

Abstract
The association and dissociation of DNA damage response (DDR) factors with damaged chromatin occurs dynamically, which is crucial for the activation of DDR signaling in a spatiotemporal manner. We previously showed that the TIP60 histone acetyltransferase complex acetylates histone H2AX, to facilitate H2AX exchange at sites of DNA damage. However, it remained unclear how the acetylation of histone H2AX by TIP60 is related to the DDR signaling. We found that the acetylation but not the phosphorylation of H2AX is essential for the turnover of NBS1 on damaged chromatin. The loss of H2AX acetylation at Lys 5 by TIP60 in cells disturbed the accumulation of NBS1 at sites of DNA damage. Although the phosphorylation of H2AX is also reportedly required for the retention of NBS1 at damage sites, our data indicated that the acetylation-dependent NBS1 turnover by TIP60 on damaged chromatin restricts the dispersal of NBS1 foci from the sites of DNA damage. These findings indicate the importance of the acetylation-dependent dynamic binding of NBS1 to damaged chromatin, created by histone H2AX exchange, for the proper accumulation of NBS1 at DNA damage sites.
INTRODUCTION
The DNA damage response (DDR) pathway is one of the most important metabolic processes to prevent genome instability, which may enhance carcinogenesis. In the response to DNA damage within chromatin, caused by agents such as ionizing radiation (IR), UV exposure, oxidative stress, and chemotherapeutic agents, the first step involves the detection of the site of DNA damage and the second requires the recruitment of DNA repair factors that can mend the DNA damage in the chromatin context (1, 2).
NBS1, encoded by the Nijmegen breakage syndrome (NBS) gene, forms a complex with Mre11 and Rad50 (MRN complex) (3, 4). The MRN complex functions as the primary sensor of double-strand DNA breaks (DSBs) and recruits signaling proteins, including ATM, to DSB sites (5). Histone H2AX, a histone H2A variant that is one of the numerous targets of ATM, becomes phosphorylated upon DSB formation (6). This phosphorylation is required for the formation of MRN complex foci at the damaged sites, which create a positive feedback loop for H2AX phosphorylation propagation (6–8). It was previously suggested that NBS1 directly interacts with the phosphorylated H2AX (9). However, recent evidence has shown that the interaction between NBS1 and phosphorylated H2AX is not direct but is mediated by MDC1 (10). MDC1 is a constitutive component of the MRN complex regardless of DNA damage and functions as a mediator to recruit several repair factors to the damaged chromatin region (including ATM, NBS1, 53BP1, BRCA1, etc.) (10). It has been proposed that MDC1 directly binds the phosphorylated histone H2AX through its tandem BRCT domains for the accumulation of the MRN complex through MDC1 on damaged chromatin (11).
Recent findings have shown that chromatin remodeling machineries, including histone modifications, nucleosome sliding by ATP-dependent chromatin remodeling factors, and histone/histone variant exchange, play integral roles in the association of checkpoint and/or sensor proteins (for example, NBS1) with chromatin to activate DDR signaling (1, 12–15). The TIP60/ESA1 histone acetyltransferase reportedly forms multiple protein complexes involved in DNA repair and DDR signaling (16–18) and regulates the formation of NBS1 foci at the sites of DNA damage (19–21).
Progress in imaging analysis technology, such as fluorescence recovery after photobleaching (FRAP), has provided informative insights into the dynamics of DDR factors or chromatin components by allowing their turnover rates to be measured at DNA damage sites (22–24). We and another group have previously shown that histone H2AX is acetylated by TIP60 after the induction of DNA damage and that its acetylation at the lysine at position 5 (K5) (Ac-H2AX) accelerates histone H2AX exchange at DSB sites, as determined by a FRAP analysis in combination with microirradiation (25, 26). It appears that histone H2AX exchange by TIP60 actively contributes to DDR signaling or DNA repair. Moreover, a previous study indicated that NBS1 is dynamically exchanged at the sites of DNA damage, which might be vital for interactions with distinct checkpoint or DNA repair pathways to transmit γ-H2AX-dependent DDR signaling (27, 28). However, it remains unclear how the acetylation of H2AX at K5 is related to the process of the focus formation or the dynamic exchange of NBS1 at the sites of DNA damage.
To address these issues, we performed biochemical analyses using the purified TIP60 and H2AX complex in combination with bioimaging. We found that the acetylation of H2AX at K5 by TIP60 is not required for the initial recruitment of NBS1 but is necessary for the accumulation of NBS1 at the damage site. Since the phosphorylation of histone H2AX at S139 is required for the accumulation of NBS1 at the sites of DNA damage (29), we examined the relationship between the acetylation of histone H2AX and its phosphorylation at S139 for NBS1 accumulation at the sites of DNA damage. The depletion of histone H2AX acetylation at K5 in cells or the expression of the TIP60 HAT mutant (TIPM) gene, which results in acetyltransferase-defective TIP60, revealed the formation of γ-H2AX and MDC1 foci after the induction of DNA damage, indicating that the phosphorylation and acetylation of histone H2AX occurred independently of each other upon DDR. Our data revealed that not only MDC1 accumulation and γ-H2AX formation but also H2AX acetylation by TIP60 are required for the accumulation of NBS1 at DNA damage sites. Actually, the acetylation seemed to have a rather suppressive role on the propagation of γ-H2AX-dependent DNA damage signaling. Furthermore, the FRAP analysis in combination with microirradiation showed that the acetylation of histone H2AX at K5 by TIP60 but not the phosphorylation of histone H2AX at S139 is required for the dynamic exchange of NBS1 at the sites of DNA damage. These findings indicate the importance of the dynamic exchange of NBS1, mediated by TIP60-dependent histone H2AX exchange, in the accumulation of NBS1 and tuning of DDR signaling at DNA damage sites.
MATERIALS AND METHODS
Plasmids.
FLAG-hemagglutinin (HA)-tagged H2AX (epitope-tagged H2AX (eH2AX); wild type [WT] and K5R [mutation that replaces K5 with arginine] and S139A mutants), TIP60 (eTIP60), or TIPM (eTIPM; acetylated mutant of TIP60) were reported previously (25, 30). Point and deletion mutants were created with a QuikChange XL site mutagenesis kit (Agilent Technologies). The DNA fragments encoding human H2AX, H2AX K5R, H2AX S139A, TIP60, or TIPM were PCR amplified and cloned into the EcoRI and BglII sites of the pCAGGS vector (31), using an In-Fusion HD cloning kit (Clontech). The NBS1-GFP (green fluorescent protein) plasmid was a kind gift from Nancy Maizeis (Washington University) (32). The pSuper retro control vector was a gift from H. Hosokawa (Kyoto University).
Cell culture and transfection.
HeLa cells (kind gift from Y. Nakatani, Dana-Faber Cancer Institute), mouse embryo fibroblasts (MEFs), GM0637 cells, and U2OS cells were cultured in Dulbecco's modified Eagle's medium supplemented with 10% fetal calf serum. Plasmid transfection was performed according to the manufacturer's protocol, using the GeneJuice transfection reagent (Novagen). H2AX knockout (KO) MEFs were a kind gift from H. Honda (Hiroshima University). For analysis using TIP60- or TIPM-expressing cells, MEFs with pSuper-retro-shTIP60 (plasmid encoding small hairpin RNA [shRNA] against TIP60 [shTIP60]) or pSuper-retro-control (mock RNA) control stably expressed by a retroviral vector were constructed. The cells were then infected with the TIP60 genes on retroviral plasmids, pOZ-eTIP60 or pOZ-eTIPM, and stably expressing cell lines were established. The plasmid construction was previously reported (25, 30).
Antibodies.
Immunoblotting analyses were performed with anti-TIP60 (kind gift from H. Hosokawa, Kyoto University), anti-acetylated H2AX (K5) (Millipore), anti-TRRAP (Santa Cruz), anti-glyceraldehyde-3-phosphate dehydrogenase (anti-GAPDH) (Abcam), anti-H2AX (Sigma), anti-phospho-H2AX (S139) (Millipore), anti-NBS1 (Millipore; Y112), and anti-HA (Roche) antibodies.
Affinity purification of H2AX.
Affinity purification of H2AX complexes was performed as previously described (25, 30). Protein sequences were determined by tandem mass spectrometry (MS/MS) using an Ultima API quadrupole time of flight (Q-TOF) instrument (Waters, Milford, MA). Peptide peak picking was performed by using Mascot Distiller, version 2.3 (Matrix Science, London, United Kingdom). Protein identification was done by a Mascot database search (version 2.3; Matrix Science) of all tandem mass spectra against the Human MS International Protein Index protein sequence database, version 3.53.
Immunohistochemistry analysis.
After the induction of IR or microirradiation, the cells were fixed with methanol-acetone for 20 min. Anti-γ-H2AX (1:2,000) and anti-NBS1 (1:50) antibodies in 1% bovine serum albumin–1× phosphate-buffered saline (PBS) were used. Cy3-conjugated AffiniPure sheep anti-rabbit IgG(H+L) (product code 711-165-152; Jackson ImmunoResearch) was used as the secondary antibody. Nuclei were stained with DAPI (4′,6-diamidino-2-phenylindole) (catalog number 10 236 276 001; Roche). Samples were examined by immunofluorescence microscopy using a Keyence BZ-9000 microscope. To score foci, BZII image software (Keyence) was used.
Microirradiation and FRAP.
The 405-nm line of the UV laser (Leica) was used for the microirradiation. For the microirradiation followed by immunofluorescence, the images were captured with the BZ-9000, and quantification was performed as follows. First, the background signal where the cells were absent was measured. Next, the average fluorescent signals from part of the undamaged nuclear area and the total microirradiated area were measured, and the background signal was subtracted (see Fig. S2A in the supplemental material). In total, 6 to 17 cells were subjected to the measurements, using the BZII image software (Keyence), and the results were plotted on graphs. Statistical comparisons were made using the Student t test. FRAP analyses with UVA microirradiation were performed as previously described (25). We used NBS1 C-terminally tagged with a single GFP and stably expressed in pCAGGS-TIP60 or -TIPM-expressing U2OS cells. Cells that expressed similar amounts of NBS1-GFP were subjected to the FRAP analysis, and the average intensities of the initial NBS1-GFP signal are shown in Fig. S4 in the supplemental material. To quantify the fluorescence recovery, single optical sections were collected at 3-s intervals for the periods of time indicated below. However, to enhance the fluorescent signal, we merged the images from two continuous time frames, and the merged images were subjected to measurement by using Image J. The background fluorescence intensity (BG) and the average fluorescence intensity in the UV-microirradiated and bleached region at each time point (It) were measured as described previously (25). For each time point, the relative intensity at each time point (Irel;t) was calculated as Irel;t = (It − BG)/(I0 − BG), where I0 is the average intensity of the region of interest before bleaching. The percent recovery after each time point in the FRAP analysis (Precovery;t) was calculated and normalized as follows: Precovery;t = 100 × (Irel;t)/(Inorm;t), where Inorm;t is the relative fluorescence intensity, calculated by the same method as Irel;t, in the UV-microirradiated but unbleached region. Mean residence time was calculated as previously described (28). Statistical comparisons were made using the Student t test.
RESULTS
The soluble H2AX complex includes TIP60 histone acetyltransferase and NBS1 only after induction of DNA damage.
To analyze the protein network via histone H2AX after the induction of DNA damage, we purified histone H2AX from the nuclear extracts of HeLa cells stably expressing a C-terminally FLAG-HA epitope-tagged H2AX (eH2AX) before and after DNA damage (25). The eH2AX was purified by affinity chromatography on anti-FLAG antibody-conjugated agarose, and the bound polypeptides were eluted with the FLAG peptide under native conditions. As a control, we performed a mock purification using the soluble nuclear extract of untransfected HeLa cells. The FLAG affinity-purified materials were further purified by immunoaffinity chromatography with anti-HA antibody-conjugated agarose (Fig. 1A) (30). The purified H2AX complex was subjected to mass spectrometry analysis, and MDC1, Rad50, RUVBL1/L2, and TRRAP were identified in the purified H2AX complex (see Table S1 in the supplemental material). We confirmed these findings by Western blotting using anti-MDC1 and anti-TRRAP antibodies, together with antibodies against TIP60 (25) and NBS1, which is reported to interact with MDC1 and Rad50 (11, 33), and found that the binding of these proteins to the histone H2AX complex increased after the induction of DNA damage (Fig. 1B).
FIG 1.
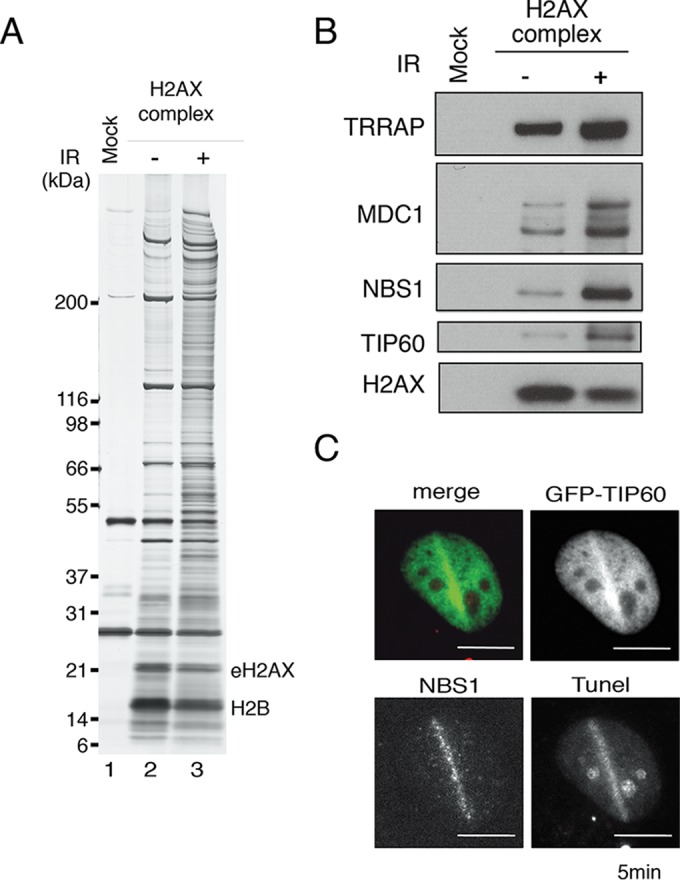
Identification of NBS1 in the H2AX complex. (A) The mock control and eH2AX complexes were immunoaffinity purified from the nuclear soluble fraction of HeLa cells that were not (−) or were (+) treated with 12 Gy IR, followed by a 5-min recovery. (B) Immunoblotting analyses were performed with anti-TRRAP, anti-MDC1, anti-NBS1, anti-TIP60, and anti-H2AX antibodies. (C) Accumulation of TIP60-GFP in GM02063 cells at sites containing DSBs after a 2-min recovery from laser UVA microirradiation. NBS1 staining was performed. Terminal deoxynucleotidyltransferase-mediated dUTP-biotin nick end labeling (TUNEL) staining was performed to detect DSBs induced by microirradiation. TIP60-GFP and TUNEL signals are shown in green and red, respectively, in the merged image. Scale bars, 10 μm.
In parallel, we purified the TIP60 complex from the nuclear extracts of HeLa cells before and after DNA damage (as for mass spectrometry results after DNA damage; see Table S2 in the supplemental material). Western blotting was performed, and NBS1, MDC1, and TRRAP were found within the purified TIP60 complex regardless of DNA damage (see Fig. S1A in the supplemental material). We further analyzed the localization of NBS1 and TIP60 in GM0637 cells expressing GFP-tagged TIP60. DSBs were introduced using the microirradiation method. Both TIP60-GFP and NBS1 accumulated at DNA damage sites along the microirradiated lines within 5 min after microirradiation (Fig. 1C), suggesting a regulatory relationship between NBS1 and TIP60.
The acetyltransferase activity of TIP60 is not required for the initial recruitment of NBS1, but it is required for the restriction of NBS1 foci at the sites of DNA damage.
Previous studies showed that the phosphorylation of histone H2AX is not required for the initial recruitment of NBS1, but it is required for its accumulation at the sites of DNA damage (29). However, it still remains unclear whether the acetylation of H2AX or the acetyltransferase activity of TIP60 is required for the recruitment or the accumulation of NBS1 at the sites of DNA damage. To explore this, first we confirmed that the mutant bearing the TIPM mutation that results in acetyltransferase-defective TIP60 lacked activity in vitro (30). In this analysis, bacterially produced and purified His-TIP60 or TIPM proteins were subjected to an in vitro acetylation assay with histone H4. TIP60 but not TIPM produced acetylated histone 4 (H4) in a dose-dependent manner (Fig. 2A). Thus, we concluded that TIPM displays defective catalytic activity. We then established MEF lines that stably expressed shTIP60 under a retroviral system and simultaneously expressed the shRNA-resistant wild-type TIP60 tagged with FLAG-HA at the N terminus (eTIP60) or the shRNA-resistant eTIP60 derivative lacking histone acetyltransferase activity (eTIPM) from a retroviral vector (Fig. 2B). In these cells, the amount of NBS1 was unaffected (Fig. 2B). We also observed the damage-induced increase in the acetylation of H2AX K5 in shTIP60-expressing cells reconstituted with shRNA-resistant TIP60 (TIP60-reconstituted cells), and the increase was compromised in TIPM-reconstituted cells (see Fig. S1B in the supplemental material). The N-terminal acetylation of bulk histone H4 was unchanged under both nonirradiated and irradiated conditions in TIP60-reconstituted cells. The expression of TIPM did not affect the acetylation status of H4, suggesting that, for H4 acetylation, acetyltransferases other than TIP60 can compensate for this function (see Fig. S1B).
FIG 2.

Acetyltransferase activity of TIP60 is required for proper NBS1 localization to DNA damage sites. (A) His-TIP60 or His-TIPM was expressed in Escherichia coli, purified, and subjected to an in vitro H4 acetylation assay. (B) eTIP60 and eTIPM were expressed in MEFs stably expressing shTIP60. The expression of endogenous TIP60 (endoTIP60), reconstituted eTIP60 (Flag-HA-TIP60) and NBS1 was detected by anti-TIP60, anti-HA, and anti-NBS1 antibody, respectively. (C) Left, examples of microirradiated cells incubated for 4 h before fixation. Microirradiation experiment using eTIP60- or eTIPM-reconstituted MEFs was followed by immunohistochemistry analysis. Top, anti-NBS1 and anti-γ-H2AX antibodies; bottom, anti-MDC1 and anti-γ-H2AX antibodies. Right, the average fluorescence intensities from the undamaged regions or the damaged regions were quantified and plotted. The average intensities of NBS1 immunofluorescence were derived from 11 cells for TIP60-reconstituted cells and 10 cells for TIPM-reconstituted cells, and the average intensities for MDC1 immunofluorescence were derived from 8 cells for TIP60-reconstituted cells and 6 cells for TIPM-reconstituted cells. Examples of measured regions are shown in Fig. S2A in the supplemental material. See Materials and Methods for quantification details. (D) Immunohistochemistry analysis of MEFs reconstituted with TIP60 (left) or TIPM (right) following IR (16 h after 8 Gy). Indirect immunofluorescence analyses were performed using either anti-NBS1 and anti-γ-H2AX or anti-MDC1 and anti-γ-H2AX antibodies. (E) Percentages of cells that formed more than 10 foci of NBS1, MDC1, or γ-H2AX are shown. Cont, control; bar, 10 μm. Graphs show standard deviations.
In order to assess the effect of TIP60 on the localization of NBS1 to DNA damage sites, we first performed the microirradiation experiment using MEFs expressing shTIP60 or mock RNA. Immunohistochemistry analysis using antibodies against NBS1 and γ-H2AX was performed, and the fluorescent signals were quantified (see Fig. S2A in the supplemental material). One hour after microirradiation, a slight decrease in NBS1 accumulation was observed in the MEFs expressing shTIP60 RNA but not in those expressing mock RNA (see Fig. S2B). The decrease of the NBS1 signal in the MEFs expressing shTIP60 became evident at 4 h after microirradiation (see Fig. S2C).
We next performed the microirradiation analysis using MEFs reconstituted by eTIP60 or eTIPM to determine the effect of the histone acetyltransferase activity of TIP60 on the recruitment of NBS1 to the damage sites at 1 h after the microirradiation. An immunohistochemistry analysis of these cells, using an anti-NBS1 antibody, indicated that the initial recruitment (1 h after microirradiation) of NBS1 to the microirradiated area in the TIP60 HAT mutant (TIPM)-reconstituted cells was comparable to that in the TIP60 WT-reconstituted cells (see Fig. S2D in the supplemental material). This result indicated that the acetyltransferase activity of TIP60 is not required for the recruitment of NBS1 to the sites of DNA damage at 1 h after the microirradiation. However, prolonged incubation after the microirradiation revealed that the localization of NBS1 to the microirradiated area in the TIPM-reconstituted cells was diffused and weaker than in the TIP60-reconstituted cells (Fig. 2C). These results indicated that the catalytic activity of TIP60 is dispensable for the initial recruitment of NBS1 but is indispensable for its following accumulation. NBS1 localization to DNA damage sites is reportedly dependent on MDC1 (28). However, importantly, the MDC1 signal was present at 1 h and 4 h after microirradiation in cells expressing shTIP60 (see Fig. S2B and C) and in eTIPM-reconstituted cells (Fig. 2C; see also Fig. S2D), suggesting that the NBS1 localization defect was not conferred by MDC1 dislocalization in the cells with defective TIP60.
The defect in NBS1 accumulation in shTIP60-expressing cells and TIPM-reconstituted cells was confirmed by an analysis using ionizing radiation (IR). An immunohistochemistry analysis using anti-NBS1, anti-MDC1, and anti-γ-H2AX antibodies was performed after the induction of DSBs by IR (2 h and 16 h after irradiation with 8 Gy of IR or 2 h after 2 Gy of IR). Consistent with previous studies (9, 28, 29), although some of the NBS1 colocalized with γ-H2AX, most of the NBS1 signal was dispersed in the nuclei in the shTIP60-expressing cells (2 h or 16 h after 8 Gy of IR and 2 h after 2 Gy; see Fig. S2E to G in the supplemental material) and TIPM-reconstituted MEFs (2 h or 16 h after 8 Gy of IR [Fig. 2D and E]; 2 h after 2 Gy [see Fig. S2H]) than in the mock RNA-expressing cells and TIP60 WT-reconstituted cells. Again, the localization of MDC1 was not altered in TIP60 knockdown MEFs (see Fig. S2E, F, and G) or eTIPM-reconstituted cells (Fig. 2D and E; see also Fig. S2H). It should be noted that TIP60 knockdown cells and eTIPM-reconstituted cells presented stronger accumulation of γ-H2AX than did mock RNA-expressing cells and TIP60-reconstituted cells (Fig. 2C and D; see also Fig. S2).
The acetylation of K5 of H2AX by TIP60 is required for NBS1 assembly at the sites of DNA damage.
We previously reported that TIP60 acetylates K5 of histone H2AX after the induction of DNA damage (25). Thus, we next examined the relationship between H2AX K5 acetylation by TIP60 and NBS1 accumulation at the sites of DNA damage. We used MEFs in which H2AX was knocked out (H2AX KO) (34, 35) and was reconstituted with either the H2AX wild-type histone, a mutant H2AX in which K5 is replaced with arginine (H2AX K5R), or a mutant H2AX in which S139 is replaced with alanine (H2AX S139A) (Fig. 3A) (36). These GFP-tagged mutant H2AXs, as well as H2AX WT-GFP, were confirmed to be incorporated efficiently into mitotic chromatin, suggesting that they retain their basal functions (see Fig. S3A in the supplemental material).
FIG 3.

Cells expressing H2AX K5R are defective in proper NBS1 localization to DNA damage sites. (A) Expression of reconstituted H2AX WT or mutants in MEFs in which the endogenous H2AX was knocked out. (B) Left, examples of microirradiated cells incubated for 4 h before fixation. Microirradiation experiment followed by immunohistochemistry analysis. Top, anti-NBS1 and anti-γ-H2AX antibodies; bottom, anti-MDC1 and anti-γ-H2AX antibodies. Right, fluorescence intensities from the undamaged regions or the damaged regions were quantified and plotted. The average intensities of NBS1 immunofluorescence were derived from 13 cells for H2AX WT-reconstituted cells and 9 cells for H2AX K5R-reconstituted cells, and the average intensities of MDC1 immunofluorescence were derived from 8 cells for H2AX WT-reconstituted cells and 8 cells for H2AX K5R-reconstituted cells. (C) Immunohistochemistry analyses of MEFs reconstituted with H2AX WT, K5R, or S139A following IR (8 Gy, 16 h post-IR). Left, anti-NBS1 and anti-γ-H2AX antibodies; right, anti-MDC1 and anti-γ-H2AX antibodies. (D) The cells with more than 10 foci of NBS1, MDC1, or γ-H2AX were counted among cells irradiated with 8 Gy of IR. Cells were subjected to immunofluorescence analyses either 2 h or 8 h after irradiation. Bar, 10 μm. Graphs show standard deviations.
The H2AX KO cells and the cells reconstituted with H2AX S139A are reportedly proficient in the recruitment but deficient in the sequestration of NBS1 to DNA damage sites (29). As reported previously, we have observed rapid recruitment (15 min) of NBS1 at microirradiated regions (see Fig. S3B in the supplemental material) (29). The defect in the sequestration of NBS1 was confirmed. When the cells were exposed to 8 Gy of IR, while WT MEFs retained the NBS1 foci after 16 h of IR, the H2AX KO MEFs and the MEFs reconstituted with H2AX S139A showed diffuse NBS1 localization (see Fig. S3B).
We examined the accumulation of NBS1 in the H2AX K5R mutant after microirradiation by performing immunohistochemistry analysis using an anti-NBS1 antibody. As expected, NBS1 localization at the microirradiated area was observed after 1 h of irradiation in the cells expressing both H2AX WT and K5R (see Fig. S3C in the supplemental material). However, diffused and decreased localization was observed in the H2AX K5R-reconstituted cells but not in H2AX WT-reconstituted cells after 4 h of irradiation (Fig. 3B).
Next, the localization of NBS1 upon IR-induced DSB formation (2 h and 16 h after irradiation with 8 Gy of IR or 2 h after 2 Gy of IR) was monitored. After the induction of DSBs, although some of the NBS1 colocalized with γ-H2AX, most of the NBS1 signal was dispersed in the nuclei in the histone H2AX K5R-reconstituted MEFs compared to that in the H2AX WT-reconstituted cells (Fig. 3C and D; see also Fig. S3D in the supplemental material). It should be noted that NBS1 foci were also defective in the H2AX S139A-reconstituted cells. Also, MDC1 was present in H2AX K5R-reconstituted cells (Fig. 3C and D; see also Fig. S3D). Taken together with the findings described above, the acetylation of histone H2AX at K5 by TIP60 is required for the efficient accumulation of NBS1 onto damaged chromatin, although MDC1 is present at DNA damage sites.
γ-H2AX focus formation is finely tuned by H2AX acetylation by TIP60 after DNA damage induction.
Consistent with the previous study (37), we detected increased H2AX phosphorylation in eTIPM-reconstituted cells (Fig. 2C and D). The increase was evident in the dose-dependent IR exposure experiments, where the status of H2AX phosphorylation was monitored in eTIP60 WT or eTIP60 HAT mutant (eTIPM)-reconstituted cells (Fig. 4A and B). In contrast, the cellular ATM activity, monitored by the autophosphorylation of Ser1981, indicated only a slight increase in ATM activation in cells expressing eTIPM compared to its activation in eTIP60 WT-reconstituted cells (Fig. 4C and D). This finding is inconsistent with the previous study; however, it may be caused by the irradiation dose or the culture conditions, as we used gamma radiation instead of bleomycin to induce DNA damage (38).
FIG 4.

Acetylation of H2AX K5 restricts γ-H2AX expansion. (A and B) γ-H2AX was monitored in eTIP60- or eTIPM-expressing MEFs (a), and the signals were quantified using ImageJ software (B). (C and D) Phospho-Ser1981 was monitored in eTIP60- or eTIPM-reconstituted MEFs (C) and quantified (D). (E) Ac-H2AX and γ-H2AX were monitored in cell lysates of H2AX KO MEFs reconstituted with H2AX WT, K5R, or S139A. Western blotting was performed with anti-Ac-H2AX(K5), anti-γ-H2AX, or anti-H2AX antibody. (F) H2AX KO MEFs were reconstituted with H2AX or H2AX K5R and subjected to Western blotting. Samples were prepared from cells irradiated with the indicated doses and incubated for 2 to 10 h. (G) Intensity ratios of the γ-H2AX to H2AX signals from the experiment whose results are shown in panel F.
Next, we determined whether the acetylation of H2AX K5 affected the amount of γ-H2AX. To address this issue, we detected acetylation of K5 (Ac-H2AX) and phosphorylation of S139 (γ-H2AX) of H2AX in cell lysates of H2AX KO MEFs reconstituted with H2AX WT, H2AX K5, or H2AX S139A. Ac-H2AX was detected in H2AX S139A-reconstituted cells and γ-H2AX in H2AX K5-reconstituted cells after IR (Fig. 4E). These results indicate that the acetylation and phosphorylation of H2AX occur independently. Importantly, γ-H2AX was increased even in the absence of IR (Fig. 4E), and this become evident in the results of dose-dependent IR exposure experiments using H2AX K5R-reconstituted MEFs, in which the endogenous H2AX was knocked out (Fig. 4F and G).
These experiments led to two conclusions. First, the acetylation of histone H2AX at K5 by TIP60 suppressed γ-H2AX. We speculate that the acetylation could confine the γ-H2AX-dependent DNA damage signaling to the DNA damage sites and does not allow it to expand, possibly, toward undamaged regions. Second, the acetylation of H2AX could play an important role in NBS1 accumulation, besides the phosphorylation of H2AX.
The dynamic exchange of NBS1-GFP requires the acetylation but not phosphorylation of H2AX upon DNA damage.
DDR and chromatin regulatory factors are not stably associated with chromatin but exchange dynamically, particularly at sites of damage (22). Indeed, the persistence of some DDR factors on damaged chromatin may impede the subsequent binding of downstream factors, resulting in the disruption of DDR signaling or DNA repair (39). It has been reported that NBS1 dynamically exchanges at the sites of DNA damage (27). However, the role of the dynamic exchange of NBS1 in DDR and how its dynamism is achieved at the sites of DNA damage remain unclear. Intriguingly, we have previously shown that the acetylation of H2AX by TIP60 facilitates the dynamic exchange of H2AX at DNA damage sites (25).
If NBS1 accumulation is dependent on the acetylation of H2AX, then it should be affected by the TIPM or the H2AX K5R mutation. Thus, we investigated the effects of the expression of the TIPM, H2AX K5R, and S139A mutants on the dynamic exchange of NBS1 at the DNA damage sites by performing a FRAP experiment followed by microirradiation (Fig. 5A and B) (25). We established stable expression of NBS1 tagged with green fluorescent protein (GFP) at the C terminus (NBS1-GFP) in checkpoint-proficient human U2OS cells that also expressed TIP60 or TIPM (Fig. 5C). Using these cells, we performed a microirradiation analysis to investigate whether the acetyltransferase activity of TIP60 is required for the recruitment of NBS1-GFP at the damage sites in vivo. The recruitment of NBS1-GFP to the sites of DNA damage in cells expressing TIPM was comparable to that in the cells expressing TIP60 (data not shown). Therefore, we examined the dynamics of NBS1-GFP at the microirradiated areas in these cells, using FRAP in combination with UVA microirradiation, and then compared the fluorescence recovery curves of NBS1-GFP in cells expressing TIP60 and TIPM. The mean residence times reflected by the curves, but not the percentages, of the recovery of fluorescence intensity after photobleaching of NBS1-GFP were monitored. Increased residence time of NBS1 on DNA damage sites was reported previously (27), and we also observed a slight increase in the residence time of NBS1 at DNA damage sites in TIP60 WT-expressing cells (Fig. 5E).
FIG 5.

H2AX K5 acetylation by TIP60 is required for efficient GFP-NBS1 turnover on damaged chromatin. (A and B) Schematics of FRAP followed by microirradiation for the undamaged area (A) and for the microirradiated area (B). (C) Expression of NBS1-GFP in U2OS cells expressing TIP60 or TIPM from the pCAGGS vector. Results for control cells expressing neither TIP60 nor NBS1-GFP are in the Empty vector lane. (D) Fluorescence recovery curves of NBS1-GFP after bleaching an unirradiated area (left; n = 12 for TIP60 and n = 13 for TIPM) or UVA-microirradiated area (right; n = 14 for TIP60 and n = 12 for TIPM). U2OS cells stably expressing TIP60 or TIPM were used. (E) Mean residence times from the analysis whose results are shown in panel D. (F) Expression of NBS1-GFP in U2OS cells expressing H2AX WT, K5R, or S139A from pCAGGS. Results for control cells expressing neither H2AXs nor NBS-GFP are in the Empty vector lane. (G) Fluorescence recovery curves of NBS1-GFP after bleaching an unirradiated area (left; n = 12 for WT, n = 10 for K5R, and n = 11 for S139A) or a UVA-microirradiated area (right; n = 12 for WT, n = 10 for K5R, and n = 10 for S139A). (H) Mean residence times from the analysis whose results are shown in panel G. Mean residence times were calculated as previously reported (28) and statistically analyzed by using the Student t test. P values are indicated. Graphs show standard deviations.
In TIPM-expressing cells, the retention times of NBS1-GFP were significantly longer than those in TIP60 WT-expressing cells (Fig. 5D and E) (28). Importantly, the prolonged residence times were observed exclusively in the microirradiated areas.
We performed further examinations in cells expressing H2AX WT, K5R, or S139A (Fig. 5F). In H2AX S139A cells, it can be speculated that faster NBS1 kinetics occurs on DNA damage sites, as the MDC1 knock down decreases the residence time of NBS1-GFP (28), and we also observed a slight decrease in the residence time of NBS1-GFP at DNA damage sites in H2AX S139A-expressing cells compared to that in H2AX WT-expressing cells (Fig. 5H).
Importantly, the dynamics of NBS1-GFP at the DNA damage sites in cells expressing H2AX K5R showed a longer residence time than those in cells expressing H2AX WT or H2AX S139A (Fig. 5G and H). These results in turn imply that NBS1 accumulation occurs in tight cooperation with the acetylation of H2AX, consistent with our finding that NBS1 associates with the H2AX complex.
DISCUSSION
Tip60 enhances the proper assembly of NBS1 at DNA damage sites.
In this study, we found that the acetylation of histone H2AX at K5 by TIP60 is required for the accumulation of NBS1 at the sites of DNA damage. We have also determined that NBS1 is included in the TIP60 complex regardless of DNA damage (see Fig. S1A in the supplemental material). We previously showed that the TIP60 complex facilitates histone H2AX exchange at DNA damage sites via the acetylation of histone H2AX on K5 but not via the phosphorylation of H2AX on S139 (25). Considering the data showing that soluble H2AX-H2B can bind to NBS1 only after the induction of DNA damage (Fig. 1B), it is quite possible that the histone H2AX exchange promoted by TIP60 induces the dynamic binding, the association and dissociation, of NBS1 to the sites of DNA damage. If so, then the acetylation-dependent binding of NBS1 to the H2AX evicted from damaged chromatin will increase the chances for NBS1 to bind to the DNA damage sites, in accordance with the incorporation of the evicted histone H2AX into the nucleosomes around the damaged region.
Acetylation-dependent NBS1 assembly is independent of MDC1 and γ-H2AX.
A previous study has shown that the phosphorylation of histone H2AX is also reportedly dispensable for the initial recruitment of NBS1 but indispensable for its accumulation at the sites of DNA damage (29). We found that the acetylation and phosphorylation of histone H2AX after the induction of DNA damage occurred independently. Importantly, MDC1 foci were observed in the eTIPM- or H2AX K5R-expressing cells after DNA damage.
Therefore, our results indicate the importance of the H2AX acetylation-dependent accumulation of NBS1 at the sites of DNA damage, which occurs independently of MDC1 and γ-H2AX.
Possible role of H2AX acetylation in modulating DNA damage signaling and DNA repair regulation.
The checkpoint mediator MDC1 reportedly recognizes H2AX phosphorylated at S139 for the subsequent recruitment of MRN and ATM to DNA damage sites, and then ATM phosphorylates histone H2AX at these sites (7, 10, 28). This positive feedback loop between phosphorylated H2AX, MDC1, and ATM is crucial for the amplification and spreading of γ-H2AX to form the nuclear foci in the region of DNA damage (7). In this study, we found that defective acetylation of histone H2AX at K5 by TIP60 produced increased H2AX phosphorylation foci after DNA damage. We interpret this result to mean that H2AX acetylation restricts the increase of damaged chromatin signaling. Alternatively, as H2AX K5R-reconstituted cells showed a weak sensitivity to IR (25), the fact that the cells are vulnerable to IR could cause the increase of γ-H2AX. However, it is still unclear how this occurs, and these points should be addressed in the future.
NBS1, which also plays a key role in promoting homologous recombination (40, 41), could accumulate at DNA damage sites via a mechanism coupled with histone exchange. In fact, we previously showed that the TIP60 complex is involved in the focus formation of Rad51, which is a key player in homologous recombination repair, at DNA damage sites (25). Homologous recombination repair requires chromatin opening to increase the accessibility of nucleases at DNA damage sites (37, 42–45). Thus, it is quite possible that the histone exchange by the TIP60 complex, via the acetylation of histone H2AX at K5, couples NBS1 assembly and chromatin opening at DNA damage sites.
The association of NBS1 with TIP60 acetyltransferase and H2AX suggests that NBS1 could be incorporated in the vicinity of DSBs for its accumulation through the H2AX exchange (Fig. 6). The accumulation of NBS1 follows the initial recruitment (H2AX-indepent phase), when the other mechanisms or possibly the nature of the DSB itself could attract NBS1. Further accumulation of NBS1 would possibly require the H2AX-dependent mechanism (H2AX-dependent phase). The acetylation of H2AX K5 would continuously incorporate the NBS1 onto damaged chromatin and maintain it in a rather dynamic state. The subsequent cooperation with γ-H2AX would permit NBS1 accumulation. In conclusion, we propose a model featuring the importance of the histone exchange-driven mechanism that couples DNA damage signaling, DNA metabolism, and chromatin dynamics in the eukaryotic system (Fig. 6).
FIG 6.

Histone H2AX acetylation by TIP60 regulates the dynamic binding of NBS1 at DNA damage sites for its accumulation. Schematic model explains the NBS1 recruitment and accumulation. See the text for details.
Supplementary Material
ACKNOWLEDGMENTS
We thank M. Takata, T. Matsumoto, T. Habu, J. Kobayashi, K. Komatsu (RBC, Kyoto University), and Y. Agata (Shiga Univiversity of Medical Science) for discussions and comments on the manuscript and H. Suzuki and A. Kinomura (Hiroshima University) for technical assistance.
This work was supported by Grants-in-Aid from the Ministry of Education, Culture, Sports, Science, and Technology of Japan.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/MCB.00757-15.
REFERENCES
- 1.Soria G, Polo SE, Almouzni G. 2012. Prime, repair, restore: the active role of chromatin in the DNA damage response. Mol Cell 46:722–734. doi: 10.1016/j.molcel.2012.06.002. [DOI] [PubMed] [Google Scholar]
- 2.Smeenk G, van Attikum H. 2013. The chromatin response to DNA breaks: leaving a mark on genome integrity. Annu Rev Biochem 82:55–80. doi: 10.1146/annurev-biochem-061809-174504. [DOI] [PubMed] [Google Scholar]
- 3.Matsuura S, Tauchi H, Nakamura A, Kondo N, Sakamoto S, Endo S, Smeets D, Solder B, Belohradsky BH, Der Kaloustian VM, Oshimura M, Isomura M, Nakamura Y, Komatsu K. 1998. Positional cloning of the gene for Nijmegen breakage syndrome. Nat Genet 19:179–181. doi: 10.1038/549. [DOI] [PubMed] [Google Scholar]
- 4.Tauchi H, Matsuura S, Kobayashi J, Sakamoto S, Komatsu K. 2002. Nijmegen breakage syndrome gene, NBS1, and molecular links to factors for genome stability. Oncogene 21:8967–8980. doi: 10.1038/sj.onc.1206136. [DOI] [PubMed] [Google Scholar]
- 5.Iijima K, Komatsu K, Matsuura S, Tauchi H. 2004. The Nijmegen breakage syndrome gene and its role in genome stability. Chromosoma 113:53–61. doi: 10.1007/s00412-004-0298-0. [DOI] [PubMed] [Google Scholar]
- 6.Scully R, Xie A. 2013. Double strand break repair functions of histone H2AX. Mutat Res 750:5–14. doi: 10.1016/j.mrfmmm.2013.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lou Z, Minter-Dykhouse K, Franco S, Gostissa M, Rivera MA, Celeste A, Manis JP, van Deursen J, Nussenzweig A, Paull TT, Alt FW, Chen J. 2006. MDC1 maintains genomic stability by participating in the amplification of ATM-dependent DNA damage signals. Mol Cell 21:187–200. doi: 10.1016/j.molcel.2005.11.025. [DOI] [PubMed] [Google Scholar]
- 8.Altmeyer M, Lukas J. 2013. To spread or not to spread—chromatin modifications in response to DNA damage. Curr Opin Genet Dev 23:156–165. doi: 10.1016/j.gde.2012.11.001. [DOI] [PubMed] [Google Scholar]
- 9.Kobayashi J, Tauchi H, Sakamoto S, Nakamura A, Morishima K, Matsuura S, Kobayashi T, Tamai K, Tanimoto K, Komatsu K. 2002. NBS1 localizes to gamma-H2AX foci through interaction with the FHA/BRCT domain. Curr Biol 12:1846–1851. doi: 10.1016/S0960-9822(02)01259-9. [DOI] [PubMed] [Google Scholar]
- 10.Stucki M, Clapperton JA, Mohammad D, Yaffe MB, Smerdon SJ, Jackson SP. 2005. MDC1 directly binds phosphorylated histone H2AX to regulate cellular responses to DNA double-strand breaks. Cell 123:1213–1226. doi: 10.1016/j.cell.2005.09.038. [DOI] [PubMed] [Google Scholar]
- 11.Stucki M, Jackson SP. 2006. gammaH2AX and MDC1: anchoring the DNA-damage-response machinery to broken chromosomes. DNA Repair (Amst) 5:534–543. doi: 10.1016/j.dnarep.2006.01.012. [DOI] [PubMed] [Google Scholar]
- 12.Gospodinov A, Herceg Z. 2013. Chromatin structure in double strand break repair. DNA Repair 12:800–810. doi: 10.1016/j.dnarep.2013.07.006. [DOI] [PubMed] [Google Scholar]
- 13.Swygert SG, Peterson CL. 2014. Chromatin dynamics: interplay between remodeling enzymes and histone modifications. Biochim Biophys Acta 1839:728–736. doi: 10.1016/j.bbagrm.2014.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.van Attikum H, Gasser SM. 2009. Crosstalk between histone modifications during the DNA damage response. Trends Cell Biol 19:207–217. doi: 10.1016/j.tcb.2009.03.001. [DOI] [PubMed] [Google Scholar]
- 15.Ikura T, Ogryzko VV. 2003. Chromatin dynamics and DNA repair. Front Biosci 8:s149–s155. doi: 10.2741/1013. [DOI] [PubMed] [Google Scholar]
- 16.Xu Y, Price BD. 2011. Chromatin dynamics and the repair of DNA double strand breaks. Cell Cycle 10:261–267. doi: 10.4161/cc.10.2.14543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sapountzi V, Logan IR, Robson CN. 2006. Cellular functions of TIP60. Int J Biochem Cell Biol 38:1496–1509. doi: 10.1016/j.biocel.2006.03.003. [DOI] [PubMed] [Google Scholar]
- 18.Squatrito M, Gorrini C, Amati B. 2006. Tip60 in DNA damage response and growth control: many tricks in one HAT. Trends Cell Biol 16:433–442. doi: 10.1016/j.tcb.2006.07.007. [DOI] [PubMed] [Google Scholar]
- 19.Chailleux C, Tyteca S, Papin C, Boudsocq F, Puget N, Courilleau C, Grigoriev M, Canitrot Y, Trouche D. 2010. Physical interaction between the histone acetyl transferase Tip60 and the DNA double-strand breaks sensor MRN complex. Biochem J 426:365–371. doi: 10.1042/BJ20091329. [DOI] [PubMed] [Google Scholar]
- 20.Sun Y, Jiang X, Price BD. 2010. Tip60: connecting chromatin to DNA damage signaling. Cell Cycle 9:930–936. doi: 10.4161/cc.9.5.10931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Robert F, Hardy S, Nagy Z, Baldeyron C, Murr R, Dery U, Masson JY, Papadopoulo D, Herceg Z, Tora L. 2006. The transcriptional histone acetyltransferase cofactor TRRAP associates with the MRN repair complex and plays a role in DNA double-strand break repair. Mol Cell Biol 26:402–412. doi: 10.1128/MCB.26.2.402-412.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Polo SE, Jackson SP. 2011. Dynamics of DNA damage response proteins at DNA breaks: a focus on protein modifications. Genes Dev 25:409–433. doi: 10.1101/gad.2021311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Walter J, Cremer T, Miyagawa K, Tashiro S. 2003. A new system for laser-UVA-microirradiation of living cells. J Microsc 209:71–75. doi: 10.1046/j.1365-2818.2003.01117.x. [DOI] [PubMed] [Google Scholar]
- 24.Tobias F, Lob D, Lengert N, Durante M, Drossel B, Taucher-Scholz G, Jakob B. 2013. Spatiotemporal dynamics of early DNA damage response proteins on complex DNA lesions. PLoS One 8:e57953. doi: 10.1371/journal.pone.0057953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ikura T, Tashiro S, Kakino A, Shima H, Jacob N, Amunugama R, Yoder K, Izumi S, Kuraoka I, Tanaka K, Kimura H, Ikura M, Nishikubo S, Ito T, Muto A, Miyagawa K, Takeda S, Fishel R, Igarashi K, Kamiya K. 2007. DNA damage-dependent acetylation and ubiquitination of H2AX enhances chromatin dynamics. Mol Cell Biol 27:7028–7040. doi: 10.1128/MCB.00579-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kusch T, Florens L, Macdonald WH, Swanson SK, Glaser RL, Yates JR III, Abmayr SM, Washburn MP, Workman JL. 2004. Acetylation by Tip60 is required for selective histone variant exchange at DNA lesions. Science 306:2084–2087. doi: 10.1126/science.1103455. [DOI] [PubMed] [Google Scholar]
- 27.Lukas C, Falck J, Bartkova J, Bartek J, Lukas J. 2003. Distinct spatiotemporal dynamics of mammalian checkpoint regulators induced by DNA damage. Nat Cell Biol 5:255–260. doi: 10.1038/ncb945. [DOI] [PubMed] [Google Scholar]
- 28.Lukas C, Melander F, Stucki M, Falck J, Bekker-Jensen S, Goldberg M, Lerenthal Y, Jackson SP, Bartek J, Lukas J. 2004. Mdc1 couples DNA double-strand break recognition by Nbs1 with its H2AX-dependent chromatin retention. EMBO J 23:2674–2683. doi: 10.1038/sj.emboj.7600269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Celeste A, Fernandez-Capetillo O, Kruhlak MJ, Pilch DR, Staudt DW, Lee A, Bonner RF, Bonner WM, Nussenzweig A. 2003. Histone H2AX phosphorylation is dispensable for the initial recognition of DNA breaks. Nat Cell Biol 5:675–679. doi: 10.1038/ncb1004. [DOI] [PubMed] [Google Scholar]
- 30.Ikura T, Ogryzko VV, Grigoriev M, Groisman R, Wang J, Horikoshi M, Scully R, Qin J, Nakatani Y. 2000. Involvement of the TIP60 histone acetylase complex in DNA repair and apoptosis. Cell 102:463–473. doi: 10.1016/S0092-8674(00)00051-9. [DOI] [PubMed] [Google Scholar]
- 31.Niwa H, Yamamura K, Miyazaki J. 1991. Efficient selection for high-expression transfectants with a novel eukaryotic vector. Gene 108:193–199. doi: 10.1016/0378-1119(91)90434-D. [DOI] [PubMed] [Google Scholar]
- 32.Yabuki M, Fujii MM, Maizels N. 2005. The MRE11-RAD50-NBS1 complex accelerates somatic hypermutation and gene conversion of immunoglobulin variable regions. Nat Immunol 6:730–736. doi: 10.1038/ni1215. [DOI] [PubMed] [Google Scholar]
- 33.Spycher C, Miller ES, Townsend K, Pavic L, Morrice NA, Janscak P, Stewart GS, Stucki M. 2008. Constitutive phosphorylation of MDC1 physically links the MRE11-RAD50-NBS1 complex to damaged chromatin. J Cell Biol 181:227–240. doi: 10.1083/jcb.200709008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Celeste A, Petersen S, Romanienko PJ, Fernandez-Capetillo O, Chen HT, Sedelnikova OA, Reina-San-Martin B, Coppola V, Meffre E, Difilippantonio MJ, Redon C, Pilch DR, Olaru A, Eckhaus M, Camerini-Otero RD, Tessarollo L, Livak F, Manova K, Bonner WM, Nussenzweig MC, Nussenzweig A. 2002. Genomic instability in mice lacking histone H2AX. Science 296:922–927. doi: 10.1126/science.1069398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bassing CH, Chua KF, Sekiguchi J, Suh H, Whitlow SR, Fleming JC, Monroe BC, Ciccone DN, Yan C, Vlasakova K, Livingston DM, Ferguson DO, Scully R, Alt FW. 2002. Increased ionizing radiation sensitivity and genomic instability in the absence of histone H2AX. Proc Natl Acad Sci U S A 99:8173–8178. doi: 10.1073/pnas.122228699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rogakou EP, Pilch DR, Orr AH, Ivanova VS, Bonner WM. 1998. DNA double-stranded breaks induce histone H2AX phosphorylation on serine 139. J Biol Chem 273:5858–5868. doi: 10.1074/jbc.273.10.5858. [DOI] [PubMed] [Google Scholar]
- 37.Sharma GG, So S, Gupta A, Kumar R, Cayrou C, Avvakumov N, Bhadra U, Pandita RK, Porteus MH, Chen DJ, Cote J, Pandita TK. 2010. MOF and histone H4 acetylation at lysine 16 are critical for DNA damage response and double-strand break repair. Mol Cell Biol 30:3582–3595. doi: 10.1128/MCB.01476-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sun Y, Jiang X, Chen S, Fernandes N, Price BD. 2005. A role for the Tip60 histone acetyltransferase in the acetylation and activation of ATM. Proc Natl Acad Sci U S A 102:13182–13187. doi: 10.1073/pnas.0504211102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mortusewicz O, Ame JC, Schreiber V, Leonhardt H. 2007. Feedback-regulated poly(ADP-ribosyl)ation by PARP-1 is required for rapid response to DNA damage in living cells. Nucleic Acids Res 35:7665–7675. doi: 10.1093/nar/gkm933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tauchi H, Kobayashi J, Morishima K, van Gent DC, Shiraishi T, Verkaik NS, vanHeems D, Ito E, Nakamura A, Sonoda E, Takata M, Takeda S, Matsuura S, Komatsu K. 2002. Nbs1 is essential for DNA repair by homologous recombination in higher vertebrate cells. Nature 420:93–98. doi: 10.1038/nature01125. [DOI] [PubMed] [Google Scholar]
- 41.Bressan DA, Baxter BK, Petrini JH. 1999. The Mre11-Rad50-Xrs2 protein complex facilitates homologous recombination-based double-strand break repair in Saccharomyces cerevisiae. Mol Cell Biol 19:7681–7687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tashiro S, Walter J, Shinohara A, Kamada N, Cremer T. 2000. Rad51 accumulation at sites of DNA damage and in postreplicative chromatin. J Cell Biol 150:283–291. doi: 10.1083/jcb.150.2.283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Murr R, Loizou JI, Yang YG, Cuenin C, Li H, Wang ZQ, Herceg Z. 2006. Histone acetylation by Trrap-Tip60 modulates loading of repair proteins and repair of DNA double-strand breaks. Nat Cell Biol 8:91–99. doi: 10.1038/ncb1343. [DOI] [PubMed] [Google Scholar]
- 44.van Attikum H, Gasser SM. 2005. ATP-dependent chromatin remodeling and DNA double-strand break repair. Cell Cycle 4:1011–1014. doi: 10.4161/cc.4.8.1887. [DOI] [PubMed] [Google Scholar]
- 45.Sinha M, Peterson CL. 2009. Chromatin dynamics during repair of chromosomal DNA double-strand breaks. Epigenomics 1:371–385. doi: 10.2217/epi.09.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.