

Abstract
Cyclic AMP-responsive element-binding protein 3-like 3, hepatocyte specific (CREBH), is a hepatic transcription factor that functions as a key regulator of energy homeostasis. Here, we defined a regulatory CREBH posttranslational modification process, namely, lysine-specific acetylation, and its functional involvement in fasting-induced hepatic lipid metabolism. Fasting induces CREBH acetylation in mouse livers in a time-dependent manner, and this event is critical for CREBH transcriptional activity in regulating hepatic lipid homeostasis. The histone acetyltransferase PCAF-mediated acetylation and the deacetylase sirtuin-1-mediated deacetylation coexist to maintain CREBH acetylation states under fasting conditions. Site-directed mutagenesis and functional analyses revealed that the lysine (K) residue at position 294 (K294) within the bZIP domain of the CREBH protein is the site where fasting-induced acetylation/deacetylation occurs. Introduction of the acetylation-deficient (K294R) or acetylation-mimicking (K294Q) mutation inhibited or enhanced CREBH transcriptional activity, respectively. Importantly, CREBH acetylation at lysine 294 was required for the interaction and synergy between CREBH and peroxisome proliferator-activated receptor α (PPARα) in activating their target genes upon fasting or glucagon stimulation. Introduction of the CREBH lysine 294 mutation in the liver leads to hepatic steatosis and hyperlipidemia in animals under prolonged fasting. In summary, our study reveals a molecular mechanism by which fasting or glucagon stimulation modulates lipid homeostasis through acetylation of CREBH.
INTRODUCTION
The liver is the central metabolic organ that plays primary roles in regulating lipid and glucose homeostasis upon alterations of metabolic conditions (1). Under fasting conditions, the liver boosts metabolic pathways of energy utilization, and the energy source is shifted from carbohydrates to triglycerides to meet the demands of the energy supplies. The major changes in liver metabolic pathways upon fasting include (i) glycogen utilization and gluconeogenesis to increase blood glucose levels and (ii) the catalysis of triglycerides (TG) in adipose tissue or liver to fatty acids (FA) and glycerol when the glycogen in liver is depleted. The released FA are directly oxidized as an energy source or metabolized to ketone bodies by the liver to supply energy to the brain, while glycerol is converted into glucose by the liver (2). Defects in these metabolic pathways may cause hepatic steatosis and abnormal levels of glucose and lipids in the blood. Notably, prolonged fasting results in the rapid mobilization of fat and the abnormal accumulation of FA and TG in the liver, leading to hepatic steatosis (3).
Transcriptional regulation plays a major role in controlling energy metabolism. Increasing evidence suggests that the regulation of transcription factors via posttranslational modifications, such as phosphorylation, ubiquitination, glycosylation, and acetylation, plays an important role in transcriptional activity as well as protein localization and interaction with other cellular partners. Histone acetylation is regulated by lysine acetyltransferases (KATs), which acetylate histones and promote transcription, whereas histone deacetylases (HDACs) remove acetyl groups and silence transcription (4). The balance between these two activities is a key to the fine-tuned regulation of cellular responses to signals. Several families of KATs, such as PCAF/GCN5, p300/CBP, TAF250, SRC1, and TIP60, have been identified (4). CBP and its related protein, p300 (CBP/p300), as well as PCAF (p300-CBP-associated factor), are well-characterized histone acetyltransferases that acetylate transcription factors and act as coactivators of transcription factors (5). The deacetylase sirtuin-1 (SIRT1) is known to mediate the nutritional and hormonal modulation of hepatic energy metabolism through the deacetylation of metabolic regulators. Upon fasting, SIRT1 mediates the deacetylation of key metabolic regulators, such as PGC1α, Foxo-1, and peroxisome proliferator-activated receptor γ (PPARγ), to maintain lipid and glucose homeostasis (6–10).
Cyclic AMP-responsive element-binding protein 3-like 3, hepatocyte specific (CREBH), is a liver-specific, endoplasmic reticulum (ER)-localized transcription factor of the CREB/ATF family (11). We and others have defined CREBH to be a stress-inducible transcriptional activator that is critically involved in inflammation and metabolism (12–16). Metabolic stress and acute liver injuries can induce CREBH cleavage, mediated by the site 1 protease (SP1) and site 2 protease (SP2), the same enzymes that process sterol regulatory element binding proteins upon the demands of lipid or sterol biosynthesis (12). The cleaved N-terminal fragment of CREBH enters into the nucleus and acts as a potent transcription factor that activates the expression of genes involved in the hepatic acute-phase response, gluconeogenesis, lipogenesis, FA oxidation, and lipolysis (12–15, 17, 18). Notably, CREBH interacts with PPARα to synergistically activate the metabolic hormone fibroblast growth factor 21 (FGF21) to regulate lipolysis, FA oxidation, and ketogenesis upon fasting or under an atherogenic high-fat (AHF) diet (15). Defects in CREBH lead to nonalcoholic steatohepatitis (NASH) and hyperlipidemia under an AHF diet or fasting conditions (13–15).
In this study, we investigated the regulatory mechanism and physiological role of CREBH acetylation in hepatic lipid metabolism upon fasting or glucagon stimulation. Our study demonstrates that PCAF and SIRT1 are the acetyltransferase and deacetylase, respectively, which modulate the acetylation states of CREBH under fasting conditions. We also reveal the lysine residue responsible for the histone acetylation and deacetylation of CREBH and the impact of CREBH acetylation on the interaction between CREBH and PPARα. Pathophysiologically, we demonstrate that modulation of CREBH acetylation can significantly affect CREBH transcriptional activity and lead to the altered lipid homeostasis associated with hepatic steatosis and hyperlipidemia.
MATERIALS AND METHODS
Materials.
Polyclonal anti-acetylated lysine–peroxidase, monoclonal anti-β-actin, and monoclonal anti-Flag–peroxidase antibodies, as well as anti-Flag affinity gel, were purchased from Sigma (St. Louis, MO). Mouse monoclonal anti-PPARα antibody was from Millipore (Billerica, MA). Polyclonal rabbit anti-CREBH antibody was generated as previously described (15). Monoclonal mouse anti-PCAF antibody and monoclonal anti-lamin B1 antibody were from Santa Cruz Biotech (Dallas, TX). Monoclonal anti-Myc antibody conjugated with horseradish peroxidase was from Cell Signaling Technologies (Danvers, MA). Monoclonal anti-SIRT1 antibody was from Abcam (Cambridge, MA). The plasmid vector expressing Flag-SIRT1 H363Y was from Addgene (Cambridge MA). The Flag-tagged CBP and PCAF expression vectors were purchased from Addgene. The SIRT1 silencer-select small interfering RNA (siRNA) and the control siRNA were purchased from Invitrogen (Carlsbad, CA).
Mouse experiments.
CREBH-null mice in which exons 4 to 7 of the CrebH gene were deleted were previously described (17). CREBH-null and wild-type control mice (age, approximately 3 months) on a C57BL/6J background were used for the experiments. For fasting, CREBH-null and wild-type control mice were housed in cages containing Pure-o'Cel bedding without food (water was provided) for 6, 12, or 24 h. For adenovirus injection experiments, recombinant adenovirus expressing the full-length human CREBH protein (wild type) or the acetylation-deficient (K294R) mutant protein was injected into CREBH-null mice through the tail vein. Approximately 1 × 1010 PFU of adenovirus in 0.25 ml phosphate-buffered saline (PBS) was intravenously injected into a mouse with a body weight of approximately 20 g. Three days after the injection, the animals were subjected to fasting for the periods of time indicated below before they were euthanized for tissue and blood sample collection. All the animal experiments were approved by the Wayne State University IACUC and carried out under the institutional guidelines for ethical animal use.
Site-directed mutagenesis.
Site-directed mutations were introduced into the putative acetylation site (Lys-294) of human CREBH using a QuikChange II site-directed mutagenesis kit (Stratagene). The K294R mutation (where AAG was changed to AGG [where the changed nucleotide is underlined]) or the K294Q mutation (where AAG was changed to CAG) was achieved by site-directed mutagenesis PCR using a Myc-tagged human CREBH expression plasmid as the template and the primers described in Table S1 in the supplemental material. All constructs were confirmed by sequencing analysis.
Luciferase assay.
A reporter plasmid carrying the FGF21 gene promoter was described previously (15). For luciferase activity measurement, a dual-luciferase reporter assay system (Promega) was used. Hepa1-6 cells were transfected with the FGF21 promoter-driven reporter construct and a plasmid carrying thymidine kinase (TK) promoter-Renilla luciferase, which was used as a transfection control. Luciferase activities were measured from cell lysates after 48 h of transfection. Expression was calculated as the relative firefly luciferase activities normalized by the activities of the transfection control, Renilla luciferase.
Protein IP and Western blot analysis.
The interactions among CREBH, PPARα, SIRT1, and PCAF were determined by immunoprecipitation (IP)-Western blot analysis. Mouse liver tissues or cultured cells were homogenized and lysed with NP-40 lysis buffer containing protease inhibitor cocktail, as previously described (19). For IP, 200 μg of the protein lysates was incubated with 3 μg of protein-specific antibody overnight at 4°C. Immunoprecipitated proteins were resolved by SDS-PAGE, followed by immunoblotting using the specific antibodies.
Subcellular protein fractionation.
To determine the levels and subcellular localization of specific proteins in mouse liver tissues, cell fractionation was carried out using a subcellular protein fractionation kit (Thermo Scientific). Liver tissue weights were measured, and liver was cut into small pieces. The tissue pieces were fractionated according to the instructions supplied with the kit, and nuclear and cytoplasmic fractions were used for Western blotting or IP-Western blot analysis.
Measurement of lipid metabolites.
Liver tissue and blood plasma samples were isolated from mice that had received normal chow or that had been fasted. To determine hepatic TG levels, approximately 30 mg liver tissue was homogenized in PBS followed by centrifugation. The supernatant was mixed with 10% Triton X-100 in PBS for measurement of TG levels using a commercial kit (BioAssay Systems). Mouse hepatic TG levels were determined by normalization to the mass of the liver tissue used for measurement of TG levels. Mouse blood plasma samples were subjected to analysis for quantitation of TG and free FA levels using commercial kits (BioAssay Systems).
Statistics.
Experimental results are shown as the means ± standard errors of the means (SEMs) (to determine the variations between animals or experiments). All in vitro experiments were repeated with biological triplicates at least three times. Mean values for biochemical data from the experimental groups were compared by paired, 2-tailed Student's t tests. Statistical analyses of multiple comparisons were performed by analysis of variance and were analyzed by ad hoc statistical tests when necessary. Statistical tests with P values of <0.05 were considered significant.
RESULTS
Fasting induces CREBH acetylation in the liver in a time-dependent manner.
Recent works have established the roles of the liver-enriched transcription factor CREBH in maintaining energy homeostasis upon metabolic alterations (12–15, 18). However, the regulatory mechanism underlying stress-induced CREBH transcriptional activities remains to be determined. To determine whether CREBH is regulated through posttranslational modification, we investigated CREBH activation and acetylation in the livers of wild-type and CREBH-null mice in response to fasting for 6, 12, or 24 h. Western blot analysis indicated that activation of CREBH, indicated by the levels of cleaved CREBH protein, was increased in the livers of mice upon fasting in a time-dependent manner (Fig. 1A). Upon cellular stress, cleaved CREBH protein transits into the nucleus, where CREBH is possibly posttranslationally modified before it functions as a transcriptional activator (12). To test whether the activated CREBH protein is acetylated upon fasting, nuclear protein fractions isolated from the liver tissues of mice after 6-, 12-, and 24-h fasts were subjected to IP-Western blot analyses to determine the levels of acetylated CREBH protein in the nucleus of cells of fasted mouse liver tissues (Fig. 1B). After normalization to the levels of total CREBH proteins, the levels of lysine-acetylated CREBH protein in the liver cell nuclear fractions were increased upon fasting in a time-dependent manner, indicating fasting-induced acetylation of CREBH in the liver cell nucleus.
FIG 1.

CREBH is acetylated in the livers of mice upon fasting. Wild-type (WT) and CREBH-null (knockout [KO]) mice were subjected to feeding or fasting for 6, 12, or 24 h before they were euthanized for liver tissue collection. IP and Western blot analysis were performed to determine the levels of acetylated CREBH protein. (A) (Left) Western blot analysis with mouse liver protein lysates to determine the levels of the CREBH precursor and its cleaved form. Levels of β-actin as a loading control were determined. (Right) Quantification of the CREBH precursor and activated proteins in the liver tissues of wild-type mice in the fed state or under conditions of fasting for 6, 12, and 24 h. The CREBH protein signals, determined by Western blotting densitometry, were normalized to the β-actin signal. The fold changes in the levels of the precursor or activated form of CREBH were determined by comparing the protein signals to those obtained under the fed condition (defined as 1). (B) (Left) The nuclear protein fractions were prepared from the pooled liver tissues of wild-type and CREBH-null mice (n = 3 mice per group) under fed or fasting conditions as described above. The nuclear proteins were immunoprecipitated with the anti-CREBH antibody to pull down the endogenous CREBH protein complex, followed by immunoblotting (IB) with antibody against acetylated lysine (Ace-K) or CREBH. The two panels at the bottom show the results of regular Western blot (WB) analyses that determined the levels of CREBH and lamin B1 (a nuclear protein marker) in the liver cell nuclear protein fractions. (Right) Quantification of the acetylated CREBH protein signals in the pooled liver cell nuclear protein fractions of wild-type mice under the fed or fasting condition. The acetylated CREBH protein signals, determined by Western blotting densitometry, were normalized to the signal of the total CREBH protein in the immunoprecipitated protein fractions. The fold changes in acetylated CREBH protein levels were determined by comparison to the protein signals under the fed condition (defined as 1). (C) Quantitative real-time PCR analysis of expression levels of CrebH mRNA in the livers of wild-type mice under the fed condition or conditions of fasting for 6, 12, and 24 h. Expression levels were normalized to β-actin mRNA levels. The fold changes in CrebH mRNA levels were shown by comparison to the CrebH mRNA level in one of the mice under the fed condition. Each bar represents the mean ± SEM (n = 3). *, P < 0.05. (D) (Left) Hepa1-6 cells were infected with recombinant adenovirus overexpressing the activated form of CREBH. Hepa1-6 cells overexpressing the activated CREBH were treated with either PBS or glucagon (25 nM) for the indicated times. The nuclear protein fractions isolated from the cells were subjected to IP-Western blot analysis or Western blot analysis to determine the levels of acetylated CREBH, total CREBH, and lamin B1. (Right) Quantification of the acetylated CREBH protein signals after normalization to the total CREBH protein signals in the immunoprecipitated protein fractions. The fold changes in acetylated CREBH protein levels were determined by comparison to the protein signals after PBS treatment (defined as 1).
Because fasting increases the expression of the CrebH mRNA and its protein (Fig. 1A and C) (14, 15, 18), we wondered whether CREBH acetylation is a fasting-regulated event that occurs independently of the increased expression of total CREBH protein upon fasting. To address this question, cells of the mouse liver hepatoma cell line Hepa1-6 were infected with recombinant adenovirus overexpressing the activated form of CREBH and then challenged with the fasting hormone glucagon or PBS as a control (Fig. 1D). The adenovirus-based overexpression system enables similar levels of the activated CREBH protein to be expressed in the nucleus under glucagon or PBS treatment. IP-Western blot analysis with the cell nuclear protein fractions expressing activated CREBH indicated that CREBH acetylation was increased upon glucagon stimulation in a time-dependent manner (Fig. 1D). This result confirmed that the increase in CREBH acetylation was regulated by glucagon stimulation but was not due to increased CREBH expression.
CREBH is acetylated at lysine 294 of its bZIP domain, and this event is critical for CREBH transcriptional activity.
To identify putative acetylation sites in CREBH proteins, we referenced the PhosphoSitePlus database for potential posttranslational modifications (20). Based on the information in the database, we identified a conserved lysine (K) acetylation residue in the bZIP domains of human, rat, and mouse CREBH proteins (Fig. 2A). This acetylation site is located at K294 in the human CREBH protein or K290 in the mouse and rat CREBH proteins. It is known that replacement of the lysine by glutamine results in mimicking of the acetylation state obtained with lysine, while replacement of the lysine by arginine is able to block acetylation (21, 22). To validate the CREBH acetylation site, we introduced two site-directed mutations into the human CREBH protein: (i) K294R, which created a CREBH acetylation-deficient mutant in which the K acetylation residue was changed to R, and (ii) K294Q, which created a CREBH acetylation-mimicking mutant in which the K acetylation residue was changed to Q to maintain the constitutive acetylation state. We coexpressed Myc-tagged wild-type human CREBH or the CREBH K294R or K294Q mutant with Flag-tagged SIRT1 in Huh7 cells in the presence or absence of an HDAC inhibitor cocktail containing trichostatin A (TsA) and nicotinamide (Nam). IP-Western blot analysis detected lysine-acetylated CREBH in the cells expressing the wild-type CREBH protein but not the CREBH acetylation-mimicking K294Q mutant or the CREBH acetylation-deficient K294R mutant (Fig. 2B). In the presence of the HDAC inhibitors, the levels of acetylated CREBH were increased in the cells expressing the wild-type CREBH protein, while the acetylated CREBH protein was still not detectable in the cells expressing the K294Q or K294R mutant (Fig. 2B). Upon overexpression of the deacetylase SIRT1, the levels of acetylated lysine proteins in cells expressing wild-type CREBH were decreased. However, neither the CREBH mutations nor deacetylation modulation (by SIRT1 expression or with HDAC inhibitors) altered the total amount of CREBH or its cleavage activation process (Fig. 2B, bottom two panels). Taken together, these results confirm that the K294 residue is the CREBH acetylation site.
FIG 2.
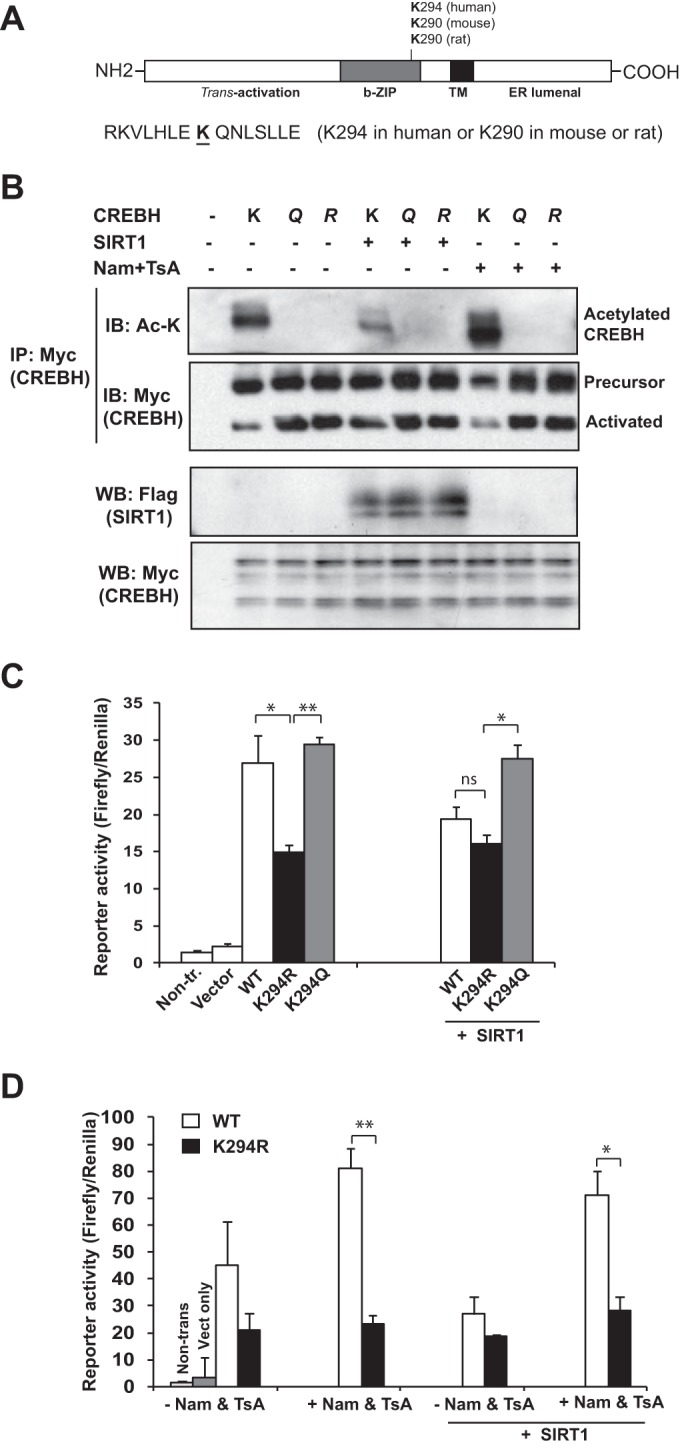
CREBH acetylation at the K294 residue is critical for its transcriptional activity. (A) Prediction of putative acetylation sites in the CREBH proteins of human, rat, and mouse species on the basis of information in the PhosphoSitePlus database (http://www.phosphosite.org/). The conserved acetylated lysine (K) residue in the bZIP domain of the CREBH proteins is underlined. This acetylation site is located at K294 in the human CREBH protein or K290 in the mouse and rat CREBH proteins. (B) IP-Western blot analysis of CREBH with an acetylated lysine (Ac-K) and total CREBH and Western blot analyses of SIRT1 and CREBH in Huh7 cells coexpressing wild-type human CREBH (K), acetylation-deficient mutant K294R (R), or acetylation-mimic mutant K294Q (Q) with SIRT1 in the presence or absence of HDAC inhibitors (Nam plus TsA). Huh-7 cells were transfected with Myc-tagged CREBH or its derivatives with Flag-tagged SIRT1. The cells were treated with vehicle or the HDAC inhibitors (2 μM TsA plus 5 mM Nam) for 2 h. Cell protein lysates were immunoprecipitated with the anti-Myc antibody to pull down CREBH, followed by immunoblotting with anti-acetyl-lysine or anti-Myc antibody to determine the levels of acetylated or total CREBH proteins, respectively. The bottom two panels show the results of Western blot analyses for determination of the levels of Flag-tagged SIRT1 and Myc-tagged CREBH in total protein lysates. (C) Luciferase reporter analysis of trans-activation activities of the human FGF21 gene promoter by CREBH or its mutants. Hepa1-6 cells were cotransfected with a plasmid vector expressing wild-type (WT) CREBH or the K294R or K294Q CREBH mutant and the plasmid vector expressing SIRT1 or the vehicle. A Renilla reporter plasmid was included in the cotransfection for normalization of luciferase reporter activities. Non-tr., nontransfected. (D) Reporter analysis of trans-activation activities of the FGF21 promoter by wild-type CREBH or K294R in the presence or absence of the HDAC inhibitors. Hepa1-6 cells were cotransfected with the vector expressing wild-type CREBH or the K294R mutant. At 48 h after transfection, the cells were treated with vehicle or HDAC inhibitors (2 μM TsA plus 5 mM Nam) for 2 h. Non-trans, nontransfected; Vect, vector. In panels C and D, each bar denotes the mean ± SEM (n = 3 biological repeats). ns, nonsignificant; *, P < 0.05; **, P < 0.01.
FGF21 is a CREBH target gene that contains a conserved CREBH-binding element in the promoter region (15). To assess the functional involvement of the K294 acetylation site in CREBH transcriptional activity, we performed reporter gene expression analysis with wild-type CREBH or the K294Q or K294R CREBH mutant and a luciferase reporter driven by the human FGF21 gene promoter. Compared to the activity of wild-type CREBH, the K294R mutant exhibited significantly reduced activity in driving expression of the FGF21 reporter (Fig. 2C). However, expression of the CREBH acetylation-mimicking K294Q mutant resulted in maximal activation of the FGF21 gene promoter compared to that achieved by expression of wild-type CREBH or the CREBH K294R mutant. Upon coexpression of the deacetylase SIRT1, wild-type CREBH but not the CREBH K294R mutant displayed significantly reduced activity in activating the FGF21 promoter (Fig. 2C). In contrast, the transcriptional activity of the K294Q acetylation-mimicking mutant was only slightly affected by the coexpression of SIRT1. These results suggest that acetylation of CREBH at K294 is critical for CREBH to exert its transcriptional activity.
Further, we confirmed the functional significance of CREBH acetylation at K294 by evaluating the effect of the K294 mutation on CREBH activity in the presence of the HDAC inhibitors. Compared to the activity of wild-type CREBH, the K294R mutant displayed significantly reduced activity in activating the FGF21 promoter in the presence of the HDAC inhibitors (Fig. 2D). However, in the absence of the HDAC inhibitors or in the presence of SIRT1 overexpression, the difference in the transcriptional activities of wild-type CREBH and the K294R mutant became insignificant, thus confirming the critical role of CREBH acetylation at K294 in CREBH transcriptional activity. The enhanced acetylation of CREBH in the presence of the HDAC inhibitors or in the absence of SIRT1 overexpression implies that CREBH acetylation is a dynamic process in which SIRT1-mediated deacetylation is involved.
The histone acetyltransferase PCAF but not CBP mediates CREBH acetylation.
Acetylation is regulated by two groups of enzymes, lysine acetyltransferases and HDACs. CBP/p300 and PCAF are representative lysine acetyltransferases that mediate the histone acetylation of transcription factors (4). To identify the acetyltransferase that acetylates CREBH, we coexpressed Flag-tagged CBP or PCAF and Myc-tagged wild-type CREBH or the K294R CREBH mutant in Huh7 cells. After immunoprecipitation of CREBH, we could detect acetylated CREBH in the cells expressing PCAF but not in cells expressing CBP in the presence of the HDAC inhibitors using an anti-acetyl-lysine antibody (Fig. 3A). When the acetylation mutation K294R was introduced, overexpression of PCAF failed to mediate CREBH acetylation. Moreover, IP-Western blot analysis showed that both wild-type CREBH and the K294R mutant could complex with PCAF but not CBP, although the K294 mutant showed a reduced affinity of binding to PCAF (Fig. 3A). These results suggest that it is PCAF and not CBP that interacts with CREBH and mediates CREBH acetylation at K294. Note that in the absence of the HDAC inhibitors, we detected lower levels of acetylated CREBH in cells expressing PCAF, confirming that CREBH acetylation is a dynamic process in which deacetylation coexists. To validate the physiological involvement of PCAF in CREBH acetylation in the liver in response to fasting, we examined the interaction between CREBH and PCAF in the livers of wild-type and CREBH-null mice in response to fasting for 6, 12, or 24 h. IP-Western blot analyses indicated that the interaction between the endogenous PCAF and CREBH was detectable in the livers of the wild-type mice but not those of the CREBH-null mice upon fasting (Fig. 3B). Importantly, the time-dependent pattern of the PCAF-CREBH interaction in the livers in the fasting state is consistent with that of fasting-induced CREBH acetylation in the liver (Fig. 1B).
FIG 3.
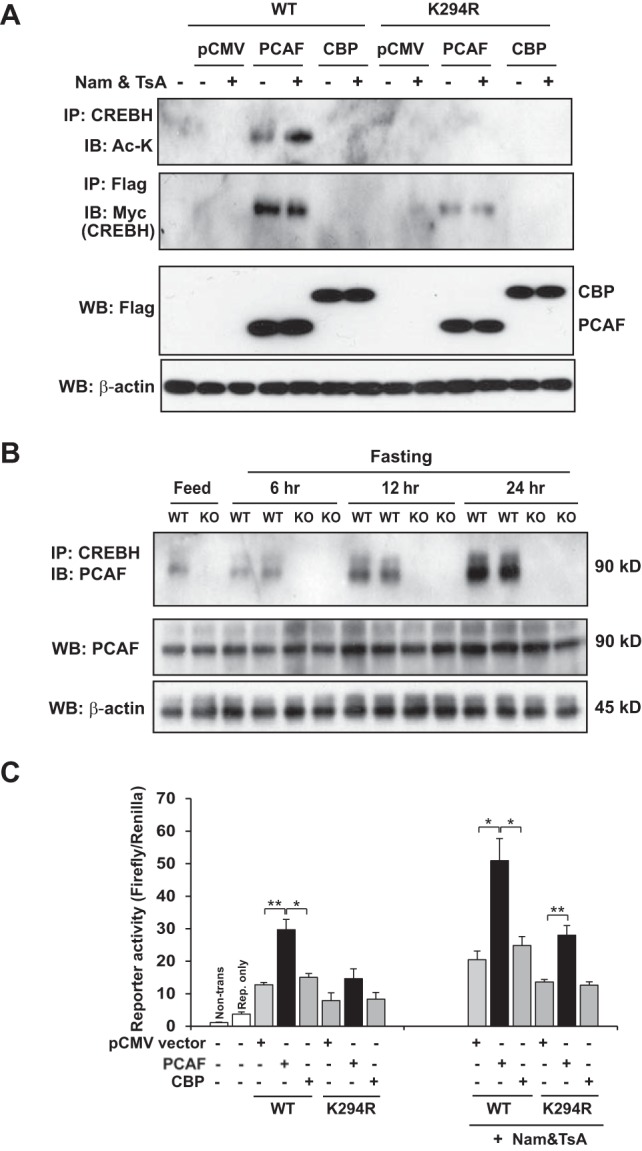
PCAF but not CBP mediates CREBH acetylation. (A) IP-Western blot analysis of acetylated CREBH as well as the CREBH interaction with PCAF or CBP. Huh-7 cells were cotransfected with the plasmid vector expressing Myc-tagged wild-type (WT) CREBH or the K294R CREBH mutant and the vector expressing Flag-tagged PCAF or CBP or the vector control (pCMV). The cells were treated with the vehicle (PBS) or HDAC inhibitors (TsA plus Nam) for 2 h. The proteins from the cell lysates were immunoprecipitated with the CREBH or Flag antibody followed by immunoblotting with acetyl-lysine antibody (to detect acetylated CREBH) or the Myc antibody (to detect the CREBH interaction with PCAF or CBP). The bottom two panels showed the results of Western blot analyses for determination of the levels of PCAF or CBP (by Flag blotting) and β-actin (loading control). (B) Wild-type and CREBH-null (knockout [KO]) mice were subjected to feeding or fasting for 6, 12, or 24 h before they were euthanized for liver tissue collection. Liver protein lysates were immunoprecipitated with the CREBH antibody followed by immunoblotting with the PCAF antibody to determine the interaction between CREBH and PCAF. Liver protein lysates were subjected to Western blot analysis to determine the levels of PCAF and β-actin. (C) Luciferase reporter analysis of the trans-activation activities of the human FGF21 gene promoter by wild-type CREBH or the K294R mutant under conditions of the expression of PCAF or CBP. Hepa1-6 cells were cotransfected with the plasmid vector expressing wild-type CREBH or the K294Q mutant and the plasmid vector expressing PCAF or CBP or the vehicle control (pCMV). A reporter plasmid for Renilla luciferase was included in the cotransfection for normalization of luciferase reporter activities. Non-trans, nontransfected; Rep., reporter. Each bar denotes the mean ± SEM (n = 3 biological repeats). *, P < 0.05; **, P < 0.01.
Next, we evaluated the functional involvement of PCAF or CBP in CREBH transcriptional activity through an FGF21 gene promoter reporter assay. Coexpression of PCAF with wild-type CREBH significantly induced expression of the luciferase reporter driven by the FGF21 promoter (Fig. 3C). In contrast, coexpression of CBP with either wild-type CREBH or the K294R CREBH mutant exerted only marginal effects on activation of the FGF21 promoter. These results confirm that PCAF but not CBP mediates CREBH acetylation at K294 and that PCAF-mediated acetylation is critical for CREBH transcriptional activity. Interestingly, the mutation at K294 did not completely abolish the activity of CREBH under conditions with the exogenous expression of PCAF and in the presence of the HDAC inhibitors (Fig. 3C). It is possible that PCAF targets alternative CREBH acetylation sites under these conditions, although K294 is the primary acetylation site for CREBH. This is an interesting question to be elucidated in the future.
SIRT1 mediates CREBH deacetylation by interacting with CREBH through its N terminus.
The histone deacetylase SIRT1 is known to modulate energy metabolism through deacetylation of metabolic regulators (6, 23). To determine the involvement of SIRT1 in CREBH acetylation states, we investigated the interaction between CREBH and SIRT1 in fasted mouse livers by IP-Western blot analysis. The interaction between CREBH and SIRT1 was detectable in the nucleus of fasted mouse liver tissues, and the interaction was increased upon fasting for 6 and 12 h but decreased after fasting for 24 h (Fig. 4A). Interestingly, SIRT1 was localized to both the nucleus and the cytoplasm after a 24-h fast, when the interaction between CREBH and SIRT1 was decreased. The decreased association of SIRT1 with CREBH after a 24-h fast was concomitant with the significantly increased levels of acetylated CREBH (Fig. 1B), suggesting that CREBH acetylation states may be modulated by an acetylation and deacetylation regulatory loop.
FIG 4.

SIRT1 mediates CREBH deacetylation by interacting with CREBH. (A) Wild-type (WT) and CREBH-null (knockout [KO]) mice were subjected to feeding or fasting for 6, 12, or 24 h before they were euthanized for liver tissue collection. IP-Western blot analyses with liver cell nuclear protein fractions were performed to determine the interaction between CREBH and SIRT1. The nuclear protein fractions were also subjected to Western blot analysis to determine the levels of SIRT1, lamin B1, and β-actin. (B) The nuclear protein fractions were prepared from the pooled liver tissues of wild-type and CREBH-null mice in the fed state or after a 6-, 12-, or 24-h fast (3 mice per group). The nuclear proteins were immunoprecipitated with the anti-CREBH antibody to pull down the CREBH protein complex, followed by immunoblotting with antibody against PPARα, SIRT1, or CREBH. The levels of lamin B1 as the loading control were determined by Western blotting. (C) IP-Western blot analysis for determination of acetylated CREBH levels in the dominant negative SIRT1 mutant or control Hepa1-6 cells. Hepa1-6 cells were cotransfected with a plasmid vector expressing Myc-tagged wild-type CREBH and a vector expressing Flag-tagged wild-type SIRT1 (wild-type) or the dominant negative SIRT1 H363Y mutant (DN). At 48 h after transfection, the cells were treated with the vehicle (PBS) or glucagon (25 nM) for 8 h. The proteins from the cell lysates were immunoprecipitated with the Myc antibody (to pull down CREBH) or Flag antibody (to pull down SIRT1), followed by immunoblotting with the acetyl-lysine antibody to detect acetylated CREBH or the Myc antibody to detect the CREBH-SIRT1 interaction. The bottom three panels show the results of Western blot analyses for determination of the levels of Flag-tagged SIRT1, Myc-tagged CREBH, and β-actin. (D) Hepa1-6 cells were cotransfected with plasmid vectors expressing Myc-tagged CREBH, Flag-tagged SIRT1, and Sirt1 siRNA (siRNA) or control siRNA (Ctl). At 48 h after the transfections, the cells were treated with glucagon (25 nM) or PBS for 12 h. The Hepa1-6 cell lysates were immunoprecipitated with anti-Myc antibody to pull down CREBH and then probed with anti-acetyl-lysine or anti-Myc antibody to determine the levels of acetylated or total CREBH proteins, respectively. The bottom three panels show the results of Western blot analyses with total cell lysates to determine the levels of SIRT1, CREBH, and β-actin. (E) Huh7 cells were cotransfected with a plasmid vector expressing CREBH and a plasmid vector expressing Flag-tagged full-length SIRT1 (SIRT1-FL), the SIRT1 N-terminal domain (SIRT1-N), the SIRT1 N-terminal and HDAC domains (SIRT1-NH), or the SIRT1 HDAC and C-terminal domains (SIRT1-HC). Cell protein lysates were immunoprecipitated with the Flag antibody to pull down SIRT1, followed by immunoblotting with the Myc antibody to determine the CREBH-SIRT1 interaction. (Top) A schematic representation of SIRT1 and its truncated forms, indicating that SIRT1 contains a nuclear localization signal (NLS), an HDAC domain, and a C-terminal coiled-coil-like domain (CC like). (Bottom) Interactions of SIRT1 and its mutants with CREBH. The bottom two panels show the results of Western blot analyses for determination of the levels of Flag-tagged SIRT1 and Myc-tagged CREBH.
We previously demonstrated that CREBH interacts with PPARα to form a functional complex upon fasting (15). We wondered whether CREBH, PPARα, and SIRT1 can be immunoprecipitated in the same protein complex from the liver under fasting states. To address this question, the CREBH-binding complex from the nucleus of cells from the livers of mice in the fed state or mice that had been fasting for 6, 12, or 24 h was pulled down by anti-CREBH antibody and then probed with the antibody against PPARα, SIRT1, or CREBH to detect the presence of PPARα, SIRT1, and CREBH in the CREBH-binding complex, respectively (Fig. 4B). While an interaction between CREBH, PPARα, and SIRT1 in the livers of mice was barely detectable under the fed condition, the interactions between CREBH, PPARα, and SIRT1 in the liver were significantly increased upon fasting for 6 or 12 h. However, after a prolonged fast for 24 h, SIRT1 was dissociated with the CREBH-PPARα complex, while the CREBH-PPARα interaction was further enhanced (Fig. 4B). This is consistent with the pattern of the presence of SIRT1 in liver cell nuclei after different periods of fasting (Fig. 4A) and the time-dependent CREBH acetylation profile upon fasting (Fig. 1B).
To test whether SIRT1 plays a major role in modulating CREBH acetylation states, we measured the levels of acetylated CREBH in Hepa1-6 cells expressing a wild-type human SIRT1 or a dominant negative SIRT1 mutant (the H363Y mutant) in response to the stimulation of the fasting hormone glucagon. Expression of SIRT1 H363Y, in which a critical histidine in the deacetylase domain of SIRT1 is replaced by a tyrosine residue, can effectively suppress endogenous SIRT1 activity in both human and mouse cells (24, 25). IP-Western blot analysis showed that the levels of acetylated CREBH were increased in the Hepa1-6 cells expressing the dominant negative SIRT1 H363Y mutant compared to those in cells expressing wild-type SIRT1 (Fig. 4C). While the levels of acetylated CREBH were increased in both wild-type SIRT1- and SIRT1 H363Y mutant-expressing cells upon stimulation with the fasting hormone glucagon, the fold increase in the amount of acetylated CREBH in cells expressing H363Y was significantly higher than that in wild-type SIRT1-expressing cells (Fig. 4C), suggesting that SIRT1 plays a major role in the deacetylation of CREBH. Additionally, IP-Western blot analysis indicated that both wild-type SIRT1 and SIRT1 H363Y were able to interact with CREBH and that the interactions were increased upon glucagon stimulation (Fig. 4C). To confirm the specific effect of SIRT1 on CREBH acetylation, the SIRT1 in Hepa1-6 cells expressing Myc-tagged CREBH was knocked down by siRNA, and then the cells were treated with glucagon or PBS (Fig. 4D). Consistent with the findings obtained by the approach with dominant negative SIRT1, the knockdown of SIRT1 increased the levels of acetylated CREBH, especially upon challenge with glucagon, thus further confirming the role of SIRT1 in the deacetylation of CREBH.
Next, we determined the domain of the SIRT1 protein that mediates the SIRT1-CREBH interaction. Flag-tagged full-length or truncated SIRT1 was coexpressed with Myc-tagged CREBH. IP-Western blot analysis indicated that the N terminus of SIRT1 is responsible for the interaction with CREBH and the interaction is enhanced by including the HDAC domain of SIRT1 (Fig. 4E). However, the HDAC domain together with the C terminus of SIRT1 is not able to interact with CREBH. These results suggest that the N-terminal region of SIRT1 is primarily responsible for the interaction between SIRT1 and CREBH and that the presence of the HDAC domain can maximize the interaction.
CREBH acetylation is required for the interaction between CREBH and PPARα upon fasting or glucagon stimulation.
CREBH is known to interact with PPARα to function as a transcriptional complex to activate FGF21 expression in the liver in the fasting state (15). Since the K294 acetylation residue is located in the bZIP domain of the CREBH protein, we wondered whether fasting-induced acetylation modulates CREBH activity by influencing the interaction between CREBH and PPARα. To test this possibility, we evaluated the fasting- or glucagon-stimulated interaction between CREBH and PPARα and the involvement of acetylation at K294 in the CREBH-PPARα interaction. First, the endogenous CREBH-PPARα complex was detected in the livers of mice after a 14-h fast (Fig. 5A). Second, glucagon treatment significantly increased the interaction between CREBH and PPARα in Hepa1-6 cells (Fig. 5B). Next, we examined the interaction of PPARα with wild-type CREBH, the acetylation-deficient K294R mutant, or the acetylation-mimicking K294Q mutant in Hepa1-6 cells under glucagon stimulation. The interaction between the acetylation-deficient K294R mutant and PPARα compared to that between wild-type CREBH and PPARα was diminished in response to glucagon stimulation (Fig. 5C). The acetylation-mimicking K294Q mutant exhibited an affinity of binding to PPARα comparable to or slightly higher than that of wild-type CREBH. These results suggest that CREBH acetylation at K294 is critical for the optimal interaction between CREBH and PPARα in the fasting state. To test for the requirement for CREBH acetylation in the fasting-induced CREBH-PPARα interaction in the liver in vivo, we expressed Myc-tagged wild-type CREBH or the K294R mutant in mouse livers using an adenovirus-based delivery system. After a 24-h fast, a strong interaction between CREBH and PPARα was detected in mouse livers expressing wild-type CREBH (Fig. 5D). In contrast, the fasting-induced interaction between CREBH and PPARα in the liver was significantly repressed by the K294R mutation. This result confirms the requirement for CREBH acetylation in the fasting-induced CREBH-PPARα interaction in vivo.
FIG 5.

CREBH acetylation is required for CREBH-PPARα interaction upon fasting or glucagon stimulation. (A) IP-Western blot analysis of the CREBH and PPARα interaction in the livers of mice in the fed state or after a 14-h fast. Whole protein lysates were prepared from mouse liver tissues and subjected to IP to pull down the endogenous CREBH protein complex using CREBH antibody. The precipitates were analyzed by immunoblotting with antibody against PPARα or CREBH. The bottom three panels show the results of Western blot analyses of liver protein lysates to determine the levels of CREBH, PPARα, and β-actin. (B) IP-Western blot analysis of the CREBH and PPARα interaction in Hepa1-6 cells under glucagon stimulation. Hepa1-6 cells were cotransfected with the plasmid vectors expressing Myc-tagged CREBH and PPARα. The cells were treated with the vehicle (PBS) or glucagon (25 nM) for 8 h. The proteins from the cell lysates were immunoprecipitated with the Myc antibody to pull down the CREBH protein complex, followed by immunoblotting with the anti-PPARα or anti-Myc antibody to determine the CREBH-PPARα interaction or the total CREBH protein levels in the immunoprecipitated fractions, respectively. The bottom three panels show the results of Western blot analyses for determination of the levels of Myc-tagged CREBH, PPARα, and β-actin. The graphs on the right sides of panels A and B are quantifications of CREBH-PPARα interaction signals in the livers of wild-type mice in the fed state or after a 14-h fast (A) or in Hepa1-6 cells with PBS or glucagon stimulation (B). The CREBH-PPARα interaction signals, determined by Western blotting densitometry, were normalized to the signal for the immunoprecipitated total CREBH protein or PPARα in total protein lysates. The fold changes in the CREBH-PPARα interaction signals were determined by comparison to the signal for one of the mice under the fed condition or the PBS-treated samples. Each bar represents the mean ± SEM (n = 3). *, P < 0.05; **, P < 0.01. Glucag., glucagon. (C) IP-Western blot analysis of interactions between PPARα and CREBH or its derivatives in Hepa1-6 cells under glucagon stimulation. Hepa1-6 cells were transfected with a plasmid vector expressing Myc-tagged wild-type CREBH or the K294R (R) or K294Q (Q) mutant. The cells were treated with the vehicle (PBS) or glucagon (25 nM) for 8 h. The proteins from the cell lysates were immunoprecipitated with Myc antibody to pull down the CREBH protein complex, followed by immunoblotting with PPARα or Myc antibody. The bottom three panels show the results of Western blot analyses for determination of the levels of PPARα, Myc-tagged CREBH, and β-actin. (D) IP-Western blot analysis of interactions between PPARα and CREBH or the K294R mutant in the livers of mice under the fasting condition. Recombinant adenovirus expressing the GFP control, a Myc-tagged full-length human CREBH protein (wild type), or the acetylation-deficient mutant (the K294R mutant) were injected into CREBH-null mice through the tail vein. After a 24-h fast, pooled liver protein lysates from the mice (n = 3 per group) were immunoprecipitated with Myc antibody to pull down the CREBH protein complex, followed by immunoblotting with PPARα or Myc antibody. The bottom three panels show the results of Western blot analyses for determination of the levels of PPARα, Myc-tagged CREBH, and β-actin. (E) Hepa1-6 cells were cotransfected with plasmid vectors expressing Myc-tagged CREBH, SIRT1, PPARα, and the Sirt1 siRNA or control (Ct) siRNA. At 48 h after the transfections, the cells were treated with glucagon (25 nM) or PBS for the indicated times. Hepa1-6 cell lysates were immunoprecipitated with anti-Myc antibody to pull down CREBH and then probed with anti-PPARα or anti-Myc antibody to determine the CREBH-PPARα interaction and CREBH levels, respectively. The bottom four panels show the results of Western blot analyses of total cell lysates for determination of the levels of SIRT1, CREBH, PPARα, and β-actin. (F) Luciferase reporter analysis of trans-activation activities of the human FGF21 gene promoter induced by CREBH or PPARα alone or by the combination of PPARα and CREBH or its mutants. Hepa1-6 cells were cotransfected with plasmid vectors expressing the CREBH wild-type, the K294R or K294Q mutant, and/or PPARα. Nontransfected cells or cells transfected with the reporter plasmid only were included as controls. A reporter plasmid carrying Renilla luciferase was included in the cotransfection for normalization of luciferase reporter activities. Each bar denotes the mean ± SEM (n = 3 biological repeats). *, P < 0.05; **, P < 0.01; NS, nonsignificant.
As shown above, CREBH, PPARα, and SIRT1 form a complex upon fasting (Fig. 4B) and SIRT1 plays a major role in modulating CREBH acetylation states (Fig. 4C and D). To determine whether SIRT1 modulates the interaction between PPARα and CREBH, we knocked down SIRT1 in Hepa1-6 cells expressing PPARα and CREBH through the use of siRNA and then challenged the cells with glucagon (Fig. 5E). While glucagon stimulation increased the CREBH-PPARα interaction in control cells in a time-dependent manner, the CREBH-PPARα interaction was further enhanced in SIRT1-knockdown cells upon glucagon stimulation. This result is consistent with the role of SIRT1 in the deacetylation of CREBH and the requirement for CREBH acetylation in the interaction between CREBH and PPARα upon fasting/glucagon treatment.
CREBH and PPARα are known to act in synergy to activate the expression of their target gene, FGF21 (15). To evaluate the impact of CREBH acetylation on the synergy between CREBH and PPARα in activating FGF21, we performed FGF21 gene reporter analysis by the use of coexpression of wild-type CREBH or the K294R or K294Q mutant with PPARα. In comparison to the effect of expression of PPARα or CREBH alone, coexpression of PPARα and wild-type CREBH or the acetylation-mimicking K294Q mutant exerted stronger effects on activation of the FGF21 promoter (Fig. 5F). In contrast, coexpression of PPARα and the acetylation-deficient mutant K294R failed to increase the level of expression of the FGF21 reporter compared to that achieved by expression of PPARα or wild-type CREBH alone (Fig. 5F), suggesting the requirement for CREBH acetylation at K294 for the synergy between CREBH and PPARα for activation of their target gene.
Lysine acetylation of CREBH prevents fasting-induced hepatic steatosis and hyperlipidemia.
To determine the pathophysiological significance of the fasting-induced acetylation of CREBH in the liver in vivo, we investigated whether hepatic expression of the CREBH mutant that lacks the acetylation state (the K294R mutant) has an impact on lipid homeostasis in mice in the fasting state. First, we evaluated the lipid-associated phenotypes of CREBH-null and wild-type control mice upon fasting. While the control mice displayed modest hepatic steatosis after a prolonged 24-h fast, the CREBH-null mice developed severe hepatic steatosis after a 24-h fast (Fig. 6A). The CREBH-null mice also exhibited hyperlipidemia, as indicated by significantly increased levels of serum TG under the fasting condition (Fig. 6D). These results were consistent with the role of CREBH in regulating lipolysis, as we previously revealed (13–15). Next, we expressed wild-type CREBH or the K294R mutant in the livers of CREBH-null mice using an adenovirus-based overexpression system (Fig. 6B and C). After a 24-h fast, while the CREBH-null mice expressing wild-type CREBH exhibited modest hepatic steatosis, the mice expressing the CREBH K294R acetylation mutant developed severe hepatic steatosis, as evidenced by oil red O staining of hepatic lipids and significantly increased hepatic TG levels (Fig. 6B and D). Moreover, significant increases in serum TG levels were observed in the mice expressing the CREBH K294R mutant compared to the levels seen in the mice expressing wild-type CREBH (Fig. 6E). The lipid-associated phenotype of animals with the defect in CREBH acetylation was comparable to that of CREBH-null mice under fasting conditions, thus supporting the critical role of the acetylation of CREBH at K294 in regulating fasting-induced hepatic lipid metabolism.
FIG 6.

CREBH acetylation is required to prevent fasting-induced hepatic steatosis and hyperlipidemia. (A) Oil red O staining of hepatic lipid droplets in the livers of CREBH-null and wild-type control mice in the fed state or after a 24-h fast. (B) Oil red O staining of lipid contents in the livers of CREBH-null mice expressing GFP, wild-type CREBH, or the K294R mutant after a 24-h fast. Recombinant adenovirus expressing the GFP control (Ad-GFP), wild-type Myc-tagged full-length human CREBH protein (Ad-WT), or the acetylation-deficient mutant (Ad-K294R) was injected into CREBH-null mice through the tail vein. At 3 days after the injection, the mice were subjected to fasting for 24 h. (C) IP-Western blot analysis of acetylated and total CREBH proteins in the livers of CREBH-null mice expressing GFP, wild-type CREBH, or the K294R mutant after fasting as described in the legend to panel B. After a 24-h fast, pooled liver protein lysates from the mice (n = 3 per group) were immunoprecipitated with Myc antibody to pull down the CREBH protein complex, followed by immunoblotting with anti-acetyl-lysine antibody. The bottom two panels show the results of Western blot analyses for determination of the levels of Myc-tagged CREBH and β-actin. (D) Enzymatic analysis of hepatic TG levels in the livers of CREBH-null mice, wild-type control mice, and CREBH-null mice expressing GFP, wild-type CREBH, or the CREBH K294R mutant after a 24-h fast. Each bar denotes the mean ± SEM (n = 3). *, P < 0.05; **, P < 0.01. (E) Serum TG levels in CREBH-null mice, wild-type control mice, and CREBH-null mice expressing GFP, wild-type CREBH, or the CREBH K294R mutant after a 24-h fast. Each bar denotes the mean ± SEM (n = 3). *, P < 0.05; **, P < 0.01. (F and G) Levels of mRNAs encoding the key enzymes or regulators in multiple metabolic pathways, as indicated, in the livers of CREBH-null mice expressing GFP control, wild-type CREBH, or the CREBH K294R mutant after a 24-h fast. Total RNAs isolated from mouse liver tissues were subjected to quantitative real-time PCR analysis. The levels of expression were normalized to the levels of β-actin mRNA expression. Fold changes in mRNA levels are shown by comparison to the levels of expression in one of the mice expressing wild-type CREBH. Each bar denotes the mean ± SEM (n = 3). *, P < 0.05; **, P < 0.01; red asterisks, P values for CREBH-null and wild-type mice versus CREBH-null mice and CREBH-null mice expressing GFP; black asterisks, P values for CREBH-null mice and mice expressing the K294R mutant versus CREBH-null and wild-type mice. Definitions of the abbreviations for the genes mentioned in panels F and G are provided in the supplemental material.
Further, we examined the expression of the CREBH target genes involved in TG and FA metabolism in the livers of CREBH-null and wild-type control mice as well as in the livers of CREBH-null mice expressing green fluorescent protein (GFP) as a control, wild-type CREBH, or the K294R mutant. Consistent with the roles of CREBH in regulating TG and FA metabolism (13–15), the levels of expression of most of the CREBH-targeted genes encoding key regulators or enzymes involved in lipolysis, FA oxidation, ketogenesis, FA elongation and conversion, TG and cholesterol metabolism, gluconeogenesis, and the metabolic hormone FGF21 in the livers of mice expressing K294R were significantly decreased compared to those in the livers of mice expressing wild-type CREBH upon fasting (Fig. 6F and G). These results indicate the importance of CREBH acetylation at K294 to the transcriptional activity of CREBH in regulating hepatic lipid homeostasis in the fasting state. Interestingly, the levels of expression of some of the CREBH-targeted genes involved in TG, FA, or cholesterol metabolism, including Cpt2, AccI, Scd1, Hmgcs2, Dgat1, and Dgat2, in the livers of mice expressing the K294R mutant was not significantly reduced compared to those in the livers of mice expressing wild-type CREBH. This observation suggests that the transcriptional function of CREBH on some of its target genes may not require lysine acetylation, an interesting possibility to be further elucidated in the future.
DISCUSSION
Our studies have characterized a regulatory posttranslational modification of CREBH, namely, lysine acetylation at position 294, and its functional involvement in fasting-induced hepatic lipid metabolism (Fig. 7). Our major findings include the following: (i) CREBH is acetylated in the liver upon fasting in a time-dependent manner, (ii) CREBH is acetylated at lysine 294 within the CREBH bZIP domain, and acetylation at this site is required for CREBH transcriptional activity, (iii) fasting-associated acetylation and deacetylation of CREBH are catalyzed by the lysine acetyltransferase PCAF and the histone deacetylase SIRT1, respectively, (iv) CREBH acetylation is required for the interaction and synergy between CREBH and PPARα in activating their target gene upon fasting or glucagon stimulation, and (v) CREBH acetylation at lysine 294 is critical to maintain hepatic lipid homeostasis in fasting states. Our study revealed a novel regulatory mechanism through which lysine acetylation modulates the lipid metabolism that is associated with hepatic steatosis and hyperlipidemia (Fig. 7). The functional relationships between CREBH, SIRT1, and PPARα in regulating hepatic lipid metabolism contribute to our understanding of the regulatory paradigm of energy homeostasis at the posttranslational level. Additionally, our study identified a conserved acetylation site within CREBH that acts as a critical regulator of hepatic lipid metabolism, and this information will be informative to pharmaceutical interventions directed toward controlling metabolic disorders by targeting CREBH acetylation.
FIG 7.
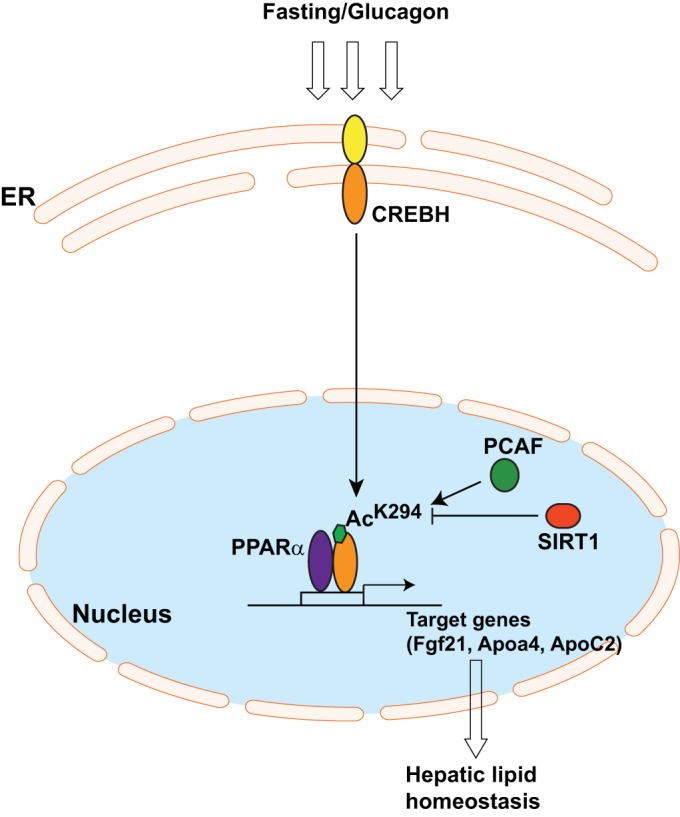
Model for regulation of CREBH activity by acetylation in fasted states. Upon fasting or glucagon stimulation, the CREBH protein is cleaved to release its activated form, which transits into the nucleus. In the nucleus, the histone lysine acetyltransferase PCAF mediates CREBH acetylation at K294, which is located within the bZIP domain. Meanwhile, the histone deacetylase SIRT1 modulates CREBH acetylation states through deacetylation activity and thereby fine-tunes the activity of CREBH. CREBH acetylation at K294 is critical for the formation of the CREBH-PPARα transcriptional complex, which regulates the expression of the genes involved in hepatic lipid homeostasis in fasting states.
CREBH functions as a key transcriptional regulator of hepatic lipid metabolism by activating expression of the genes involved in lipogenesis, lipolysis, FA elongation and oxidation, and ketogenesis (13, 15). While the roles of CREBH in hepatic lipid metabolism have been established, the fine-tuned regulation of this powerful regulator has remained elusive. In this study, we show that fasting or glucagon stimulation induces CREBH protein expression, cleavage, and acetylation (Fig. 1 and 5). We demonstrate that lysine acetylation at position 294 is a stress-inducible, rate-limiting step that is critical for CREBH function. The K294R mutant blocked CREBH acetylation and thus dramatically repressed the transcriptional activities of CREBH, while the K294Q mutant could mimic the CREBH acetylation state and increase CREBH activities (Fig. 2 and 3). The functional importance of CREBH acetylation was demonstrated in animal models. CREBH acetylation at K294 is required to maintain lipid homeostasis in fasting states, as animals expressing the K294R mutant in the liver exhibited severe hepatic steatosis and hyperlipidemia under conditions of prolonged fasting (Fig. 6). These findings not only reveal the critical role of acetylation in CREBH transcriptional activities but also indicate a potential target site in the CREBH protein that may be utilized to modulate CREBH activities.
A critical question is about the mechanism by which acetylation modulates CREBH activities. The bZIP domain of CREBH, where the CREBH acetylation site (K294) is located, contains the DNA-binding and multimerization regions. Acetylation at the bZIP domains may lead to subtle allosteric or electrostatic changes that favor heterodimerization and DNA binding of transcription factors (26, 27). In this study, we demonstrated that CREBH acetylation at K294 is required for the interaction between CREBH and PPARα in glucagon-stimulated hepatocytes or in mouse livers under fasting conditions (Fig. 5A to D). Consistent with this, the defect in CREBH acetylation led to diminished synergy between CREBH and PPARα in activating their common target gene, FGF21 (Fig. 5F). Therefore, the lack of the CREBH-PPARα functional complex may partially account for the hepatic steatosis and hyperlipidemia phenotypes observed in animals with defects in CREBH acetylation at K294 (Fig. 6).
Our study implicates CREBH acetylation as a dynamic process in which the lysine acetyltransferase PCAF and the deacetylase SIRT1 are involved. In the presence of the HDAC inhibitors, PCAF-mediated acetylation and the transcriptional activities of CREBH were significantly increased (Fig. 3), suggesting that CREBH acetylation and deacetylation activities coexist. This notion was supported by the time-dependent CREBH acetylation profile as well as CREBH-PCAF and CREBH-SIRT1 interaction dynamics in the livers of mice in the fasting state (Fig. 1, 3, and 4). We observed that SIRT1 is localized to both the nucleus and the cytoplasm in the fed state or after a 24-h fast but that it accumulated in the nucleus and interacted with CREBH in the liver upon a 6- or 12-h fast. The acetylation of CREBH and the CREBH-PCAF interaction peaked after a 24-h fast, when the CREBH-SIRT1 interaction was concomitantly decreased (Fig. 1, 3, and 4). Therefore, the dynamics of the acetylation/deacetylation cycle mediated through PCAF and SIRT1 likely play a key role in modulating CREBH acetylation states and transcriptional activities in fasting states. Importantly, our study demonstrates that a functional CREBH-PPARα-SIRT1 complex exists in the liver upon fasting (Fig. 4B). While the interactions between CREBH, PPARα, and SIRT1 were significantly increased upon fasting for 6 or 12 h, SIRT1 was dissociated with the CREBH-PPARα complex after a 24-h fast, whereas the CREBH-PPARα interaction was further enhanced. This observation is consistent with the presence and function of SIRT1 in the nucleus of liver cells after the different fasting times as well as the time-dependent CREBH acetylation and CREBH-PPARα interaction profiles upon fasting. SIRT1 may play an important role in maintaining hepatic energy homeostasis through modulation of CREBH acetylation states and the activity of the CREBH-PPARα functional complex under metabolic stress conditions. Several important questions about CREBH acetylation remain to be elucidated in future research: is the CREBH acetylation state associated with the hyperlipidemia that develops under a high-fat diet? Does enhancement of CREBH acetylation protect from the progression of hepatic steatosis? Is CREBH acetylation repressed in human patients with NASH and/or hyperlipidemia symptoms? Answers to these questions will be important to the understanding and treatment of metabolic disorders associated with CREBH activity.
Supplementary Material
ACKNOWLEDGMENTS
This work was partially supported by National Institutes of Health (NIH) grants DK090313 and ES017829 and American Heart Association grants 0635423Z and 09GRNT2280479 (to K.Z.).
K.Z. designed and conducted the experiments, analyzed the data, and wrote and edited the manuscript; H.K. designed and conducted the experiments, analyzed the data, and edited the manuscript; R.M. conducted the experiments and analyzed the data; X.C. conducted the experiments; D.F. conducted the experiments and edited the manuscript.
No potential conflicts of interest relevant to this article were reported.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/MCB.00665-15.
REFERENCES
- 1.Bechmann LP, Hannivoort RA, Gerken G, Hotamisligil GS, Trauner M, Canbay A. 2012. The interaction of hepatic lipid and glucose metabolism in liver diseases. J Hepatol 56:952–964. doi: 10.1016/j.jhep.2011.08.025. [DOI] [PubMed] [Google Scholar]
- 2.Cahill GF., Jr 2006. Fuel metabolism in starvation. Annu Rev Nutr 26:1–22. doi: 10.1146/annurev.nutr.26.061505.111258. [DOI] [PubMed] [Google Scholar]
- 3.Rouvinen-Watt K, Mustonen AM, Conway R, Pal C, Harris L, Saarela S, Strandberg U, Nieminen P. 2010. Rapid development of fasting-induced hepatic lipidosis in the American mink (Neovison vison): effects of food deprivation and re-alimentation on body fat depots, tissue fatty acid profiles, hematology and endocrinology. Lipids 45:111–128. doi: 10.1007/s11745-009-3377-4. [DOI] [PubMed] [Google Scholar]
- 4.Kouzarides T. 1999. Histone acetylases and deacetylases in cell proliferation. Curr Opin Genet Dev 9:40–48. doi: 10.1016/S0959-437X(99)80006-9. [DOI] [PubMed] [Google Scholar]
- 5.Verdone L, Agricola E, Caserta M, Di Mauro E. 2006. Histone acetylation in gene regulation. Brief Funct Genomic Proteomic 5:209–221. doi: 10.1093/bfgp/ell028. [DOI] [PubMed] [Google Scholar]
- 6.Picard F, Kurtev M, Chung N, Topark-Ngarm A, Senawong T, Machado De Oliveira R, Leid M, McBurney MW, Guarente L. 2004. Sirt1 promotes fat mobilization in white adipocytes by repressing PPAR-gamma. Nature 429:771–776. doi: 10.1038/nature02583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lagouge M, Argmann C, Gerhart-Hines Z, Meziane H, Lerin C, Daussin F, Messadeq N, Milne J, Lambert P, Elliott P, Geny B, Laakso M, Puigserver P, Auwerx J. 2006. Resveratrol improves mitochondrial function and protects against metabolic disease by activating SIRT1 and PGC-1alpha. Cell 127:1109–1122. doi: 10.1016/j.cell.2006.11.013. [DOI] [PubMed] [Google Scholar]
- 8.Feige JN, Lagouge M, Canto C, Strehle A, Houten SM, Milne JC, Lambert PD, Mataki C, Elliott PJ, Auwerx J. 2008. Specific SIRT1 activation mimics low energy levels and protects against diet-induced metabolic disorders by enhancing fat oxidation. Cell Metab 8:347–358. doi: 10.1016/j.cmet.2008.08.017. [DOI] [PubMed] [Google Scholar]
- 9.von Meyenn F, Porstmann T, Gasser E, Selevsek N, Schmidt A, Aebersold R, Stoffel M. 2013. Glucagon-induced acetylation of Foxa2 regulates hepatic lipid metabolism. Cell Metab 17:436–447. doi: 10.1016/j.cmet.2013.01.014. [DOI] [PubMed] [Google Scholar]
- 10.Choudhary C, Weinert BT, Nishida Y, Verdin E, Mann M. 2014. The growing landscape of lysine acetylation links metabolism and cell signalling. Nat Rev Mol Cell Biol 15:536–550. doi: 10.1038/nrm3841. [DOI] [PubMed] [Google Scholar]
- 11.Omori Y, Imai J, Watanabe M, Komatsu T, Suzuki Y, Kataoka K, Watanabe S, Tanigami A, Sugano S. 2001. CREB-H: a novel mammalian transcription factor belonging to the CREB/ATF family and functioning via the box-B element with a liver-specific expression. Nucleic Acids Res 29:2154–2162. doi: 10.1093/nar/29.10.2154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhang K, Shen X, Wu J, Sakaki K, Saunders T, Rutkowski DT, Back SH, Kaufman RJ. 2006. Endoplasmic reticulum stress activates cleavage of CREBH to induce a systemic inflammatory response. Cell 124:587–599. doi: 10.1016/j.cell.2005.11.040. [DOI] [PubMed] [Google Scholar]
- 13.Zhang C, Wang G, Zheng Z, Maddipati KR, Zhang X, Dyson G, Williams P, Duncan SA, Kaufman RJ, Zhang K. 2012. Endoplasmic reticulum-tethered transcription factor cAMP responsive element-binding protein, hepatocyte specific, regulates hepatic lipogenesis, fatty acid oxidation, and lipolysis upon metabolic stress in mice. Hepatology 55:1070–1082. doi: 10.1002/hep.24783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lee JH, Giannikopoulos P, Duncan SA, Wang J, Johansen CT, Brown JD, Plutzky J, Hegele RA, Glimcher LH, Lee AH. 2011. The transcription factor cyclic AMP-responsive element-binding protein H regulates triglyceride metabolism. Nat Med 17:812–815. doi: 10.1038/nm.2347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kim H, Mendez R, Zheng Z, Chang L, Cai J, Zhang R, Zhang K. 2014. Liver-enriched transcription factor CREBH interacts with peroxisome proliferator-activated receptor alpha to regulate metabolic hormone FGF21. Endocrinology 155:769–782. doi: 10.1210/en.2013-1490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nakagawa Y, Satoh A, Yabe S, Furusawa M, Tokushige N, Tezuka H, Mikami M, Iwata W, Shingyouchi A, Matsuzaka T, Kiwata S, Fujimoto Y, Shimizu H, Danno H, Yamamoto T, Ishii K, Karasawa T, Takeuchi Y, Iwasaki H, Shimada M, Kawakami Y, Urayama O, Sone H, Takekoshi K, Kobayashi K, Yatoh S, Takahashi A, Yahagi N, Suzuki H, Yamada N, Shimano H. 2014. Hepatic CREB3L3 controls whole-body energy homeostasis and improves obesity and diabetes. Endocrinology 155:4706–4719. doi: 10.1210/en.2014-1113. [DOI] [PubMed] [Google Scholar]
- 17.Luebke-Wheeler J, Zhang K, Battle M, Si-Tayeb K, Garrison W, Chhinder S, Li J, Kaufman RJ, Duncan SA. 2008. Hepatocyte nuclear factor 4alpha is implicated in endoplasmic reticulum stress-induced acute phase response by regulating expression of cyclic adenosine monophosphate responsive element binding protein H. Hepatology 48:1242–1250. doi: 10.1002/hep.22439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lee MW, Chanda D, Yang J, Oh H, Kim SS, Yoon YS, Hong S, Park KG, Lee IK, Choi CS, Hanson RW, Choi HS, Koo SH. 2010. Regulation of hepatic gluconeogenesis by an ER-bound transcription factor, CREBH. Cell Metab 11:331–339. doi: 10.1016/j.cmet.2010.02.016. [DOI] [PubMed] [Google Scholar]
- 19.Zheng Z, Zhang C, Zhang K. 2011. Measurement of ER stress response and inflammation in the mouse model of nonalcoholic fatty liver disease. Methods Enzymol 489:329–348. doi: 10.1016/B978-0-12-385116-1.00019-4. [DOI] [PubMed] [Google Scholar]
- 20.Hornbeck PV, Kornhauser JM, Tkachev S, Zhang B, Skrzypek E, Murray B, Latham V, Sullivan M. 2012. PhosphoSitePlus: a comprehensive resource for investigating the structure and function of experimentally determined post-translational modifications in man and mouse. Nucleic Acids Res 40:D261–D270. doi: 10.1093/nar/gkr1122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dormeyer W, Ott M, Schnolzer M. 2005. Probing lysine acetylation in proteins: strategies, limitations, and pitfalls of in vitro acetyltransferase assays. Mol Cell Proteomics 4:1226–1239. doi: 10.1074/mcp.M500047-MCP200. [DOI] [PubMed] [Google Scholar]
- 22.Yang XJ, Seto E. 2008. Lysine acetylation: codified crosstalk with other posttranslational modifications. Mol Cell 31:449–461. doi: 10.1016/j.molcel.2008.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rodgers JT, Puigserver P. 2007. Fasting-dependent glucose and lipid metabolic response through hepatic sirtuin 1. Proc Natl Acad Sci U S A 104:12861–12866. doi: 10.1073/pnas.0702509104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Brunet A, Sweeney LB, Sturgill JF, Chua KF, Greer PL, Lin Y, Tran H, Ross SE, Mostoslavsky R, Cohen HY, Hu LS, Cheng HL, Jedrychowski MP, Gygi SP, Sinclair DA, Alt FW, Greenberg ME. 2004. Stress-dependent regulation of FOXO transcription factors by the SIRT1 deacetylase. Science 303:2011–2015. doi: 10.1126/science.1094637. [DOI] [PubMed] [Google Scholar]
- 25.Ghosh HS, Spencer JV, Ng B, McBurney MW, Robbins PD. 2007. Sirt1 interacts with transducin-like enhancer of split-1 to inhibit nuclear factor kappaB-mediated transcription. Biochem J 408:105–111. doi: 10.1042/BJ20070817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gu W, Roeder RG. 1997. Activation of p53 sequence-specific DNA binding by acetylation of the p53 C-terminal domain. Cell 90:595–606. doi: 10.1016/S0092-8674(00)80521-8. [DOI] [PubMed] [Google Scholar]
- 27.Hung HL, Kim AY, Hong W, Rakowski C, Blobel GA. 2001. Stimulation of NF-E2 DNA binding by CREB-binding protein (CBP)-mediated acetylation. J Biol Chem 276:10715–10721. doi: 10.1074/jbc.M007846200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.