

Abstract
The THAP11 and ZNF143 transcription factors recognize overlapping DNA sequences and are reported to exhibit signs of both competitive and cooperative binding. HCFC1 serves as a scaffold protein, bridging interactions between transcription factors, including THAP11 and ZNF143, and transcriptional coregulators. The exact mechanism of how DNA sequences guide the recruitment of the THAP11/ZNF143/HCFC1 complex to chromatin is still controversial. In this study, we use chromosomally integrated synthetic constructs and clustered regularly interspaced short palindromic repeat (CRISPR)-Cas9-mediated approaches in intact cells to elucidate the role of the DNA sequence in the recruitment of this complex and to establish its biological relevance. We show that the ACTACA submotif, shared by both THAP11 and ZNF143, directs the recruitment of THAP11 and HCFC1 to ZNF143-occupied loci. Importantly, its position, spacing, and orientation relative to the ZNF143 core motif are critical for this action. CRISPR-Cas9-mediated alterations of the ACTACA submotif at endogenous promoters recapitulated results obtained with synthetic constructs and resulted in altered gene transcription and histone modifications at targeted promoters. Our in vivo approaches provide strong evidence for the molecular role of the ACTACA submotif in THAP11, ZNF143, and HCFC1 cooperative recruitment to chromatin and its biological role in target gene expression.
INTRODUCTION
Host cell factor 1 (HCFC1) is an atypical transcriptional coregulator that is translated as a single 2,035-amino-acid peptide and undergoes proteolytic cleavage at the centrally located PRO repeats (1, 2), and the resulting N and C termini noncovalently reassociate via two pairs of self-association sequences (3, 4). The N-terminal fragment of HCFC1 contains a six-Kelch-repeat β-propeller (Kelch domain) and a basic region, both of which facilitate protein-protein association. The Kelch domain recognizes a 4-amino-acid ([E/D]HXY) HCFC1 binding motif (HBM) (5) found in a large number of transcription factors and cofactors (6), including LZIP (5), Set1 (7), E2F4 (8), and the THAP family of proteins (9). Recently reported findings suggest that HCFC1 also associates with ZNF143 via its Kelch domain (10), but the mechanism of this interaction remains unclear, because ZNF143 lacks the HBM. The basic region of HCFC1 mediates associations between a distinct set of proteins, such as GABP (11), Sin3 (7), and Sp1 (12), but can also bind proteins associated with the Kelch domain, as exemplified by E2F4 (8).
HCFC1 is conserved in metazoans and has been implicated in playing critical roles in cell cycle regulation and proliferation (13–18). A single-point mutation in the HCFC1 Kelch domain in the temperature-sensitive hamster cell line tsBN67 causes HCFC1 dissociation from chromatin at a nonpermissive temperature, leading to cell cycle arrest in G1 phase as well as defects in cytokinesis (14, 17, 18). Similar cell cycle aberrations were noted for HeLa cells upon small interfering RNA (siRNA)-mediated knockdown of HCFC1 (13). Initially, the role of HCFC1 in cell proliferation and cell cycle progression had been attributed to its association with members of the E2F family of transcription factors, E2F1 and E2F4, and recruitment to promoters of cell cycle control genes (19, 20). Nonetheless, a recent study in our laboratory revealed that HCFC1 recruitment to the promoters of these genes is largely mediated by the coordinated action of the THAP domain-containing protein 11 (THAP11) and zinc finger protein 143 (ZNF143) transcription factors and is independent of chromatin occupancy by E2F proteins (21).
ZNF143 is a well-studied zinc finger protein that recognizes a TCCCANNNNNCNNNGCG DNA sequence (SBS1; “N” represents a nucleotide with low information content in the motif) (Fig. 1A) (22, 23) and participates in the regulation of RNA polymerase II- and RNA polymerase III-mediated transcription of protein-coding and noncoding mRNA as well as small nuclear RNA (snRNA) (24, 25). Recent reports indicate that ZNF143 may participate in chromatin looping (26, 27), suggesting that THAP11 and HCFC1 may participate in this activity as well. Genome-wide studies revealed that an accessory ACTACA DNA sequence can frequently (∼42%) be found 5′ of the ZNF143-associated SBS1 motif (22, 23, 28), forming the SBS2 motif ACTACANNTCCCANNNNNCNNNGCG (Fig. 1A). In vitro, this sequence does not confer increased DNA binding affinity over the SBS1 motif (28, 29); however, ZNF143 chromatin immunoprecipitation sequencing (ChIP-seq) read tag counts are significantly enriched at sites containing the SBS2 motif (28), suggesting that it provides, directly or indirectly, some stabilizing chromatin association effects in vivo.
FIG 1.
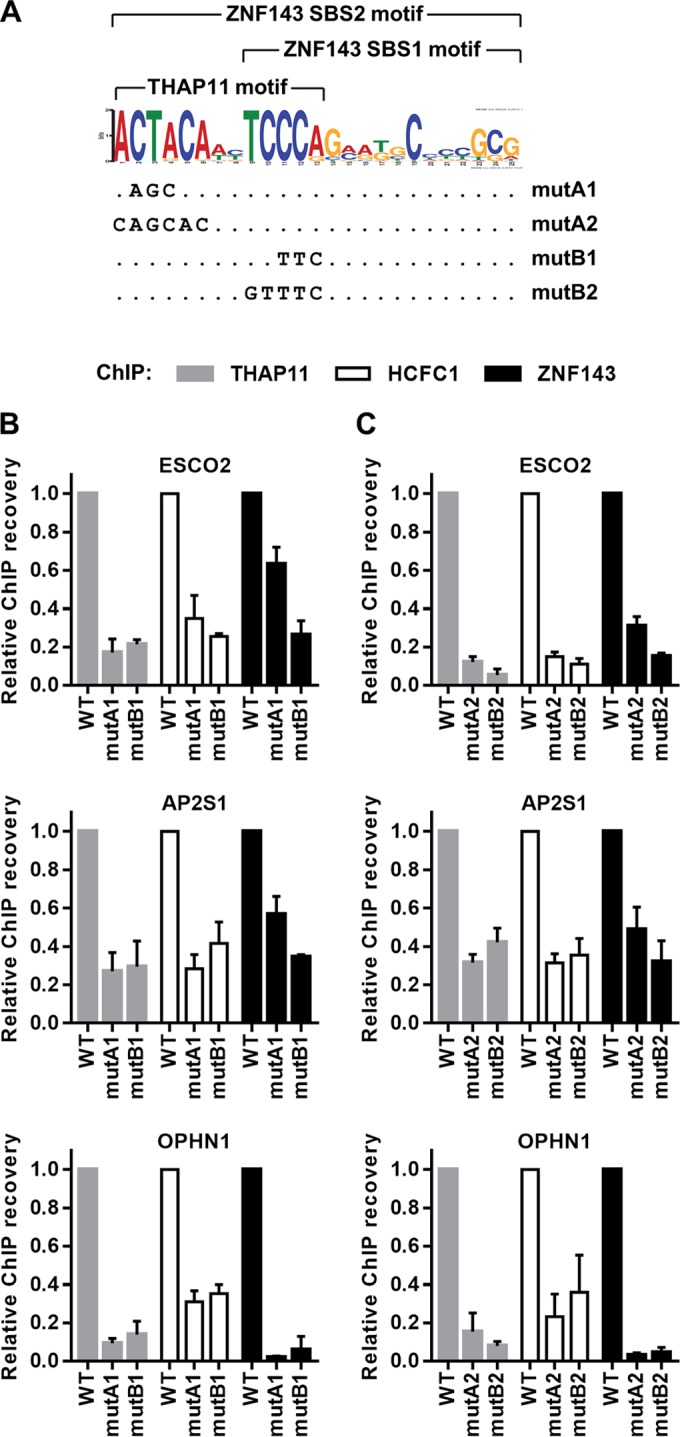
The ACTACA submotif is critical for THAP11, HCFC1, and ZNF143 chromatin occupancy. (A) DNA sequence logo of the ZNF143 SBS2 motif derived from ChIP-seq data (28). The motif is composed of the ZNF143 core motif (SBS1) and the 5′-adjacent ACTACA sequence. THAP11 is associated with the THAP11 motif. mutA1, mutA2, mutB1, and mutB2 utilized in this paper are indicated at the bottom. (B and C) ESCO2, AP2S1, and OPHN1 chromosomally integrated promoter constructs harboring either the 3-nucleotide transversion mutA1 or mutB1 (B) or transversions of the complete ACTACA (mutA2) or TCCCA (mutB2) sequence (C) (or no mutation [WT]) (as described for panel A) were assayed by ChIP using THAP11, HCFC1, and ZNF143 antibodies. Bar graphs indicate means ± standard deviations of data from 3 replicate ChIP experiments.
THAP11 is a member of the THAP domain-containing family of zinc finger transcription factors (30). Early studies of THAP11 in mouse embryonic stem (ES) cells identified its role in embryogenesis and the maintenance of ES cell pluripotency (31). Intriguingly, THAP11 ChIP-seq data from a follow-up study (32) indicated that THAP11 chromatin binding is highly associated with the ACTACANNTCCCA sequence (THAP11 motif; “N” represents a nucleotide with low information content in the motif) (Fig. 1A), which corresponds to the 5′ end of the extended ZNF143-associated motif. Thus far, data from in vitro experiments have suggested that THAP11 and ZNF143 compete for binding to these DNA sequences (28). In contrast to these results, observations in our laboratory suggest that, in vivo, THAP11, ZNF143, and HCFC1 are mutually dependent on chromatin associations at gene promoters containing the SBS2 motif and cooccupy the same genomic loci (21). Thus, the mechanism of THAP11 binding to chromatin and the exact target sequence that its THAP domain recognizes remain uncertain.
To resolve these contrasting in vitro and in vivo results and to determine the molecular underpinnings of how THAP11, ZNF143, and HCFC1 bind chromatin, we performed in vivo experiments using synthetic, chromosomally integrated DNA templates and clustered regularly interspaced short palindromic repeat (CRISPR)-Cas9 approaches to determine THAP11/ZNF143/HCFC1 complex occupancy upon loss-of-function and gain-of-function mutations at native binding sites. Our studies show that the sequence, orientation, and spacing of the target DNA motif strongly dictate cooperative chromatin occupancy by these factors in cells and are key biological determinants of the transcriptional output of target genes.
MATERIALS AND METHODS
Plasmid construction.
Promoters of interest were amplified from HeLa genomic DNA by using primers encompassing 260 to 550 bp of the promoter and 70 to 300 bp of the 5′ noncoding untranslated region (UTR) (see Table S1 in the supplemental material for primer sequences and genome coordinates). The ZNF143/THAP11 DNA binding motif was located on either the 5′ or 3′ end of the amplicon with at least 20 bp of additional genomic DNA before the end of the amplicon. The amplified promoters were cloned into the pGL4.10 vector (catalog number E6651; Promega) by using KpnI and EcoRV sites and sequenced. Mutagenesis was performed by using a QuikChange Lightning site-directed mutagenesis kit (catalog number 210518; Agilent Technologies) and verified by sequencing. The resulting promoters were PCR amplified by using the same primers as those described above but containing different sets of restriction sites and cloned into the pRetroX-Tight-Pur-Luc plasmid (control plasmid from kit 631059; Clontech Laboratories, Inc.) digested with BamHI and BglII to remove the PTight promoter. The resulting plasmids were sequenced prior to transduction into cells.
Cell culture and viral transduction.
SW620 (ATCC CCL-227) and 293T/17 (ATCC CRL-11268) cells were maintained in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum (FBS) at 37°C in 5% CO2. Virus for transductions was generated in 293T/17 cells as described previously (33). SW620 cells were transduced with a 1:10 dilution of viral supernatant into fresh DMEM containing 10% FBS and Polybrene at a final concentration of 8 μg/ml. Transductions were carried out at 37°C overnight. On the following day, medium was changed to fresh DMEM with 10% FBS. At 2 days postransduction, cells were split into fresh medium supplemented with 1 μg/ml puromycin and maintained in this medium for ∼1 week, changing the medium every 3 days.
Chromatin immunoprecipitation.
SW620 cells were harvested at confluence, and chromatin immunoprecipitation (ChIP) was performed essentially as previously described (33). A total of 100 to 300 μg of chromatin (as determined by a bicinchoninic acid [BCA] assay) per immunoprecipitation was used with antibodies to HCFC1 (0.5 μg) (catalog number A301-399A; Bethyl Laboratories, Inc.), THAP11 (1 μg) (catalog number MAB5727; R&D Systems), and ZNF143 (1 μg) (catalog number H00007702-M01; Abnova).
qPCR of chromosomally integrated constructs.
Quantitative PCR (qPCR) was performed by using primers specifically targeting the chromosomally integrated retroviral constructs and spanning or adjacent to the SBS1 or SBS2 site. One primer from each set was designed to target the plasmid backbone, while the other primer was designed against the genomic DNA sequence that was cloned into the retroviral plasmid (see Table S1 in the supplemental material for primer sequences). Power SYBR green PCR master mix (catalog number 4368702; Life Technologies) was used for qPCRs.
CRISPR-Cas9 guide RNA design and validation.
Guide RNAs (gRNAs) were designed by using a CRISPR design tool (see http://crispr.mit.edu/). gRNAs designed to target genomic regions within 15 bp of the SBS1 or SBS2 motif at endogenous AP2S1, OPHN1, DNAJC10, and TFAM promoters and predicted to have the highest specificity were selected and cloned into the pSpCas9(BB)-2A-GFP vector (catalog number 48138; Addgene) (34). The constructs were transfected into 293T/17 cells by using Lipofectamine 2000 reagent (catalog number 11668019; Invitrogen). Cells were harvested at 48 h posttransfection, and genomic DNA was extracted by using a Qiagen DNeasy blood and tissue kit (catalog number 69504; Qiagen). Primers surrounding the sequence targeted by the gRNAs and spanning ∼400 bases were used for PCR amplification from the genomic DNA. The PCR product was denatured at 95°C for 2 min, reannealed by slowly decreasing the temperature in a thermocycler, and subjected to T7 endonuclease I (T7EI) (catalog number M0302S; NEB) digestion for 30 min at 37°C. The digestion product was run out on an agarose gel. The efficiency of gRNA-mediated CRISPR-Cas9 genomic DNA cleavage was estimated by the amount of digestion by the T7EI endonuclease (protocol adapted from reference 35). gRNAs showing the best cleavage efficiency for each promoter were chosen for further experiments (see Table S1 in the supplemental material).
CRISPR-Cas9-mediated genome editing in SW620 cells.
SW620 cells were transfected with 2 μg of the pSpCas9(BB)-2A-GFP gRNA constructs and 40 pmol of the corresponding 150-nucleotide (nt)-long single-stranded oligonucleotide bearing the ACTACA mutation and 60- to 84-nt-long homology arms to the adjacent genomic sequence (see Table S1 in the supplemental material). Transfection of the gRNA construct targeting the OPHN1 locus was performed by using the Neon electroporation system (Invitrogen) with the following parameters: 5 × 106 cells electroporated with a single pulse set to 1,550 V and 20-ms width. Transfection of gRNAs targeting the AP2S1, DNAJC10, and TFAM promoters was performed by using the Nucleofector 2b electroporation system (Lonza) with kit T and program O-017 according to the manufacturer's recommendations. Following electroporation, cells were grown for 36 h under normal conditions and selected by fluorescence-activated cell sorter (FACS) analysis for green fluorescent protein (GFP)-positive cells. Single cells were sorted into 96-well plates, grown to confluence, and duplicated by splitting. One set of plates was lysed in ThermoPol buffer [20 mM Tris-HCl, 10 mM (NH4)2SO4, 10 mM KCl, 2 mM MgSO4, 0.1% Triton X-100] supplemented with 50 μg/ml proteinase K and 1.7 μM SDS at 50°C for 1 h (see http://research.mssm.edu/soriano/lab/ES_screening_by_PCR.html). For OPHN1 mutants, 1 μl of the lysate was transferred into a 25-μl Taq polymerase PCR mixture containing primers specific for the mutated OPHN1 promoter sequence. Positive PCR amplifications indicate the presence of homology-directed repair (HDR) and were used as screening selection criteria. For AP2S1, DNAJC10, and TFAM mutants, the lysate was diluted 6-fold in water, and 1 μl of the resulting solution was used in SYBR qPCR mixtures with primers specific for the mutated sequence. A melting curve analysis was used to select mutation-positive clones.
SW620 mutant clone genotyping.
Genomic DNA from SW620 cells positive for the ACTACA mutation was amplified by using the same primers as those used for the T7EI assay. The resulting PCR product was submitted for sequencing. For clones where direct sequencing of the PCR product yielded multiple sequencing peaks per base (OPHN1 and TFAM) (see Fig. 5A and 8A, respectively), the PCR product was TA cloned by using the pGEM-T Easy vector kit (catalog number A1360; Promega). Several pGEM-T clones were picked for each mutation-positive SW620 clone and submitted for sequencing. In each case, sequences for two different alleles in each of the heterozygous SW620 clones were recovered (see Fig. 5B and 8B). For further experiments, we selected two clones homozygous for the targeted ACTACA mutation in each AP2S1 and DNAJC10 promoter (see Fig. 5C and 8C). Since the initial transfected SW620 population was not clonal, we used morphological differences between clones to ensure that the two selected clones were not derived from the same transfected parent cell. For TFAM mutants, we were not able to obtain homozygous mutations. We selected two distinct clones harboring the desired ACTACA mutation on one allele and an indel (insertion/deletion) upstream of the ZNF143 binding site on the other allele (see Fig. 8C). For the OPHN1 clone, we were able to obtain only one heterozygous clone containing the desired ACTACA mutation on one allele (mutA) and a TCCCA deletion on the other allele (delB) (see Fig. 5C).
FIG 5.

CRISPR-Cas9-mediated mutagenesis of AP2S1 and OPHN1 promoters in SW620 cells. (A and B) Chromatograms obtained by Sanger sequencing of AP2S1 and OPHN1 promoter PCR products (A) and TA-cloned OPHN1 promoter fragments representing individual alleles (B). (C) Genotypes of OPHN1 and AP2S1 clones selected for further experiments. (D) Specificities of primer sets targeting either OPHN1 delB or mutA alleles were verified as described in Materials and Methods. Results for qPCR amplification of delB allele- or mutA allele-containing pGEM-T plasmids are expressed as fold changes over pGEM-T plasmid backbone amplification. Primer sets are indicated on the x axis. PCR templates are indicated by different bar colors. AP2S1 mutA1 and mutA2 represent two independent clonal isolates. n.d., not determined.
FIG 8.

CRISPR-Cas9-mediated mutagenesis of DNAJC10 and TFAM promoters in SW620 cells. (A and B) Chromatograms obtained by Sanger sequencing of DNAJC10 and TFAM promoter PCR products (A) and TA-cloned TFAM (insA1 clone) promoter fragments representing individual alleles (B). (C) Genotypes of DNAJC10 and TFAM clones selected for further experiments. insA1 and insA2 represent two independent clonal isolates. HDR indicates alleles mutated by homology-directed repair. Indel indicates alleles mutated by nonhomologous end joining and containing random insertions/deletions.
Allele-specific ChIP-qPCR.
Cell culture, harvesting, and immunoprecipitation were performed as described above. qPCR was performed by using allele-specific primer sets. The primer sets targeting the wild-type (WT) or mutant alleles (see Table S1 in the supplemental material) were designed with 4 to 6 bases at the 3′ end of the forward primer overlapping the ACTACA motif to provide specificity. The mutant and WT primer sets share the same reverse primer to reduce variations in qPCR amplification. Primer set specificities were verified by qPCR amplification of synthetic sequences containing the desired mutations (Fig. 5D).
Gene expression quantification.
mRNA from SW620 cells was purified by using a Quick-RNA MiniPrep kit (catalog number R1055; Zymo Research) according to the manufacturer's instructions. Five hundred nanograms of total mRNA was reverse transcribed in a 10-μl qScript cDNA SuperMix (catalog number 95048; Quanta Biosciences) reaction mixture according to the manufacturer's instructions. qPCR was performed with the primers indicated in Table S1 in the supplemental material. The beta-2-microglobulin gene was used as a housekeeping control for normalization.
RESULTS
The ACTACA submotif is critical for THAP11, HCFC1, and ZNF143 chromatin occupancy.
In order to address the apparent inconsistency between in vitro and in vivo observations (21, 28, 36) and to elucidate the role of the ACTACA submotif found adjacent to the ZNF143 core motif, we established an in vivo system that allowed us to analyze the binding of the THAP11/ZNF143/HCFC1 complex on a chromosomally integrated DNA template, the sequence of which we could efficiently manipulate. We constructed retroviral plasmids that incorporate 260 to 550 nucleotides of native ZNF143 target gene promoters, including the transcriptional start site and ∼70 to 300 nucleotides of the 5′ UTR. The constructs were delivered retrovirally into SW620 cells, which we have used in previous studies (33), and stably integrated into chromatin. As representatives of gene promoters harboring the SBS2 motif, we selected the AP2S1, ESCO2, and OPHN1 genes. These promoters contain a single SBS2 motif; are bound by THAP11, ZNF143, and HCFC1 at readily detectable levels; and were previously studied in our laboratory (21, 33) and other laboratories (29).
Previous in vitro experiments demonstrated that transversion of 3 nucleotides in the TCCCA but not the ACTACA sequences of the SBS2 motif (mutB1 and mutA1, respectively) (Fig. 1A) in the ESCO2 promoter is sufficient to abrogate the ZNF143-DNA interaction (29). However, mutation of the ACTACA sequence in the ESCO2 promoter has a negative impact on luciferase expression (29) albeit not as significant as mutation of the TCCCA sequence. Thus, to evaluate the relative contributions of these submotifs in vivo, we mutated AP2S1, ESCO2, and OPHN1 promoters in the retroviral vector constructs by introducing 3 nucleotide transversions in the ACTACA and TCCCA sequences (mutA1 and mutB1, respectively) (Fig. 1A). The mutated and wild-type constructs were independently transduced into SW620 cells, and THAP11, ZNF143, and HCFC1 chromatin occupancy was analyzed by using ChIP-qPCR. As expected, the mutB1 construct resulted in a dramatic loss of ZNF143 binding at the integrated promoter (Fig. 1B). This observation is consistent with in vitro electrophoretic mobility shift assays (EMSAs) indicating that mutations in the TCCCA motif interfere with ZNF143 DNA binding activity. Additionally, we observed significant depletion of THAP11 and HCFC1 chromatin occupancy, consistent with our previous observation of THAP11/HCFC1 subcomplex dependency on ZNF143 (21). An AGC mutation in the ACTACA submotif (mutA1) had a similar effect on THAP11 and HCFC1 occupancy; however, ZNF143 binding was affected less severely at the ESCO2 and AP2S1 promoters than with the TTC mutation (mutB1) (Fig. 1B). Mutations at the OPHN1 promoter, however, showed overall more dramatic reductions in ZNF143 binding and were similar for both AGC and TTC mutations.
In order to determine whether the ACTACA and TCCCA sequences are critical, we proceeded to mutate these submotifs by introducing transversions of all significant nucleotides (mutA2 and mutB2) (Fig. 1A). Interestingly, mutation of all residues had a minimal additional effect on THAP11, ZNF143, and HCFC1 binding (Fig. 1C) compared to mutA1 and mutB1 (Fig. 1B). Together, these results indicate that the TCCCA submotif is critical for chromatin occupancy of all three proteins, while the ACTACA and TCCCA submotifs are equally important for THAP11 and HCFC1 binding. Unlike in vitro EMSA data, our cell-based results suggest that the ACTACA motif, in some contexts, also plays important roles in ZNF143 recruitment to chromatin.
The ACTACA submotif is sufficient to recruit the THAP11/HCFC1 subcomplex to ZNF143 SBS1 sites.
While genome-wide THAP11 chromatin occupancy is associated strictly with the ACTACAnnTCCCA sequence (10, 32), ZNF143 ChIP-seq peaks have an even distribution of SBS1 and SBS2 motifs (28). Thus, we wondered whether the introduction of the ACTACA sequence at a position adjacent to an existing and ZNF143-bound SBS1 sequence, effectively transforming it into an SBS2 motif, could influence THAP11, HCFC1, and ZNF143 chromatin occupancy. To this end, we cloned DNAJC10, RNU6-2 (previously referred to as U6), and TFAM gene promoters into the retroviral vector. At the endogenous loci of these genes, ZNF143 occupancy is readily detectable and coincides with the SBS1 motif, but there is relatively little detectable THAP11 or HCFC1 binding (Fig. 2). The retroviral constructs were mutated to insert the ACTACA sequence 2 bases 5′ of the SBS1 motif (insA) (Fig. 3A), as found at endogenous THAP11/ZNF143/HCFC1-occupied promoters, such as AP2S1, ESCO2, and OPHN1. Insertion of the ACTACA sequence promoted a dramatic increase in THAP11 and HCFC1 binding at all 3 promoters (Fig. 3B to D). Conversely, ZNF143 occupancy was not significantly affected at the DNAJC10 and RNU6-2 promoters (Fig. 3B and D) but was increased >2-fold at the TFAM promoter (Fig. 3C). Importantly, the addition of the ACTACA sequence and increased recruitment of the THAP11/HCFC1 subcomplex did not negatively impact ZNF143 binding. In order to determine if the ACTACA motif alone (in the absence of the adjacent TCCCA motif) could recruit THAP11 and HCFC1 to chromatin, we proceeded to mutate the SBS1 motif in the DNAJC10, RNU6-2, and TFAM promoters containing the additional ACTACA sequence (insA/mutB). As expected, transversion of the TCCCA sequence completely reverted the THAP11 and HCFC1 gain in chromatin occupancy (Fig. 3B to D). Additionally, this mutation eliminated ZNF143 binding at all three promoters. Taken together, our data suggest that the ACTACA motif is sufficient to recruit THAP11 and HCFC1 to ZNF143-occupied promoters and, in some cases (e.g., TFAM gene), plays a supporting role in ZNF143 binding.
FIG 2.
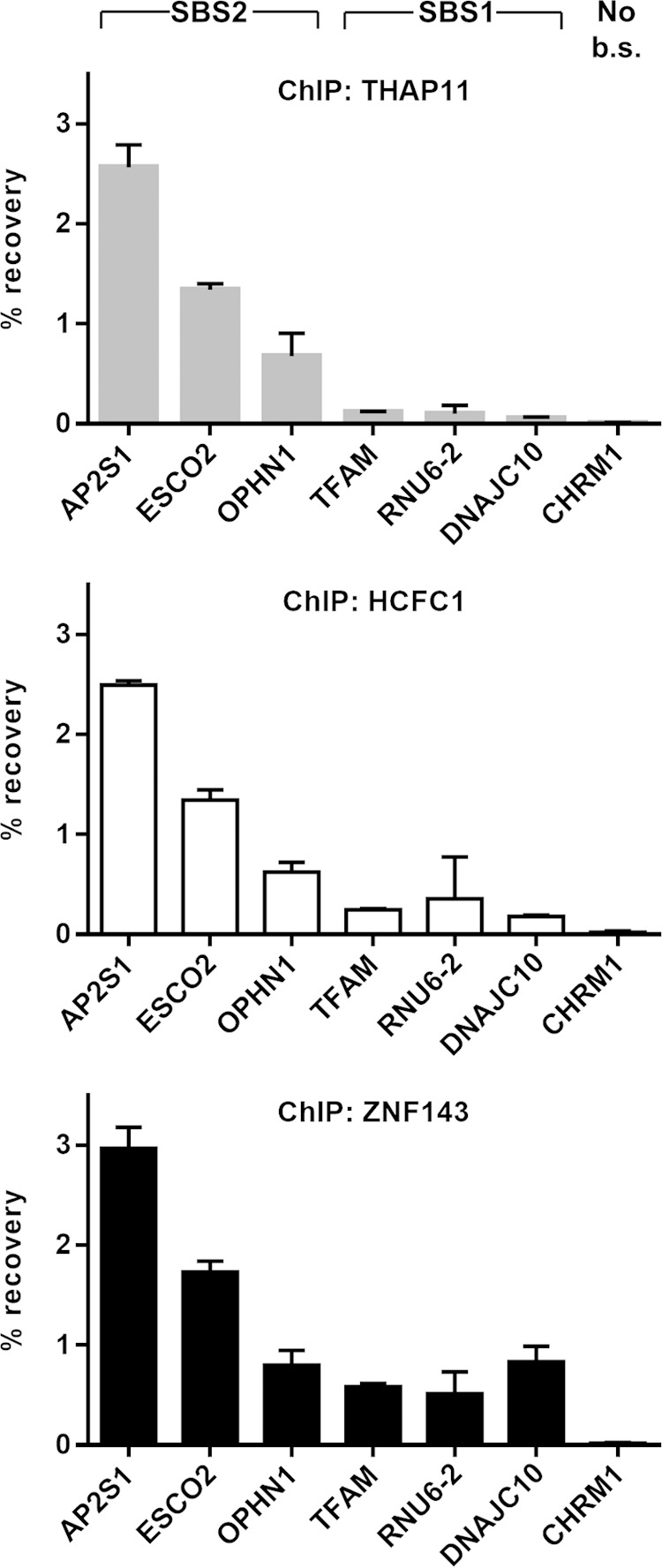
THAP11 and HCFC1 occupancy at SBS1-only-containing promoters is relatively low. THAP11, HCFC1, and ZNF143 proteins were chromatin immunoprecipitated from SW620, and their presence at SBS2-containing (AP2S1, ESCO2, and OPHN1) and SBS1-containing (TFAM, RNU6-2, and DNAJC10) promoters was assayed by qPCR. The CHRM1 promoter, which does not contain the SBS1 or SBS2 binding site (No b.s.), was used as a negative control. Bar graphs indicate means ± standard deviations of data from 3 replicate ChIP experiments.
FIG 3.

The ACTACA submotif is sufficient to recruit the THAP11/HCFC1 subcomplex to ZNF143 SBS1 sites. (A) Diagram of mutations introduced for this set of experiments. The ACTACA sequence was inserted 2 bases 5′ of existing and ZNF143-bound SBS1 motifs (insA). The TCCCA sequence in the resulting construct was then mutated to destroy the ZNF143 binding site (insA/mutB). (B to D) Mutated and chromosomally integrated DNAJC10 (B), TFAM (C), and RNU6-2 (D) promoters were assayed by ChIP using THAP11, HCFC1, and ZNF143 antibodies. Bar graphs indicate means ± standard deviations of data from 3 replicate ChIP experiments.
Two-nucleotide spacing, position, and orientation of the ACTACA sequence are critical for THAP11/HCFC1 recruitment.
Given the significant impact that the ACTACA sequence has on THAP11/HCFC1 recruitment, we wondered whether the conserved spacing between the ACTACA submotif and the SBS1 motif had a strict requirement of 2 bases or if it could be altered with a minimal effect on THAP11/HCFC1 binding. To this end, we altered the location at which the ACTACA submotif was inserted into the DNAJC10 promoter (Fig. 4A, top). Two-nucleotide spacing corresponds to the naturally occurring configuration of the SBS2 motif and, as demonstrated in Fig. 4A, results in de novo THAP11 and HCFC1 recruitment. Altering the spacing to 1, 3, 7, or 12 nucleotides completely abolished the de novo recruitment of THAP11 and HCFC1 to the ZNF143-bound DNAJC10 promoter. ZNF143 binding was not negatively or positively affected by the variable spacing compared to the wild-type promoter.
FIG 4.
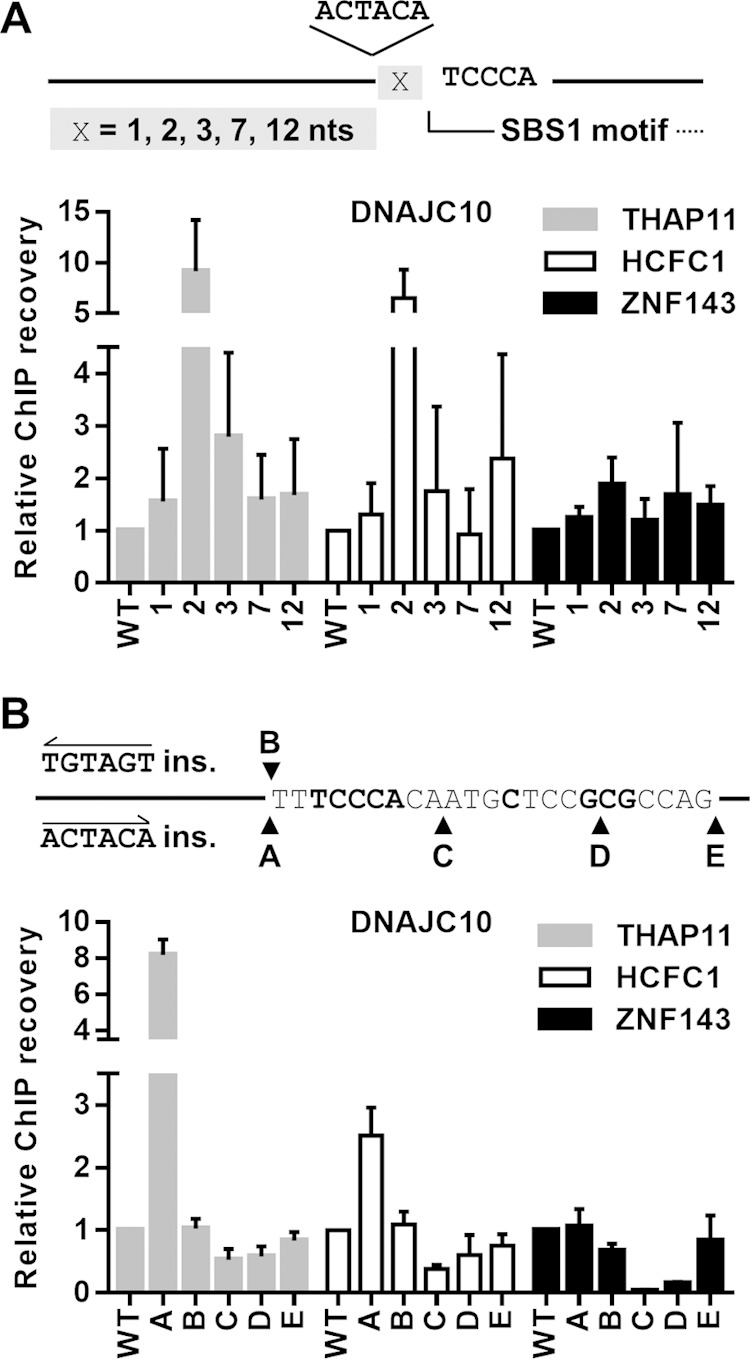
Two-nucleotide spacing, position, and orientation of the ACTACA sequence are critical for THAP11/HCFC1 recruitment. (A) Diagram depicting the location of the ACTACA sequence insertion relative to the SBS1 motif in the DNAJC10 promoter. X represents a single nucleotide. The ACTACA sequence was inserted 1, 2, 3, 7, or 12 nucleotides 5′ of the SBS1 motif. x-axis labels for bar graphs correspond to the numbers of nucleotides between the SBS1 motif and the ACTACA sequence. (B) Diagram depicting the locations and sequences that were inserted into the wild-type DNACJ10 retroviral construct. The labels shown for each insertion also correspond to the x-axis labels for bar graphs. The TGTAGT sequence inserted at the “B” position is the reverse complement of the ACTACA sequence and corresponds to the everted arrangement of the ACTACA sequence and SBS1 motif. Mutated and chromosomally integrated DNAJC10 promoters were assayed by ChIP using THAP11, HCFC1, and ZNF143 antibodies. Bar graphs indicate means ± standard deviations of data from 3 replicate ChIP experiments.
Similarly, we wanted to determine if the orientation of the ACTACA sequence in relation to the SBS1 motif had any effect on THAP11/HCFC1 recruitment. Again, we used the DNAJC10 promoter and inserted the ACTACA motif in the direct (as found in native SBS2 motifs) (A samples) or reverse (B samples) orientation (Fig. 4B) relative to the SBS1 motif. Unlike the direct orientation, insertion of the ACTACA sequence in the reverse orientation failed to recruit THAP11 or HCFC1 to chromatin (compare WT, A, and B samples). Additionally, we assayed whether positioning of the ACTACA sequence on the opposite side of the SBS1 motif would have any effect on chromatin occupancy. We inserted the ACTACA sequence at various distances 3′ of the core TCCCA sequence (C, D, and E samples) (Fig. 4B). Insertion of the ACTACA sequence at any of these positions, including the area 3′ of the full SBS1 motif (E samples), failed to recruit THAP11 or HCFC1 to chromatin. However, we observed a dramatic loss of ZNF143 occupancy when the ACTACA sequence was inserted before the 3′-GCG sequence (samples C and D), disrupting the natural ZNF143 binding site. Collectively, our results provide new direct evidence that the relative position, spacing, and orientation of the ACTACA sequence in relation to the SBS1 motif play critical roles in THAP11 and HCFC1 recruitment to chromatin.
ACTACA submotif mutation affects recruitment of the THAP11/ZNF143/HCFC1 complex to endogenous gene promoters and alters their expression.
In order to provide biological relevance to the mechanistic studies of theTHAP11 and ZNF143 DNA motifs and to determine if our observations of ectopic loci are also valid for native gene promoters, we utilized a CRISPR-Cas9 endonuclease approach coupled with homology-directed repair (HDR) to introduce transversion mutations into the ACTACA sequence of the endogenous THAP11/ZNF143/HCFC1-bound SBS2 motifs in the AP2S1 and OPHN1 promoters (as done previously for the retroviral constructs) (Fig. 1C) in SW620 cells. We isolated two independent clones homozygous for the ACTACA transversion at the endogenous AP2S1 promoter (mutA1 and mutA2) (Fig. 5A and C) (see Materials and Methods) and one clone heterozygous for the ACTACA transversion at the endogenous OPHN1 promoter (mutA) (Fig. 5A to C) (see Materials and Methods). In the OPHN1 mutA clone, we identified two alleles containing either a 5-nucleotide deletion of the TCCCA submotif or a transversion of the ACTACA sequence (Fig. 5C). The genotypes of all clones were confirmed by Sanger sequencing and are shown in Fig. 5A and B. The distinct nature of the mutations in the OPHN1 mutA clone allowed us to design allele-specific primers (Fig. 5D) and perform ChIP experiments to independently investigate the differences in THAP11/ZNF143/HCFC1 recruitment at both alleles. For consistency, similar primers, targeting either the WT or mutated sequences, were designed for the AP2S1 promoter.
We performed ChIP for THAP11, ZNF143, and HCFC1 in wild-type and mutant SW620 cells. Immunoprecipitated DNA was amplified with primers specifically targeting wild-type or mutated alleles. In agreement with our chromosomally integrated promoter constructs, we observed a dramatic decrease in chromatin occupancy of all three proteins at the ACTACA transversion as well as the TCCCA deletion mutant OPHN1 promoters, compared to wild-type cells (Fig. 6A), indicating that both ACTACA and TCCCA sequences are indeed critical for THAP11/ZNF143/HCFC1 complex recruitment to the endogenous OPHN1 promoter. We show that the lack of chromatin occupancy by these factors is specific for the mutated OPHN1 promoter, since the complex remains bound at the nontargeted THAP11/ZNF143/HCFC1 target gene promoters RBL1, UBXN8, and ZNF32 (Fig. 6A). Interestingly, while we observed a ∼60% loss of THAP11/HCFC1/ZNF143 at the mutA2 retrovirally integrated AP2S1 promoter (Fig. 1C), depletion of these factors at the endogenous promoter upon ACTACA mutation was weaker for all three factors (Fig. 6B) (see Discussion).
FIG 6.

ACTACA submotif mutation affects recruitment of the THAP11/ZNF143/HCFC1 complex to endogenous gene promoters. (A and B) THAP11, HCFC1, and ZNF143 ChIPs were performed on wild-type (WT) SW620 cells or OPHN1 (A) or AP2S1 (B) mutant clonal cells. (A) For the OPHN1 promoter, qPCR was performed in an allele-specific manner using mutA allele-specific and delB allele-specific primer sets. (B) mutA1 and mutA2 represent two independent AP2S1-mutant clonal isolates. Bar graphs indicate means ± standard deviations of data from 3 replicate ChIP experiments.
To integrate chromatin occupancy results with biological output data, we checked OPHN1 and AP2S1 mRNA expression levels. OPHN1 expression was virtually undetectable in mutA cells (>200-fold decrease compared to wild-type cells), while the expression of other THAP11/ZNF143/HCFC1 target genes, like RBL1, UBXN8, and ZNF32, remained unchanged (Fig. 7A). It is worth noting that both OPHN1 alleles, found on the duplicated X chromosome in male SW620 cells (37–39), are expected to be actively transcribed, due to the lack of X chromosome inactivation in male cancers (40); thus, the dramatic decrease in total mRNA levels is most likely reflective of changes in transcriptional activity from both alleles. In accordance, RNA polymerase II (RNAPII) levels and histone modifications associated with active gene transcription (trimethylated histone H3 lysine 4 [H3K4me3] and acetylated histone H3 lysine 9 [H3K9ac]) were drastically decreased in mutant cells at both alleles (Fig. 7B). Additionally, we observed increases in total histone H3 occupancy and trimethylated histone H3 lysine 9 (H3K9me3) marks in mutant cells (Fig. 7B), consistent with a more condensed, silent chromatin state. Contrary to OPHN1 expression levels and despite relatively small decreases in THAP11/ZNF143/HCFC1 recruitment to the AP2S1 mutant promoter, mRNA expression was enhanced in AP2S1 mutant cells (Fig. 7C). Expression levels of the nontargeted genes RBL1, UBXN8, and ZNF32 also did not change in these cells, indicating the specific nature of AP2S1 upregulation. Taken together, these in vivo observations validate our integrated retroviral construct assays and provide evidence for the important role that the ACTACA and TCCCA motifs play in direct regulation of target gene transcription.
FIG 7.

ACTACA and TCCCA submotif mutations affect endogenous gene expression and epigenomic states. (A) mRNA levels of OPHN1 and other THAP11/ZNF143/HCFC1 target genes in wild-type (WT) or mutant (mut) clonal cells. (B) ChIP was performed on WT or mutant clonal cells by using RNAPII, total histone H3 (H3), H3K4me3, H3K9ac, and H3K9me3 antibodies. The occupancy of each protein was detected by using allele-specific qPCR (delB and mutA). (C) mRNA levels of AP2S1 and other THAP11/ZNF143/HCFC1 target genes in WT or mutant clonal cells. Bar graphs indicate means ± standard deviations of data from 3 replicate experiments.
The ACTACA submotif is sufficient to direct de novo THAP11/HCFC1 recruitment to endogenous ZNF143-bound SBS1-containing promoters.
We next wanted to test whether the ACTACA motif can recruit THAP11/HCFC1 to ZNF143-bound and SBS1-containing promoters at endogenous loci. We used the CRISPR-Cas9 system to introduce the ACTACA motif at the DNAJC10 and TFAM promoters. We obtained 2 homozygous SW620 clones (insA1 and insA2) (Fig. 8A and C) where the ACTACA sequence was introduced 2 nucleotides upstream of the SBS1 motif in the DNAJC10 promoter. For the TFAM promoter mutants, we isolated 2 heterozygous clones (insA1 and insA2) (Fig. 8), where one allele of the TFAM promoter in each clone was mutated to contain the ACTACA sequence 2 nucleotides upstream of the SBS1 motif and the other allele in each clone contained an indel at the site of CRISPR-Cas9 cleavage (GT deletion and G insertion in the insA1 and insA2 mutants, respectively). We designed allele-specific primers targeting only the ACTACA-containing alleles for all mutants and performed ChIP using THAP11, ZNF143, and HCFC1 antibodies. As with retrovirally integrated promoters, we observed a dramatic increase in THAP11 recruitment to ACTACA-containing promoters compared to WT cells (Fig. 9, left). HCFC1 recruitment to these promoters was also markedly enhanced (Fig. 9, middle). In agreement with the data for the retroviral constructs, ZNF143 recruitment to the TFAM promoter was potentiated by the addition of the ACTACA sequence (Fig. 9B, right); however, unlike our previous observations (Fig. 3B), we also saw an increase in ZNF143 recruitment to the endogenous DNAJC10 promoter containing the ACTACA insertion (Fig. 9A, right). Thus, the ACTACA sequence serves as a potent mediator of THAP11/ZNF143/HCFC1 binding to chromatin.
FIG 9.

The ACTACA submotif is sufficient to recruit THAP11/HCFC1 to endogenous ZNF143-bound promoters. THAP11, HCFC1, and ZNF143 ChIPs were performed on WT SW620 cells or DNAJC10 (A) or TFAM (B) mutant clonal cells. Bar graphs indicate means ± standard deviations of data from 3 replicate ChIP experiments.
DISCUSSION
The role of the ACTACA motif, which frequently appears in an position adjacent to the core ZNF143 binding sequence, has remained unclear for nearly a decade. It is associated with increased ZNF143 chromatin occupancy (28) but does not demonstrate any affinity for ZNF143 in in vitro assays (28, 29). In this study, we utilized ectopic chromosomally integrated promoter constructs and CRISPR-Cas9 approaches targeting endogenous loci to analyze the role of this sequence. We show that the ACTACA submotif, in some contexts, plays a role in ZNF143 recruitment (e.g., OPHN1). Furthermore, we demonstrate that the ACTACA submotif is necessary and sufficient to direct THAP11 and HCFC1 recruitment to existing ZNF143 binding sites containing only the core ZNF143 DNA sequence. Together, the data in this study provide new evidence for the significance of the ACTACA submotif and cooperative binding of THAP11, ZNF143, and HCFC1.
Previous in vitro experiments using EMSAs indicated that the ACTACA extension of the ZNF143 core motif is not required for ZNF143 binding (29), nor does it enhance ZNF143 affinity for DNA (28). Furthermore, EMSAs using THAP11 and ZNF143 DNA binding domains did not show collaborative DNA binding, leading to the conclusion that THAP11 and ZNF143 binding is competitive (28). The dispensability of the ACTACA submotif for ZNF143 binding is not surprising, since only ∼42% of ZNF143 peaks contain this sequence; however, these ChIP-seq peaks show a significant increase in ZNF143 occupancy (28). Additionally, previous work in our laboratory demonstrated that THAP11, ZNF143, and HCFC1 occupancies at SBS2-containing promoters are mutually dependent in intact cells (21). Thus, there appears to be a distinction between ZNF143 DNA binding properties in vitro and in vivo. A similar discrepancy between in vitro and in vivo DNA associations was recently reported for the MYC transcription factor (41). A mutation in the MYC protein inhibiting its interaction with WDR5 was sufficient to abrogate MYC recruitment to chromatin but showed no effect on its ability to bind DNA in EMSAs. There are a number of factors that could account for the disparity of in vitro and in vivo ZNF143 DNA binding. EMSA is performed on naked DNA probes devoid of the natural chromatin environment. Additionally, by virtue of their in vitro nature, EMSAs may not include all the cofactors necessary to observe native DNA binding behavior. Our ChIP assays on chromosomally integrated promoters attempt to bridge this apparent gap and probe the role of ACTACA in a more biologically accurate environment.
Unlike EMSAs, our results with prototypical target genes indicate that the ACTACA motif indeed plays an important role in ZNF143 recruitment to chromatin. Mutation of this motif at some of the SBS2-containing promoters, such as OPHN1, causes a loss of ZNF143; however, other promoters, like AP2S1 and ESCO2, demonstrate only partial dependence on this motif. Furthermore, the addition of the ACTACA motif at the TFAM (both retroviral constructs and endogenous loci) and DNAJC10 (endogenous loci only) promoters resulted in >2-fold-higher ZNF143 occupancy. This is consistent with the binding-enhancing role of the ACTACA motif observed in ChIP-seq experiments (28). Nonetheless, not all of the promoters demonstrate this behavior. Introduction of the ACTACA sequence at the RNU6-2 promoters showed no significant increase in ZNF143 occupancy. It is possible that this promoter already contains other sequences or cofactors that provide maximal ZNF143 binding, thus masking any effect from the ACTACA submotif. Consistent with this hypothesis, the ZNF143 binding site at the RNU6-2 promoter was previously shown to contain an Oct1 binding sequence and to be cooperatively bound by Oct1 and ZNF143 (42). The OPHN1 promoter shows similar levels of depletion for mutations in both the ACTACA and TCCCA submotifs, which is in agreement with the dramatic loss of ZNF143 chromatin occupancy at this promoter upon THAP11 or HCFC1 knockdown (J. B. Parker, unpublished data).
In addition to the ZNF143 occupancy-enhancing role of the ACTACA submotif, this sequence is essential for THAP11 and HCFC1 recruitment. Introduction of the ACTACA sequence into existing SBS1 motifs, effectively converting them to SBS2 sequences, resulted in de novo recruitment of THAP11 and HCFC1. This was the case for not only RNA polymerase II-transcribed genes (DNAJC10 and TFAM genes) but also RNU6-2, which is transcribed by RNA polymerase III. Importantly, increased THAP11/HCFC1 recruitment did not result in decreased ZNF143 binding; conversely, ZNF143 binding was increased at the TFAM and DNAJC10 promoters. Thus, unlike the competitive binding of THAP11 and ZNF143 demonstrated in EMSAs (28), the data presented here, along with our previously reported observations (21), provide strong evidence that THAP11 and HCFC1 bind cooperatively with ZNF143 in vivo in an ACTACA sequence-dependent manner. Such cooperative binding may account for the higher number of ZNF143 ChIP-seq read counts at SBS2-containing peaks than at SBS1-only peaks (28). In support of our findings, the unusual structure of the THAP11 DNA-interacting L4 loop, which is shorter than those of other members of the THAP family of proteins (43, 44), and data from isothermal titration calorimetry (ITC) experiments (36) suggest that THAP11 may not have a strong, specific association with DNA and could require partnering with other transcription factors for stable chromatin binding.
The ACTACA sequence has been detected in bioinformatic (23) and ChIP-seq (28, 45) analyses as an extension of the core ZNF143 binding motif and not as a separate, associated motif, suggesting that the ACTACA sequence does not tend to occur at various distances from the SBS1 motif in vivo and, thus, may have a strict spacing requirement. By manipulating the position, spacing, and orientation of the ACTACA sequence and the SBS1 motif, we provide direct evidence that all of these factors are critical for THAP11 and HCFC1 binding. Positioning of the ACTACA sequence anywhere other than 2 nucleotides 5′ of the SBS1 motif led to a failure in the recruitment of THAP11 and HCFC1 to chromatin. We hypothesize that ZNF143 makes direct protein-DNA contacts with the SBS1 motif, while THAP11 and HCFC1 are recruited by ZNF143 and establish additional protein-DNA interactions with the ACTACA submotif. Due to the THAP11 protein's atypical THAP domain and its predicted weaker affinity for DNA, THAP11 chromatin occupancy may be greatly dependent on ZNF143. In agreement with this hypothesis, we observe that mutation of either the ACTACA submotif, which we predict to directly impact THAP11 binding, or the TCCCA sequence in the SBS1 motif, which would affect ZNF143 chromatin occupancy, has the same detrimental effect on THAP11 and HCFC1 recruitment.
Finally, to investigate the biological role of the ACTACA submotif in vivo and to determine the relevance of our chromosomally integrated promoter model, we mutated the ACTACA sequence in the SBS2 motifs at the endogenous OPHN1 and AP2S1 promoters using a CRISPR-Cas9 approach. Importantly, we introduced a 6-nucleotide mutation, without introducing any additional exogenous sequences, into the surrounding genomic context and were able to assay the direct effects of our mutation in a native environment. Similarly to our chromosomally integrated promoter constructs, we observed a dramatic depletion of THAP11, HCFC1, and ZNF143 for both alleles in OPHN1 mutant cells. Importantly, both of these mutations resulted in a near-complete loss of OPHN1 expression along with a loss of histone marks associated with active transcription. Interestingly, we did not observe a potent loss of the THAP11/ZNF143/HCFC1 complex at the AP2S1 promoter upon mutation of the ACTACA motif. Closer examination of the locus revealed a potential cryptic SBS2 binding site ∼100 bp away from our target sequence, which contains 2-nucleotide mismatches in the ACTACA submotif but may be sufficient to maintain THAP11/ZNF143/HCFC1 chromatin occupancy at this locus. It is also possible that the ACTACA requirement for THAP11/ZNF143/HCFC1 complex binding is gene specific. This hypothesis would be consistent with the dispensability of the ACTACA motif for ZNF143 binding on a genome-wide scale, based on previously reported ChIP-seq observations (28), since this motif is present at only half of the ZNF143-bound sites but is critical for ZNF143 binding at the OPHN1 promoter. It remains unclear why mutation of the ACTACA motif on the retrovirally integrated constructs yielded a more potent loss of THAP11/ZNF143/HCFC1, since the cryptic SBS2 site was also present in this sequence. We believe that this observation further supports the need to investigate transcription factor binding not only within the cellular environment but also in the correct genomic context. Nonetheless, even with a marginal loss of THAP11/HCFC1/ZNF143, we observed an increase in AP2S1 gene expression, suggesting that proper THAP11/ZNF143/HCFC1 recruitment is important for transcriptional regulation. Notably, the CRISPR-Cas9 approach allowed us to assay, as a biological readout, the transcriptional state of the OPHN1 and AP2S1 genes after THAP11/ZNF143/HCFC1 complex depletion (or partial depletion in the case of AP2S1) without the potential global secondary effects of knocking down THAP11, ZNF143, or HCFC1.
In summary, our in vivo approaches identified the ACTACA sequence as a necessary and sufficient motif for recruiting THAP11 and HCFC1 to ZNF143-occupied promoters. Additionally, our observations provide further evidence for cooperative THAP11, ZNF143, and HCFC1 chromatin occupancy and demonstrate a biological need for the DNA sequence and chromatin occupancy by these proteins in the direct transcriptional regulation of genes such as the OPHN1 gene. The studies presented here demonstrate the unique nature and regulation of individual genes and provide the basis for future work involving genome editing and chromosomally integrated constructs, in order to develop more general and unique conclusions about THAP11/ZNF143/HCFC1 complex recruitment to chromatin and its biological effects using additional target genes. We expect that similar studies will be performed with other systems to determine the necessity and sufficiency of requirements of DNA target sequences for in vivo chromatin recruitment of transcriptional regulatory factors.
Supplementary Material
ACKNOWLEDGMENTS
Flow cytometry work was performed at the Robert H. Lurie Comprehensive Cancer Center of Northwestern University Flow Cytometry Core Facility, which is supported by Cancer Center support grant NCI CA060553. This work was supported in part by NCI grant R01 CA133755 (D.C.), the Robert H. Lurie Comprehensive Cancer Center, and the Department of Obstetrics and Gynecology at Northwestern University.
A.V., J.B.P., and D.C. conceived the research plan, designed experiments, and analyzed data. A.V. performed research and wrote the draft manuscript. All authors reviewed and edited the manuscript.
We declare that we have no competing financial interests.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/MCB.00477-15.
REFERENCES
- 1.Wilson AC, Peterson MG, Herr W. 1995. The HCF repeat is an unusual proteolytic cleavage signal. Genes Dev 9:2445–2458. doi: 10.1101/gad.9.20.2445. [DOI] [PubMed] [Google Scholar]
- 2.Lazarus MB, Jiang J, Kapuria V, Bhuiyan T, Janetzko J, Zandberg WF, Vocadlo DJ, Herr W, Walker S. 2013. HCF-1 is cleaved in the active site of O-GlcNAc transferase. Science 342:1235–1239. doi: 10.1126/science.1243990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wilson AC, Boutros M, Johnson KM, Herr W. 2000. HCF-1 amino- and carboxy-terminal subunit association through two separate sets of interaction modules: involvement of fibronectin type 3 repeats. Mol Cell Biol 20:6721–6730. doi: 10.1128/MCB.20.18.6721-6730.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Park J, Lammers F, Herr W, Song J-J. 2012. HCF-1 self-association via an interdigitated Fn3 structure facilitates transcriptional regulatory complex formation. Proc Natl Acad Sci U S A 109:17430–17435. doi: 10.1073/pnas.1208378109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Freiman RN, Herr W. 1997. Viral mimicry: common mode of association with HCF by VP16 and the cellular protein LZIP. Genes Dev 11:3122–3127. doi: 10.1101/gad.11.23.3122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Luciano RL, Wilson AC. 2003. HCF-1 functions as a coactivator for the zinc finger protein Krox20. J Biol Chem 278:51116–51124. doi: 10.1074/jbc.M303470200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wysocka J, Myers MP, Laherty CD, Eisenman RN, Herr W. 2003. Human Sin3 deacetylase and trithorax-related Set1/Ash2 histone H3-K4 methyltransferase are tethered together selectively by the cell-proliferation factor HCF-1. Genes Dev 17:896–911. doi: 10.1101/gad.252103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Knez J, Piluso D, Bilan P, Capone JP. 2006. Host cell factor-1 and E2F4 interact via multiple determinants in each protein. Mol Cell Biochem 288:79–90. doi: 10.1007/s11010-006-9122-x. [DOI] [PubMed] [Google Scholar]
- 9.Mazars R, Gonzalez-de-Peredo A, Cayrol C, Lavigne A-C, Vogel JL, Ortega N, Lacroix C, Gautier V, Huet G, Ray A, Monsarrat B, Kristie TM, Girard J-P. 2010. The THAP-zinc finger protein THAP1 associates with coactivator HCF-1 and O-GlcNAc transferase: a link between DYT6 and DYT3 dystonias. J Biol Chem 285:13364–13371. doi: 10.1074/jbc.M109.072579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Michaud J, Praz V, Faresse NJ, JnBaptiste CK, Tyagi S, Schütz F, Herr W. 2013. HCFC1 is a common component of active human CpG-island promoters and coincides with ZNF143, THAP11, YY1, and GABP transcription factor occupancy. Genome Res 23:907–916. doi: 10.1101/gr.150078.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Vogel JL, Kristie TM. 2000. The novel coactivator C1 (HCF) coordinates multiprotein enhancer formation and mediates transcription activation by GABP. EMBO J 19:683–690. doi: 10.1093/emboj/19.4.683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gunther M, Laithier M, Brison O. 2000. A set of proteins interacting with transcription factor Sp1 identified in a two-hybrid screening. Mol Cell Biochem 210:131–142. doi: 10.1023/A:1007177623283. [DOI] [PubMed] [Google Scholar]
- 13.Julien E, Herr W. 2003. Proteolytic processing is necessary to separate and ensure proper cell growth and cytokinesis functions of HCF-1. EMBO J 22:2360–2369. doi: 10.1093/emboj/cdg242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wysocka J, Reilly PT, Herr W. 2001. Loss of HCF-1-chromatin association precedes temperature-induced growth arrest of tsBN67 cells. Mol Cell Biol 21:3820–3829. doi: 10.1128/MCB.21.11.3820-3829.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mangone M, Myers MP, Herr W. 2010. Role of the HCF-1 basic region in sustaining cell proliferation. PLoS One 5:e9020. doi: 10.1371/journal.pone.0009020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Reilly PT, Wysocka J, Herr W. 2002. Inactivation of the retinoblastoma protein family can bypass the HCF-1 defect in tsBN67 cell proliferation and cytokinesis. Mol Cell Biol 22:6767–6778. doi: 10.1128/MCB.22.19.6767-6778.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Goto H, Motomura S, Wilson AC, Freiman RN, Nakabeppu Y, Fukushima K, Fujishima M, Herr W, Nishimoto T. 1997. A single-point mutation in HCF causes temperature-sensitive cell-cycle arrest and disrupts VP16 function. Genes Dev 11:726–737. doi: 10.1101/gad.11.6.726. [DOI] [PubMed] [Google Scholar]
- 18.Reilly PT, Herr W. 2002. Spontaneous reversion of tsBN67 cell proliferation and cytokinesis defects in the absence of HCF-1 function. Exp Cell Res 277:119–130. doi: 10.1006/excr.2002.5551. [DOI] [PubMed] [Google Scholar]
- 19.Tyagi S, Chabes AL, Wysocka J, Herr W. 2007. E2F activation of S phase promoters via association with HCF-1 and the MLL family of histone H3K4 methyltransferases. Mol Cell 27:107–119. doi: 10.1016/j.molcel.2007.05.030. [DOI] [PubMed] [Google Scholar]
- 20.Tyagi S, Herr W. 2009. E2F1 mediates DNA damage and apoptosis through HCF-1 and the MLL family of histone methyltransferases. EMBO J 28:3185–3195. doi: 10.1038/emboj.2009.258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Parker JB, Yin H, Vinckevicius A, Chakravarti D. 2014. Host cell factor-1 recruitment to E2F-bound and cell-cycle-control genes is mediated by THAP11 and ZNF143. Cell Rep 9:967–982. doi: 10.1016/j.celrep.2014.09.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Anno Y-N, Myslinski E, Ngondo-Mbongo RP, Krol A, Poch O, Lecompte O, Carbon P. 2011. Genome-wide evidence for an essential role of the human Staf/ZNF143 transcription factor in bidirectional transcription. Nucleic Acids Res 39:3116–3127. doi: 10.1093/nar/gkq1301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Myslinski E, Gérard M-A, Krol A, Carbon P. 2006. A genome scale location analysis of human Staf/ZNF143-binding sites suggests a widespread role for human Staf/ZNF143 in mammalian promoters. J Biol Chem 281:39953–39962. doi: 10.1074/jbc.M608507200. [DOI] [PubMed] [Google Scholar]
- 24.Schaub M, Myslinski E, Schuster C, Krol A, Carbon P. 1997. Staf, a promiscuous activator for enhanced transcription by RNA polymerases II and III. EMBO J 16:173–181. doi: 10.1093/emboj/16.1.173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schuster C, Krol A, Carbon P. 1998. Two distinct domains in Staf to selectively activate small nuclear RNA-type and mRNA promoters. Mol Cell Biol 18:2650–2658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bailey SD, Zhang X, Desai K, Aid M, Corradin O, Cowper-Sal Lari R, Akhtar-Zaidi B, Scacheri PC, Haibe-Kains B, Lupien M. 2015. ZNF143 provides sequence specificity to secure chromatin interactions at gene promoters. Nat Commun 2:6186. doi: 10.1038/ncomms7186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Heidari N, Phanstiel DH, He C, Grubert F, Jahanbanian F, Kasowski M, Zhang MQ, Snyder MP. 2014. Genome-wide map of regulatory interactions in the human genome. Genome Res 24:1905–1917. doi: 10.1101/gr.176586.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ngondo-Mbongo RP, Myslinski E, Aster JC, Carbon P. 2013. Modulation of gene expression via overlapping binding sites exerted by ZNF143, Notch1 and THAP11. Nucleic Acids Res 41:4000–4014. doi: 10.1093/nar/gkt088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nishihara M, Yamada M, Nozaki M, Nakahira K, Yanagihara I. 2010. Transcriptional regulation of the human establishment of cohesion 1 homolog 2 gene. Biochem Biophys Res Commun 393:111–117. doi: 10.1016/j.bbrc.2010.01.094. [DOI] [PubMed] [Google Scholar]
- 30.Roussigne M, Kossida S, Lavigne A-C, Clouaire T, Ecochard V, Glories A, Amalric F, Girard J-P. 2003. The THAP domain: a novel protein motif with similarity to the DNA-binding domain of P element transposase. Trends Biochem Sci 28:66–69. doi: 10.1016/S0968-0004(02)00013-0. [DOI] [PubMed] [Google Scholar]
- 31.Dejosez M, Krumenacker JS, Zitur LJ, Passeri M, Chu L-F, Songyang Z, Thomson JA, Zwaka TP. 2008. Ronin is essential for embryogenesis and the pluripotency of mouse embryonic stem cells. Cell 133:1162–1174. doi: 10.1016/j.cell.2008.05.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dejosez M, Levine SS, Frampton GM, Whyte WA, Stratton SA, Barton MC, Gunaratne PH, Young RA, Zwaka TP. 2010. Ronin/Hcf-1 binds to a hyperconserved enhancer element and regulates genes involved in the growth of embryonic stem cells. Genes Dev 24:1479–1484. doi: 10.1101/gad.1935210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Parker JB, Palchaudhuri S, Yin H, Wei J, Chakravarti D. 2012. A transcriptional regulatory role of the THAP11–HCF-1 complex in colon cancer cell function. Mol Cell Biol 32:1654–1670. doi: 10.1128/MCB.06033-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, Zhang F. 2013. Genome engineering using the CRISPR-Cas9 system. Nat Protoc 8:2281–2308. doi: 10.1038/nprot.2013.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kim HJ, Lee HJ, Kim H, Cho SW, Kim J-S. 2009. Targeted genome editing in human cells with zinc finger nucleases constructed via modular assembly. Genome Res 19:1279–1288. doi: 10.1101/gr.089417.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gervais V, Campagne S, Durand J, Muller I, Milon A. 2013. NMR studies of a new family of DNA binding proteins: the THAP proteins. J Biomol NMR 56:3–15. doi: 10.1007/s10858-012-9699-1. [DOI] [PubMed] [Google Scholar]
- 37.Abdel-Rahman WM, Katsura K, Rens W, Gorman PA, Sheer D, Bicknell D, Bodmer WF, Arends MJ, Wyllie AH, Edwards PAW. 2001. Spectral karyotyping suggests additional subsets of colorectal cancers characterized by pattern of chromosome rearrangement. Proc Natl Acad Sci U S A 98:2538–2543. doi: 10.1073/pnas.041603298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Knutsen T, Padilla-Nash HM, Wangsa D, Barenboim-Stapleton L, Camps J, McNeil N, Difilippantonio MJ, Ried T. 2010. Definitive molecular cytogenetic characterization of 15 colorectal cancer cell lines. Genes Chromosomes Cancer 49:204–223. doi: 10.1002/gcc.20730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Melcher R, Steinlein C, Feichtinger W, Müller CR, Menzel T, Lührs H, Scheppach W, Schmid M. 2000. Spectral karyotyping of the human colon cancer cell lines SW480 and SW620. Cytogenet Cell Genet 88:145–152. doi: 10.1159/000015508. [DOI] [PubMed] [Google Scholar]
- 40.Weakley SM, Wang H, Yao Q, Chen C. 2011. Expression and function of a large non-coding RNA gene XIST in human cancer. World J Surg 35:1751–1756. doi: 10.1007/s00268-010-0951-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Thomas LR, Wang Q, Grieb BC, Phan J, Foshage AM, Sun Q, Olejniczak ET, Clark T, Dey S, Lorey S, Alicie B, Howard GC, Cawthon B, Ess KC, Eischen CM, Zhao Z, Fesik SW, Tansey WP. 2015. Interaction with WDR5 promotes target gene recognition and tumorigenesis by MYC. Mol Cell 58:440–452. doi: 10.1016/j.molcel.2015.02.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Schaub M, Myslinski E, Krol A, Carbon P. 1999. Maximization of selenocysteine tRNA and U6 small nuclear RNA transcriptional activation achieved by flexible utilization of a Staf zinc finger. J Biol Chem 274:25042–25050. doi: 10.1074/jbc.274.35.25042. [DOI] [PubMed] [Google Scholar]
- 43.Campagne S, Saurel O, Gervais V, Milon A. 2010. Structural determinants of specific DNA-recognition by the THAP zinc finger. Nucleic Acids Res 38:3466–3476. doi: 10.1093/nar/gkq053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sabogal A, Lyubimov AY, Corn JE, Berger JM, Rio DC. 2010. THAP proteins target specific DNA sites through bipartite recognition of adjacent major and minor grooves. Nat Struct Mol Biol 17:117–123. doi: 10.1038/nsmb.1742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wang H, Zou J, Zhao B, Johannsen E, Ashworth T, Wong H, Pear WS, Schug J, Blacklow SC, Arnett KL, Bernstein BE, Kieff E, Aster JC. 2011. Genome-wide analysis reveals conserved and divergent features of Notch1/RBPJ binding in human and murine T-lymphoblastic leukemia cells. Proc Natl Acad Sci U S A 108:14908–14913. doi: 10.1073/pnas.1109023108. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.