotide building blocks. This decrease, coupled with increased xanthine levels, a marker of purine catabolism (Brynildsen et al., 2013; Chittezham Thomas et al., 2013; Xi et al., 2000), supports the notion that antibiotics may accelerate nucleotide turnover which may be indicative of higher levels of DNA damage (Baek et al., 2011; Dwyer et al., 2014; 2015; Zhao et al., 2015). This interpretation is consistent with previous work reporting on the sensitivity ofrecA mutants to bactericidal antibiotics (Chittezham Thomas et al., 2013; Kohanski et al., 2007; Liu et al., 2010; Rosato et al., 2014; Wright, 2012), and studies linking guanine pool oxidation to antibiotic lethality (Baek et al., 2011; Cohen et al., 2013; Foti et al., 2012).
We observed specific evidence of increased oxidative stress by a marked effect on the tightly regulated glutathione pools. This is consistent with previous observations that pretreating bacteria with glutathione, a natural antioxidant in both eukaryotes and bacteria (Allison et al., 2011; Masip et al., 2006; Smirnova et al., 2012), provides significant protection from bactericidal antibiotics (Dwyer et al., 2014; Goswami et al., 2006). Generally, a depressed ratio of reduced glutathione (GSH) to oxidized glutathione (GSSG) is considered an indicator of oxidative stress (Kohanski et al., 2007; Masip et al., 2006). Consistent with this, we observed modest increases in the levels of GSH, coupled with much larger increases in GSSG (Figure 3E). The levels of opthalmate, a product of glutathione synthase and analogue of glutathione (Evans et al., 2009; Soga et al., 2006), were also elevated in response to antibiotic treatment. Together, these changes in metabolite levels suggest increased GSH biosynthesis to compensate for ongoing turnover and consumption by antioxidant activities.
Collectively, these data provide direct evidence of complex metabolic changes induced by lethal antibiotic stress. Furthermore, our results indicate that despite disparate cellular targets, functionally diverse antibiotics induce broadly overlapping, early metabolite alterations characterized by increased levels of central carbon metabolites, active breakdown of the nucleotide pool, reduced lipid levels, and an elevated redox state.
Antibiotic treatment leads to a buildup of toxic metabolic by-products
These metabolic profiling data advance the view that bactericidal antibiotics significantly perturb bacterial metabolism, including changes suggestive of intracellular oxidative stress. This connection with oxidative stress is in agreement with previous observations that bactericidal antibiotics induce the production of reactive oxygen species, ROS (Allison et al., 2011; Belenky and Collins, 2011; Dwyer et al., 2007; 2012; 2014; 2015; Foti et al., 2012; Kohanski et al., 2007; 2008; 2010b; Wang et al., 2010); see Dwyer et al. (2015) for an extensive review of prior work on this topic.
Metabolomic measurements such as those conducted in this study are a powerful tool for identifying global changes in metabolism; however, because of their static nature, they cannot be used to directly determine metabolic flux. Thus, these data are most effective when used in conjunction with additional experimental measurements. Based on the strong metabolomic-based indication of an activation of oxidative stress by bactericidal antibiotics, we performed additional metabolomics-independent measurements of cellular oxidation.
ROS that can damage cellular components include superoxide, H2O2, hydroxyl radicals, and peroxyl radicals. While H2O2 reacts poorly or not at all with most biological molecules (Winterbourn, 2013), highly reactive hydroxyl radicals are generated from H2O2 iron-dependent Fenton chemistry (Imlay, 2013; Winterbourn, 2013). Due to the difficulty of determining whether the reactive species in biological contexts is the ferrous peroxide (Fe[IV]) intermediate, the ferryl-oxo intermediate, or the ultimate hydroxyl radical, it has been suggested that all these highly reactive species be simply referred to as Fenton oxidants (Winterbourn, 2013). Hydroxyl radicals react with biomolecules at diffusion-controlled rates in a reaction volume that is less than 2 nm in cells (Cadet and Wagner, 2013). However, much longer-lived and diffusible peroxyl radicals generated by the Fenton oxidants (Dedon, 2008; Lim et al., 2004), can induce damage to cellular components far beyond the site of radical generation and contribute to a buildup of damage.
It is challenging to measure most ROS because of the unstable nature of these reactive molecules (Halliwell, 2007; Roots and Okada, 1975). One commonly used approach is to employ reactive dyes that fluoresce upon oxidation. Using a variety of dyes based on different chemistries, we have previously reported that well-aerated wild-type cells treated with Amp, Kan, or Nor exhibit statistically significant increases in fluorescence (Dwyer et al., 2014), compared with no-dye controls for antibiotic treatment-related autofluorescence and morphology changes (Paulander et al., 2014; Renggli et al., 2013). While consistent with antibiotics inducing oxidative stress, the interpretation of these dye-based observations is nevertheless subject to certain caveats (Imlay, 2015; Kalyanaraman et al., 2012; Wardman, 2007). We therefore chose to implement an approach of simultaneously examining the effects of antibiotic treatment on three well-established biomarkers for oxidative stress – protein carbonylation, malondialdehyde adducts, and the presence of 8-oxo-guanine in DNA and RNA (Fedorova et al., 2014; Haghdoost et al., 2006; Halliwell, 2007; Kasai, 2002; Valenzuela, 1991). Unlike dye assays, the assays for these biomarkers involve no perturbation of the system and so are not subject to the same caveats. These biomarkers have the additional advantage that the fingerprint generated when a reactive species attacks a biological molecule can be used to infer the reactive species that was involved (Halliwell, 2007).
One of the key roles of glutathione is to buffer the oxidation state of cellular proteins (Masip et al., 2006). Thus, the changes to the glutathione pool we detected (Figure 3E) may be indicative of an ongoing antioxidant response to antibiotic-dependent ROS induction and protein damage. The carbonylation of proteins is a stable form of oxidative damage that can lead to protein dysfunction and so is commonly used as a biomarker indicating oxidative cellular injury (Curtis et al., 2012; Fedorova et al., 2014). Bacterial protein oxidation and resulting aggregation can lead to significant cellular toxicity. For example, in M. tuberculosis, the sequestration of irreversibly oxidized protein by chaperone activity was found to protect cells from oxidative stress and antibiotic treatment (Vaubourgeix et al., 2015). To probe oxidative damage to the bacterial protein pool, we utilized a commercially available protein carbonyl ELISA assay to measure the amount of protein carbonylation in cells treated with Amp, Kan, or Nor in comparison to H2O2 (a positive control) or untreated cells. We found that the three bactericidal antibiotics and H2O2 led to significant increases in protein carbonylation, up to 10 times above basal level in the case of Kan and Nor (Figure 4A).
Figure 4. Bactericidal antibiotics induce protein carbonylation and modification of protein by MDA.

(A, B) ELISA-based determination of protein carbonylation and levels of MDA modified protein. E. coli were treated with 10 mM H2O2, 10 μg/mL Amp, 10 μg/mL Kan or 250 ng/mL Nor for 60 minutes. Statistical significance is shown (*: p ≤ 0.05) using the untreated control at 60 minutes for determination.
Protein carbonyl derivatives are not formed by direct reaction with superoxide or H2O2, but rather by direct metal-catalyzed oxidative attack on the amino acid side chain of proline, arginine, lysine, and histidine (Nyström, 2005). Additionally, carbonyl derivatives on lysine, cysteine, and histidine can be formed by secondary reactions with reactive carbonyl compounds on carbohydrates, lipids, and advanced glycation/lipoxidation end-products (Curtis et al., 2012; Nyström, 2005; Stadtman and Levine, 2000; Zhou et al., 2005). Notably, in our metabolic profiling data, we detected changes to lipid precursors. While it is difficult to connect wide-ranging metabolomic changes to any one event, it is possible that these perturbations are associated with oxidative lipid damage. E. coli contain monounsaturated lipids that can undergo oxidative degradation when exposed to certain ROS (Porter et al., 1995; Pradenas et al., 2013). Additionally, manipulating the saturation state of bacterial lipids has been found to impact susceptibility to various forms of oxidizing stress (Harley et al., 1978; Overath et al., 1970; Pradenas et al., 2012). This altered sensitivity can result from elevated propensity for lipid peroxidation and reduced membrane fluidity (Chamberlain and Moss, 1987; Koningsl et al., 1984).
Malondialdehyde (MDA) is widely used in eukaryotes as a marker of oxidative stress as it is generated from polyunsaturated fatty acids in phospholipids in a reaction initiated by oxy radicals such as hydroxyl radicals (Halliwell, 2007; Marnett, 2002). While bacteria do not synthesize polyunsaturated fatty acids, HPLC and TLC separations have been used to show that MDA can also arise from deoxyribose by an alternative pathway that is initiated by a Fenton oxidant abstracting a hydrogen atom at the 4’-position to form a carbon-centered radical that adds molecular oxygen at diffusion-controlled rates (Dedon, 2008; Zhou et al., 2005). To ascertain MDA levels in antibiotic-treated bacteria, we utilized an ELISA assay with an antibody specific for protein modification with the toxic MDA aldehyde. We found that treatment with bactericidal antibiotics led to increased levels of MDA modification on bacterial proteins compared to the no-treatment group, with Nor showing the greatest effect at an increase of almost three-fold (Figure 4B); increases were also observed following treatment with H2O2. Our detection of MDA adducts suggests that Fenton oxidants are being generated in cells exposed to bactericidal antibiotics. Because bacteria are capable of incorporating linoleic acid, a polyunsaturated fatty acid, into their phospholipids if it is available and linoleic acid can yield malondialdehyde in the presence of Fenton oxidants (Zhou et al., 2005), it is possible that this form of lipid oxidation could also contribute to antibiotic-induced MDA production.
Our metabolomic data set also identified significant changes to the nucleotide pool in antibiotic-treated bacteria that are suggestive of nucleotide degradation and possible guanine pool oxidation. The modification of DNA and RNA is a core consequence of oxidative stress, and the oxidation of guanine is most likely because of its low redox potential (Neeley and Essigmann, 2006). The presence of 8-oxo-7,8-dihydroguanine (8-oxo-guanine) in DNA or RNA is widely used as a biomarker of oxidative stress and can result from either oxidation at the level of the nucleic acid or oxidation at the level of the nucleoside triphosphate followed by incorporation. The presence of 8-oxo-dG in DNA has many deleterious consequences, including mismatched base pairing, mutagenesis, and the generation of lethal DNA double-strand breaks (Cadet, 2003; Foti et al., 2012; Haghdoost et al., 2006). Additionally, the induction of 8-oxo-G in RNA can lead to protein mistranslation, protein aggregation and resulting cellular damage (Tanaka et al., 2007). Protein mistranslation and aggregation have been proposed to play important roles in antibiotic-induced death (Kohanski et al., 2008; Ling et al., 2012). To quantitate the levels of both 8-oxo-dG and 8-oxo-G, we utilized two different ELISA assays specific for these modifications. The DNA pool showed an increase of approximately 40% in the levels of 8-oxo-dG across all antibiotic treatments and upon the addition of H2O2, compared to untreated cells (Figure 5A). Our results are consistent with independent prior evidence that antibiotics increase the levels of 8-oxo-dG in DNA (Yeom et al., 2010). We found that the basal levels of 8-oxo-G on the RNA pool were about three times higher than the levels of 8-oxo-dG on DNA (Figure 5B). In addition, the RNA pool showed increased 8-oxo-G content in all treatment conditions ranging from a 113% increase with Kan to a 175% increase with Amp; the H2O2 positive control (Liu et al., 2012) had the largest increase at 478%.
Figure 5. Bactericidal antibiotics induce DNA and RNA oxidation.
(A, B), ELISA-based determination of 8-oxo-dG and 8-oxo-G on total DNA and RNA, respectively. E. coli were treated with 10 mM H2O2, 10 μg/mL Amp, 10 μg/mL Kan, or 250 ng/mL Nor for 60 minutes. Statistical significance is shown (*: p ≤ 0.05) using the untreated control at 60 minutes for determination.
Our detection of increased levels of 8-oxo-guanine in DNA and RNA is a particularly powerful indicator that Fenton oxidants are being generated in response to antibiotics, as neither superoxide nor H2O2 reacts significantly with nucleic acids or nucleotides (Cadet and Wagner, 2013; Imlay et al., 1988; Imlay, 2003; Winterbourn, 2013). Rather, 8-oxo-guanine formation is likely initiated by Fenton oxidants and proceeds through 8-hydroxy-7,8-dihydroguan-8-yl radicals (Cadet and Wagner, 2013; Neeley and Essigmann, 2006). Our observations are consistent with reports that toxic DNA damage by H2O2 is mediated through the Fenton reaction (Imlay et al., 1988) and that aerobically grown E. coli generates sufficient H2O2 to create toxic levels of DNA damage (Park et al., 2005). DNA, RNA, dNTPs, and NTPs are anionic ligands that promote the Fenton reaction with micromolar H2O2 (Park et al., 2005; Rush et al., 1990). During such reactions, Fenton oxidants are produced in the immediate vicinity of the nucleic acids or nucleotides, so these molecules are at especially high risk of high oxidation compared to many other biological molecules. Such localization also favors the oxidation of deoxyribose to generate MDA and other toxic products (Dedon, 2008). Interestingly, 8-oxo-guanine can also be generated by peroxyl radicals (Lim et al., 2004; Simandan et al., 1998) that have been produced by the initial action of Fenton oxidants. There is also particularly compelling evidence to suggest the participation of vicinal pyrimidine peroxyl radicals in the formation of 8-oxo-dG (Bergeron et al., 2010; Douki et al., 2002). Further, it should be noted that peroxyl radicals are much longer-lived than hydroxyl radicals (Lim et al., 2004; Simandan et al., 1998) and hence 8-oxo-guanine formation elicited by antibiotics could potentially be affected by the presence of antioxidants such as glutathione that react with peroxyl radicals (Platt and Gieseg, 2003).
The lower levels of oxidation observed in the DNA pool (compared to the RNA pool) are likely to be in part because the 8-oxo-dG signal on DNA is suppressed in E. coli by the activity of DNA repair enzymes such as MutM and MutY (Foti et al., 2012). On the other hand, RNA does not undergo such repair and is more accessible to ROS because of its single-stranded nature and more transient association with proteins. Multiple studies have shown that levels of oxidative damage are higher and more stable on RNA than on DNA. Damage to the RNA and DNA pool is likely to have deleterious downstream effects on translation and DNA stability, respectively.
It only takes a single unrepaired double-stand break (DSB) to kill a bacterial cell (Bonura and Smith, 1975; Kouzminova and Kuzminov, 2012); consequently, it can take very little 8-oxo-dG in DNA and/or chromosomal fragmentation to kill a cell (Mahaseth and Kuzminov, 2015). For example, recent work has provided evidence that even the very low endogenous levels of 8-oxo-dG in DNA can prove lethal if cells hyper-initiate DNA replication (Charbon et al., 2014). Also, simply increasing the levels of DinB (DNA pol IV) can prove lethal due to the increased incorporation of 8-oxo-dG into DNA (Dwyer et al., 2014; Foti et al., 2012). In each case, the action of glycosylases that recognize base pairs containing 8-oxo-dG contributes to cell death, suggesting that single-strand breaks resulting from incomplete base excision repair give rise to lethal DSBs during replication. Because MutM and MutY each have an associated lyase that catalyzes a β or a β,δ-elimination, they generate ends that require further processing to expose the 3’-OH needed for polymerase action (Manuel et al., 2004).
DSBs could also be generated by glycosylase action at closely spaced lesions (Foti et al., 2012). Closely spaced lesions, including tandem lesions containing 8-oxo-dG that are generated by the reaction of pyrimidine peroxyl radicals (Bergeron et al., 2010), can interfere with the process of base excision repair (Bergeron et al., 2010; Cunniffe et al., 2014; Eccles et al., 2010) and could block replication. Alternatively, incomplete base excision repair intermediates could stall DNA polymerase and lead to a lethal collision between two replication forks or stall RNA polymerase leading to a collision between a replication fork and stalled transcription apparatus (Charbon et al., 2014; Merrikh et al., 2012). Also, as 8-oxo-guanine has a lower redox potential than any of the four normal bases, it can be further oxidized to more toxic products, including spiroiminodihydantoin and guanidinohydantoin (Neeley and Essigmann, 2006).
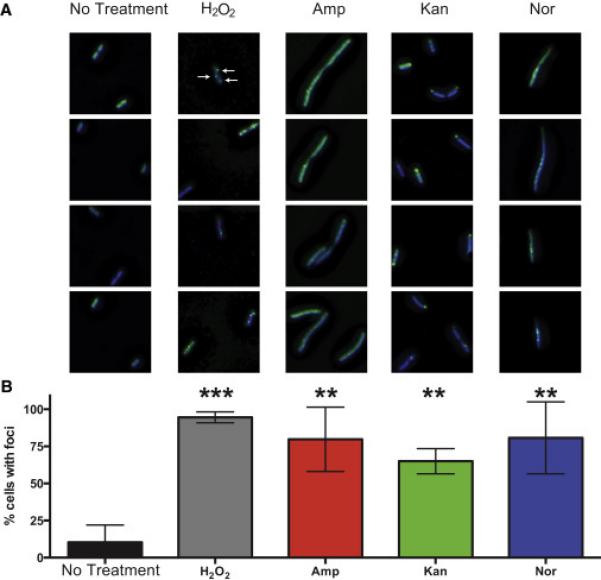
Motivated by our nucleotide results and to test further the hypothesis that bactericidal antibiotics induce DSBs in the bacterial chromosome, we took advantage of a novel engineered fluorescent protein-based probe (Shee et al., 2013). This method relies on a fusion of GFP to the Gam protein from phage Mu that robustly binds to DSBs when expressed in E. coli. To visually profile the formation of DSB foci, we treated cells expressing Gam-GFP with H2O2, which is known to induce DSBs (Friedberg et al., 1996), or with Amp, Kan, or Nor. Expression of Gam-GFP allows reliable detection and enumeration of DSBs, which appear as fluorescent GFP foci (Shee et al., 2013). As some antibiotics cause protein misfolding and polar aggregation, cells were stained with DAPI prior to imaging, and only foci co-localized with DAPI in the deconvolved three-dimensional image were quantified as DSB foci.
Consistent with previous work (Shee et al., 2013), we found that approximately 10% of cells growing in the absence of drug exhibited foci. All of the treatment conditions led to robustly elevated levels of DSBs (Figure 6) as evidenced by a statistically significant increase in the percentage of cells exhibiting foci. Treatment with Amp, Kan, Nor, or H2O2 also induced the formation of multiple foci per cells, with an average of 75.4%, 68.1%, 35.3%, and 81.1% multifocal cells, respectively, compared to just 4.7% of untreated cells (Figure S4). Two of the antibiotics, Amp, and Kan, induced significant morphological changes that can increase both the size of the bacterial cell and DNA content in cells that failed to undergo division. These morphological changes may result in somewhat elevated foci counts in larger cells but cannot explain the more the 10-fold elevation in foci counts. Despite the somewhat divergent GamGFP fluorescent signatures, these data suggest that antibiotic treatment leads to the formation of DSBs in E. coli and are consistent with a prior study showing that antibiotic exposure leads to chromosomal DSBs (Dwyer et al., 2012). They are also consistent with multiple lines of evidence indicating that introduction of 8-oxo-dG into DNA plays a causal contributing role to antibiotic lethality, including the observation that bactericidal antibiotics cause MutM- and MutY-dependent TUNEL staining that precedes cell death (Foti et al., 2012).
Figure 6. Bactericidal antibiotics induce double-strand breaks in E. coli.

(A) Selected images showing Gam-GFP distribution (green) in untreated cells or cells treated for 2 hours with 10 mM H2O2, 2.5 μg/mL Amp, 10 μg/mL Kan, or 125 ng/mL Nor. Bacterial DNA is stained with DAPI (blue). Arrows indicate gam-GFP foci, which occur at double-strand breaks. GFP foci not colocalized with DAPI were excluded from the analysis. (B) Percent of cells with GFP foci in the indicated treatment group. Bars represent the average of three independent experiments in which 50 to 150 cells were quantitated for each condition. Error bars represent SEM. Statistical significance using Sidak's multiple comparisons test: *, p>0.05; **, p<0.01; ***, p<0.001.
Discussion
β-lactams, aminoglycosides, and quinolones have well-established primary mechanisms of action. However, recent work has indicated that in addition to these distinct mechanisms, subsequent metabolic changes that occur downstream of the interaction of the antibiotics with their targets also play an important role in antibiotic lethality (Cohen et al., 2013; Dwyer et al., 2014; 2015; Gomez and McKinney, 2004; Van Acker et al., 2014). In this work, we evaluated the downstream effects of bactericidal antibiotics on bacterial metabolism by profiling the metabolome of E. coli following treatment with ampicillin, kanamycin, or norfloxacin. We found that the initial metabolic response to these different antibiotics, is similar soon after treatment, but diverges after longer exposure to the antibiotics. This similarity in the initial metabolic response to antibiotics closely parallels previous studies that found common transcriptional and physiological responses to bactericidal antibiotics (Dwyer et al., 2014; Kohanski et al., 2007). These same studies found that ROS production and metabolic activity contribute to antibiotic lethality (Lobritz et al., 2015). Consistent with these observations, the present study showed that the bactericidal antibiotic treatments altered glutathione metabolism and reduced the GSH/GSSG ratio, indicating an ongoing antioxidant response consistent with the generation of oxidants.
Previous studies have proposed that elevated cellular respiration leads to the production of ROS in response to bactericidal antibiotics (Dwyer et al., 2014; Kohanski et al., 2007). Consistent with these observations, we found higher levels of TCA cycle intermediates and altered NAD+ metabolism indicative of altered TCA cycle activity and oxidative phosphorylation (Kohanski et al., 2010b). We also detected a more general elevation in concentrations of carbohydrate, cofactor/vitamin, and energy metabolites. These data indicate that bacteria treated with bactericidal antibiotics may experience a metabolically activated state that correlates with cell death. This notion is supported by numerous studies that found that perturbations to metabolic capacity can impact the bactericidal action of antibiotics (Baek et al., 2011; Brynildsen et al., 2013; Chittezham Thomas et al., 2013; Gomez and McKinney, 2004; Rosato et al., 2014; Van Acker et al., 2014). In addition, β-lactam antibiotics were recently found to induce an energy-expensive, futile biosynthetic cycle of peptidoglycan polymerization and degradation (Cho et al., 2014), and even more recently, it has been reported that metabolic futile cycles can increase H2O2 production and sensitivity to oxidants (Adolfsen and Brynildsen, 2015). Taken together with our observations, these reports suggest the possibility that futile cycles associated with antibiotic-induced metabolic changes could account, at least in part, for the myriad of observations (Dwyer et al., 2015) indicating that bacterial antibiotics elicit oxidative stress that contributes causally to bacterial mutagenesis and cell death.
Metabolomic data sets can be used to detect both primary changes and secondary effects arising from cellular responses to an applied stress. For example, we observed significant changes to glutathione pools, indicating antioxidant responses, as well as a reduction in nucleotides and lipid precursors. One possible explanation of this profile is that antibiotic-induced oxidants damage cellular components such as proteins, lipids, and nucleotides that are then turned over by the cell. To test this hypothesis, we examined protein carbonylation, malondialdehyde adducts, and the presence of 8-oxo-guanine in DNA and RNA. These toxic downstream metabolic by-products of ROS production are more stable than ROS and can be detected using well-established techniques. Consistent with the above hypothesis, we found that antibiotics significantly increased the levels of these well-established biomarkers of oxidative stress. Additionally, oxidation of DNA and free nucleotides can induce mismatched pairing, mutagenesis, and DSBs (Cadet, 2003; Foti et al., 2012; Haghdoost et al., 2006). We detected significantly elevated rates of DSBs that support previous reports that detected higher mutagenesis rates and DNA damage during antibiotic exposure (Dwyer et al., 2012; Foti et al., 2012; Kohanski et al., 2010a; Méhi et al., 2014; Nair et al., 2013).
The implication that oxidation of the biomarkers elicited by antibiotic treatment involves Fenton oxidants is particularly significant because metabolic alterations can increase the lethality of H2O2 by potentiating Fenton chemistry. For example, increases in intracellular cysteine levels increase the killing by H2O2 in aerobically grown cells by more than 100-fold (Park et al., 2005), an effect attributed to cysteine increasing the amount of H2O2 undergoing Fenton chemistry by reducing Fe+3 to Fe+2 (Berglin et al., 1982; Park et al., 2005). Interestingly, we observed a significant increase in intracellular cysteine levels after 30 minutes exposure to all three bactericidal antibiotics (Supplemental Data S1). Additionally, numerous other metabolic changes such as increased keto-acids (Winterbourn, 2013), changes in heme synthesis (Mancini and Imlay, 2015), and H2S production (Shatalin et al., 2011), could also influence the overall amount of Fenton chemistry taking place in response to an antibiotic challenge. Thus, antibiotic-induced metabolic changes could impact oxidation of cellular components by up-regulating oxidative metabolism and/or by shifting the rate of the Fenton reaction. Taken together, these considerations suggest that when considering the potential of intracellularly generated H2O2 to kill cells (Imlay, 2015), it is not sufficient to focus simply on the amount of unreacted H2O2 that is present. Rather, when considering drug-induced, ROS-associated cell death, one must also take into account the amount of Fenton oxidant that is being produced, the metabolic changes induced by antibiotics, and the rate of biomolecule oxidation, among other factors.
Fenton oxidants are highly reactive, have extremely short half lives, and have very limited distances of diffusion (Roots and Okada, 1975). Hydroxyl radicals will essentially react with the first susceptible molecule they encounter. Our identification of toxic metabolic by-products following antibiotic treatment provides a possible mechanism as to how localized ROS production could impact multiple and distributed cellular components, propagating the toxic effects of these molecules. Based on our data and recent publications, we speculate that the initial antibiotic-triggered metabolic changes, such as elevated rates of oxidative phosphorylation (Dwyer et al., 2014; Lobritz et al., 2015), at the bacterial membrane, lead to higher rates of ROS production; these oxidants then react locally with membrane lipids and proteins to form toxic products, as well as forming longer-lived peroxyl radicals that can diffuse to impact the entire bacterial cell. The result is widespread cellular damage and dysfunction, which contribute to cellular death (Dwyer et al., 2014; Foti et al., 2012; Lobritz et al., 2015). In addition, oxidative damage to cellular components is more stable than the initial Fenton oxidants and might act as a toxic sink to maintain a lower steady-state level of molecules like H2O2, while at the same time allowing for the buildup of cellular damage.
Bacteria live very close to the edge with respect to their ability to tolerate oxidative damage. For example, reducing the levels of superoxide dismutase or MutT, the 8-oxo-dGTP sanitizer, by only a factor of two (Setoyama et al., 2011) results in changes to mutation frequency. In addition, 89% of spontaneous base-pair mutations in aerobically grown E. coli have been attributed to ROS (Sakai et al., 2006). Bacteria often react to stresses by increasing their mutation rate to help them adapt and indeed exposure of aerobically grown E. coli to sublethal levels of bactericidal antibiotics increases the mutation rate (Kohanski et al., 2010a). In the intensively studied case of exposure to subinhibitory levels of ampicillin, the underlying molecular mechanism is thought to involve incorporation of oxidized nucleotides into nascent DNA coupled with a temporary suppression of mismatch repair so that they are not removed (Gutierrez et al., 2013). Because bacteria are most frequently exposed to sublethal doses of antibiotics in natural settings, the evolution of a system that increases ROS-dependent mutagenesis under the stress of low levels of antibiotics would have been beneficial. Thus, the ROS-dependent component of killing by clinically used concentrations of antibiotics that is due to DNA damage could be a deleterious consequence of a strategy that is normally beneficial for adaptation under less stressful conditions (Dwyer et al., 2015).
Taken together, our findings lend support to the suggestion that targeting microbial metabolism to enhance the bactericidal activity of antibiotics may be an effective means of reducing the burden of infections (Allison et al., 2011; Brynildsen et al., 2013). More broadly, our results suggest that, for other stresses besides antibiotics, induced metabolic changes may also result in the generation of increased Fenton oxidants, thus helping to explain the common involvement of ROS toxicity in a long list of physiologically diverse stresses (Dong et al., 2015; Imlay, 2015).
Experimental Procedures
Bacterial strain
MG1655 E. coli (ATCC# 700926) was used for all studies.
Metabolic profiling and computational analysis
Metabolic profiling was performed by Metabolon, Inc. (Durham, USA) and statistical analyses were performed in MATLAB (MATLAB 8.1, The MathWorks Inc., Natick, MA, 2013). A complete description of the metabolic profiling and computational analysis can be found in Supplemental Experimental Procedures.
MG1655 collection and lysis for ELISA
Overnight cultures were diluted 1:250 in 25 mL LB, in 250 mL baffled flasks and grown to an OD600 of 0.2 to 0.3. Cells treated with antibiotics for one hour, were washed in PBS and stored at −80°C in 200 μL B-PER II with 100 μg/mL lysozyme and 5 U/mL DNase I (Thermo Scientific). A BCA assay (Pierce) was used to quantify protein concentration. Please see Supplemental Experimental Procedures for a detailed experimental protocol.
ELISA
MDA was quantified using an in-house designed ELISA protocol. Protein carbonylation was measured with a Protein Carbonyl ELISA kit (Enzo Life Sciences). 8-oxo-dG levels and 8-oxo-G levels were quantified using OxiSelect Oxidative DNA Damage ELISA kit and the OxiSelect Oxidative RNA Damage ELISA kit (Cell Biolabs), respectively. Please see Supplemental Experimental Procedures for a detailed experimental protocol.
GamGFP assay
Imaging was performed on a Zeiss Axio Observer Z1 microscope, using a 63× water lens and 2.5× optovar, and quantification of GFP foci was performed using Imaris software. Please see Supplemental Experimental Procedures for a detailed experimental protocol.
Supplementary Material
Acknowledgements
We thank Susan M. Rosenberg for generously providing the GamGFP strain used in this study. This work was supported by the National Institutes of Health Director's Pioneer Award Program (J.J.C.), the Howard Hughes Medical Institute, the Wyss Institute for Biologically Inspired Engineering, a Defense Threat Reduction Agency grant HDTRA1-15-1-0051 (J.J.C) and National Institutes of Health grant R01 CA021615 (G.C.W.). G.C.W. is an American Cancer Society Professor.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Adolfsen KJ, Brynildsen MP. Futile cycling increases sensitivity toward oxidative stress in Escherichia coli. Metab. Eng. 2015;29:26–35. doi: 10.1016/j.ymben.2015.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allison KR, Brynildsen MP, Collins JJ. Metabolite-enabled eradication of bacterial persisters by aminoglycosides. Nature. 2011;473:216–220. doi: 10.1038/nature10069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baek S-H, Li AH, Sassetti CM. Metabolic regulation of mycobacterial growth and antibiotic sensitivity. PLoS Biol. 2011;9:e1001065. doi: 10.1371/journal.pbio.1001065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belenky P, Collins JJ. Antioxidant Strategies to Tolerate Antibiotics. Science. 2011;334:915–916. doi: 10.1126/science.1214823. [DOI] [PubMed] [Google Scholar]
- Bergeron F, Auvré F, Radicella JP, Ravanat J-L. HO* radicals induce an unexpected high proportion of tandem base lesions refractory to repair by DNA glycosylases. Proc. Natl. Acad. Sci. U.S.a. 2010;107:5528–5533. doi: 10.1073/pnas.1000193107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berglin EH, Edlund MB, Nyberg GK, Carlsson J. Potentiation by L- cysteine of the bactericidal effect of hydrogen peroxide in Escherichia coli. J. Bacteriol. 1982;152:81–88. doi: 10.1128/jb.152.1.81-88.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonura T, Smith KC. Enzymatic production of deoxyribonucleic acid double-strand breaks after ultraviolet irradiation of Escherichia coli K-12. J. Bacteriol. 1975;121:511–517. doi: 10.1128/jb.121.2.511-517.1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brynildsen MP, Winkler JA, Spina CS, MacDonald IC, Collins JJ. Potentiating antibacterial activity by predictably enhancing endogenous microbial ROS production. Nat Biotechnol. 2013;31:160–165. doi: 10.1038/nbt.2458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cadet J. Oxidative damage to DNA: formation, measurement and biochemical features. Mutation Research/Fundamental and Molecular Mechanisms of Mutagenesis. 2003;531:5–23. doi: 10.1016/j.mrfmmm.2003.09.001. [DOI] [PubMed] [Google Scholar]
- Cadet J, Wagner JR. DNA base damage by reactive oxygen species, oxidizing agents, and UV radiation. Cold Spring Harb Perspect Biol. 2013;5 doi: 10.1101/cshperspect.a012559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chamberlain J, Moss SH. Lipid peroxidation and other membrane damage produced in Escherichia coli K1060 by near-UV radiation and deuterium oxide. Photochem. Photobiol. 1987;45:625–630. doi: 10.1111/j.1751-1097.1987.tb07389.x. [DOI] [PubMed] [Google Scholar]
- Charbon G, Bjørn L, Mendoza-Chamizo B, Frimodt-Møller J, Løbner-Olesen A. Oxidative DNA damage is instrumental in hyperreplication stress-induced inviability of Escherichia coli. Nucleic Acids Res. 2014;42:13228–13241. doi: 10.1093/nar/gku1149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chittezham Thomas V, Kinkead LC, Janssen A, Schaeffer CR, Woods KM, Lindgren JK, Peaster JM, Chaudhari SS, Sadykov M, Jones J, et al. A dysfunctional tricarboxylic acid cycle enhances fitness of Staphylococcus epidermidis during β-lactam stress. MBio. 2013;4 doi: 10.1128/mBio.00437-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho H, Uehara T, Bernhardt TG. Beta-lactam antibiotics induce a lethal malfunctioning of the bacterial cell wall synthesis machinery. Cell. 2014;159:1300–1311. doi: 10.1016/j.cell.2014.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen NR, Lobritz MA, Collins JJ. Microbial persistence and the road to drug resistance. Cell Host Microbe. 2013;13:632–642. doi: 10.1016/j.chom.2013.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunniffe S, O'Neill P, Greenberg MM, Lomax ME. Reduced repair capacity of a DNA clustered damage site comprised of 8-oxo-7,8-dihydro-2′- deoxyguanosine and 2-deoxyribonolactone results in an increased mutagenic potential of these lesions. Mutat. Res. 2014;762:32–39. doi: 10.1016/j.mrfmmm.2014.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curtis JM, Hahn WS, Long EK, Burrill JS, Arriaga EA, Bernlohr DA. Protein carbonylation and metabolic control systems. Trends Endocrinol. Metab. 2012;23:399–406. doi: 10.1016/j.tem.2012.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dedon PC. The chemical toxicology of 2-deoxyribose oxidation in DNA. Chem. Res. Toxicol. 2008;21:206–219. doi: 10.1021/tx700283c. [DOI] [PubMed] [Google Scholar]
- Dong TG, Dong S, Catalano C, Moore R, Liang X, Mekalanos JJ. Generation of reactive oxygen species by lethal attacks from competing microbes. Proc. Natl. Acad. Sci. U.S.a. 2015;112:2181–2186. doi: 10.1073/pnas.1425007112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Douki T, Riviere J, Cadet J. DNA tandem lesions containing 8-oxo-7,8-dihydroguanine and formamido residues arise from intramolecular addition of thymine peroxyl radical to guanine. Chem. Res. Toxicol. 2002;15:445–454. doi: 10.1021/tx0155909. [DOI] [PubMed] [Google Scholar]
- Dwyer DJ, Belenky PA, Yang JH, MacDonald IC, Martell JD, Takahashi N, Chan CTY, Lobritz MA, Braff D, Schwarz EG, et al. Antibiotics induce redox-related physiological alterations as part of their lethality. Proc. Natl. Acad. Sci. U.S.a. 2014;111:E2100–E2109. doi: 10.1073/pnas.1401876111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dwyer DJ, Camacho DM, Kohanski MA, Callura JM, Collins JJ. Antibiotic-induced bacterial cell death exhibits physiological and biochemical hallmarks of apoptosis. Molecular Cell. 2012;46:561–572. doi: 10.1016/j.molcel.2012.04.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dwyer DJ, Collins JJ, Walker GC. Unraveling the physiological complexities of antibiotic lethality. Annu. Rev. Pharmacol. Toxicol. 2015;55:313–332. doi: 10.1146/annurev-pharmtox-010814-124712. [DOI] [PubMed] [Google Scholar]
- Dwyer DJ, Kohanski MA, Hayete B, Collins JJ. Gyrase inhibitors induce an oxidative damage cellular death pathway in Escherichia coli. Mol. Syst. Biol. 2007;3:91. doi: 10.1038/msb4100135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eccles LJ, Lomax ME, O'Neill P. Hierarchy of lesion processing governs the repair, double-strand break formation and mutability of three-lesion clustered DNA damage. Nucleic Acids Res. 2010;38:1123–1134. doi: 10.1093/nar/gkp1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans AM, DeHaven CD, Barrett T, Mitchell M, Milgram E. Integrated, nontargeted ultrahigh performance liquid chromatography/electrospray ionization tandem mass spectrometry platform for the identification and relative quantification of the small-molecule complement of biological systems. Anal. Chem. 2009;81:6656–6667. doi: 10.1021/ac901536h. [DOI] [PubMed] [Google Scholar]
- Fedorova M, Bollineni RC, Hoffmann R. Protein carbonylation as a major hallmark of oxidative damage: update of analytical strategies. Mass Spectrom Rev. 2014;33:79–97. doi: 10.1002/mas.21381. [DOI] [PubMed] [Google Scholar]
- Foti JJ, Devadoss B, Winkler JA, Collins JJ, Walker GC. Oxidation of the guanine nucleotide pool underlies cell death by bactericidal antibiotics. Science. 2012;336:315–319. doi: 10.1126/science.1219192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedberg EC, Walker GC, Siede W. DNA repair and mutagenesis. American Society for Microbiology (ASM); 1996. [Google Scholar]
- Gomez JE, McKinney JD. M. tuberculosis persistence, latency, and drug tolerance. Tuberculosis (Edinb) 2004;84:29–44. doi: 10.1016/j.tube.2003.08.003. [DOI] [PubMed] [Google Scholar]
- Goswami M, Mangoli SH, Jawali N. Involvement of reactive oxygen species in the action of ciprofloxacin against Escherichia coli. Antimicrob Agents Chemother. 2006;50:949–954. doi: 10.1128/AAC.50.3.949-954.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutierrez A, Laureti L, Crussard S, Abida H, Rodríguez-Rojas A, Blázquez J, Baharoglu Z, Mazel D, Darfeuille F, Vogel J, et al. β-Lactam antibiotics promote bacterial mutagenesis via an RpoS-mediated reduction in replication fidelity. Nat Commun. 2013;4:1610. doi: 10.1038/ncomms2607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haghdoost S, Sjölander L, Czene S, Harms-Ringdahl M. The nucleotide pool is a significant target for oxidative stress. Free Radic. Biol. Med. 2006;41:620–626. doi: 10.1016/j.freeradbiomed.2006.05.003. [DOI] [PubMed] [Google Scholar]
- Halliwell B. Free Radicals in Biology and Medicine. Oxford University Press; 2007. [Google Scholar]
- Harley JB, Santangelo GM, Rasmussen H, Goldfine H. Dependence of Escherichia coli hyperbaric oxygen toxicity on the lipid acyl chain composition. J. Bacteriol. 1978;134:808–820. doi: 10.1128/jb.134.3.808-820.1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imlay JA, Chin SM, Linn S. Toxic DNA damage by hydrogen peroxide through the Fenton reaction in vivo and in vitro. Science. 1988;240:640–642. doi: 10.1126/science.2834821. [DOI] [PubMed] [Google Scholar]
- Imlay JA. Pathways of oxidative damage. Annu Rev Microbiol. 2003;57:395–418. doi: 10.1146/annurev.micro.57.030502.090938. [DOI] [PubMed] [Google Scholar]
- Imlay JA. The molecular mechanisms and physiological consequences of oxidative stress: lessons from a model bacterium. Nat Rev Microbiol. 2013;11:443–454. doi: 10.1038/nrmicro3032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imlay JA. Diagnosing oxidative stress in bacteria: not as easy as you might think. Curr. Opin. Microbiol. 2015;24:124–131. doi: 10.1016/j.mib.2015.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalyanaraman B, Darley-Usmar V, Davies KJA, Dennery PA, Forman HJ, Grisham MB, Mann GE, Moore K, Roberts LJ, Ischiropoulos H. Measuring reactive oxygen and nitrogen species with fluorescent probes: challenges and limitations. Free Radic. Biol. Med. 2012;52:1–6. doi: 10.1016/j.freeradbiomed.2011.09.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasai H. Chemistry-based studies on oxidative DNA damage: formation, repair, and mutagenesis. Free Radic. Biol. Med. 2002;33:450–456. [PubMed] [Google Scholar]
- Kohanski MA, DePristo MA, Collins JJ. Sublethal antibiotic treatment leads to multidrug resistance via radical-induced mutagenesis. Molecular Cell. 2010a;37:311–320. doi: 10.1016/j.molcel.2010.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohanski MA, Dwyer DJ, Collins JJ. How antibiotics kill bacteria: from targets to networks. Nat Rev Microbiol. 2010b;8:423–435. doi: 10.1038/nrmicro2333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohanski MA, Dwyer DJ, Hayete B, Lawrence CA, Collins JJ. A common mechanism of cellular death induced by bactericidal antibiotics. Cell. 2007;130:797–810. doi: 10.1016/j.cell.2007.06.049. [DOI] [PubMed] [Google Scholar]
- Kohanski MA, Dwyer DJ, Wierzbowski J, Cottarel G, Collins JJ. Mistranslation of membrane proteins and two-component system activation trigger antibiotic-mediated cell death. Cell. 2008;135:679–690. doi: 10.1016/j.cell.2008.09.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koningsl AWT, Gipp JJ, Yatvin MB. Radio- and thermosensitivity of E. coli K1060 after thiol depletion by diethylmaleate. Radiat Environ Biophys. 1984;23:245–253. doi: 10.1007/BF01407596. [DOI] [PubMed] [Google Scholar]
- Kouzminova EA, Kuzminov A. Chromosome demise in the wake of ligase-deficient replication. Mol. Microbiol. 2012;84:1079–1096. doi: 10.1111/j.1365-2958.2012.08076.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim P, Wuenschell GE, Holland V, Lee D-H, Pfeifer GP, Rodriguez H, Termini J. Peroxyl radical mediated oxidative DNA base damage: implications for lipid peroxidation induced mutagenesis. Biochemistry. 2004;43:15339–15348. doi: 10.1021/bi048276x. [DOI] [PubMed] [Google Scholar]
- Ling J, Cho C, Guo L-T, Aerni HR, Rinehart J, Söll D. Protein aggregation caused by aminoglycoside action is prevented by a hydrogen peroxide scavenger. Molecular Cell. 2012;48:713–722. doi: 10.1016/j.molcel.2012.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu A, Tran L, Becket E, Lee K, Chinn L, Park E, Tran K, Miller JH. Antibiotic sensitivity profiles determined with an Escherichia coli gene knockout collection: generating an antibiotic bar code. Antimicrob Agents Chemother. 2010;54:1393–1403. doi: 10.1128/AAC.00906-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu M, Gong X, Alluri RK, Wu J, Sablo T, Li Z. Characterization of RNA damage under oxidative stress in Escherichia coli. Biol. Chem. 2012;393:123–132. doi: 10.1515/hsz-2011-0247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lobritz MA, Belenky P, Porter CBM, Gutierrez A, Yang JH, Schwarz EG, Dwyer DJ, Khalil AS, Collins JJ. Antibiotic efficacy is linked to bacterial cellular respiration. Proc. Natl. Acad. Sci. U.S.a. 2015;112:8173–8180. doi: 10.1073/pnas.1509743112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahaseth T, Kuzminov A. Cyanide enhances hydrogen peroxide toxicity by recruiting endogenous iron to trigger catastrophic chromosomal fragmentation. Mol. Microbiol. 2015;96:349–367. doi: 10.1111/mmi.12938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mancini S, Imlay JA. The induction of two biosynthetic enzymes helps Escherichia coli sustain heme synthesis and activate catalase during hydrogen peroxide stress. Mol. Microbiol. 2015 doi: 10.1111/mmi.12967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manuel RC, Hitomi K, Arvai AS, House PG, Kurtz AJ, Dodson ML, McCullough AK, Tainer JA, Lloyd RS. Reaction intermediates in the catalytic mechanism of Escherichia coli MutY DNA glycosylase. J Biol Chem. 2004;279:46930–46939. doi: 10.1074/jbc.M403944200. [DOI] [PubMed] [Google Scholar]
- Marnett LJ. Oxy radicals, lipid peroxidation and DNA damage. Toxicology. 2002;181- 182:219–222. doi: 10.1016/s0300-483x(02)00448-1. [DOI] [PubMed] [Google Scholar]
- Masip L, Veeravalli K, Georgiou G. The many faces of glutathione in bacteria. Antioxid. Redox Signal. 2006;8:753–762. doi: 10.1089/ars.2006.8.753. [DOI] [PubMed] [Google Scholar]
- Merrikh H, Zhang Y, Grossman AD, Wang JD. Replication- transcription conflicts in bacteria. Nat Rev Microbiol. 2012;10:449–458. doi: 10.1038/nrmicro2800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Méhi O, Bogos B, Csörgő B, Pál F, Nyerges A, Papp B, Pál C. Perturbation of iron homeostasis promotes the evolution of antibiotic resistance. Mol. Biol. Evol. 2014;31:2793–2804. doi: 10.1093/molbev/msu223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nair CG, Chao C, Ryall B, Williams HD. Sub-lethal concentrations of antibiotics increase mutation frequency in the cystic fibrosis pathogen Pseudomonas aeruginosa. Lett. Appl. Microbiol. 2013;56:149–154. doi: 10.1111/lam.12032. [DOI] [PubMed] [Google Scholar]
- Nandakumar M, Nathan C, Rhee KY. Isocitrate lyase mediates broad antibiotic tolerance in Mycobacterium tuberculosis. Nat Commun. 2014;5:4306. doi: 10.1038/ncomms5306. [DOI] [PubMed] [Google Scholar]
- Neeley WL, Essigmann JM. Mechanisms of formation, genotoxicity, and mutation of guanine oxidation products. Chem. Res. Toxicol. 2006;19:491–505. doi: 10.1021/tx0600043. [DOI] [PubMed] [Google Scholar]
- Nyström T. Role of oxidative carbonylation in protein quality control and senescence. Embo J. 2005;24:1311–1317. doi: 10.1038/sj.emboj.7600599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Overath P, Schairer HU, Stoffel W. Correlation of in vivo and in vitro phase transitions of membrane lipids in Escherichia coli. Proc. Natl. Acad. Sci. U.S.a. 1970;67:606–612. doi: 10.1073/pnas.67.2.606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park S, You X, Imlay JA. Substantial DNA damage from submicromolar intracellular hydrogen peroxide detected in Hpx- mutants of Escherichia coli. Proc. Natl. Acad. Sci. U.S.a. 2005;102:9317–9322. doi: 10.1073/pnas.0502051102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paulander W, Wang Y, Folkesson A, Charbon G, Løbner-Olesen A, Ingmer H. Bactericidal antibiotics increase hydroxyphenyl fluorescein signal by altering cell morphology. PLoS ONE. 2014;9:e92231. doi: 10.1371/journal.pone.0092231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Platt AA, Gieseg SP. Inhibition of protein hydroperoxide formation by protein thiols. Redox Rep. 2003;8:81–86. doi: 10.1179/135100003125001288. [DOI] [PubMed] [Google Scholar]
- Porter NA, Caldwell SE, Mills KA. Mechanisms of free radical oxidation of unsaturated lipids. Lipids. 1995;30:277–290. doi: 10.1007/BF02536034. [DOI] [PubMed] [Google Scholar]
- Pradenas GA, Díaz-Vásquez WA, Pérez-Donoso JM, Vásquez CC. Monounsaturated fatty acids are substrates for aldehyde generation in tellurite-exposed Escherichia coli. Biomed Res Int. 2013;2013:563756. doi: 10.1155/2013/563756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pradenas GA, Paillavil BA, Reyes-Cerpa S, Pérez-Donoso JM, Vásquez CC. Reduction of the monounsaturated fatty acid content of Escherichia coli results in increased resistance to oxidative damage. Microbiology (Reading, Engl.) 2012;158:1279–1283. doi: 10.1099/mic.0.056903-0. [DOI] [PubMed] [Google Scholar]
- Renggli S, Keck W, Jenal U, Ritz D. Role of autofluorescence in flow cytometric analysis of Escherichia coli treated with bactericidal antibiotics. J. Bacteriol. 2013;195:4067–4073. doi: 10.1128/JB.00393-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roots R, Okada S. Estimation of life times and diffusion distances of radicals involved in X-ray-induced DNA strand breaks or killing of mammalian cells. 1975 [PubMed] [Google Scholar]
- Rosato RR, Fernandez R, Paz LI, Singh CR, Rosato AE. TCA Cycle-Mediated Generation of ROS Is a Key Mediator for HeR-MRSA Survival under β-Lactam Antibiotic Exposure. PLoS ONE. 2014;9:e99605. doi: 10.1371/journal.pone.0099605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rush JD, Maskos Z, Koppenol WH. Reactions of Iron(Ii) Nucleotide Complexes with Hydrogen-Peroxide. FEBS Lett. 1990;261:121–123. [Google Scholar]
- Sakai A, Nakanishi M, Yoshiyama K, Maki H. Impact of reactive oxygen species on spontaneous mutagenesis in Escherichia coli. Genes Cells. 2006;11:767–778. doi: 10.1111/j.1365-2443.2006.00982.x. [DOI] [PubMed] [Google Scholar]
- Setoyama D, Ito R, Takagi Y, Sekiguchi M. Molecular actions of Escherichia coli MutT for control of spontaneous mutagenesis. Mutat. Res. 2011;707:9–14. doi: 10.1016/j.mrfmmm.2010.12.001. [DOI] [PubMed] [Google Scholar]
- Shatalin K, Shatalina E, Mironov A, Nudler E. H2S: a universal defense against antibiotics in bacteria. Science. 2011;334:986–990. doi: 10.1126/science.1209855. [DOI] [PubMed] [Google Scholar]
- Shee C, Cox BD, Gu F, Luengas EM, Joshi MC, Chiu L-Y, Magnan D, Halliday JA, Frisch RL, Gibson JL, et al. Engineered proteins detect spontaneous DNA breakage in human and bacterial cells. Elife. 2013;2:e01222. doi: 10.7554/eLife.01222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simandan T, Sun J, Dix TA. Oxidation of DNA bases, deoxyribonucleosides and homopolymers by peroxyl radicals. Biochem J. 1998;335:233–240. doi: 10.1042/bj3350233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smirnova G, Muzyka N, Oktyabrsky O. Transmembrane glutathione cycling in growing Escherichia coli cells. Microbiol. Res. 2012;167:166–172. doi: 10.1016/j.micres.2011.05.005. [DOI] [PubMed] [Google Scholar]
- Soga T, Baran R, Suematsu M, Ueno Y, Ikeda S, Sakurakawa T, Kakazu Y, Ishikawa T, Robert M, Nishioka T, et al. Differential metabolomics reveals ophthalmic acid as an oxidative stress biomarker indicating hepatic glutathione consumption. J Biol Chem. 2006;281:16768–16776. doi: 10.1074/jbc.M601876200. [DOI] [PubMed] [Google Scholar]
- Stadtman ER, Levine RL. Protein oxidation. Ann. N. Y. Acad. Sci. 2000;899:191–208. doi: 10.1111/j.1749-6632.2000.tb06187.x. [DOI] [PubMed] [Google Scholar]
- Tanaka M, Chock PB, Stadtman ER. Oxidized messenger RNA induces translation errors. Proc. Natl. Acad. Sci. U.S.a. 2007;104:66–71. doi: 10.1073/pnas.0609737104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valenzuela A. The biological significance of malondialdehyde determination in the assessment of tissue oxidative stress. Life Sci. 1991;48:301–309. doi: 10.1016/0024-3205(91)90550-u. [DOI] [PubMed] [Google Scholar]
- Van Acker H, Van Dijck P, Coenye T. Molecular mechanisms of antimicrobial tolerance and resistance in bacterial and fungal biofilms. Trends Microbiol. 2014;22:326–333. doi: 10.1016/j.tim.2014.02.001. [DOI] [PubMed] [Google Scholar]
- Vaubourgeix J, Lin G, Dhar N, Chenouard N, Jiang X, Botella H, Lupoli T, Mariani O, Yang G, Ouerfelli O, et al. Stressed mycobacteria use the chaperone ClpB to sequester irreversibly oxidized proteins asymmetrically within and between cells. Cell Host Microbe. 2015;17:178–190. doi: 10.1016/j.chom.2014.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Zhao X, Malik M, Drlica K. Contribution of reactive oxygen species to pathways of quinolone-mediated bacterial cell death. J Antimicrob Chemother. 2010;65:520–524. doi: 10.1093/jac/dkp486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wardman P. Fluorescent and luminescent probes for measurement of oxidative and nitrosative species in cells and tissues: progress, pitfalls, and prospects. Free Radic. Biol. Med. 2007;43:995–1022. doi: 10.1016/j.freeradbiomed.2007.06.026. [DOI] [PubMed] [Google Scholar]
- Winterbourn CC. The biological chemistry of hydrogen peroxide. Meth. Enzymol. 2013;528:3–25. doi: 10.1016/B978-0-12-405881-1.00001-X. [DOI] [PubMed] [Google Scholar]
- Wright GD. Antibiotics: a new hope. Chem. Biol. 2012;19:3–10. doi: 10.1016/j.chembiol.2011.10.019. [DOI] [PubMed] [Google Scholar]
- Xi H, Schneider BL, Reitzer L. Purine catabolism in Escherichia coli and function of xanthine dehydrogenase in purine salvage. J. Bacteriol. 2000;182:5332–5341. doi: 10.1128/jb.182.19.5332-5341.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeom J, Imlay JA, Park W. Iron homeostasis affects antibiotic-mediated cell death in Pseudomonas species. J. Biol. Chem. 2010;285:22689–22695. doi: 10.1074/jbc.M110.127456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao X, Hong Y, Drlica K. Moving forward with reactive oxygen species involvement in antimicrobial lethality. J Antimicrob Chemother. 2015;70:639–642. doi: 10.1093/jac/dku463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou X, Taghizadeh K, Dedon PC. Chemical and biological evidence for base propenals as the major source of the endogenous M1dG adduct in cellular DNA. J Biol Chem. 2005;280:25377–25382. doi: 10.1074/jbc.M503079200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.