

Abstract
Acetylcholine receptor (AChR) expression in innervated muscle is limited to the synaptic region. Neuron-induced electrical activity participates in this compartmentalization by promoting the repression of AChR expression in the extrasynaptic regions. Here, we show that the corepressor CtBP1 (C-terminal binding protein 1) is present on the myogenin promoter together with repressive histone marks. shRNA-mediated downregulation of CtBP1 expression is sufficient to derepress myogenin and AChR expression in innervated muscle. Upon denervation, CtBP1 is displaced from the myogenin promoter and relocates to the cytoplasm, while repressive histone marks are replaced by activating ones concomitantly to the activation of myogenin expression. We also observed that upon denervation the p21-activated kinase 1 (PAK1) expression is upregulated, suggesting that phosphorylation by PAK1 may be involved in the relocation of CtBP1. Indeed, preventing CtBP1 Ser158 phosphorylation induces CtBP1 accumulation in the nuclei and abrogates the activation of myogenin and AChR expression. Altogether, these findings reveal a molecular mechanism to account for the coordinated control of chromatin modifications and muscle gene expression by presynaptic neurons via a PAK1/CtBP1 pathway.
INTRODUCTION
Chemical synapses are the preferred mean of intercellular communication used by neurons. Efficient transmission of the information emitted by presynaptic neurons requires a highly specialized postsynaptic network whereby membrane receptor aggregation and gene expression are coordinately regulated (1). At the cholinergic neuromuscular synapse, agrin secreted by the nerve terminal activates the postsynaptic tyrosine kinase receptor MuSK through Lrp4, triggering both acetylcholine receptor (AChR) aggregation in the postsynaptic membrane and AChR gene expression in subsynaptic nuclei (2). MuSK is associated with numerous proteins to mediate its activities, including p21-activated kinase 1 (PAK1), which plays a key role in AChR aggregation (3). AChRs accumulated in the postsynaptic membrane bind acetylcholine released by the nerve terminal, thereby initiating nerve-evoked electrical activity in muscle fibers.
Skeletal muscle fibers are multinucleated giant cells that present the unique characteristic of containing two classes of nuclei with distinct chromatin organization and different gene expression programs. Indeed, in response to neuronal agrin, the chromatin in few nuclei located beneath the nerve terminal is relaxed and genes coding for the components of the neuromuscular junction, including AChR subunits, are activated (4). On the contrary, in nonsynaptic nuclei, chromatin is more condensed, and electrical activity represses most of these genes.
The basic helix-loop-helix myogenic transcription factor myogenin plays a pivotal role during skeletal muscle differentiation prior to innervation and is an activator of AChR gene expression. Once muscle fibers are innervated, myogenin expression is repressed by electrical activity, thereby contributing to the confinement of the AChR genes expression to the subsynaptic region. In adult muscle, myogenin is therefore absent, unless muscle innervation is compromised. In such cases, myogenin expression is reactivated and participates in the upregulation of AChR expression all along the muscle fibers (5, 6). At least three mechanisms are involved in myogenin activation after denervation: (i) the downregulation of the expression of the DNA binding transcriptional repressor of myogenin MSY3 (7), (ii) the downregulation of the transcriptional repressor Dach2 that blocks activation of myogenin expression by MEF3/six (8, 9), and (iii) the downregulation of MITR/HDAC9 expression to alleviate its repressive action on MEF2 (5).
Histones play a key role in the regulation of the genome compaction and dynamic and have been shown to undergo a wide variety of posttranslational modifications associated with transcription activation or inhibition (10, 11). The development of chromatin immunoprecipitation (ChIP) techniques and of genome wide analysis of histone modifications have provided a global view of the combination of histone modifications associated with transcriptional activation and repression (12, 13), consistent with the idea that the various posttranslational modifications of histones present at specific loci form a code specifying downstream transcriptional events. In skeletal muscle, myogenin expression has previously been shown to be associated with chromatin acetylations (5).
Histone acetylation is controlled by histone acetyltransferases and histone deacetylases (HDACs) and is associated with activated transcription. Histone methylation on lysines is controlled by vast families of histone methyltransferases and histone demethylases and is more versatile than acetylation since lysines can be mono-, di-, or trimethylated and can either be linked to chromatin condensation or opening, depending on the affected histone residue. Extensive studies on the methylation of lysines 4 and 9 of histone H3 have established that lysine 4 of histone H3 (H3K4) di- and trimethylation (H3K4me2 and H3K4me3) correlate with transcriptional activation and are commonly associated with lysine 9 of histone H3 (H3K9) acetylation (H3K9ac), whereas H3K9 di- and trimethylation (H3K9me2 and H3K9me3) correlate with transcriptional repression (10, 11).
The specific combination of various histone modifications on a promoter implies the existence of mechanisms to coordinate the action of histone-modifying enzymes. These mechanisms involve large transcriptional regulatory complexes that can promote the concomitant recruitment of a combination of enzymes on a given promoter via coactivators or corepressors. The biological function and regulation of corepressors have been thoroughly described in the case of nuclear hormone receptors but remain elusive in most other cases, especially in postnatal tissues. C-terminal binding protein 1 (CtBP1) is a transcriptional corepressor that mediates its transcriptional function through interaction with various DNA-binding repressors. Biochemical approaches have identified the corepressor CtBP1 as a molecular platform able to promote H3K9 deacetylation, H3K4 demethylation, and H3K9 methylation on numerous promoters, through the recruitment of various chromatin modifiers such as HDACs, histone demethylases, and histone methyltransferases (14). CtBP1 also antagonizes the activity of the coactivator p300 (15). Recent studies have also shown that CtBP1 could also participate in the activation of gene expression (16) suggesting that, depending on the components of the regulatory complex, opposing outcomes are possible. CtBP1 was also shown to be regulated by phosphorylation on serine 158 (Ser158) by PAK1 which promotes CtBP1 cytoplasmic localization (17). CtBP1 inactivation in mice was shown to block late stages of muscle differentiation, but the precise function and regulation of CtBP1 in skeletal muscle remains unknown (18). We show here that CtBP1 is a key mediator of the action of neural factors and electrical activity on muscle gene expression by translating the activity of the kinase PAK1 into chromatin modifications on the promoters of myogenin and AChR genes.
MATERIALS AND METHODS
All data shown are representative of three or more experiments. For histograms, mean values and standard deviations are indicated. Statistical analysis (Mann-Whitney test) was performed as indicated to ensure the validity of the data. Scanning of autoradiograms and quantification were carried out using ImageJ. The tibialis anterior (TA) muscles were used in all experiments.
Preparation of muscle extracts.
TA muscles were washed in cold phosphate-buffered saline (PBS) and crushed at 4°C to make a paste in 4 volumes of hypotonic buffer (20 mM Tris-HCl [pH 8], 1 mM EDTA, 5 mM dithiothreitol [DTT], Roche protease inhibitor cocktail) and then incubated for 15 min on ice. Triton X-100 was added to a 0.1% final concentration, and the mixture was subjected to 20 strokes of Dounce homogenization (loose pestle). Nuclei were recovered by centrifugation 5 min at 5,000 rpm at 4°C. The supernatant (cytoplasmic extract) was removed and stored at −20°C. The pellet was resuspended in 4 volumes of high-salt buffer (20 mM Tris-HCl [pH 8], 20% glycerol, 0.42 M NaCl, 1.5 mM MgCl2, 0.2 mM EDTA, 0.5 mM DTT, protease inhibitors), followed by incubation for 15 min on ice. After 20 strokes of Dounce homogenization (tight pestle) and a 10-min centrifugation at 10,000 rpm at 4°C, the supernatant (nuclear extract) was diluted twice with buffer C (20 mM Tris-HCl [pH 8], 1.5 mM MgCl2, 0.2 mM EDTA, 0.5 mM DTT, protease inhibitors) and stored at −20°C.
Muscle chromatin extracts and ChIP.
Ten mouse TA muscles were cut in small pieces and incubated for 15 min on ice in buffer RBI (100 mM KCl, 5 mM MgCl2, 5 mM EDTA, 5 mM sodium pyrophosphate [pH 6.8], protease inhibitors). Muscles were washed in cold PBS and treated with 2% formaldehyde in PBS for 15 min at 37°C. Glycine at 0.125 mM (final) was then added for 5 min. The muscle samples were washed in cold PBS and crushed at 4°C to make a paste in lysis buffer A (10 mM Tris-HCl [pH 7.9], 85 mM KCl, 0.5% Nonidet P-40, protease inhibitors). After 10 min on ice, the mixture was homogenized with 20 Dounce strokes with a loose pestle. After a 5 min of spinning at 5,000 rpm at 4°C, the pellet was resuspended in sodium dodecyl sulfate (SDS) lysis buffer (50 mM Tris-HCl [pH 6.8], 10 mM EDTA, 1% SDS, protease inhibitors). After 10 min on ice, the mixture was spin 5 min at 5,000 rpm at 4°C, and the supernatant was subjected to sonication in a Bioruptor (Diagenode) for 15 min on medium power with 30-s/30-s sonication/pause cycles. The size of fragments was controlled on an agarose gel after de-cross-linking.
The protein concentration of the chromatin extract was determined with a Bio-Rad Protein assay. A total of 100 to 200 μg of extract was incubated with rotation for 3 to 12 h at 4°C with 10 to 20 μg of antibody or no antibody (used to define the basal level). Then, 10 to 20 μl of protein G-magnetic beads (EZ Magna ChIP; Millipore) was added for 2 h at 4°C, and the samples were then treated according to the protocol described by the manufacturer of the EZ Magna ChIP kit.
After elution, the samples were treated for de-cross-linking by incubation at 65°C for 4 to 12 h with the addition of 0.3 M NaCl. 10 mM EDTA, 40 mM Tris-HCl (pH 7), and 40 ng of proteinase K/μl were added, and the mixture was further incubated 1 h at 45°C. DNA was recovered by phenol-chloroform extraction and ethanol precipitation and then resuspended in 50 μl of H2O. Next, 20 μl was used for PCR analysis. Immunoprecipitated DNA was analyzed by both classical PCR and quantitative PCR (Q-PCR). The PCR analyses were performed either in a Biometra T3 thermocycler or using a Q-PCR Rotorgene system (Qiagen). Q-PCR data were analyzed with the Qiagen software, and reactions were also recovered and loaded onto an agarose gel stained with ethidium bromide to control the amplification and for scanning using the ImageJ software. The results were expressed as the enrichment from the initial sample.
Western blotting and coimmunoprecipitation.
Extracts of mouse TA muscles were prepared using the same protocol as for the ChIP extract except that the formaldehyde/glycine treatment was omitted. The supernatant of the first lysis (cytoplasmic extract) and second lysis (nuclear extract) were recovered. For Western blotting, 20 to 50 μg of extract was used for SDS-PAGE on a Mini-Protean 3 system (Bio-Rad) with 8 to 15% acrylamide gels depending on the protein to be detected. Proteins were transferred to polyvinylidene difluoride membranes using a Criterion blotter (Bio-Rad).
The coimmunoprecipitation were done using a Catch-and-Release reversible immunoprecipitation kit (Millipore). Immunoprecipitated proteins were detected by Western blotting with an ECL detection kit (GE Healthcare). Staining of the gels was performed with PageBlue protein staining solution (Fermentas).
Antibodies.
The following antibodies were used: anti-CtBP1 (BD Biosciences, catalog no. 612042), anti-PAK1 (Ozyme, catalog no. cst-2602), anti-acetyl-histone H3(Lys9) (Millipore, catalog no. 07-352), anti-dimethyl-histone H3(Lys9) (Millipore, catalog no. 07-441), anti-dimethyl-histone H3(Lys4) (Millipore, catalog no. 07-030), anti-phospho(Ser10)-acetyl(Lys14)-histone H3 (Millipore, catalog no. 07-081), anti-histone H4 (Abcam, catalog no. ab7311), anti-histone H3 (clone 6.6.2; Millipore, catalog no. 05-499), normal mouse IgG (Millipore, catalog no. 12-371), anti-RNA Pol II (Millipore, catalog no. 17-620), ECL rabbit IgG, horseradish peroxidase (HRP)-linked F(ab′)2 fragment (from donkey; GE Healthcare, catalog no. NA9340-1mL), anti-mouse IgG HRP-linked whole antibody (from sheep; GE Healthcare, catalog no. NA931-1mL), and Alexa Fluor 488–F(ab′)2 fragment of goat anti-mouse IgG(H+L) (Invitrogen, catalog no. A11017).
Oligonucleotides.
The following oligonucleotides were used: myogenin F (ChIP), 5′-AGGGGAGAAGAAGGCAATGT-3′; myogenin-R (ChIP), 5′-AGAAACCCAGAAGGGCAAAT-3′; myogenin-F (Q-PCR), 5′-CAATGCACTGGAGTTCGGTC-3′; myogenin-R (Q-PCR), 5′-ACAATCTCAGTTGGGCATGG-3′; AChR-F (Q-PCR), 5′-GAGGACCACCGTGAGATTGT-3′; AChR-R (Q-PCR), 5′-AATCGACCCATTGCTGTTTC-3′, CtBP1-F (Q-PCR), 5′-TGCCACATCCTGAACCTGTA-3′; CtBP1-R (Q-PCR), 5′-GAGGACGTTGAAGCCAAAAG-3′; PAK1-F (Q-PCR), 5′-ACACGGTTCGAGAAGATTGG-3′, PAK1-R (Q-PCR), 5′-TCCCTCATGACCAGGATCTC-3′; cyclophilin-B-F (Q-PCR), 5′-GATGGCACAGGAGGAAAG-3′; and cyclophilin-B-R (Q-PCR): 5′-AACTTTGCCGAAAACCACA-3′. The following Pak1 Stealth RNAi siRNA Mus musculus (Invitrogen) oligonucleotides were also used: PAK1MSS237494, 5′-GGAAUGGAUGGCUCGUCAAGUUAA-3′; PAK1MSS237495, 5′-UGUGCCGAGAGUGUCUACAAGCUUU-3′; and PAK1MSS237496, 5′-GGGCAUUAUGGCAAUUGAAAUGAUU-3′.
In vivo electroporation.
The operative procedure was performed using aseptic techniques and according to the local ethical committee recommendations (Comité Rhône Alpes d'Ethique pour l'Expérimentation Animale). Five-week-old OF1 female mice were anesthetized with an intraperitoneal injection of ketamine (100 mg/kg) and xylazine (10 mg/kg) to obtain a deep state of general anesthesia. A 30-μl volume of 0.45% NaCl containing 5 μg of DNA of interest or 500 pmol of Stealth siRNA (Invitrogen) and 2 μg of electroporation marker DNA (pmCherry-Nuc) was used. Injected tibialis anterior muscles were then electroporated with 1-cm2 plaque electrodes placed on each side of the leg with eight 200-V/cm pulses of 20 ms applied at 2 Hz (BTX ECM 830 electroporator). Muscles are collected 7 days after the electroporation. When denervation was required, muscles were denervated 7 days after electroporation and collected 3 days later.
Immunofluorescence.
Tibialis anterior muscle fibers were isolated as fiber bundles and fixed for 20 min in 3.7% formalin in Tris-buffered saline (150 mM NaCl in 50 mM Tris [pH 7.4]). Nuclei were stained when fibers were mounted in Vectashield with DAPI (4′,6′-diamidino-2-phenylindole; Clinisciences). Fluorescent images were visualized on a Zeiss AxioImager driven by the MetaMorph imaging software (Molecular Devices). Images were processed with Photoshop (Adobe System).
RNA preparation, reverse transcription, and real-time quantitative PCR.
After electroporation, fluorescent fibers were microdissected and homogenized in 350 μl of TRI Reagent (Sigma) in Matrix D lysis tubes (Bio 101) using a cooled (dry ice) Fast-Prep-24 (MP Biomedical) set for a 60-s duration and 6.5-m/s velocity. RNA were extracted with 100 μl of chloroform and then precipitated with 120 μl of propanol-2. The chloroform-propanol steps were performed twice to guarantee a high level of RNA purity. Q-PCR was performed on cDNA (reverse transcription-PCR [RT-PCR] using the Moloney murine leukemia virus reverse transcriptase from Fermentas) with a Qiagen Rotor-Gene Q 2plex HRM platform and a Qiagen QuantiFast SYBR green PCR kit. The data were normalized to cyclophilin B mRNA levels, which are invariant upon denervation.
DNA constructs.
The pFlag-CtBP1 vector was constructed on the basis of pME18S-HDAC2. After double digestion with EcoRI and NotI, the HDAC2 coding sequence was replaced by a CtBP1 PCR-amplified coding sequence. The pshRNA-CtBP1(5.1) vector was obtained by cloning a double-strand oligonucleotide in the pRNAT-H1.1/Neo (GenScript) vector (5′-GATCCCGTTCGTCGGTACAGGTTCAGGATTGATATCCGTCCTGAACCTGTACCGACGAATTTTTTCCAAA-3′), along with its complementary strand (5′-AGCTTTTGGAAAAAATTCGTCGGTACAGGTTCAGGACGGATATCAATCCTGAACCTGTACCGACGAACGG-3′). The nucleotides for the BamHI and HindIII restriction sites are underlined, and the loop sequence is indicated in boldface.
The expression vector for CtBP1-S158A was obtained by substitution of Ser158 by an alanine in the pFlag-CtBP1 vector using a QuikChange XL site-directed mutagenesis kit (Stratagene), as recommended by the manufacturer (5′-GGCACTCGAGTCCGAGCGGTAGAGCAGATCCGAG-3′; the codon substitution in the directed mutagenesis oligonucleotide is indicated in boldface). The expression vectors for CtBP1-YFP and CtBP1-S158A-YFP were obtained by PCR amplifying, respectively, the CtBP1 and CtBP1-S158A coding sequences using the oligonucleotides 5′-TTCAGATCTATGGGCAGCTCGCACTTGC-3′ and 5′-GTCCTCGAGCTACAACTGGTCACTGGCGT-3′, which were digested with BglII and XhoI and subcloned in BglII/SalI-digested pEYFP-C1 plasmid (Clontech).
RESULTS
CtBP1 is required for myogenin repression in innervated muscle.
Repression of the myogenin gene in innervated muscle is associated with low levels of histone H3K9 acetylation on its promoter, while its activation after denervation is associated with increased H3K9 acetylation (5). To determine whether other histone modifications are regulated by muscle innervation, the levels of histone acetylation and methylation on the myogenin promoter were evaluated by ChIP experiments on chromatin extracts from innervated or 3-day-denervated muscles (at this time point after denervation, myogenin has reached a higher level of induction). The results indicated that myogenin upregulation in denervated muscles coincided with increased levels of H3K4 dimethylation (H3K4me2) and H3K9 acetylation (H3K9ac) and a decreased level of H3K9 dimethylation (H3K9me2) on the myogenin promoter (Fig. 1).
FIG 1.

ChIP analysis of the myogenin promoter. Innervated or denervated muscle extracts were used for ChIP experiments with antibodies against H3K4me2, H3K9me2, H3ac, CtBP1, the RNA polymerase 2 largest subunit (Pol2), or mouse IgG (mIgG). Immunoprecipitated DNA was amplified by Q-PCR with primers spanning the myogenin promoter (see Materials and Methods). Inn, innervated; Den, denervated.
The corepressor CtBP1, which promotes H3K9 methylation and H3K4 demethylation while inhibiting H3K9 acetylation (14, 19), is known to be highly expressed in skeletal muscle (18). ChIP experiments were thus performed to determine whether CtBP1 was present on the myogenin promoter in its repressed state. In innervated muscle, CtBP1 was detected on the myogenin promoter, while RNA polymerase II was not (Fig. 1). Conversely, denervation triggered CtBP1 eviction and RNA polymerase II recruitment on the promoter (Fig. 1) concomitant with the induction of myogenin expression (as previously described [20–22]; see also Fig. 2C). Since myogenin activates the expression of AChR genes, we also wondered whether CtBP1 was also interacting with the promoter of these genes to repress them. However, a similar ChIP experiment did not detect CtBP1 on the promoter of AChRα (data not shown).
FIG 2.

CtBP1 regulates myogenin and AChrα expression. Each point corresponds to the analysis of a different TA muscle. The central bars, representing the means, and standard deviations are indicated. In each panel, the first lane shows the initial level of mRNA in innervated muscle. (A) Effects of CtBP1 shRNA on CtBP1 mRNA level (upper panel) or CtBP1 protein level (lower panel). (B) Effects of CtBP1 shRNA on myogenin mRNA level (upper panel) or AChRα mRNA level (lower panel). (C) Effect of overexpressed recombinant CtBP1 in denervated muscle on myogenin (upper panel) or AChRα (lower panel) mRNA levels. In panels A and B, Ctrl corresponds to the control with scramble shRNA. In panel C, Inn and Den correspond, respectively, to the control of innervated and denervated muscles treated with the empty vector.
To evaluate a possible function of CtBP1 in myogenin repression, shRNA constructs directed toward CtBP1 (or an empty vector as a control), together with the fluorescent marker pMCherry-Nuc, were electroporated into the tibialis anterior (TA) muscles of adult mice. One week later, muscles were collected, and electroporated fibers were microdissected prior to analysis by RT-PCR and Western blotting. CtBP1 mRNA (Fig. 2A, upper panel) and protein (Fig. 2A, lower panel) were found to be dramatically reduced in the fibers electroporated with the CtBP1 shRNA compared to controls. Myogenin mRNA level was increased by 50-fold in the presence of the CtBP1 shRNA (Fig. 2B, upper panel) but not with the empty vector (Ctrl). Consistently, the mRNA level of the AChRα subunit was also significantly increased by the electroporation of the CtBP1 shRNA (Fig. 2B, lower panel). In addition, electroporation of a plasmid overexpressing CtBP1 in denervated muscle (CtBP1+Den) leads to a significant reduction in the activation induced by denervation of both myogenin (Fig. 2C, upper panel) and AChRα (Fig. 2C, lower panel) promoters compared to the electroporation of an empty vector (Den).
Altogether, these results demonstrate that CtBP1 is present on the myogenin promoter in innervated muscle and that denervation induces its eviction, thereby relieving myogenin repression. We next investigated the mechanisms that control the presence of CtBP1 on the myogenin promoter.
PAK1 regulates the presence of CtBP1 in the nuclei through cytoplasmic relocation.
A possible mechanism to relieve the myogenin promoter from the CtBP1 repression could involve the downregulation of CtBP1 expression. However, neither CtBP1 mRNA levels (Fig. 3A) nor protein (Fig. 3B) levels differed significantly and reproducibly between innervated and denervated muscles. Alternatively, innervation could control CtBP1 localization. To evaluate this possibility, cytoplasmic and nuclear extracts were prepared from innervated and denervated TA muscles and analyzed by Western blotting with an anti-CtBP1 antibody (Fig. 3C). In innervated muscle (Inn), CtBP1 levels are significantly higher in the nucleus than in the cytoplasm, whereas 3 days after denervation (Den) the CtBP1 levels were significantly higher in the cytoplasmic fraction. To confirm this change in localization, an expression vector for a CtBP1-YFP fusion protein was electroporated in TA muscles. Denervation was performed 1 week after electroporation, and muscles were collected and microdissected 3 days later. The results of an analysis of the fibers are shown in Fig. 3D. Nuclei were clearly labeled in innervated muscle fibers (wt Inn), whereas in denervated fibers (wt Den) nuclear staining was weak and only present in a few nuclei, though staining was concomitantly slightly increased in the cytoplasm (Fig. 3D, wt). Therefore, denervation induces a relocalization of CtBP1 from the nucleus to the cytoplasm.
FIG 3.

Innervation regulates the nuclear localization of CtBP1. (A) CtBP1 mRNAs were quantified by quantitative RT-PCR in innervated or denervated muscles extracts. (B) CtBP1 protein levels in innervated or denervated muscle extracts quantified by Western blotting. (C) Western blot of CtBP1 in cytoplasmic and nuclear extracts of innervated or denervated muscle. Identical amounts of protein extracts, as shown by protein staining, were separated using 10% PAGE, and the protein contents were analyzed by Western blotting with CtBP1 antibody and either a histone H3 antibody (for nuclear extracts) or a tubulin antibody (for cytoplasmic extracts). (D) Detection of labeled nuclei after electroporation of a CtBP1-YFP plasmid (wt) or a CtBP1-S158A-YFP plasmid (S158A) in innervated (Inn) or denervated (Den) muscle. YFP detection, DAPI, and merge images are indicated. One representative fiber is shown in each case.
Phosphorylation on Ser158 of CtBP1 by the PAK1 kinase was previously shown to promote CtBP1 cytoplasmic localization (17). PAK1 is expressed in skeletal muscle and is localized at the neuromuscular junction, where it cooperates with MuSK for AChR aggregation (3). Extrasynaptic functions of PAK1 have not yet been investigated. To determine whether CtBP1 nuclear relocalization was associated with increased levels of PAK1, RT-PCR and Western blot analyses were performed on total muscle extracts. RT-PCR revealed that PAK1 expression was significantly increased after denervation (Fig. 4A) but PAK2 or PAK3 expression was not (see Fig. S1 in the supplemental material) (23). Similarly, PAK1 protein levels were significantly increased in denervated muscles compared to innervated muscles (Fig. 4B). Actually, a doublet was detected by the PAK1 antibody, with the highest band most likely corresponding to the phosphorylated-activated forms of PAK1 and suggesting that, unlike in innervated muscles, most PAK1 molecules were phosphorylated after denervation. All of these results suggest that CtBP1 cytoplasmic relocalization after denervation could result from the upregulation of PAK1. To test this hypothesis, a set of three PAK1 siRNAs was electroporated in TA muscle. Three days after denervation, muscles were collected, and electroporated fibers were microdissected. RT-PCR and Western blot analysis indicated that the level of PAK1 mRNA (Fig. 5A) and protein in a whole-cell extract (Fig. 5B, lane 2) were significantly reduced in fibers electroporated with the PAK1 siRNA. Western blot analysis of nuclear extracts prepared from electroporated fibers at 3 days postdenervation revealed also an enhanced nuclear accumulation of CtBP1 with respect to control siRNA (Fig. 5C, compare lanes 1 and 2), indicating that, in the absence of PAK1, CtBP1 remained in the nuclei, suggesting that PAK1 is required for the relocalization of CtBP1 in the cytoplasm after denervation. Analysis of the cytoplasmic fraction of these muscles indicates that the level of CtBP1 also increased (although to a lower extent) in the cytoplasm, suggesting that the phosphorylation of CtBP1 by PAK1 induces a degradation of CtBP1. Thus, in the absence of PAK1 phosphorylation, CtBP1 appears to be stabilized both in nuclei and in the cytoplasm.
FIG 4.
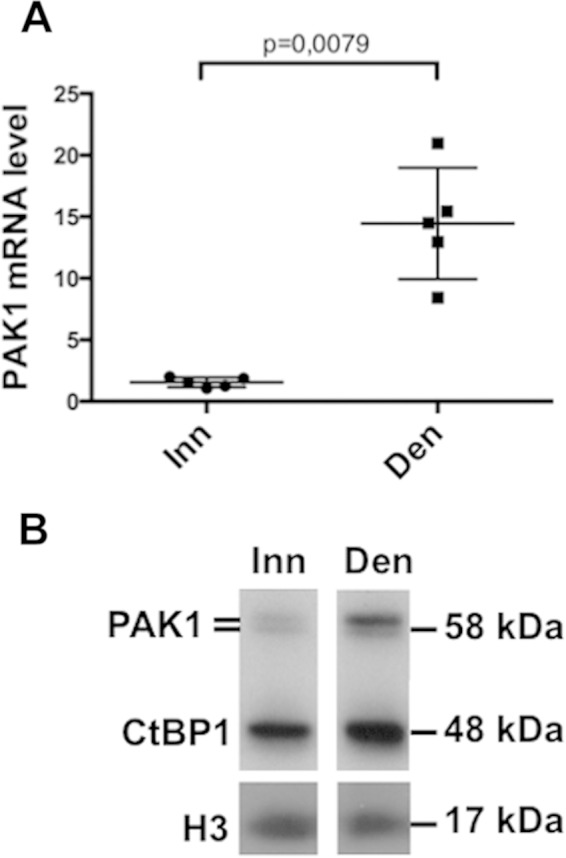
PAK1 level is regulated by innervation. The levels of PAK1 mRNA detected by RT-PCR (A) and the PAK1 protein detected by Western blotting (B) in innervated and denervated TA muscles were determined.
FIG 5.

Inhibition of PAK1 expression induces an increase in nuclear CtBP1 level. (A) Levels of PAK1 mRNA detected by quantitative RT-PCR after electroporation of PAK1 siRNA (right) or scramble siRNA (left) in innervated muscle. (B) Levels of PAK1 protein detected by Western blotting in a total TA muscle extract after coelectroporation of mCherry-Nuc with scramble siRNA in denervated muscle (lane 1) or coelectroporation of mCherry-Nuc with PAK1 siRNA in denervated muscle (lane 2). (C) Effect of the PAK1 siRNA on the levels of CtBP1 protein detected by Western blotting in TA cytoplasmic (lanes 1 and 2) or nuclear (lanes 3 and 4) extracts from denervated muscle electroporated with Scramble siRNA (lanes 1 to 3) or with PAK1 siRNA (lanes 2 to 4).
Next, the functional involvement of PAK1 in myogenin derepression was evaluated. PAK1 or scramble siRNA were electroporated in TA muscle, and myogenin and AChRα expression levels were measured by RT-PCR 3 days after denervation (Fig. 6A). As expected, myogenin (upper panel) and AChRα (lower panel) mRNA levels were highly increased after denervation in the presence of the scramble siRNA (siScrbl-Den) but not (or to a much lesser level) in the presence of the PAK1 siRNA (siPAK1-Den), showing a requirement for PAK1 in myogenin upregulation. To test the possibility that PAK1-mediated phosphorylation of CtBP1 relieves myogenin repression, CtBP1 Ser158 (the target of PAK1 phosphorylation) was mutated to alanine (CtBP1 S158A), and the resulting PAK1-insensitive CtBP1 mutant was electroporated in TA muscle. Evaluation of myogenin expression by RT-PCR after denervation revealed that overexpression of CtBP1 S158A prevented myogenin (Fig. 6B, upper panel) and AChRα (Fig. 6B, lower panel) upregulation more efficiently than the wild-type (wt) CtBP1 (4-fold versus 1.6-fold, respectively [compare Fig. 6B and 2C]).
FIG 6.

Effects of PAK1 siRNA (A) or CtBP1-S158A mutant (B) on the levels of myogenin (upper panel) and AChRα (lower panel) mRNA in denervated tibialis anterior muscle, as detected by RT-PCR. In each panel the left column shows the data for a control (innervated muscle electroporated with scramble siRNA in panel A; innervated muscle in panel B), the middle column shows the effect of denervation (including electroporation with Scramble siRNA in panel A), and the right column shows the effect of PAK1 siRNA electroporation in denervated muscle (A) or the effect of CtBP1-S158A overexpression in denervated muscle (B).
To further confirm the role of Ser158 phosphorylation, an expression vector for a CtBP1-S158A-YFP fusion protein was electroporated in TA muscles (in parallel with the wt analysis previously described) (Fig. 3D). As for the wt, denervation was performed 1 week after electroporation, and muscles were collected and microdissected 3 days later. Analysis of the fibers clearly shows that although the cytoplasmic staining in the denervated muscle (Fig. 3D, S158A-Den) increased in a similar way as the wild-type denervated muscles (wt Den), the nuclei of the CtBP1-S158A-YFP-treated muscles still contain some CtBP1, in contrast to what was observed with the wild-type denervated muscles (wt-Den). This finding is in agreement with our other observations suggesting that PAK1 phosphorylation of CtBP1-Ser158 alters the stability of CtBP1. In the absence of Ser158 phosphorylation, either by treating muscles with PAK1 siRNA or by preventing the phosphorylation by Ser158 mutation, CtBP1 appears to be stabilized both in nuclei and the cytoplasm.
Based on these observations, we expected that CtBP1 and PAK1 could interact and that this interaction should be amplified in the denervated muscle. As shown in Fig. 7A, we found that PAK1 antibody (left panel, lane 2), but not mouse IgG antibody (lane 3), can immunoprecipitate CtBP1 from a muscle extract. Similarly, a phosphorylated PAK1 (P-PAK1) antibody (right panel, lane 3) can immunoprecipitate CtBP1. We then compared the amount of CtBP1 immunoprecipitated by P-PAK1 antibody from innervated and denervated muscle extracts: as shown in Fig. 7B (left panel), although a similar amount of P-PAK1 was immunoprecipitated in both cases, a much larger amount a CtBP1 was immunoprecipitated from a denervated extract (lane 4) than from the innervated extract (lane 2), demonstrating that the PAK1-CtBP1 interaction is indeed increased in the denervated extract. Protein staining (middle panel) shows equal loading of the input material in the experiment, and a histogram (right panel) based on the scanning of the bands detected in the Western blot shows the observed differences. Altogether our data demonstrate that CtBP1 has a critical function in the repression of the myogenin promoter in the extrasynaptic nuclei of innervated fibers and that its delocalization upon denervation appears to rely on PAK1.
FIG 7.

Interaction between CtBP1 and PAK1/P-PAK1. (A) The left panel shows Western blot detection of CtBP1 immunoprecipitated by CtBP1 antibody (lane 1), PAK1 antibody (lane 2), or mouse IgG antibody (lane 3); the right panel shows Western blot detection of CtBP1 in tibialis anterior extract (lane 1) and immunoprecipitated by mouse IgG antibody (lane 2) or P-PAK1 antibody (lane 3). (B) Comparison of CtBP1 and P-PAK1 immunoprecipitation by P-PAK1 antibody in innervated versus denervated muscle extracts. The left panel shows Western blot detection of CtBP1 and P-PAK1 in the input extracts (lanes 1 and 3) and in the immunoprecipitated fractions (lanes 2 and 4) from either innervated (Inn: lanes 1 to 2) or denervated (Den, lanes 3 to 4) extracts. In the middle panel, Coomassie blue staining shows equal loading of the input extracts. In the right panel, histograms based on scanning of the Western blot bands show that more CtBP1 is immunoprecipitated from the denervated extract than from the innervated extract, while a similar amount of P-PAK1 was immunoprecipitated. Circled P, phosphorylation.
DISCUSSION
Muscle excitability is intimately linked to gene expression. Acetylcholine released by neurons binds acetylcholine receptors in the postsynaptic membrane that initiate action potentials all along muscle fibers. Nerve-evoked electrical activity globally diminishes histone acetylation (5) and represses myogenin expression, thereby inhibiting the expression of AChR genes in extrasynaptic nuclei (20–22). Here, we show that expression of the kinase PAK1, which controls the nuclear localization of the corepressor CtBP1 by phosphorylation (17), is also regulated by innervation. Denervation activates the expression of PAK1 and promotes CtBP1 relocalization in the cytoplasm through an enhanced CtBP1–P-PAK1 interaction. Activation of myogenin expression by denervation is associated with the removal of CtBP1 from the myogenin promoter, together with an increase in H3K9ac and H3K4me2, as well as a decrease in H3K9me2. Furthermore, CtBP1 inhibition with shRNA is sufficient to induce myogenin upregulation in innervated muscle, whereas PAK1 knockdown with siRNA or expression of a nonphosphorylatable CtBP1 mutant prevent myogenin upregulation in denervated muscle.
Although the expression of the nonphosphorylatable S158A CtBP1 mutant almost totally prevented myogenin upregulation after denervation, the inhibition of PAK1 expression by siRNA electroporation did not completely abolish myogenin upregulation after denervation. This could be due to the fact that PAK1 siRNA did not completely abolish the presence of PAK1 protein despite an almost complete disappearance of the mRNA (Fig. 5A and B). Alternatively, the closely related PAK2 or PAK3 kinases may have partially compensated for the inhibition of PAK1. However, these kinases are not highly expressed in muscle (23) and are not upregulated upon denervation (see Fig. S1 in the supplemental material).
PAK1 and CtBP1: coordinators of gene expression and actin cytoskeleton?
PAK1 was previously shown to be required for AChR clustering in response to agrin (3). PAK1 might also activate AChR gene expression at the neuromuscular junction. Indeed, AChR gene activation by neural factors is mediated by extracellular signal-regulated kinase (ERK) and c-Jun N-terminal kinase (JNK) signaling pathways downstream of MuSK (24). PAK1 interacts with MuSK and was shown to activate both ERK and JNK (25, 26). In addition, cdc42, which activates PAK1, participates to the activation of JNK by agrin downstream of MuSK.
PAK1 is mainly known for its role in the control of cellular cytoskeleton, especially in the nervous system (27, 28). The finding that PAK1 expression and activity is regulated by electrical activity could be highly relevant in the brain, where it could participate in the observed coupling between activity and cytoskeletal remodeling (29).
The expression of specific genes has to be coupled to a specific cytoplasmic machinery to correctly address and stabilize newly synthesized proteins. This mechanism is particularly important in cells that establish synapses, since specific proteins have to be addressed and stabilized at the synaptic membrane and eventually recycled or degraded. Like any synapse, the neuromuscular junction is linked to a specific cytoskeletal structure (30–34), and the small GTPases rac1 and cdc42 are involved in the specification of the postsynaptic actin network downstream of MuSK (3, 35). As a MuSK interactor and a rac1/cdc42 effector in actin regulation and AChR aggregation (3, 36), PAK1 probably participates to the coordination of postsynaptic organization and gene expression.
Considering the involvement of PAK1 in the control of cellular cytoskeleton, the finding that PAK1 expression is regulated by electrical activity and participates to transcriptional activation suggests that in muscle extrasynaptic regions PAK1 could couple cytoskeleton organization and gene expression to coordinate expression and trafficking of extrasynaptic AChR in denervated muscle.
Interestingly, in addition to its transcriptional function, CtBP1 has been shown to cooperate with PAK1 to regulate membrane fission and vesicular trafficking (37, 38). Therefore, CtBP1 could participate to AChR trafficking in extrasynaptic area when relocalized in the cytoplasm upon denervation. The CtBP1-PAK1 interaction that we have detected, amplified in the denervated muscle (Fig. 7), is also representative of the important connection between the two proteins.
CtBP1, a cornerstone of myogenin repression by electrical activity.
CtBP proteins are encoded by the CtBP1 and CtBP2 genes, which exhibit widespread expression during embryogenesis, including somites and limb buds (18). Unlike CtBP1, CtBP2 is mainly nuclear, does not shuttle between the nucleus and the cytoplasm, and is not phosphorylated by PAK1. CtBP1 and CtBP2 can heterodimerize in the nucleus and probably cooperate to repress transcription (14). Accordingly, the defects associated with CtBP1 inactivation in mice were accentuated in a CtBP2 heterozygous background: muscle precursors were present in CtBP1−/−:CtBP2+/− animals, but analysis of diaphragm muscles at embryonic day 15.5 of development revealed the absence of muscle fibers (18). Altogether, this suggests that in innervated muscle CtBP1 and CtBP2 probably cooperate in the nucleus to inhibit myogenin transcription.
Myogenin activation by denervation requires the downregulation of MITR and Dach2 expression to allow transcriptional activation by MEF2 and MEF3/six, respectively (5, 9), as well as the removal of the transcriptional repressor MSY3 (7). This probably explains why the inactivation of MITR or Dach2 alone was not sufficient to activate myogenin expression to levels comparable with those obtained after denervation (5, 8). The fact that CtBP1 knockdown in innervated muscle is sufficient to induce a 20-fold activation of myogenin expression suggests that it affected the different modes of repression of myogenin. Moreover, CtBP1 participates to transcriptional repression of MEF2 by MITR (39, 40). In innervated muscle, MEF2 is therefore part of a repressive complex that includes MITR and CtBP1.
The corepressor activity of CtBP1 involves the recruitment of enzymes that promote H3K9 methylation and prevent H3K4 acetylation and methylation (14). Consistently, the presence of CtBP1 on the myogenin promoter coincides with a marked methylation of H3K9, together with reduced H3K9 acetylation and H3K4 methylation. Interestingly, another mechanism whereby CtBP1 promotes repression is through inhibition of the histone acetyltransferase p300 (15). p300 is required for the activation of myogenin by MEF2 (41–43). This provides another link between CtBP1 and the inhibition of MEF2 transactivating function.
In conclusion, our study demonstrates that the corepressor CtBP1 plays a central role in myogenin regulation in innervated muscle. Figure 8 summarizes the activities involved in myogenin regulation in innervated and denervated muscle. In the future, it will be interesting to identify the enzymes recruited by CtBP and involved in the control of H3K4 and H3K9 posttranslational modifications.
FIG 8.

PAK1 and CtBP1 mediate the action of neural inputs on gene expression in muscle. Upon binding to LRP4, agrin induces the aggregation of synaptic proteins, including AChR and ErbB receptors. It also activates gene expression via the ERK and JNK intracellular signaling pathways that phosphorylate and activate Ets transcription factors. PAK1 is activated by the neural factors agrin and neuregulins via MuSK/LRP4 and ErbB receptors, respectively, and is involved in the cytoskeletal network organization. In extrasynaptic nuclei, CtBP1 is recruited on the promoter of the myogenin gene, where it participates in repression by inhibiting the p300/CBP histone acetyltransferase and by recruiting histone deacetylases, H3K4 demethylases, and H3K9 methyltransferases. In denervated muscle fibers, PAK1 expression is significantly activated in extrasynaptic regions. Increased PAK1 levels induce CtBP1 phosphorylation (indicated by a “circled P” in the figure) and cytoplasmic localization, thereby allowing the recruitment of H3K4 methyltransferases and H3K9 demethylases on the myogenin promoter, as well as the coactivation of Six and MEF2 by CBP/p300. Myogenin then activates the expression of the AChR α, β, δ, and γ subunit genes.
Supplementary Material
ACKNOWLEDGMENTS
We are grateful to Francesca Palladino and Yann-Gaël Gangloff for critical reading of the manuscript, Véronique Morel for assistance with statistical analyses, Adeline Govehovitch for technical assistance, and Alexandre Guiraud for providing the mCherry-Nuc plasmid. We acknowledge the technical support of the animal facility (PBES) and the imaging facility (PLATIM) of SFR Biosciences.
This study was supported by ANR and AFM grants and by INSERM (V.M.). C.V. received an Italian Foundation for Cancer Research Fellowships (FIRC, Milan, Italy). This study was also supported by Italian Association for Cancer Research (AIRC, Milan, Italy) grant IG 14675 to D.C.
J.-L.T., V.M., A.R.-C., A.M., and L.S. designed and performed the experiments, C.V. and D.C. designed and generated the YFP-CtBP1 plasmid, and J.-L.T, V.M., and L.S. contributed in writing the paper.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/MCB.00354-15.
REFERENCES
- 1.Burden SJ. 2011. SnapShot: neuromuscular junction. Cell 144:826–826.e1. doi: 10.1016/j.cell.2011.02.037. [DOI] [PubMed] [Google Scholar]
- 2.Wu H, Xiong WC, Mei L. 2010. To build a synapse: signaling pathways in neuromuscular junction assembly. Development 137:1017–1033. doi: 10.1242/dev.038711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Luo ZG, Wang Q, Zhou JZ, Wang J, Luo Z, Liu M, He X, Wynshaw-Boris A, Xiong WC, Lu B, Mei L. 2002. Regulation of AChR clustering by Dishevelled interacting with MuSK and PAK1. Neuron 35:489–505. doi: 10.1016/S0896-6273(02)00783-3. [DOI] [PubMed] [Google Scholar]
- 4.Ravel-Chapuis A, Vandromme M, Thomas JL, Schaeffer L. 2007. Postsynaptic chromatin is under neural control at the neuromuscular junction. EMBO J 26:1117–1128. doi: 10.1038/sj.emboj.7601572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mejat A, Ramond F, Bassel-Duby R, Khochbin S, Olson EN, Schaeffer L. 2005. Histone deacetylase 9 couples neuronal activity to muscle chromatin acetylation and gene expression. Nat Neurosci 8:313–321. doi: 10.1038/nn1408. [DOI] [PubMed] [Google Scholar]
- 6.Moresi V, Williams AH, Meadows E, Flynn JM, Potthoff MJ, McAnally J, Shelton JM, Backs J, Klein WH, Richardson JA, Bassel-Duby R, Olson EN. 2010. Myogenin and class II HDACs control neurogenic muscle atrophy by inducing E3 ubiquitin ligases. Cell 143:35–45. doi: 10.1016/j.cell.2010.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Berghella L, De Angelis L, De Buysscher T, Mortazavi A, Biressi S, Forcales SV, Sirabella D, Cossu G, Wold BJ. 2008. A highly conserved molecular switch binds MSY-3 to regulate myogenin repression in postnatal muscle. Genes Dev 22:2125–2138. doi: 10.1101/gad.468508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tang H, Goldman D. 2006. Activity-dependent gene regulation in skeletal muscle is mediated by a histone deacetylase (HDAC)-Dach2-myogenin signal transduction cascade. Proc Natl Acad Sci U S A 103:16977–16982. doi: 10.1073/pnas.0601565103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tang H, Macpherson P, Marvin M, Meadows E, Klein WH, Yang XJ, Goldman D. 2009. An HDAC4/myogenin positive feedback loop coordinates denervation-dependent gene induction and suppression. Mol Biol Cell 20:1120–1131. doi: 10.1091/mbc.E08-07-0759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cruickshank MN, Besant P, Ulgiati D. 2010. The impact of histone posttranslational modifications on developmental gene regulation. Amino Acids 39:1087–1105. doi: 10.1007/s00726-010-0530-6. [DOI] [PubMed] [Google Scholar]
- 11.Munshi A, Shafi G, Aliya N, Jyothy A. 2009. Histone modifications dictate specific biological readouts. J Genet Genomics 36:75–88. doi: 10.1016/S1673-8527(08)60094-6. [DOI] [PubMed] [Google Scholar]
- 12.Cao Y, Yao Z, Sarkar D, Lawrence M, Sanchez GJ, Parker MH, MacQuarrie KL, Davison J, Morgan MT, Ruzzo WL, Gentleman RC, Tapscott SJ. 2010. Genome-wide MyoD binding in skeletal muscle cells: a potential for broad cellular reprogramming. Dev Cell 18:662–674. doi: 10.1016/j.devcel.2010.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ho L, Crabtree GR. 2010. Chromatin remodeling during development. Nature 463:474–484. doi: 10.1038/nature08911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chinnadurai G. 2007. Transcriptional regulation by C-terminal binding proteins. Int J Biochem Cell Biol 39:1593–1607. doi: 10.1016/j.biocel.2007.01.025. [DOI] [PubMed] [Google Scholar]
- 15.Kim JH, Cho EJ, Kim ST, Youn HD. 2005. CtBP represses p300-mediated transcriptional activation by direct association with its bromodomain. Nat Struct Mol Biol 12:423–428. doi: 10.1038/nsmb924. [DOI] [PubMed] [Google Scholar]
- 16.Ray SK, Li HJ, Metzger E, Schule R, Leiter AB. 2014. CtBP and associated LSD1 are required for transcriptional activation by NeuroD1 in gastrointestinal endocrine cells. Mol Cell Biol 34:2308–2317. doi: 10.1128/MCB.01600-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Barnes CJ, Vadlamudi RK, Mishra SK, Jacobson RH, Li F, Kumar R. 2003. Functional inactivation of a transcriptional corepressor by a signaling kinase. Nat Struct Biol 10:622–628. doi: 10.1038/nsb957. [DOI] [PubMed] [Google Scholar]
- 18.Hildebrand JD, Soriano P. 2002. Overlapping and unique roles for C-terminal binding protein 1 (CtBP1) and CtBP2 during mouse development. Mol Cell Biol 22:5296–5307. doi: 10.1128/MCB.22.15.5296-5307.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chinnadurai G. 2009. The transcriptional corepressor CtBP: a foe of multiple tumor suppressors. Cancer Res 69:731–734. doi: 10.1158/0008-5472.CAN-08-3349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Duclert A, Piette J, Changeux JP. 1991. Influence of innervation of myogenic factors and acetylcholine receptor alpha-subunit mRNAs. Neuroreport 2:25–28. doi: 10.1097/00001756-199101000-00006. [DOI] [PubMed] [Google Scholar]
- 21.Eftimie R, Brenner HR, Buonanno A. 1991. Myogenin and MyoD join a family of skeletal muscle genes regulated by electrical activity. Proc Natl Acad Sci U S A 88:1349–1353. doi: 10.1073/pnas.88.4.1349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Witzemann V, Sakmann B. 1991. Differential regulation of MyoD and myogenin mRNA levels by nerve induced muscle activity. FEBS Lett 282:259–264. doi: 10.1016/0014-5793(91)80490-T. [DOI] [PubMed] [Google Scholar]
- 23.Arias-Romero LE, Chernoff J. 2008. A tale of two Paks. Biol Cell 100:97–108. doi: 10.1042/BC20070109. [DOI] [PubMed] [Google Scholar]
- 24.Lacazette E, Le Calvez S, Gajendran N, Brenner HR. 2003. A novel pathway for MuSK to induce key genes in neuromuscular synapse formation. J Cell Biol 161:727–736. doi: 10.1083/jcb.200210156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ching YP, Leong VY, Lee MF, Xu HT, Jin DY, Ng IO. 2007. P21-activated protein kinase is overexpressed in hepatocellular carcinoma and enhances cancer metastasis involving c-Jun NH2-terminal kinase activation and paxillin phosphorylation. Cancer Res 67:3601–3608. doi: 10.1158/0008-5472.CAN-06-3994. [DOI] [PubMed] [Google Scholar]
- 26.Park ER, Eblen ST, Catling AD. 2007. MEK1 activation by PAK: a novel mechanism. Cell Signal 19:1488–1496. doi: 10.1016/j.cellsig.2007.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kreis P, Rousseau V, Thevenot E, Combeau G, Barnier JV. 2008. The four mammalian splice variants encoded by the p21-activated kinase 3 gene have different biological properties. J Neurochem 106:1184–1197. doi: 10.1111/j.1471-4159.2008.05474.x. [DOI] [PubMed] [Google Scholar]
- 28.Smith SD, Jaffer ZM, Chernoff J, Ridley AJ. 2008. PAK1-mediated activation of ERK1/2 regulates lamellipodial dynamics. J Cell Sci 121:3729–3736. doi: 10.1242/jcs.027680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kreis P, Barnier JV. 2009. PAK signalling in neuronal physiology. Cell Signal 21:384–393. doi: 10.1016/j.cellsig.2008.11.001. [DOI] [PubMed] [Google Scholar]
- 30.Deconinck AE, Potter AC, Tinsley JM, Wood SJ, Vater R, Young C, Metzinger L, Vincent A, Slater CR, Davies KE. 1997. Postsynaptic abnormalities at the neuromuscular junctions of utrophin-deficient mice. J Cell Biol 136:883–894. doi: 10.1083/jcb.136.4.883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mitsui T, Kawajiri M, Kunishige M, Endo T, Akaike M, Aki K, Matsumoto T. 2000. Functional association between nicotinic acetylcholine receptor and sarcomeric proteins via actin and desmin filaments. J Cell Biochem 77:584–595. doi:. [DOI] [PubMed] [Google Scholar]
- 32.Pato C, Stetzkowski-Marden F, Gaus K, Recouvreur M, Cartaud A, Cartaud J. 2008. Role of lipid rafts in agrin-elicited acetylcholine receptor clustering. Chem Biol Interact 175:64–67. doi: 10.1016/j.cbi.2008.03.020. [DOI] [PubMed] [Google Scholar]
- 33.Sanes JR, Lichtman JW. 2001. Induction, assembly, maturation and maintenance of a postsynaptic apparatus. Nat Rev Neurosci 2:791–805. doi: 10.1038/35097557. [DOI] [PubMed] [Google Scholar]
- 34.Shi L, Butt B, Ip FC, Dai Y, Jiang L, Yung WH, Greenberg ME, Fu AK, Ip NY. 2010. Ephexin1 is required for structural maturation and neurotransmission at the neuromuscular junction. Neuron 65:204–216. doi: 10.1016/j.neuron.2010.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Weston C, Gordon C, Teressa G, Hod E, Ren XD, Prives J. 2003. Cooperative regulation by Rac and Rho of agrin-induced acetylcholine receptor clustering in muscle cells. J Biol Chem 278:6450–6455. doi: 10.1074/jbc.M210249200. [DOI] [PubMed] [Google Scholar]
- 36.Wells CM, Jones GE. 2010. The emerging importance of group II PAKs. Biochem J 425:465–473. doi: 10.1042/BJ20091173. [DOI] [PubMed] [Google Scholar]
- 37.Liberali P, Kakkonen E, Turacchio G, Valente C, Spaar A, Perinetti G, Bockmann RA, Corda D, Colanzi A, Marjomaki V, Luini A. 2008. The closure of Pak1-dependent macropinosomes requires the phosphorylation of CtBP1/BARS. EMBO J 27:970–981. doi: 10.1038/emboj.2008.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Valente C, Turacchio G, Mariggio S, Pagliuso A, Gaibisso R, Di Tullio G, Santoro M, Formiggini F, Spano S, Piccini D, Polishchuk RS, Colanzi A, Luini A, Corda D. 2012. A 14-3-3γ dimer-based scaffold bridges CtBP1-S/BARS to PI(4)KIIIβ to regulate post-Golgi carrier formation. Nat Cell Biol 14:343–354. doi: 10.1038/ncb2445. [DOI] [PubMed] [Google Scholar]
- 39.McKinsey TA, Zhang CL, Olson EN. 2002. Signaling chromatin to make muscle. Curr Opin Cell Biol 14:763–772. doi: 10.1016/S0955-0674(02)00389-7. [DOI] [PubMed] [Google Scholar]
- 40.Zhang CL, McKinsey TA, Lu JR, Olson EN. 2001. Association of COOH-terminal-binding protein (CtBP) and MEF2-interacting transcription repressor (MITR) contributes to transcriptional repression of the MEF2 transcription factor. J Biol Chem 276:35–39. doi: 10.1074/jbc.M007364200. [DOI] [PubMed] [Google Scholar]
- 41.Angelelli C, Magli A, Ferrari D, Ganassi M, Matafora V, Parise F, Razzini G, Bachi A, Ferrari S, Molinari S. 2008. Differentiation-dependent lysine 4 acetylation enhances MEF2C binding to DNA in skeletal muscle cells. Nucleic Acids Res 36:915–928. doi: 10.1093/nar/gkm1114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sartorelli V, Huang J, Hamamori Y, Kedes L. 1997. Molecular mechanisms of myogenic coactivation by p300: direct interaction with the activation domain of MyoD and with the MADS box of MEF2C. Mol Cell Biol 17:1010–1026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Serra C, Palacios D, Mozzetta C, Forcales SV, Morantte I, Ripani M, Jones DR, Du K, Jhala US, Simone C, Puri PL. 2007. Functional interdependence at the chromatin level between the MKK6/p38 and IGF1/PI3K/AKT pathways during muscle differentiation. Mol Cell 28:200–213. doi: 10.1016/j.molcel.2007.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.