

Abstract
Neurite outgrowth is key to the formation of functional circuits during neuronal development. Neurotrophins, including nerve growth factor (NGF), increase neurite outgrowth in part by altering the function and expression of Ca2+-permeable cation channels. Here we report that transient receptor potential vanilloid 2 (TRPV2) is an intracellular Ca2+-permeable TRPV channel upregulated by NGF via the mitogen-activated protein kinase (MAPK) signaling pathway to augment neurite outgrowth. TRPV2 colocalized with Rab7, a late endosome protein, in addition to TrkA and activated extracellular signal-regulated kinase (ERK) in neurites, indicating that the channel is closely associated with signaling endosomes. In line with these results, we showed that TRPV2 acts as an ERK substrate and identified the motifs necessary for phosphorylation of TRPV2 by ERK. Furthermore, neurite length, TRPV2 expression, and TRPV2-mediated Ca2+ signals were reduced by mutagenesis of these key ERK phosphorylation sites. Based on these findings, we identified a previously uncharacterized mechanism by which ERK controls TRPV2-mediated Ca2+ signals in developing neurons and further establish TRPV2 as a critical intracellular ion channel in neuronal function.
INTRODUCTION
Establishment of precise neural connections during nervous system development is essential in forming functional circuits. Neurite outgrowth allows for connection and communication between developing neurons and their targets. In the developing peripheral nervous system, nerve growth factor (NGF) is a target-derived extracellular cue necessary for outgrowth (1). Upon binding to its extracellular receptor, NGF activates the phosphoinositide 3-kinase (PI3K) signaling pathway, which is essential for the survival of developing neurons, and the mitogen-activated protein kinase (MAPK) pathway, which promotes differentiation and neurite outgrowth (2, 3). These signaling pathways have numerous downstream effectors in developing neurons, including several Ca2+-permeable transient receptor potential (TRP) channels (4–8).
Thermosensitive TRP channels from the vanilloid subfamily (thermoTRPV channels) consist of four nonselective Ca2+-permeable cation channels, TRP vanilloid 1 (TRPV1) to TRPV4, originally described as pain and temperature sensors in adult sensory neurons (9–12). Recent evidence suggests, however, that only TRPV1 functions as a molecular sensor of heat and painful stimuli in vivo, while the function of TRPV2 to -4 remains unclear (13–16). TRPV2 and TRPV4 have also been detected in developing peripheral neurons, suggesting that they may play a role in growth programs during development (17, 18). Consistent with this notion, TRPV2 has been implicated in axon outgrowth (18), and critical mutations in TRPV4 result in peripheral axonal neuropathies (19, 20). Despite these initial findings, the details by which thermoTRPV channels influence neuronal development remain unknown.
Here we explore the molecular mechanisms by which thermoTRPV channels contribute to neurotrophin-mediated peripheral neuron development. We found that among the thermoTRPV proteins, TRPV2 and TRPV4 are abundantly present in embryonic sensory neurons, and application of NGF specifically increased TRPV2 protein levels. TRPV2 upregulation occurred through the MAPK signaling pathway, which is essential for neuronal differentiation (2, 3), to enhance neurite outgrowth in a Ca2+-dependent manner. Generation of TRPV2 monoclonal antibodies (21) allowed us to determine that endogenous TRPV2 colocalized with Rab7, a late endosomal marker associated with retrograde trafficking, in embryonic dorsal root ganglion (DRG) neurons. Exposure to NGF leads to the formation of signaling endosomes containing TrkA and active MAPK components (22, 23). Consistently, we found colocalization of TRPV2 with both TrkA and phosphorylated Erk1/2 (pErk1/2), suggesting that TRPV2 populates signaling endosomes and is directly modulated by extracellular signal-regulated kinase (ERK). In line with this hypothesis, we found that TRPV2 is phosphorylated by Erk2 in vitro and identified sites on the soluble N and C termini of TRPV2 critical for phosphorylation by Erk2. Mutation of these sites reduced NGF-induced neurite growth and altered TRPV2 protein expression and Ca2+ signals, indicating that phosphorylation of TRPV2 by ERK is essential for the enhancement of Ca2+ signaling and neurite outgrowth. Thus, we propose a mechanism by which ERK regulates Ca2+ signaling via TRPV2 to augment neurite outgrowth in developing neurons.
MATERIALS AND METHODS
Chemicals and antibodies.
The following antibodies were used: anti-1D4 (24) (1 μg/ml for Western blotting and immunocytochemistry), anti-TRPV2 2D6 (2 μg/ml for Western blotting), and anti-TRPV2 17A11 (10 μg/ml for immunocytochemistry) mouse monoclonal antibodies (MAbs), which were generated in our laboratory (21); anti-β-actin mouse MAb (catalog number 3700; Cell Signaling Technology, Danvers, MA) (1:1,000 dilution for Western blotting); anti-phospho-p44/42 MAPK (Erk1/2) (Thr202/Tyr204) XP rabbit MAb (catalog number 4370; Cell Signaling Technology) (1:1,000 dilution for Western blotting and 1:200 for immunocytochemistry); anti-p44/42 MAPK (Erk1/2) mouse MAb (catalog number 9107; Cell Signaling Technology) (1:1,000 dilution for Western blotting); anti-Akt pan-mouse MAb (catalog number 2920; Cell Signaling Technology) (1:1,000 dilution for Western blotting); anti-phosphorylated Akt (pAkt) (Santa Cruz Biotechnology, Dallas, TX); anti-Na/K ATPase (catalog number 3010; Cell Signaling Technology) (1:100 dilution for Western blotting); anti-TRPV1 (UC Davis/NIH NeuromAb Facility; clone N221/17, AB_11000725) (1:500 dilution for Western blotting and 1:100 dilution for immunocytochemistry); anti-TRPV3 (UC Davis/NIH NeuromAb Facility; clone N15/4, AB_10671952) (1:1,000 dilution for Western blotting and 1:100 dilution for immunocytochemistry); anti-TRPV4 (catalog number 39260; AbCam Inc., Cambridge, MA) (1:200 dilution for Western blotting and immunocytochemistry); anti-TrkA (catalog number AB9354; Millipore, Billerica, MA) (1:100 dilution for immunocytochemistry); and anti-Rab7 (catalog number SC-6563; Santa Cruz Biotechnology) (1:100 dilution for immunocytochemistry).
NGF-7s, wortmannin, and LY294002 were purchased from Sigma (St. Louis, MO). U0126 was obtained from Cell Signaling Technology. PhosStop phosphatase inhibitor and EDTA-free complete protease inhibitor were purchased from Roche (Indianapolis, IN). Peptide-N-glycosidase F (PNGase F) was obtained from New England BioLabs (Ipswich, MA).
Cell culture and transfection.
All cells were cultured in 95% O2 and 5% CO2 at 37°C in a humidified atmosphere. Pheochromocytoma 12 (PC12) cells were a kind gift of John Mieyal (Case Western Reserve University). PC12 cells were maintained in Ham's F-12–Kaughn (F12K) nutrient mix (Life Technologies, Carlsbad, CA) with 15% horse serum (Life Technologies), 5% fetal bovine serum (FBS; GE Healthcare, Piscataway, NJ), 100 U/ml penicillin, and 100 μg/ml streptomycin (Life Technologies). For NGF treatments, cells were changed from normal growth medium to differentiation medium, which consisted of F12K nutrient mix, 1% horse serum, 0.5% FBS, and 100 ng/ml NGF. HEK293T and F11 cells were cultured and transfected as described previously (21).
Transfections of PC12 cells with plasmid DNA were performed by using Lipofectamine LTX reagent (Life Technologies) according to the manufacturer's protocol. Transfection of small interfering RNA (siRNA) was performed by using RNAiMax Lipofectamine (Life Technologies) according to the manufacturer's protocol. Control siRNA was obtained from Santa Cruz (catalog number SC-44235). siRNAs targeting TRPV2, Erk1, and Erk2 were obtained from Dharmacon (catalog numbers L-091197-02 for TRPV2, L-100592-02 for Erk1, and L-096054-02 for Erk2). In the case of experiments employing PI3K or MEK inhibitors, cells were pretreated with vehicle (dimethyl sulfoxide [DMSO]) or inhibitors for 30 min prior to NGF treatment.
Dissociation and culture of primary E18 DRG neurons.
Embryonic day 18 (E18) DRG neurons from Sprague-Dawley rats were obtained from Brain Bits (Springfield, IL). Cells were dissociated by treatment with 0.1 U collagenase and 0.8 U dispase (Roche) in Hibernate A medium without Ca2+ or Mg2+ (Brain Bits) for 1 h at 37°C. Next, the tissue was resuspended in Hibernate AB medium (Brain Bits) and triturated with a p200 pipette. The cells were centrifuged at 200 × g and resuspended in Nb4Activ (Life Technologies) containing NGF (25 ng/ml). Cells were then seeded onto poly-d-lysine (Sigma)-coated glass coverslips in 6-well plates. Cells were cultured for 5 days prior to fixation for immunofluorescence. Half of the medium was changed on day 2. In the case of experiments employing PI3K or MEK inhibitors, cells were treated with vehicle (DMSO) or inhibitors at the time of plating.
Plasmids.
Rat TRPV2 in pcDNA3.1 was obtained from David Julius (University of California—San Francisco). TRPV2 was engineered with a C-terminal 1D4 epitope (TETSQVAPA) as described previously (21). Control and TRPV2 short hairpin RNAs (shRNAs) were expressed in pGFP-C-shLenti (Origene, Rockville, MD). In addition to expressing shRNA under the control of the U6 promoter, this plasmid also allowed for expression of green fluorescent protein (GFP) under the control of the cytomegalovirus (CMV) promoter.
Site-directed mutagenesis.
To generate TRPV2 mutants, amino acid changes were introduced by using mutated oligonucleotides for E/K (E599K forward primer 5′-CTGGATGCCTCCCTAAAGCTGTTCAAGTTCACC-3′ and reverse primer 5′-GGTGAACTTGAACAGCTTTAGGGAGGCATCCAG-3′ and E609K forward primer 5′-ACCATTGGTATGGGGAAGCTGGCTTTCCAG-3′ and reverse primer 5′-CTGGAAAGCCAGCTTCCCCATACCAATGGT-3′) or S/A (S6A forward primer 5′-ATGACTTCAGCCTCCGCCCCCCCAGCTTTCAGGCTGGAG-3′ and reverse primer 5′-CTCCAGCCTGAAAGCTGGGGGGGCGGAGGCTGAAGTCAT-3′, S37A forward primer 5′-CAGGAACCGCCCCCCATGGAGGCACCATTCCAGAGGGAGGAC-3′ and reverse primer 5′-GTCCTCCCTCTGGAATGGTGCCTCCATGGGGGGCGGTTCCTG-3′, S47A forward primer 5′-CAGAGGGAGGACCGGAATTCCGCCCCTCAGATCAAAGTGAAC-3′ and reverse primer 5′-GTTCACTTTGATCTGAGGGGCGGAATTCCGGTCCTCCCTCTG-3′, and S760A forward primer 5′-CAGGTCCTCCAGGCCCCCACAGAGACC-3′ and reverse primer 5′-GGTCTCTGTGGGGGCCTGGCGGACCTG-3′) and wild-type (WT) TRPV2-tagged 1D4 (TRPV2-1D4) as a template. The TRPV2 mutant constructs were obtained by PCR using the Accuprime polymerase kit from Life Technologies. The mutants were confirmed by DNA sequencing.
Cytosolic Ca2+ measurements.
Measurements of intracellular Ca2+ concentrations were obtained by using the fluorescent Ca2+ indicator Fluo4-AM (Life Technologies). Cells were cotransfected with red fluorescent protein (RFP) to identify transfected cells. Experiments were performed with extracellular solution (ECS) containing 120 mM NaCl, 3 mM KCl, 2 mM CaCl2, 2 mM MgCl2, 30 mM glucose, 11 mM sorbitol, and 20 mM HEPES (pH 7.3). Cells were loaded with 5 μM Fluo4-AM in ECS at room temperature for 30 min, followed by two 5-min washes in ECS. All measurements were obtained at room temperature. Emitted fluorescence was measured from single cells by using a C1 Plus confocal system on a Nikon Eclipse Ti-E microscope (Nikon Instruments Inc., Melville, NY) at wavelengths of 495 nm (excitation) and 520 nm (emission) at a frequency of 1 image every 3 s. Fluorescence intensity was analyzed by using ImageJ software.
Western blot analysis.
Protein extracts were prepared in lysis buffer (50 mM Tris [pH 7.4], 150 mM NaCl, 2 mM EGTA, 1% Triton X-100, and protease and phosphatase inhibitors) as described previously (21). Fifty micrograms of protein extract was separated by SDS-PAGE using 4 to 20% Tris-glycine gels (Life Technologies) and transferred onto nitrocellulose membranes. Following a block with 10% nonfat dry milk in Tris-buffered saline–Tween (TBST), membranes were probed with primary antibody followed by IRDye-conjugated secondary antibodies (LiCor, Lincoln, NE). Immunoreactivity was detected by using the Odyssey infrared imaging system (LiCor). Quantification of band intensities was performed by using LiCor Odyssey software. For quantification of TRPV2 band intensity, both the upper and lower bands were included.
Removal of N-linked glycans.
To remove N-linked glycans, PC12 cell protein extracts were treated with PNGase F in G7 reaction buffer and 1% NP-40 (New England BioLabs) for 1 h at 37°C.
RNA extraction and cDNA synthesis.
RNA from cultured F11 and PC12 cells was extracted by using TRIzol (Life Technologies) according to the manufacturer's instructions. Briefly, 1 ml of TRIzol was added to each well of a 6-well dish, and the dish was incubated at room temperature for 5 min. Two hundred microliters of chloroform (Fisher Scientific) was added to the mixture, and the mixture was vigorously shaken for 15 s and incubated at room temperature for 5 min. Samples were centrifuged at 12,000 × g for 15 min at 4°C. The upper aqueous phase was transferred to a tube containing 0.5 ml isopropanol, and the tube was shaken by hand for 30 s and incubated at room temperature for 10 min. Following centrifugation at 12,000 × g for 15 min at 4°C, the supernatant was removed. The pellet was washed in 75% RNase-free ethanol (Fisher Scientific, Waltham, MA) and centrifuged at 7,500 × g for 5 min at 4°C. Ethanol was removed from the tube, and the pellet was air dried for 10 min on ice. The pellet containing extracted RNA was then resuspended in 100 μl diethyl pyrocarbonate (DEPC)-treated water. cDNA synthesis was completed by using SuperScript II reverse transcriptase (Life Technologies) with 1 μg of RNA as the template, according to the manufacturer's instructions.
Semiquantitative reverse transcription-PCR (RT-PCR) of TRPV2 and GAPDH.
The PCR amplification was completed with a 50-μl final volume using 5 μl of cDNA from the synthesis procedure described above, 10 μl 5× GoTaq buffer, 1 μl (1 μM final concentration) primers (glyceraldehyde-3-phosphate dehydrogenase [GAPDH] forward primer 5′-ATGGTGAAGGTCGGTGTG-3′, GAPDH reverse primer 5′-GCCTCTCTCTTGCTCTCAGT-3′, TRPV2 forward primer 5′-ATGACTTCAGCCTCCAGCC-3′, and TRPV2 reverse primer 5′-TCCAGCGCAGGTATTCTAGC-3′), 1 μl deoxynucleoside triphosphate (dNTP) mix (200 μM final concentration), 0.25 μl GoTaq2 (Promega, Madison, WI), and 29.25 μl H2O.
The PCR protocol consisted of a denaturation step at 94°C for 5 min and 34 cycles of denaturation at 94°C for 30 s, annealing at 52°C for 30 s, and extension at 68°C for 1.5 min. Twenty microliters of the PCR mixture was analyzed on 1.2% agarose gels and visualized with ethidium bromide staining. Quantification of band intensity was completed by using ImageJ.
Immunofluorescence.
Cells were fixed and stained as described previously (21). Confocal images were obtained by using a Leica TCS SP2 laser scanning confocal microscope with an HCX PL APO CS 40.0-by-1.25 oil UV objective. Argon (488-nm) and helium-neon (594-nm) lasers were used for excitation, and images were obtained by using Leica confocal software version 2.61. Brightness and contrast were adjusted by using Adobe Photoshop. Only linear changes were made. Any enhancements were applied across images in an identical manner.
Cell surface biotinylation.
Cell surface biotinylation experiments were performed as described previously (21). Briefly, PC12 cells were treated with NGF for 24 h. Cells were washed in phosphate-buffered saline (PBS) and incubated with EZ-Link sulfo-N-hydroxysuccinimide (NHS)–biotin (Thermo Scientific, Waltham, MA) for 30 min at 37°C. The reaction mixture was quenched with 100 mM glycine in PBS, followed by a wash in glycine-free PBS. Cells were then lysed and incubated with streptavidin-agarose (Life Technologies) to capture biotinylated proteins. The biotinylated fraction was analyzed by Western blotting.
Morphology analysis of PC12 cells.
PC12 cells were plated onto 6-well plates coated with 50 μg/ml rat tail collagen type I (Life Technologies). Twenty-four hours later, cells were transfected with either GFP, WT TRPV2, or a dominant negative TRPV2 construct (DN TRPV2) as described above. At 24 h posttransfection, growth medium was changed to differentiation medium, which consisted of F12K mix, 1% horse serum, 0.5% FBS, and 100 ng/ml NGF, for 72 h. To identify cells overexpressing TRPV2, cells were fixed and immunostained with 1D4 antibody. Cells expressing GFP were used as vector-transfected controls. For knockdown experiments, cells were transiently transfected with control or TRPV2 shRNA (Origene). shRNA constructs also expressed GFP, which allowed for detection of transfected cells. Cells under each condition were imaged in duplicate by using a Leica DMI 6000 B fluorescence microscope for three independent experiments. Fluorescence images from 25 random fields were collected. The number of cells bearing a neurite was determined by visual examination of the field and counting the total number of 1D4- or GFP-positive cells and 1D4- or GFP-positive cells that possessed a neurite at least twice the length of the cell body diameter. Neurite length was manually determined by using ImageJ software. Neurite length was measured for all cells in the visual field for which the entire neurite was visible and the extension did not contact another cell. Cells from aggregates were disregarded.
In vitro kinase assay.
Recombinant TRPV2 was expressed and purified as described previously (25). Five hundred nanograms of myelin basic protein (MBP) (Sigma) or recombinant TRPV2 was incubated with or without 100 ng of recombinant Erk2 (Sigma) in 1× Erk2 kinase buffer (25 mM morpholinepropanesulfonic acid [MOPS] [pH 7.2], 12.5 mM glycerol-2-phosphate, 25 mM MgCl2, 5 mM EGTA, 2 mM EDTA, 0.25 mM dithiothreitol [DTT]). The kinase reaction was initiated by the addition of 25 μM cold ATP (Promega) and 10 μCi γ-32P (Perkin-Elmer, Waltham, MA) to the mixture. The reaction was terminated 45 min after initiation by the addition of SDS-PAGE gel loading buffer (Bio-Rad, Hercules, CA) containing 50 mM β-mercaptoethanol to the mixture. Samples were run on a 4 to 20% Tris-glycine gel (Invitrogen), stained by using GelCode Blue stain (Thermo Scientific), dried overnight, and exposed to autoradiography film (Denville Scientific, South Plainfield, NJ).
Isolation of TRPV2 from HEK293T cells and mass spectrometry analysis.
HEK293T cells were transfected with 1D4-tagged TRPV2 for 48 h, followed by cell lysis and immunoprecipitation with Sepharose beads conjugated to 1D4 antibody. Captured proteins were treated with PNGase F, eluted with 1D4 peptide (10 mg/ml; Genscript), and resolved by using SDS-PAGE. The presence of the TRPV2 protein was confirmed by Western blotting. Protein was digested in-gel by using Asp-N (Promega, Madison, WI) (100 ng for 4 h at 37°C) and modified trypsin (Promega, Madison, WI) (100 ng overnight at 37°C) dual-enzyme approach. For phosphorylation analysis, the proteolytic peptide mixture was further enriched for phosphopeptides by using a TiO2 column (Pierce, Thermo Scientific).
Liquid chromatography-mass spectrometry (LC-MS) experiments were carried out on an Orbitrap Elite mass spectrometer (Thermo Electron, San Jose, CA) interfaced with a Waters nanoAquity ultraperformance liquid chromatography (UPLC) system (Waters, Taunton, MA). To analyze possible posttranslational modifications (PTMs) of the TRPV2 protein, ∼300 ng of a proteolytic peptide mixture of TPRV2 was loaded onto a trap column (180 μm by 20 mm packed with C18 Symmetry [5 μm, 100 Å]; Waters, Taunton, MA) to preconcentrate the sample and wash away excess salts. Reverse-phase separation was performed on a reversed-phase column (75-μm by 250-mm nanocolumn packed with C18 BEH130 [1.7 μm, 130 Å]; Waters, Taunton, MA), using a gradient of 2 to 45% mobile phase B (0.1% formic acid and acetonitrile [ACN]) and mobile phase A (100% water–0.1% formic acid) over a period of 60 min at 37°C at a flow rate of 300 nl/min. Peptides eluting from the column were directed to a nanoelectrospray source with a capillary voltage of 2.5 kV. All mass spectra were obtained from data-dependent experiments. MS and tandem MS (MS/MS) spectra were acquired in the positive-ion mode, with the full-scan MS recorded for eluted peptides (m/z range of 350 to 1,800) in an Fourier transform (FT) mass analyzer at a resolution (R) of 120,000, followed by MS/MS of the 20 most intense peptide ion scans in the ion trap (IT) mass analyzer.
The resulting MS/MS data were initially searched against the TRPV2 protein database by using Mass Matrix software to identify all specific sites of modification (26). In particular, MS/MS spectra were searched for nonspecific peptides of TRPV2 by using mass accuracy values of 8 ppm and 0.7 Da for MS1 and MS2 scans, respectively, with allowed variable modifications including carbamidomethylation for cysteines; oxidative modifications for methionine amino acids; phosphorylation for serine, threonine, and tyrosine; and asparagine deamidation. All detected MS2 spectra for each site of phosphorylation were manually verified.
Statistical analyses.
All results are presented as means ± standard errors of the means (SEM). Significance of differences was determined by analysis of variance (ANOVA) followed by Tukey's post hoc test for multiple comparisons unless otherwise indicated. Statistical significance was accepted at P values of <0.05, <0.01, and <0.001.
RESULTS
TRPV2 is expressed in developing neurons and regulated by NGF.
To assess the expression of thermoTRPV channels in the development of peripheral neurons, embryonic day 18 (E18) rat DRG neurons were immunostained with antibodies against TRPV1 to -4. TRPV1 and TRPV3 immunoreactivities were sparse in βIII-tubulin-positive neurons, while TRPV2 and TRPV4 immunoreactivities were more abundant (Fig. 1A). TRPV4 appeared almost exclusively in the cell bodies of neurons, while TRPV2 was present in both cell bodies and neuronal extensions (Fig. 1A). We also tested for expression of TRPV1 to -4 in NGF-sensitive PC12 cells. The neuroendocrine PC12 cell line is derived from a rat adrenal chromaffin tumor and is commonly used as a model for studying NGF signaling and neuronal cell differentiation (2, 27). We failed to detect TRPV1 protein in PC12 cell lysates in both the absence and presence of NGF by Western blot analysis. The specificity of the TRPV1 antibody was tested by using mouse kidney extracts, which produced a band at ∼100 kDa, corresponding to the predicted molecular mass of the monomeric TRPV1 protein (Fig. 1B). The TRPV3 and TRPV4 proteins were detected in PC12 cells, but their levels were unaffected by NGF treatment (Fig. 1B). In contrast, the TRPV2 protein content increased substantially after NGF treatment (Fig. 1B). An increase in phosphorylated Erk1/2 (pErk1/2) levels confirmed the activation of NGF signaling (Fig. 1B).
FIG 1.
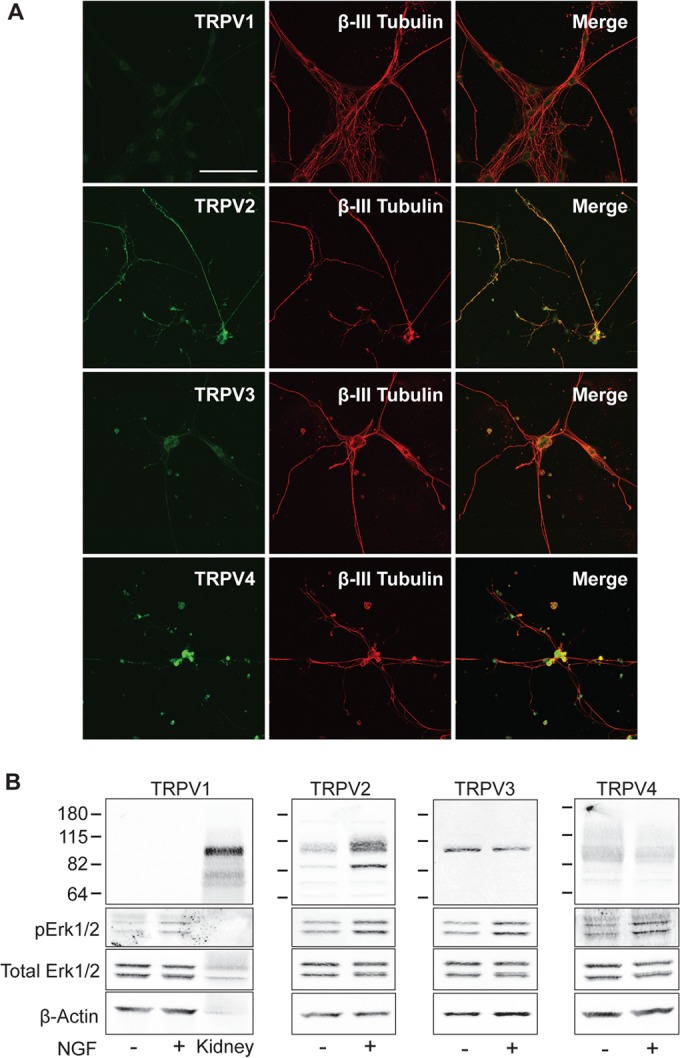
NGF upregulates TRPV2 in models of developing neurons. (A) E18 DRG neurons immunostained with antibodies against the thermoTRPV channels (TRPV1, TRPV2, TRPV3, and TRPV4) (green) and βIII-tubulin (red). Bar, 100 μm. (B) Western blot analysis of 50 μg of PC12 cell lysates using the indicated thermoTRPV antibodies plus phosphorylated Erk1/2 (pErk1/2), total Erk1/2, and β-actin. Cells were cultured in differentiation medium in the absence or presence of NGF (100 ng/ml) for 12 h. Since PC12 cells lacked TRPV1 immunoreactivity, mouse kidney lysate was loaded as a positive control.
TRPV2 was previously implicated in axon outgrowth during peripheral neuron development (18); however, the mechanisms by which TRPV2 is regulated during neuronal development remain unknown. We therefore further explored how NGF regulates TRPV2. RT-PCR confirmed the presence of TRPV2 mRNA in PC12 cells (Fig. 2A). F11 cells, which are derived from embryonic DRG neurons and endogenously express TRPV2 (28), were used as a positive control (Fig. 2A). Our TRPV2 monoclonal antibody (21) displayed immunoreactivity with two bands in PC12 cell extracts. One band corresponded to the predicted molecular mass of rat TRPV2 (∼86 kDa), while the other ran at a higher molecular mass (∼100 kDa) (Fig. 1B). The specificity of the TRPV2 antibody was confirmed by using siRNA directed against TRPV2. Cells transfected with TRPV2 siRNA displayed decreased band intensities for both the 86-kDa and 100-kDa bands compared to control cells (Fig. 2B). We had previously observed two bands in cells heterologously expressing TRPV2 and predicted that the upper band may represent glycosylated TRPV2 (21). Treatment of PC12 cell protein extracts with PNGase F, which cleaves N-linked glycans, eliminated the higher-molecular-mass band, indicating that this band represents N-glycosylated TRPV2 (Fig. 2C). These results confirm that both bands recognized by the TRPV2 antibody correspond to the TRPV2 protein.
FIG 2.

NGF treatment results in sustained upregulation of TRPV2 expression in PC12 cells. (A) mRNA expression of TRPV2 in F11 cells and PC12 cells was examined by RT-PCR using TRPV2-specific PCR primers. (B) Western blot analysis using TRPV2 monoclonal antibody of PC12 cells transfected with 200 nM control siRNA or TRPV2 siRNA for 48 h. The bar graph represents relative TRPV2 band intensities. The TRPV2 band intensity for control siRNA was defined as 1. Data represent means ± SEM (P < 0.05 by an unpaired t test). (C) PC12 cell protein extracts were treated with and without PNGase F and immunoblotted with TRPV2 and β-actin antibodies. (D) PC12 cells were treated with NGF (100 ng/ml) for the indicated times. Fifty micrograms of whole-cell lysates was then analyzed by Western blotting with TRPV2 and β-actin antibodies. The line graph represents relative TRPV2 protein levels for each time point. The β-actin-normalized TRPV2 band intensity at the 0-h time point was defined as 1. *, P < 0.05 relative to the 0-h NGF time point as determined by one-way ANOVA followed by Dunnett's post hoc test. Data represent means ± SEM for 3 independent experiments. (E) RT-PCR analysis of PC12 cells treated with NGF (100 ng/ml) for the indicated times. RNA was extracted and reverse transcribed to cDNA, followed by PCR amplification using TRPV2- and GAPDH-specific primers. The line graph represents relative TRPV2 mRNA levels for each time point. The GAPDH-normalized TRPV2 band intensity at the 0-h time point was defined as 1. Data represent means ± SEM from 3 independent experiments. *, P < 0.05 relative to the 0-h NGF time point as determined by one-way ANOVA followed by Dunnett's post hoc test. (F) PC12 cells were treated for the indicated times with NGF (100 ng/ml) and then fixed and immunostained with anti-TRPV2 antibody. Bar, 25 μm.
A time course analysis revealed that treatment of PC12 cells with NGF led to an increase in TRPV2 protein content beginning at 6 h and peaking at between 24 and 48 h (Fig. 2D). The TRPV2 protein content remained elevated for at least 72 h of NGF treatment (Fig. 2D). Semiquantitative RT-PCR experiments showed that the TRPV2 mRNA content also increased at the 6- and 12-h time points, after which mRNA levels reverted back to baseline levels (Fig. 2E). Immunofluorescence revealed a comparable time course for upregulation of the TRPV2 protein (Fig. 2F). TRPV2 localized to developing neurites after NGF treatment, similar to its distribution in embryonic DRG neurons (Fig. 2F). Overall, these results confirm the presence of TRPV2 protein in NGF-sensitive developing sensory neurons and show that treatment of PC12 cells with NGF results in a sustained increase in the amount of TRPV2 protein.
TRPV2 enhances NGF-induced neurite outgrowth.
Intracellular Ca2+ signals are involved in morphological changes mediated by NGF (29). Since NGF signaling is tightly linked to neurite outgrowth in PC12 cells, we next explored the contribution of TRPV2 activity to NGF-induced neurite extension. Activation of TRPV2 by membrane stretching was previously shown to increase intracellular Ca2+ levels and axon outgrowth in developing neurons (18). Studying endogenous TRPV2 activity and TRPV2-mediated Ca2+ changes has remained difficult due to limited specific pharmacological tools available to activate or inhibit the channel (30, 31). TRPV2 may also display channel activity in the absence of any specific agonist or activator (18, 32). Recently, probenecid was described to be a specific TRPV2 activator (33). TRPV2-null neurons, however, display Ca2+ responses to probenecid that are indistinguishable from those of wild-type (WT) neurons (14). We also did not observe probenecid-activated changes in cytosolic Ca2+ levels in cells expressing TRPV2 (data not shown). We therefore chose to employ a genetic approach to test the effects of TRPV2 activity on NGF-induced neurite outgrowth (31). A dominant negative TRPV2 construct (DN TRPV2), where two key Glu residues in the pore region of the channel were mutated to Lys (E599K and E609K), was used in this study. These Glu residues are conserved in rat and mouse TRPV2 proteins, and mutation of these residues in mouse TRPV2 to Lys has been shown to decrease Ca2+ permeation through the pore of the channel (32, 34). F11 cells provided a suitable system for testing the effect of DN TRPV2 on TRPV2-mediated changes in Ca2+ levels, since F11 cells endogenously express TRPV2 (28) and allow for efficient transfection. F11 cells transfected with DN TRPV2 displayed reduced cytosolic Ca2+ responses after treatment with 2-aminoethoxydiphenyl borate (2-APB), a TRPV2 activator (35, 36), compared to those in cells expressing the vector alone (Fig. 3A). Overexpression of WT TRPV2 in F11 cells augmented the increases in Ca2+ levels in response to 2-APB (Fig. 3A).
FIG 3.

TRPV2 activity enhances NGF-induced neurite outgrowth in PC12 cells. (A) Intracellular Ca2+ levels were measured by Fluo4 fluorescence using time-lapse microscopy. F11 cells expressing the empty vector (n = 80), WT TRPV2 (n = 72), or DN TRPV2 (n = 61) were treated with 2-APB (100 μM) at the 45-s time point. Data represent the mean normalized Fluo4 intensities ± SEM. (B) Representative micrographs of PC12 cells treated with NGF (100 ng/ml for 72 h) after transfection with GFP (control) or 1D4-tagged WT TRPV2 or DN TRPV2. After NGF treatment, cells were fixed and stained with 1D4 antibody followed by Alexa Fluor 488-labeled secondary antibody. Bar, 100 μm. (C) PC12 cells were transfected with GFP, WT TRPV2, or DN TRPV2; grown in the absence or presence of NGF (100 ng/ml for 72 h); and then imaged for fluorescence as described above for panel B. Data represent means ± SEM from 3 independent experiments. n.s., not significant. (D) Neurite lengths for PC12 cells transfected with GFP (n = 141), WT TRPV2 (n = 122), or DN TRPV2 (n = 67) and treated with NGF (100 ng/ml; 72 h). Data represent means ± SEM from 3 independent experiments.
We next tested the effects of WT TRPV2 and DN TRPV2 overexpression on NGF-mediated neurite outgrowth in PC12 cells. Overexpression of WT TRPV2 and DN TRPV2 in PC12 cells had no effect on the percentage of neurite-bearing cells in the absence of applied NGF (Fig. 3C). However, WT TRPV2 overexpression increased the percentage of cells possessing a neurite (Fig. 3C) and the length of neurites (Fig. 3B and D) compared to GFP-transfected controls after 3 days of culture in the presence of NGF. Expression of DN TRPV2 failed to increase the percentage of cells with neurites (Fig. 3C). Importantly, neurite length in the presence of NGF was significantly decreased for cells expressing DN TRPV2 compared to GFP controls (Fig. 3B and D).
We also tested the effect of silencing of TRPV2 expression on NGF-induced neurite outgrowth using shRNA. The shRNA constructs also expressed GFP, which indicated transfected cells. Transfection of TRPV2 shRNA efficiently knocked down TRPV2 overexpressed in HEK293T cells (Fig. 4A). Since a smaller percentage of PC12 cells were transfected, immunofluorescence was employed to assess the knockdown of endogenous TRPV2 protein. Transfection of TRPV2-specific shRNA reduced endogenous TRPV2 immunofluorescence in PC12 cells compared to cells transfected with control shRNA (Fig. 4B and C). Expression of TRPV2 shRNA had no effect on the percentage of neurite-bearing PC12 cells after NGF treatment (Fig. 4D); however, cells expressing TRPV2 shRNA showed a significant decrease in neurite length compared to cells expressing control shRNA (Fig. 4E). Overall, these data indicate that increased expression of functional TRPV2 significantly contributes to the enhancement of neurite length in response to NGF signaling.
FIG 4.

Silencing of TRPV2 expression impairs NGF-induced neurite outgrowth. (A) Western blot analysis with the indicated antibodies of HEK293T cells transiently transfected with rat TRPV2 and either control or TRPV2 shRNA. (B) PC12 cells were transfected with control shRNA or TRPV2 shRNA. Twenty-four hours later, cells were treated with NGF (100 ng/ml) for 72 h. Cells were then fixed and immunostained for TRPV2 (red). GFP indicates a transfected cell (green), and nuclei were stained with Hoechst dye (blue). Bar, 50 μm. (C) Mean TRPV2 fluorescence intensity ± SEM for GFP-positive cells (control shRNA, n = 30; TRPV2 shRNA, n = 19) (P < 0.001 as determined by an unpaired t test). A.U., arbitrary units. (D) PC12 cells were transfected with control or TRPV2 shRNA, followed by 72 h of treatment with NGF (100 ng/ml). Fifty random images were obtained for GFP fluorescence and analyzed for the percentage of cells with neurites. Data represent the mean percentages of neurite-bearing, GFP-positive cells ± SEM from 3 independent experiments. n.s., not significant (P = 0.94) as determined by an unpaired t test. (E) Mean neurite lengths ± SEM for PC12 cells transfected with control shRNA (n = 362) or TRPV2 shRNA (n = 454) and treated with NGF for 72 h (P < 0.001 as determined by an unpaired t test).
NGF-induced increase in TRPV2 protein levels is mediated by MAPK signaling.
Binding of NGF to its extracellular receptor TrkA initiates intracellular signaling cascades that promote cell survival and differentiation. The PI3K signaling pathway is necessary for NGF-induced survival of PC12 cells, while the MAPK signaling pathway is essential for differentiation and neurite outgrowth in PC12 cells (2, 3). Previous studies suggested that the function of TRPV2 is regulated by growth factors via the PI3K signaling pathway (32, 37, 38). We therefore tested the effects of the PI3K inhibitors LY294002 and wortmannin on NGF-regulated TRPV2 expression. After 24 h of treatment of PC12 cells with NGF, an increase in the TRPV2 protein level was observed; PI3K inhibitors did not have a significant effect on NGF-induced upregulation of TRPV2 protein levels (Fig. 5A). Both LY294002 and wortmannin reduced the phosphorylation of Akt (pAkt) in both the absence and presence of NGF, indicating suppression of PI3K activity (Fig. 5A). Inhibition of MAPK signaling with the MEK1/2 inhibitor U0126 significantly reduced the NGF-induced upregulation of TRPV2 protein levels in PC12 cells (Fig. 5B). U0126 decreased pErk1/2 levels, confirming the inhibition of the MAPK pathway (Fig. 5B). Treatment with U0126 also decreased TRPV2 mRNA levels in PC12 cells after 6 h of treatment with NGF compared to vehicle controls (Fig. 5C). Consistent with data from previous reports (39, 40), U0126 decreased the percentage of neurite-bearing cells and neurite length after NGF treatment compared to the vehicle control and LY294002 (Fig. 5D).
FIG 5.

MAPK signaling mediates upregulation of TRPV2. (A) PC12 cells were pretreated with DMSO, LY294002 (5 μM), or wortmannin (20 nM) for 30 min, followed by treatment with NGF (100 ng/ml for 24 h), as indicated. TRPV2, pAkt, total Akt, and β-actin protein levels were determined by Western blotting. The β-actin-normalized TRPV2 band intensity for DMSO-treated cells in the absence of NGF was defined as 1. Data in the bar graph represent means ± SEM from 3 independent experiments. (B) PC12 cells were pretreated with DMSO or U0126 (10 μM) for 30 min, followed by treatment with NGF (100 ng/ml for 24 h), as indicated. TRPV2, pErk1/2, total Erk1/2, and β-actin protein levels were determined by Western blotting. The β-actin-normalized TRPV2 band intensity for DMSO-treated cells in the absence of NGF was defined as 1. Data in the bar graph represent means ± SEM from 3 independent experiments. (C) PC12 cells were pretreated with DMSO or U0126 (10 μM) for 30 min, followed by treatment with NGF (100 ng/ml for 6 h), as indicated. TRPV2 and GAPDH mRNA levels were determined by RT-PCR. The GAPDH-normalized TRPV2 band intensity for DMSO-treated cells in the absence of NGF was defined as 1. Data in the bar graph represent means ± SEM from 3 independent experiments. (D) Bar graphs representing the percentages of cells with neurites and neurite lengths for PC12 cells cultured in the presence of NGF and either the vehicle (DMSO), LY294002 (5 μM), or U0126 (10 μM). Data represent means ± SEM from 3 independent experiments. (E) PC12 cells were transfected with control or Erk1/2 siRNA for 48 h, followed by 12 h of treatment with NGF, as indicated. TRPV2, pErk1/2, total Erk1/2, and β-actin protein levels were determined by Western blotting. The β-actin-normalized TRPV2 or total Erk1/2 band intensities for control siRNA-transfected cells in the absence of NGF were defined as 1. Data in bar graphs represent means ± SEM from 3 independent experiments. (F) PC12 cells were transfected and treated as described above for panel E, and TRPV2 and GAPDH mRNA levels were determined by RT-PCR. The GAPDH-normalized TRPV2 band intensity for control siRNA-transfected cells in the absence of NGF was defined as 1. Data in the bar graph represent means ± SEM from 4 independent experiments. (G) Bar graphs representing percentages of cells with neurites and neurite lengths for PC12 cells transfected with control or Erk1/2 siRNA and cultured in the presence of NGF for 72 h. Data represent means ± SEM from 5 independent experiments. (H) PC12 cells were treated with NGF (100 ng/ml) for the indicated times, followed by Western blot analysis with TRPV2, pErk1/2, total Erk1/2, and β-actin antibodies. Data in line graphs represent relative β-actin-normalized TRPV2 and total Erk1/2-normalized pErk1/2 levels. Band intensities at the 0-h NGF time point were defined as 1. Data represent means ± SEM from 3 independent experiments. *, P < 0.05 relative to the 0-h NGF time point as determined by one-way ANOVA followed by Dunnett's post hoc test. (I) E18 DRG neurons were cultured in the presence of DMSO, LY294002 (25 μM), or U1026 (10 μM) for 5 days and then immunostained for TRPV2 (green) and βIII-tubulin (red). All images were obtained with identical microscope settings. Bar, 100 μm.
The major effector of neurite outgrowth mediated by MAPK downstream of MEK1/2 is Erk1/2. To determine if Erk1/2 specifically regulates the upregulation of TRPV2, we employed siRNA to reduce Erk1/2 expression. Transfection of PC12 cells with Erk1/2 siRNA reduced Erk1/2 protein levels ∼40 to 50% compared to control siRNA (Fig. 5E). Similar to the MEK1/2 inhibitor, transfection of Erk1/2 siRNA attenuated NGF-induced upregulation of both TRPV2 protein (Fig. 5E) and TRPV2 mRNA (Fig. 5F) in PC12 cells. Additionally, Erk1/2 siRNA reduced NGF-induced neurite outgrowth compared to control siRNA (Fig. 5G). Activation of MAPK/ERK signaling by NGF in PC12 cells peaks after minutes of exposure (41). We observed increases in pErk1/2 levels after 10 min of treatment with NGF, whereas TRPV2 protein levels did not significantly increase until 6 h of exposure to NGF (Fig. 5H). This shows that activation of MAPK/ERK signaling precedes the upregulation of TRPV2 expression, and TRPV2 is regulated downstream of Erk1/2.
Importantly, we also observed a decrease in TRPV2 immunofluorescence in primary embryonic DRG neurons cultured in the presence of U1026 for 5 days compared to cells cultured with DMSO or LY294002 (Fig. 5I). These results indicate that the MAPK signaling pathway, which promotes neurite outgrowth, contributes to the NGF-induced upregulation of TRPV2 expression in both PC12 cells and primary developing sensory neurons.
NGF does not induce TRPV2 translocation to the plasma membrane in PC12 cells.
Growth factors such as insulin-like growth factor 1 (IGF-1) were initially thought to induce the translocation of TRPV2 to the plasma membrane through the PI3K signaling pathway, where it mediates a Ca2+ current that contributes to growth factor signaling (37). Using our monoclonal TRPV2 antibody, however, we previously found that IGF-1 had no effect on the plasma membrane translocation of overexpressed and endogenous TRPV2 (21). Since we observed that PI3K signaling had no effect on the upregulation of TRPV2, we next tested if NGF treatment increased plasma membrane levels of TRPV2 in PC12 cells. Cell surface biotinylation experiments failed to detect TRPV2 at the surface of PC12 cells in both the absence and presence of NGF (Fig. 6A). In contrast, TRPV3 was present in the biotinylated fraction in both the absence and presence of NGF, and surface levels of TRPV3 did not change in response to NGF treatment (Fig. 6B). Additionally, we found that TRPV2 colocalized with Rab7, a late endosomal marker, after NGF treatment (Fig. 6C), further indicating that TRPV2 does not translocate to the cell surface after NGF stimulation. Consistent with results from our laboratory and others showing that IGF-1 and other growth factors have no effect on the surface levels of TRPV2 (21, 32, 42), these data show that while NGF treatment increases TRPV2 content, it does not induce the translocation of the channel to the cell surface. The absence of TRPV2 in the biotinylated fraction indicates that TRPV2 predominantly localizes to intracellular membranes in PC12 cells.
FIG 6.
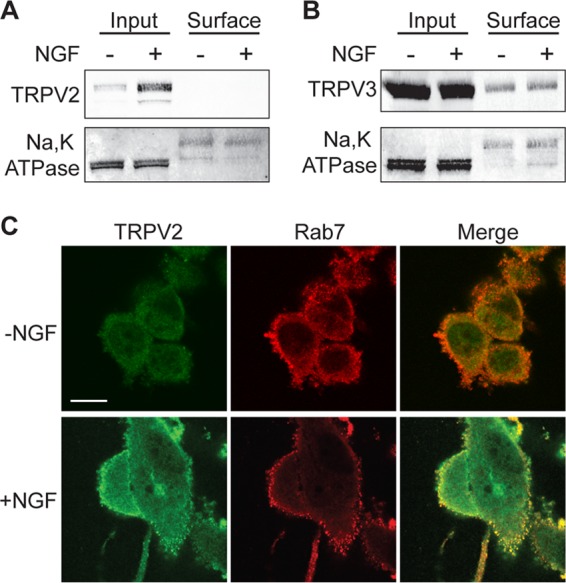
NGF does not induce translocation of TRPV2 to the plasma membrane in PC12 cells. (A) PC12 cells were cultured in the absence or presence of NGF (100 ng/ml) for 24 h. Surface proteins were biotinylated in intact cells at 37°C. Cells were then lysed, and biotinylated proteins were captured with streptavidin = agarose. Captured proteins were resolved by SDS-PAGE and detected by Western blotting with TRPV2 and Na/K ATPase antibodies. Na/K ATPase was used as a positive control. Surface proteins represent the biotinylated fraction, and input represents 5% of the total extracted protein. (B) Cell surface biotinylation experiment performed as described above for panel A. TRPV3 and Na/K ATPase antibodies were used for Western blot analysis. (C) PC12 cells were cultured in the absence or presence of NGF (100 ng/ml) for 24 h, followed by fixation and staining for TRPV2 (green) and Rab7 (red). Bar, 10 μm.
ERK phosphorylates TRPV2 to enhance neurite outgrowth.
When NGF binds and activates TrkA at the terminals of developing neurites, the NGF-TrkA complex is internalized into endosomes and transported along microtubules toward the cell body in a retrograde manner, where it continues to signal through PI3K and MAPK (43). Activated Erk1/2 is closely associated with these TrkA-containing endosomes, where it can facilitate local signaling throughout the developing neuronal processes (22). Since we observed that TRPV2 colocalized with an endosomal marker in PC12 cells, and TRPV2 immunoreactivity is present in both the cell body and βIII-tubulin-containing neurites of embryonic DRG neurons (Fig. 1A), we next tested if TRPV2 is associated with these NGF signaling endosomes in embryonic DRG neurons. TRPV2 immunoreactivity showed substantial colocalization with Rab7 (Fig. 7A), which is essential for retrograde transport of Trk-containing signaling endosomes in developing neurons (23). Partial colocalization between TRPV2 and both TrkA (Fig. 7B) and pErk1/2 (Fig. 7C) in embryonic DRG neurons was also observed. These results suggested that ERK might directly modulate TRPV2 protein within neurites of developing neurons.
FIG 7.
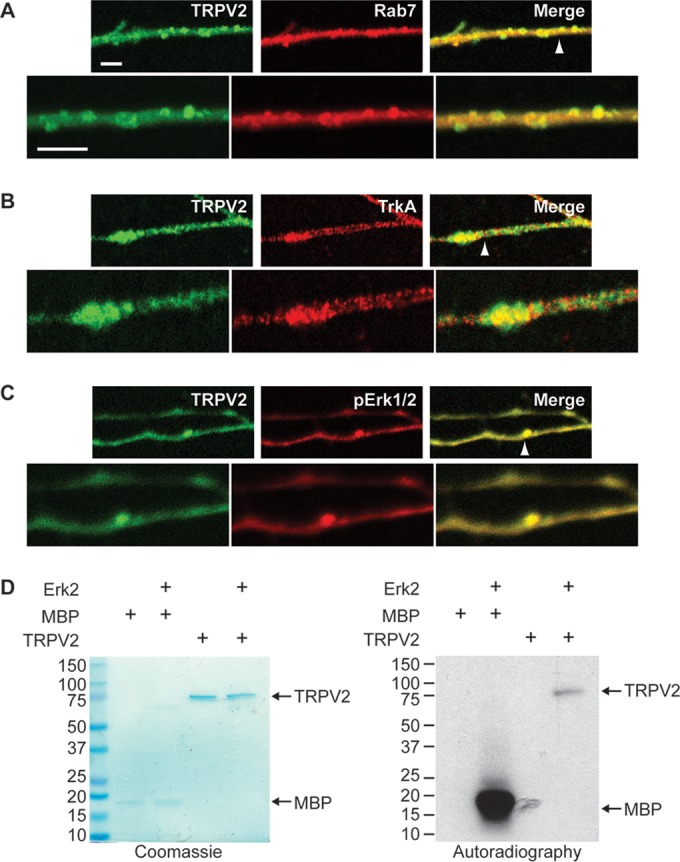
TRPV2 is phosphorylated by ERK. (A to C) E18 DRG neurons were cultured for 5 days in the presence of NGF and then fixed and immunostained for TRPV2 (green) and Rab7 (red) (A), TrkA (red) (B), or pErk1/2 (red) (C). The arrowheads point to regions of zoomed images represented in the bottom panels. Bars, 5 μm. (D) Recombinant MBP or TRPV2 (500 ng) was incubated in the absence or presence of recombinant active Erk2 (100 ng) and [γ-32P]ATP, as indicated. Phosphorylation of MBP or TRPV2 was detected by autoradiography. The presence of recombinant proteins was confirmed by Coomassie staining.
To test if ERK phosphorylates TRPV2, recombinant activated Erk2 was incubated with [γ-32P]ATP and either myelin basic protein (MBP), a known ERK substrate, or pure tetrameric TRPV2 (25). TRPV2 and MBP were both phosphorylated in the presence but not in the absence of activated Erk2 (Fig. 7D), suggesting that TRPV2 is a viable ERK substrate and that ERK may directly phosphorylate TRPV2 through NGF signaling.
Posttranslational modifications of TRPV2 have not been extensively characterized. To directly test if phosphorylation of TRPV2 by ERK affects NGF-induced neurite outgrowth, we first needed to determine the possible TRPV2 residues that ERK phosphorylates. Mass spectrometry analysis of TRPV2 expressed in HEK293T cells revealed several phosphorylation sites. Four identified Ser residues were within proline-directed motifs (Ser/Thr-Pro) and predicted to act as ERK substrates (Ser6, Ser37, Ser47, and Ser760) (Fig. 8A to D). All four sites are within flexible regions of the distal N and C termini of the protein (Fig. 8E). Mutation of all four sites to Ala (S6A, S37A, S47A, and S760A) (TRPV2Δ4S) substantially reduced the phosphorylation of TRPV2 by Erk2 in vitro (Fig. 9A). In vitro phosphorylation experiments for single Ser-to-Ala mutants yielded inconclusive results, suggesting that multiple Ser residues are phosphorylated by ERK (data not shown).
FIG 8.

Mass spectrometry analysis of heterologously expressed TRPV2 to determine phosphorylation sites. (A) MS/MS spectra of the phosphorylated form of the TSASSPPAFR peptide (positions 2 to 11) observed as a doubly protonated ion at m/z 550.74. The presence of y6-y8, b5-98, and b9-98 fragment ions with a 79.97-Da mass shift shows that modification of this peptide occurred at S6. Asterisks indicate the fragment ions that were modified by 79.97 Da. (B) MS/MS spectra of the modified form of the QEPPPMESPFQRE peptide (positions 30 to 42) observed as a doubly protonated ion at m/z 834.34. The presence of y6-y7 fragment ions with a 79.97-Da mass shift and y8-y11 ions with 79.97-Da and 16-Da mass shifts shows that phosphorylation and oxidation of this peptide occurred at positions S37 and M35, respectively. Asterisks indicate the fragment ions that were modified by 79.97 Da. (C) MS/MS spectra of the phosphorylated form of the DRNSSPQIKVNL peptide (positions 43 to 54) observed as a doubly protonated ion at m/z 725.85. The presence of y8-y10 and b5-b10 fragment ions with a 79.97-Da mass shift shows that modification of this peptide occurred at S47. Asterisks indicate the fragment ions that were modified by 79.97 Da. (D) MS/MS spectra of the phosphorylated form of the DHLPLQVLQS peptide (positions 751 to 760) observed as a doubly protonated ion at m/z 615.30. The presence of y1-y7 fragment ions with a 79.97-Da mass shift and unmodified b5-b9 fragment ions shows that modification of this peptide occurred at S760. Asterisks indicate the fragment ions that were modified by 79.97 Da. (E) Domain structure of a TRPV2 monomer. TRPV2 consists of large soluble N and C termini. The N terminus contains six ankyrin repeats that form the ankyrin repeat domain. The four predicted ERK phosphorylation sites (indicated by arrows) reside on the distal N and C termini.
FIG 9.

Phosphorylation of TRPV2 by ERK enhances NGF-induced neurite outgrowth in PC12 cells. (A) One microgram of recombinant TRPV2, WT TRPV2, or TRPV2Δ4S isolated from HEK293T cells was incubated with activated Erk2 and [γ-32P]ATP. Phosphorylation of the TRPV2 proteins was determined by autoradiography, and the presence of protein was confirmed by Coomassie staining. (B) Representative images of PC12 cells transfected with GFP plus either the empty vector or 1D4-tagged WT TRPV2 or TRPV2Δ4S and treated with NGF (100 ng/ml) in either the absence or presence of U0126 (10 μM) for 72 h. (C) Percentage of cells with neurites, for PC12 cells imaged in panel B. Data represent means ± SEM from 3 independent experiments. (D) Mean neurite lengths ± SEM for cells imaged in panel B (vector control plus DMSO, n = 265; vector control plus U0126, n = 126; WT TRPV2 plus DMSO, n = 181; WT TRPV2 plus U0126, n = 93; TRPV2Δ4S plus DMSO, n = 195; TRPV2Δ4S plus U0126, n = 119). (E) Western blot analysis of PC12 cells transfected and treated as described above for panels B to D with the indicated antibodies. Data in the bar graph represent mean band intensities ± SEM from 3 independent experiments. The β-actin-normalized TRPV2-1D4 band intensity for WT-TRPV2-expressing, DMSO-treated cells was defined as 1. (F) Western blot analysis with the indicated antibodies for PC12 cells transfected with control or Erk1/2 siRNA for 24 h, followed by 24 h of transfection with GFP plus the vector or 1D4-tagged WT TRPV2 or TRPV2Δ4S and 72 h of treatment with NGF. Data in the bar graph represent mean band intensities ± SEM from 3 independent experiments. The β-actin-normalized TRPV2-1D4 band intensity for cells transfected with control siRNA and WT TRPV2 was defined as 1. (G) Bar graph representing the percentage of PC12 cells with neurites, transfected and treated as described above for panel F. Data represent means ± SEM from 3 independent experiments. (H) Mean neurite length ± SEM for cells transfected and treated as described above for panel F (vector plus control siRNA, n = 145; vector plus Erk1/2 siRNA, n = 151; WT TRPV2 plus control siRNA, n = 162; WT TRPV2 plus Erk1/2 siRNA, n = 123; TRPV2Δ4S plus control siRNA, n = 138; TRPV2Δ4S plus Erk1/2 siRNA, n = 103). (I) Intracellular Ca2+ transients measured by Fluo4 fluorescence using time-lapse microscopy. F11 cells expressing WT TRPV2 or TRPV2Δ4S were treated with 2-APB (100 μM) at the 45-s time point (indicated by arrows). Data represent mean normalized Fluo4 intensities. Data in the bar graph represent peaks in normalized Fluo4 fluorescence intensity determined under each condition (WT TRPV2 plus DMSO, n = 177; WT TRPV2 plus U0126, n = 129; TRPV2Δ4S plus DMSO, n = 77; TRPV2Δ4S plus U0126, n = 125). Data represent the means ± SEM.
We next tested if the phosphorylation of TRPV2 by ERK affected NGF-induced neurite outgrowth in PC12 cells. We employed a protocol in which cells were transfected with the vector control, WT TRPV2, or TRPV2Δ4S followed by a 30-min pretreatment with either the vehicle or U0126 and a 72-h treatment with NGF. U0126 significantly reduced the percentage of cells possessing a neurite under all transfection conditions (Fig. 9B and C). Overexpression of TRPV2Δ4S did not significantly affect the percentage of cells possessing a neurite compared to WT TRPV2; however, the neurite length for cells transfected with TRPV2Δ4S was significantly reduced compared to that for cells transfected with WT TRPV2 in the absence of U0126 (Fig. 9B and D). Interestingly, U0126 reduced neurite length for vector control- and WT TRPV2-transfected cells but not cells expressing TRPV2Δ4S (Fig. 9D). Pretreatment with U0126 reduced the expression of exogenously expressed WT TPRV2 in PC12 cells after NGF exposure (Fig. 9E). Additionally, the expression level of TRPV2Δ4S was significantly lower than that of WT TRPV2 in the absence of the inhibitor, and levels of TRPV2Δ4S were unaffected by U0126 (Fig. 9E). Silencing of Erk1/2 had similar effects on neurite outgrowth mediated by WT TRPV2 and TRPV2Δ4S; namely, Erk1/2 siRNA reduced protein levels (Fig. 9F) and NGF-induced neurite outgrowth for exogenously expressed WT TRPV2 but not TRPV2Δ4S (Fig. 9G and H). Erk1/2 siRNA efficiently reduced Erk1/2 protein levels under these conditions (vector, 68.2% ± 0.66% of control siRNA; WT TRPV2, 69.6% ± 0.61% of control siRNA; TRPV2Δ4S, 66.1% ± 0.44% of control siRNA). These results indicate that while inhibition of ERK signaling decreases the expression of WT TRPV2 and reduces neurite outgrowth mediated by WT TRPV2, it has little to no effect on TRPV2Δ4S. This is consistent with the hypothesis that ERK phosphorylates TRPV2 at these N- and C-terminal Ser residues downstream of NGF to enhance neurite outgrowth.
Since we previously associated TRPV2-mediated Ca2+ changes with neurite outgrowth, we predicted that expression of TRPV2Δ4S might decrease TRPV2-mediated Ca2+ transients. Ca2+ imaging analysis showed that the response of TRPV2Δ4S to 2-APB was significantly reduced compared to that of WT TRPV2 in F11 cells (Fig. 9I). Additionally, U0126 reduced the response of WT TRPV2-expressing cells to 2-APB but not that of cells expressing TRPV2Δ4S (Fig. 9I). Overall, these results indicate that phosphorylation of TRPV2 by ERK enhances TRPV2-mediated expression and Ca2+ signals to increase neurite outgrowth downstream of NGF.
DISCUSSION
TRPV2 was discovered as a growth factor-regulated Ca2+-permeable ion channel in mouse fibroblasts (37). The channel was also originally proposed to act as a noxious temperature detector in sensory neurons (9). TRPV2 knockout mice, however, display normal sensory transduction, including temperature sensation, suggesting that TRPV2 does not function as a noxious heat sensor in vivo (14, 16). Additionally, the mechanisms by which growth factors regulate endogenous TRPV2 function remain unclear (30, 42).
TRPV2 is ubiquitously expressed, in contrast to the other thermoTRPV proteins (44), further signifying that it plays some other important physiological role. Endogenously expressed TRPV2 has been detected primarily in intracellular membranes, as opposed to the cell surface, including endosomes (45–47). It has been proposed that TRPV2 displays constitutive basal activity, and to date, there are no confirmed endogenous channel modulators, while exogenous TRPV2 activators and inhibitors are promiscuous Ca2+ channel modulators (30). The channel has been implicated in numerous different physiological and pathological processes, including growth factor signaling, macrophage migration, cancer cell metastasis, heart disease, and neuronal cell development (18, 30, 34, 37, 38, 44, 48). Nonetheless, the function of endogenous TRPV2 remains controversial due to the lack of reliable tools available to specifically modulate and detect the channel protein (30).
Generation of monoclonal TRPV2 antibodies has allowed us to begin exploring the endogenous function of TRPV2 (21). Strong evidence points toward a role for TRPV2 in the development of peripheral neurons (14, 18). Global knockout of TRPV2 in mice resulted in perinatal lethality (14). Furthermore, TRPV2 mRNA was detected in the mouse spinal cord at E9.5, before the expression of other thermoTRPV channels (18). Using our newly generated antibodies, we observed TRPV2 protein in embryonic DRG neurons and PC12 cells, both of which are well-established models for studying peripheral neuron development and neurotrophin signaling (49). Treatment of PC12 cells with NGF, which is essential for the survival and differentiation of developing peripheral neurons, led to a sustained increase in TRPV2 protein content without affecting plasma membrane levels of TRPV2. Since evidence indicates that TRPV2 displays constitutive basal activity (18, 32), upregulation of TRPV2 protein content is likely a major mechanism of functional regulation of the channel by NGF.
Ca2+ is an important second messenger in NGF-mediated neuronal cell differentiation (29). Increases in cytoplasmic Ca2+ levels are associated with cytoskeletal rearrangements leading to morphological changes, alterations in gene transcription, and changes in cell excitability (29). Here we observed that overexpression of a Ca2+-impermeable TRPV2 mutant in PC12 cells reduced NGF-induced neurite outgrowth. Silencing of TRPV2 expression also decreased neurite length in PC12 cells after NGF treatment, further indicating that NGF-induced upregulation of functional TRPV2 likely alters intracellular Ca2+ levels in developing neurons, leading to increases in neurite extension. Inhibiting or silencing of TRPV2 did not completely impair neurite outgrowth in PC12 cells, consistent with evidence indicating that multiple Ca2+-permeable channels are involved in neurite outgrowth (50). These data are in line with data from a previous report showing that silencing or inhibition of TRPV2 reduces axon growth in primary DRG neurons (18). Abnormal peripheral neuron development may account for the embryonic abnormalities and eventual perinatal lethality observed for TRPV2-null mice (14).
We further explored the molecular details by which TRPV2 is regulated by NGF. NGF signaling begins at the axon terminal of developing neurons. Binding of NGF to its receptor TrkA at the cell surface causes receptor dimerization, autophosphorylation, and activation of signaling pathways, including PI3K and MAPK (43). After NGF binding, the NGF-TrkA complex is internalized into endosomes, where TrkA can continue to signal as it is transported along microtubules toward the neuronal cell body (22, 43). Although PI3K signaling has previously been linked to TRPV2 trafficking and activity (32, 37), we found no connection between PI3K activity and NGF-induced upregulation of TRPV2 protein levels. In contrast, pharmacological inhibition of MAPK signaling significantly reduced TRPV2 protein levels in both primary developing neurons and PC12 cells. Additionally, siRNA-mediated inhibition of MAPK signaling reduced TRPV2 expression in PC12 cells.
TrkA-containing endosomes are thought to be long-lived and enriched in NGF signaling components such as MAPKs (22, 43). These endosomal platforms allow for NGF-TrkA to meet local signaling needs within neuronal extensions and affect gene transcription once the endosomes reach the nucleus in the cell body (51). We found that the Ca2+-permeable protein TRPV2 displays a punctate staining pattern in the extensions of embryonic DRG neurons. TRPV2 colocalized with Rab7, a marker for late endosomes, in addition to both TrkA and pErk1/2 in neurite extensions, suggesting that these important neurotrophin signaling components populate similar endosomal pools as TRPV2. Additionally, we observed that Erk2 phosphorylates TRPV2 in vitro, suggesting that ERK may directly modulate TRPV2 and, thus, intracellular Ca2+ signaling within developing neurites.
Posttranslational modifications of TRPV2 are not well characterized, although we identified four Ser residues on its distal N and C termini of the channel predicted to be ERK substrates (52). Two of these predicted residues (Ser6 and Ser760) were identified as phosphorylation sites for TRPV2 from nervous system tissues in a phosphoproteomic study (53), while two other sites (Ser37 and Ser47) were identified in macrophages (54). Mutation of these predicted ERK sites reduced the phosphorylation of TRPV2 by Erk2 in vitro as well as NGF-mediated neurite outgrowth, TRPV2 protein expression, and 2-APB-induced Ca2+ transients. Overall, these data are consistent with a mechanism by which ERK phosphorylates TRPV2, increasing channel expression and Ca2+ signals to enhance neurite outgrowth in developing neurons.
Based on our studies, we propose that MAPK signaling regulates TRPV2 downstream of NGF by altering the expression of TRPV2 and directly through phosphorylation of the channel by ERK. Our data indicate that TRPV2 localizes to Rab7-positive late endosomes, which are involved in retrograde trafficking on NGF and TrkA (23). TRPV2 activity within endosomes would mediate Ca2+ flux from the endosomal lumen to the cytoplasm of the neurite. The close association between TRPV2 and NGF-TrkA within signaling endosomes allows for direct modulation of the TRPV2 protein by MAPK signaling components within developing neuronal processes (22). ERK directly phosphorylates TRPV2 on the cytoplasmic N and C termini of the channel, leading to increased TRPV2 protein content, alterations in local Ca2+ signaling within neurites, and augmented neurite outgrowth (Fig. 10). Localization of TRPV2 to signaling endosomes provides an efficient and versatile mechanism for regulating Ca2+ signaling within long neurites of developing neurons without directly changing neuronal excitability (55). Downstream effects of TRPV2 activity remain to be explored, although one possibility is that TRPV2-mediated Ca2+ signals derived from endosomes might mediate endosomal maturation and fusion (56).
FIG 10.
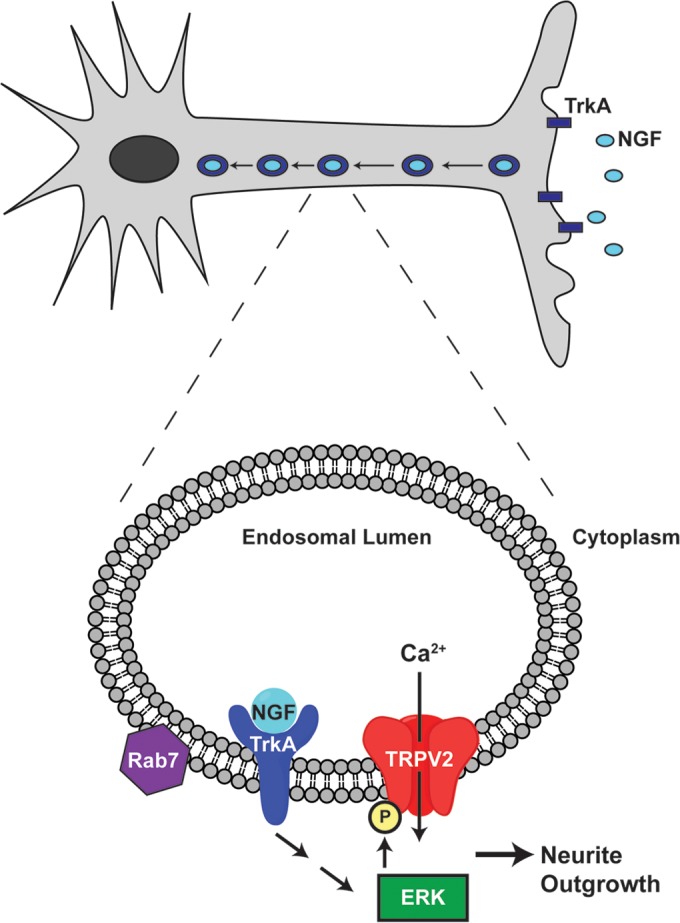
Model depicting the proposed mechanism by which NGF-activated MAPK signaling affects TRPV2 to enhance neurite outgrowth. Target-derived NGF binds to TrkA at the end of neurites. The complex is internalized into signaling endosomes and transported toward the nucleus in the cell body. At the nucleus, ERK signaling can increase TRPV2 expression. Within endosomes in the neurite shaft, TrkA activates ERK, which can phosphorylate TRPV2. Changes in Ca2+ due to increased TRPV2 protein expression and activity then enhance neurite outgrowth.
In addition, TRPV2 expression has been shown to be upregulated in DRG neurons after peripheral nerve injury (57, 58). Injury is thought to activate intrinsic neuronal growth programs, including neurotrophin signaling, to promote axon regrowth (59). Neurotrophin release in response to injury also leads to inflammatory signaling, hyperalgesia, and aberrant sprouting in peripheral sensory neurons, resulting in potentially debilitating neuropathic pain (60–67). Increased expression of TRPV2 was thought to contribute directly to pain sensation after nerve injury (58). Nevertheless, based on recent TRPV2 genetic deletion studies (14) coupled with our current findings, it is possible that TRPV2 functions directly in the regeneration of sensory neurons after injury. Further studies to delineate the role of TRPV2 in neuronal sprouting and growth are needed to determine if TRPV2 represents a molecular player and potential therapeutic target in neurite outgrowth related to injury-induced neuropathic pain.
ACKNOWLEDGMENTS
This work was supported by American Heart Association NCRP scientist development grant 11SDG5280029 and National Institute of Diabetes and Digestive and Kidney Diseases grant DK097223.
We thank Shasta Sabo and Karlie Fedder for assistance with Ca2+ imaging experiments and Denice Major and Dawn Smith for assistance with hybridoma culture and F11 cell culture (Case Western Tissue Culture and Hybridoma Core is supported by National Institutes of Health core grant EY11373).
REFERENCES
- 1.Reichardt LF. 2006. Neurotrophin-regulated signalling pathways. Philos Trans R Soc Lond B Biol Sci 361:1545–1564. doi: 10.1098/rstb.2006.1894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Vaudry D, Stork PJ, Lazarovici P, Eiden LE. 2002. Signaling pathways for PC12 cell differentiation: making the right connections. Science 296:1648–1649. doi: 10.1126/science.1071552. [DOI] [PubMed] [Google Scholar]
- 3.Klesse LJ, Meyers KA, Marshall CJ, Parada LF. 1999. Nerve growth factor induces survival and differentiation through two distinct signaling cascades in PC12 cells. Oncogene 18:2055–2068. doi: 10.1038/sj.onc.1202524. [DOI] [PubMed] [Google Scholar]
- 4.Shim S, Goh EL, Ge S, Sailor K, Yuan JP, Roderick HL, Bootman MD, Worley PF, Song H, Ming GL. 2005. XTRPC1-dependent chemotropic guidance of neuronal growth cones. Nat Neurosci 8:730–735. doi: 10.1038/nn1459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kumar S, Chakraborty S, Barbosa C, Brustovetsky T, Brustovetsky N, Obukhov AG. 2012. Mechanisms controlling neurite outgrowth in a pheochromocytoma cell line: the role of TRPC channels. J Cell Physiol 227:1408–1419. doi: 10.1002/jcp.22855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Heiser JH, Schuwald AM, Sillani G, Ye L, Muller WE, Leuner K. 2013. TRPC6 channel-mediated neurite outgrowth in PC12 cells and hippocampal neurons involves activation of RAS/MEK/ERK, PI3K, and CAMKIV signaling. J Neurochem 127:303–313. doi: 10.1111/jnc.12376. [DOI] [PubMed] [Google Scholar]
- 7.Wang GX, Poo MM. 2005. Requirement of TRPC channels in netrin-1-induced chemotropic turning of nerve growth cones. Nature 434:898–904. doi: 10.1038/nature03478. [DOI] [PubMed] [Google Scholar]
- 8.Heo DK, Chung WY, Park HW, Yuan JP, Lee MG, Kim JY. 2012. Opposite regulatory effects of TRPC1 and TRPC5 on neurite outgrowth in PC12 cells. Cell Signal 24:899–906. doi: 10.1016/j.cellsig.2011.12.011. [DOI] [PubMed] [Google Scholar]
- 9.Caterina MJ, Rosen TA, Tominaga M, Brake AJ, Julius D. 1999. A capsaicin-receptor homologue with a high threshold for noxious heat. Nature 398:436–441. doi: 10.1038/18906. [DOI] [PubMed] [Google Scholar]
- 10.Caterina MJ, Schumacher MA, Tominaga M, Rosen TA, Levine JD, Julius D. 1997. The capsaicin receptor: a heat-activated ion channel in the pain pathway. Nature 389:816–824. doi: 10.1038/39807. [DOI] [PubMed] [Google Scholar]
- 11.Guler AD, Lee H, Iida T, Shimizu I, Tominaga M, Caterina M. 2002. Heat-evoked activation of the ion channel, TRPV4. J Neurosci 22:6408–6414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Smith GD, Gunthorpe MJ, Kelsell RE, Hayes PD, Reilly P, Facer P, Wright JE, Jerman JC, Walhin JP, Ooi L, Egerton J, Charles KJ, Smart D, Randall AD, Anand P, Davis JB. 2002. TRPV3 is a temperature-sensitive vanilloid receptor-like protein. Nature 418:186–190. doi: 10.1038/nature00894. [DOI] [PubMed] [Google Scholar]
- 13.Huang SM, Li X, Yu Y, Wang J, Caterina MJ. 2011. TRPV3 and TRPV4 ion channels are not major contributors to mouse heat sensation. Mol Pain 7:37. doi: 10.1186/1744-8069-7-37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Park U, Vastani N, Guan Y, Raja SN, Koltzenburg M, Caterina MJ. 2011. TRP vanilloid 2 knock-out mice are susceptible to perinatal lethality but display normal thermal and mechanical nociception. J Neurosci 31:11425–11436. doi: 10.1523/JNEUROSCI.1384-09.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Caterina MJ, Leffler A, Malmberg AB, Martin WJ, Trafton J, Petersen-Zeitz KR, Koltzenburg M, Basbaum AI, Julius D. 2000. Impaired nociception and pain sensation in mice lacking the capsaicin receptor. Science 288:306–313. doi: 10.1126/science.288.5464.306. [DOI] [PubMed] [Google Scholar]
- 16.Vriens J, Nilius B, Voets T. 2014. Peripheral thermosensation in mammals. Nat Rev Neurosci 15:573–589. doi: 10.1038/nrn3784. [DOI] [PubMed] [Google Scholar]
- 17.Jang Y, Jung J, Kim H, Oh J, Jeon JH, Jung S, Kim KT, Cho H, Yang DJ, Kim SM, Kim IB, Song MR, Oh U. 2012. Axonal neuropathy-associated TRPV4 regulates neurotrophic factor-derived axonal growth. J Biol Chem 287:6014–6024. doi: 10.1074/jbc.M111.316315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shibasaki K, Murayama N, Ono K, Ishizaki Y, Tominaga M. 2010. TRPV2 enhances axon outgrowth through its activation by membrane stretch in developing sensory and motor neurons. J Neurosci 30:4601–4612. doi: 10.1523/JNEUROSCI.5830-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Deng HX, Klein CJ, Yan J, Shi Y, Wu Y, Fecto F, Yau HJ, Yang Y, Zhai H, Siddique N, Hedley-Whyte ET, Delong R, Martina M, Dyck PJ, Siddique T. 2010. Scapuloperoneal spinal muscular atrophy and CMT2C are allelic disorders caused by alterations in TRPV4. Nat Genet 42:165–169. doi: 10.1038/ng.509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Landoure G, Zdebik AA, Martinez TL, Burnett BG, Stanescu HC, Inada H, Shi Y, Taye AA, Kong L, Munns CH, Choo SS, Phelps CB, Paudel R, Houlden H, Ludlow CL, Caterina MJ, Gaudet R, Kleta R, Fischbeck KH, Sumner CJ. 2010. Mutations in TRPV4 cause Charcot-Marie-tooth disease type 2C. Nat Genet 42:170–174. doi: 10.1038/ng.512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cohen MR, Huynh KW, Cawley D, Moiseenkova-Bell VY. 2013. Understanding the cellular function of TRPV2 channel through generation of specific monoclonal antibodies. PLoS One 8:e85392. doi: 10.1371/journal.pone.0085392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Delcroix JD, Valletta JS, Wu C, Hunt SJ, Kowal AS, Mobley WC. 2003. NGF signaling in sensory neurons: evidence that early endosomes carry NGF retrograde signals. Neuron 39:69–84. doi: 10.1016/S0896-6273(03)00397-0. [DOI] [PubMed] [Google Scholar]
- 23.Deinhardt K, Salinas S, Verastegui C, Watson R, Worth D, Hanrahan S, Bucci C, Schiavo G. 2006. Rab5 and Rab7 control endocytic sorting along the axonal retrograde transport pathway. Neuron 52:293–305. doi: 10.1016/j.neuron.2006.08.018. [DOI] [PubMed] [Google Scholar]
- 24.Molday RS, MacKenzie D. 1983. Monoclonal antibodies to rhodopsin: characterization, cross-reactivity, and application as structural probes. Biochemistry 22:653–660. doi: 10.1021/bi00272a020. [DOI] [PubMed] [Google Scholar]
- 25.Huynh KW, Cohen MR, Chakrapani S, Holdaway HA, Stewart PL, Moiseenkova-Bell VY. 2014. Structural insight into the assembly of TRPV channels. Structure 22:260–268. doi: 10.1016/j.str.2013.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Xu H, Freitas MA. 2007. A mass accuracy sensitive probability based scoring algorithm for database searching of tandem mass spectrometry data. BMC Bioinformatics 8:133. doi: 10.1186/1471-2105-8-133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Greene LA, Tischler AS. 1976. Establishment of a noradrenergic clonal line of rat adrenal pheochromocytoma cells which respond to nerve growth factor. Proc Natl Acad Sci U S A 73:2424–2428. doi: 10.1073/pnas.73.7.2424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jahnel R, Bender O, Munter LM, Dreger M, Gillen C, Hucho F. 2003. Dual expression of mouse and rat VRL-1 in the dorsal root ganglion derived cell line F-11 and biochemical analysis of VRL-1 after heterologous expression. Eur J Biochem 270:4264–4271. doi: 10.1046/j.1432-1033.2003.03811.x. [DOI] [PubMed] [Google Scholar]
- 29.Sutherland DJ, Pujic Z, Goodhill GJ. 2014. Calcium signaling in axon guidance. Trends Neurosci 37:424–432. doi: 10.1016/j.tins.2014.05.008. [DOI] [PubMed] [Google Scholar]
- 30.Peralvarez-Marin A, Donate-Macian P, Gaudet R. 2013. What do we know about the transient receptor potential vanilloid 2 (TRPV2) ion channel? FEBS J 280:5471–5487. doi: 10.1111/febs.12302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lev S, Minke B. 2010. Constitutive activity of TRP channels methods for measuring the activity and its outcome. Methods Enzymol 484:591–612. doi: 10.1016/B978-0-12-381298-8.00029-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Penna A, Juvin V, Chemin J, Compan V, Monet M, Rassendren FA. 2006. PI3-kinase promotes TRPV2 activity independently of channel translocation to the plasma membrane. Cell Calcium 39:495–507. doi: 10.1016/j.ceca.2006.01.009. [DOI] [PubMed] [Google Scholar]
- 33.Bang S, Kim KY, Yoo S, Lee SH, Hwang SW. 2007. Transient receptor potential V2 expressed in sensory neurons is activated by probenecid. Neurosci Lett 425:120–125. doi: 10.1016/j.neulet.2007.08.035. [DOI] [PubMed] [Google Scholar]
- 34.Iwata Y, Katanosaka Y, Arai Y, Shigekawa M, Wakabayashi S. 2009. Dominant-negative inhibition of Ca2+ influx via TRPV2 ameliorates muscular dystrophy in animal models. Hum Mol Genet 18:824–834. doi: 10.1093/hmg/ddn408. [DOI] [PubMed] [Google Scholar]
- 35.Hu HZ, Gu Q, Wang C, Colton CK, Tang J, Kinoshita-Kawada M, Lee LY, Wood JD, Zhu MX. 2004. 2-Aminoethoxydiphenyl borate is a common activator of TRPV1, TRPV2, and TRPV3. J Biol Chem 279:35741–35748. doi: 10.1074/jbc.M404164200. [DOI] [PubMed] [Google Scholar]
- 36.Juvin V, Penna A, Chemin J, Lin YL, Rassendren FA. 2007. Pharmacological characterization and molecular determinants of the activation of transient receptor potential V2 channel orthologs by 2-aminoethoxydiphenyl borate. Mol Pharmacol 72:1258–1268. doi: 10.1124/mol.107.037044. [DOI] [PubMed] [Google Scholar]
- 37.Kanzaki M, Zhang YQ, Mashima H, Li L, Shibata H, Kojima I. 1999. Translocation of a calcium-permeable cation channel induced by insulin-like growth factor-I. Nat Cell Biol 1:165–170. doi: 10.1038/11086. [DOI] [PubMed] [Google Scholar]
- 38.Link TM, Park U, Vonakis BM, Raben DM, Soloski MJ, Caterina MJ. 2010. TRPV2 has a pivotal role in macrophage particle binding and phagocytosis. Nat Immunol 11:232–239. doi: 10.1038/ni.1842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Das KP, Freudenrich TM, Mundy WR. 2004. Assessment of PC12 cell differentiation and neurite growth: a comparison of morphological and neurochemical measures. Neurotoxicol Teratol 26:397–406. doi: 10.1016/j.ntt.2004.02.006. [DOI] [PubMed] [Google Scholar]
- 40.Obara Y, Yamauchi A, Takehara S, Nemoto W, Takahashi M, Stork PJ, Nakahata N. 2009. ERK5 activity is required for nerve growth factor-induced neurite outgrowth and stabilization of tyrosine hydroxylase in PC12 cells. J Biol Chem 284:23564–23573. doi: 10.1074/jbc.M109.027821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Santos SD, Verveer PJ, Bastiaens PI. 2007. Growth factor-induced MAPK network topology shapes Erk response determining PC-12 cell fate. Nat Cell Biol 9:324–330. doi: 10.1038/ncb1543. [DOI] [PubMed] [Google Scholar]
- 42.Bezzerides VJ, Ramsey IS, Kotecha S, Greka A, Clapham DE. 2004. Rapid vesicular translocation and insertion of TRP channels. Nat Cell Biol 6:709–720. doi: 10.1038/ncb1150. [DOI] [PubMed] [Google Scholar]
- 43.Harrington AW, Ginty DD. 2013. Long-distance retrograde neurotrophic factor signalling in neurons. Nat Rev Neurosci 14:177–187. doi: 10.1038/nrn3253. [DOI] [PubMed] [Google Scholar]
- 44.Uhlen M, Fagerberg L, Hallstrom BM, Lindskog C, Oksvold P, Mardinoglu A, Sivertsson A, Kampf C, Sjostedt E, Asplund A, Olsson I, Edlund K, Lundberg E, Navani S, Szigyarto CA, Odeberg J, Djureinovic D, Takanen JO, Hober S, Alm T, Edqvist PH, Berling H, Tegel H, Mulder J, Rockberg J, Nilsson P, Schwenk JM, Hamsten M, von Feilitzen K, Forsberg M, Persson L, Johansson F, Zwahlen M, von Heijne G, Nielsen J, Ponten F. 2015. Proteomics tissue-based map of the human proteome. Science 347:1260419. doi: 10.1126/science.1260419. [DOI] [PubMed] [Google Scholar]
- 45.Saito M, Hanson PI, Schlesinger P. 2007. Luminal chloride-dependent activation of endosome calcium channels: patch clamp study of enlarged endosomes. J Biol Chem 282:27327–27333. doi: 10.1074/jbc.M702557200. [DOI] [PubMed] [Google Scholar]
- 46.Dong XP, Wang X, Xu H. 2010. TRP channels of intracellular membranes. J Neurochem 113:313–328. doi: 10.1111/j.1471-4159.2010.06626.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wainszelbaum MJ, Proctor BM, Pontow SE, Stahl PD, Barbieri MA. 2006. IL4/PGE2 induction of an enlarged early endosomal compartment in mouse macrophages is Rab5-dependent. Exp Cell Res 312:2238–2251. doi: 10.1016/j.yexcr.2006.03.025. [DOI] [PubMed] [Google Scholar]
- 48.Iwata Y, Katanosaka Y, Arai Y, Komamura K, Miyatake K, Shigekawa M. 2003. A novel mechanism of myocyte degeneration involving the Ca2+-permeable growth factor-regulated channel. J Cell Biol 161:957–967. doi: 10.1083/jcb.200301101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhou FQ, Snider WD. 2006. Intracellular control of developmental and regenerative axon growth. Philos Trans R Soc Lond B Biol Sci 361:1575–1592. doi: 10.1098/rstb.2006.1882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Jacques-Fricke BT, Seow Y, Gottlieb PA, Sachs F, Gomez TM. 2006. Ca2+ influx through mechanosensitive channels inhibits neurite outgrowth in opposition to other influx pathways and release from intracellular stores. J Neurosci 26:5656–5664. doi: 10.1523/JNEUROSCI.0675-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Marlin MC, Li G. 2015. Biogenesis and function of the NGF/TrkA signaling endosome. Int Rev Cell Mol Biol 314:239–257. doi: 10.1016/bs.ircmb.2014.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Xue Y, Liu Z, Cao J, Ma Q, Gao X, Wang Q, Jin C, Zhou Y, Wen L, Ren J. 2011. GPS 2.1: enhanced prediction of kinase-specific phosphorylation sites with an algorithm of motif length selection. Protein Eng Des Sel 24:255–260. doi: 10.1093/protein/gzq094. [DOI] [PubMed] [Google Scholar]
- 53.Lundby A, Secher A, Lage K, Nordsborg NB, Dmytriyev A, Lundby C, Olsen JV. 2012. Quantitative maps of protein phosphorylation sites across 14 different rat organs and tissues. Nat Commun 3:876. doi: 10.1038/ncomms1871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Weintz G, Olsen JV, Fruhauf K, Niedzielska M, Amit I, Jantsch J, Mages J, Frech C, Dolken L, Mann M, Lang R. 2010. The phosphoproteome of Toll-like receptor-activated macrophages. Mol Syst Biol 6:371. doi: 10.1038/msb.2010.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Howe CL, Mobley WC. 2004. Signaling endosome hypothesis: a cellular mechanism for long distance communication. J Neurobiol 58:207–216. doi: 10.1002/neu.10323. [DOI] [PubMed] [Google Scholar]
- 56.Abe K, Puertollano R. 2011. Role of TRP channels in the regulation of the endosomal pathway. Physiology (Bethesda) 26:14–22. doi: 10.1152/physiol.00048.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Frederick J, Buck ME, Matson DJ, Cortright DN. 2007. Increased TRPA1, TRPM8, and TRPV2 expression in dorsal root ganglia by nerve injury. Biochem Biophys Res Commun 358:1058–1064. doi: 10.1016/j.bbrc.2007.05.029. [DOI] [PubMed] [Google Scholar]
- 58.Gaudet AD, Williams SJ, Hwi LP, Ramer MS. 2004. Regulation of TRPV2 by axotomy in sympathetic, but not sensory neurons. Brain Res 1017:155–162. doi: 10.1016/j.brainres.2004.05.045. [DOI] [PubMed] [Google Scholar]
- 59.Richner M, Ulrichsen M, Elmegaard SL, Dieu R, Pallesen LT, Vaegter CB. 2014. Peripheral nerve injury modulates neurotrophin signaling in the peripheral and central nervous system. Mol Neurobiol 50:945–970. doi: 10.1007/s12035-014-8706-9. [DOI] [PubMed] [Google Scholar]
- 60.Amaya F, Shimosato G, Nagano M, Ueda M, Hashimoto S, Tanaka Y, Suzuki H, Tanaka M. 2004. NGF and GDNF differentially regulate TRPV1 expression that contributes to development of inflammatory thermal hyperalgesia. Eur J Neurosci 20:2303–2310. doi: 10.1111/j.1460-9568.2004.03701.x. [DOI] [PubMed] [Google Scholar]
- 61.Ji RR, Samad TA, Jin SX, Schmoll R, Woolf CJ. 2002. p38 MAPK activation by NGF in primary sensory neurons after inflammation increases TRPV1 levels and maintains heat hyperalgesia. Neuron 36:57–68. doi: 10.1016/S0896-6273(02)00908-X. [DOI] [PubMed] [Google Scholar]
- 62.Lankford KL, Waxman SG, Kocsis JD. 1998. Mechanisms of enhancement of neurite regeneration in vitro following a conditioning sciatic nerve lesion. J Comp Neurol 391:11–29. doi:. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Shortland P, Kinman E, Molander C. 1997. Sprouting of A-fibre primary afferents into lamina II in two rat models of neuropathic pain. Eur J Pain 1:215–227. doi: 10.1016/S1090-3801(97)90107-5. [DOI] [PubMed] [Google Scholar]
- 64.Fukuoka T, Tokunaga A, Tachibana T, Dai Y, Yamanaka H, Noguchi K. 2002. VR1, but not P2X(3), increases in the spared L4 DRG in rats with L5 spinal nerve ligation. Pain 99:111–120. doi: 10.1016/S0304-3959(02)00067-2. [DOI] [PubMed] [Google Scholar]
- 65.Hudson LJ, Bevan S, Wotherspoon G, Gentry C, Fox A, Winter J. 2001. VR1 protein expression increases in undamaged DRG neurons after partial nerve injury. Eur J Neurosci 13:2105–2114. doi: 10.1046/j.0953-816x.2001.01591.x. [DOI] [PubMed] [Google Scholar]
- 66.Kim HY, Park CK, Cho IH, Jung SJ, Kim JS, Oh SB. 2008. Differential changes in TRPV1 expression after trigeminal sensory nerve injury. J Pain 9:280–288. doi: 10.1016/j.jpain.2007.11.013. [DOI] [PubMed] [Google Scholar]
- 67.Yang Q, Wu Z, Hadden JK, Odem MA, Zuo Y, Crook RJ, Frost JA, Walters ET. 2014. Persistent pain after spinal cord injury is maintained by primary afferent activity. J Neurosci 34:10765–10769. doi: 10.1523/JNEUROSCI.5316-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]