

Abstract
Rifampin chemoprophylaxis against Neisseria meningitidis infections led to the onset of rifampin resistance in clinical isolates harboring point mutations in the rpoB gene, coding for the RNA polymerase β chain. These resistant strains are rare in medical practice, suggesting their decreased fitness in the human host. In this study, we isolated rifampin-resistant rpoB mutants from hypervirulent serogroup C strain 93/4286 and analyzed their different properties, including the ability to grow/survive in different culture media and in differentiated THP-1 human monocytes and to compete with the wild-type strain in vitro. Our results demonstrate that different rpoB mutations (H553Y, H553R, and S549F) may have different effects, ranging from low- to high-cost effects, on bacterial fitness in vitro. Moreover, we found that the S549F mutation confers temperature sensitivity, possibly explaining why it is observed very rarely in clinical isolates. Comparative high-throughput RNA sequencing analysis of bacteria grown in chemically defined medium demonstrated that the low-cost H553Y substitution resulted in global transcriptional changes that functionally mimic the stringent response. Interestingly, many virulence-associated genes, including those coding for meningococcal type IV pili, porin A, adhesins/invasins, IgA protease, two-partner secretion system HrpA/HrpB, enzymes involved in resistance to oxidative injury, lipooligosaccharide sialylation, and capsular polysaccharide biosynthesis, were downregulated in the H553Y mutant compared to their level of expression in the wild-type strain. These data might account for the reduced capacity of this mutant to grow/survive in differentiated THP-1 cells and explain the rarity of H553Y mutants among clinical isolates.
INTRODUCTION
Neisseria meningitidis is a leading cause of bacterial meningitis and sepsis worldwide. People who have been in contact with patients affected by meningococcal disease are at a greatly increased risk for contracting the disease. In many European countries, chemoprophylaxis with rifampin is one of the major preventive measures used to avoid further spread and possible epidemics. The efficacy of rifampin chemoprophylaxis during disease outbreaks is demonstrated by the reduction of the rate of meningococcal carriage by from 82% to 98% at 7 to 14 days of follow-up (1–3). Resistant strains have been found to emerge among 10% and 27% (4, 5) of treated carriers. Nevertheless, although rifampin has been used for more than 30 years in the management of contacts and mutator strains with an elevated frequency of mutation to rifampin resistance (Rifr) are rather common in hypervirulent lineages (6, 7), only a few cases of meningococcal disease caused by rifampin-resistant invasive isolates have been reported so far (8, 9), suggesting a decreased biological fitness of resistant isolates. Indeed, the frequency of resistance to rifampin seems to be very low even in noninvasive (carriage) isolates, although fewer data are available on this subject (8, 10–12). However, as resistance leads to the failure of chemoprophylaxis (13), even those few cases of disease due to resistant strains must be considered with the most attention. For instance, a cluster of meningococcal disease caused by rifampin-resistant serogroup C meningococci of clonal complex sequence type 11 (ST-11) has recently been reported in France (14).
Rifampin binds to bacterial RNA polymerase and prevents the productive initiation of transcription, but it does not inhibit transcription after promoter clearance. In all bacteria, most of the mutations conferring Rifr change the amino acids directly involved in antibiotic binding to RNA polymerase and are clustered within three distinct sites, clusters I, II, and III, in the central segment of the β chain of the RNA polymerase (15). As these mutations affect evolutionarily conserved residues (Fig. 1A), they are expected to compromise transcription efficiency and, hence, the physiology and fitness of the organism. Indeed, a direct relationship between the fitness cost of rpoB mutations and their effects on transcription was demonstrated in Escherichia coli. In particular, Rifr RNA polymerases have altered properties in transcriptional elongation and/or termination (15).
FIG 1.

Map of Rifr-conferring rpoB mutations, growth, and stationary-phase survival of rifampin-resistant strains in complex medium. (A) Map of the N. meningitidis RNA polymerase (RpoB) subunit with the locations of rif clusters N, I, II, and III (top) and the amino acid sequence alignment of rif cluster I from N. meningitidis, E. coli, S. aureus, B. subtilis, M. tuberculosis, and Streptomyces coelicolor A3 (2) (bottom). Numbering begins at the first amino acid (Aa) of the RpoB sequence. Circles above the N. meningitidis sequence, the amino acid substitutions (H553Y, H553R, and S549F) analyzed in this study; asterisks, evolutionarily conserved amino acid residues. (B and C) Growth (B) and stationary-phase survival (C) curves of the mutants and the wild-type strain in GC broth at 37°C and 32°C. The experiments were performed in triplicate with three independent cultures, and statistical significance was examined by the Student t test. Results are indicated as means ± SDs. Asterisks indicate statistical significance (*, P < 0.05; **, P < 0.01; ***, P < 0.001).
However, a fitness burden is not always associated with rpoB mutations. Using very accurate competition assays to quantify the relative fitness cost of all of the known mutations in rpoB conferring Rifr, Brandis and coworkers (16) have recently found that mutations in some alleles conferring Rifr may be neutral in terms of the fitness cost on growth. The burden on fitness depends, of course, not only on the amino acid affected but also on the nature of the substitution. Substitution of the conserved histidine residue in cluster I of rpoB is extremely frequent among clinical Rifr isolates in many bacterial species, reflecting the low fitness cost imposed by amino acid substitutions at this position. In fact, the substitution H481N (Fig. 1A) in the RpoB protein of Staphylococcus aureus was not demonstrably associated with a cost of resistance in vitro (17). In contrast, the substitution H526Y in E. coli and the corresponding substitution H481Y in S. aureus (Fig. 1A) gave an appreciable, although modest, fitness burden (18).
In clinical isolates, the initial fitness cost is often mitigated by compensatory mutations. For instance, in Mycobacterium tuberculosis the RpoB S531L substitution, which is the most frequent rifampin resistance-conferring substitution in clinical strains worldwide, was initially associated with the lowest fitness cost in laboratory strains and no apparent fitness defect in clinical strains (19). Later, Brandis and coworkers (20) studied the S531L substitution in isogenic backgrounds using high-resolution competition assays and found a large fitness cost that could be compensated for by secondary mutations in the rpoA, rpoB, and rpoC genes.
As in other bacterial species, high-level resistance to rifampin in N. meningitidis is caused by mutation mapping in rifampin resistance-determining cluster I (21, 22). Most frequently, residue H553 (H526 in E. coli) is involved, and the substitution H553Y (Fig. 1A) is rather frequent in resistant isolates causing secondary cases of meningococcal disease in contact persons following rifampin prophylaxis (23). By using 3 pairs of linked cases (i.e., primary cases due to a rifampin-susceptible isolate and secondary cases due to a resistant isolate in a contact person) of meningococcal disease caused by serogroup B, A, and C rifampin-susceptible and rifampin-resistant isolates harboring the same H553Y substitution, Taha et al. (23) demonstrated that the virulence of the serogroup B rifampin-resistant isolate was substantially attenuated compared to that of the corresponding susceptible isolate in BALB/c mice infected intraperitoneally. In contrast, attenuation was not significant for the other two pairs of less virulent serogroup C and serogroup A strains.
In this study, we isolated rifampin-resistant mutants from the hypervirulent serogroup C strain 93/4286 (electrophoretic type 37 [ET-37]) and analyzed a number of properties of these mutants, including the ability to grow and survive in different culture media and in differentiated human THP-1 monocytes. Then, we focused on a mutant harboring the RpoB H553Y substitution, which showed a reduced capacity to grow/survive in differentiated THP-1 cells but not in the culture media tested. RNA sequencing (RNA-seq) analysis was used to determine the effects of the RpoB H553Y substitution on the global transcript profile with the aim of identifying the molecular mechanisms underlying the phenotypic traits associated with this substitution.
MATERIALS AND METHODS
Bacterial strains and growth conditions.
The meningococcal strains used in this study were the serogroup C 93/4286 isolate and its rifampin-resistant mutants. The 93/4286 strain, belonging to the ET-37 hypervirulent lineage, was kindly provided by Novartis Vaccine and Diagnostics, Siena, Italy. Meningococci were cultured on GC agar or broth (Oxoid) supplemented with 1% (vol/vol) Polyvitox (Oxoid) at 37°C with 5% CO2. When needed, rifampin (Sigma-Aldrich) was added to a final concentration of 36 μg/ml. Meningococci were also cultured in the chemically defined medium MCDA (24, 25) with different sodium ion concentrations [Na+]: 21.6 mM (MCDA-1), 41.6 mM, 61.6 mM (MCDA-2), 101.6 mM, and 121.6 mM (26). The pH of all media was adjusted to 7.4. To evaluate the fitness of each strain, at every stage of growth, serial dilutions were plated on GC agar in the presence or absence of rifampin and incubated at 37°C with 5% CO2 for 24 to 48 h. After growth, cell viable counts were determined by the CFU method. The same procedure was performed to assess the growth and survival of wild-type and mutant strains in MCDA medium at different [Na+]. To verify the effect of temperature on the growth of the meningococci, the wild-type and mutant strains were also grown at 32°C using the above-mentioned protocol. All experiments were performed in triplicate with three independent cultures, the results obtained were analyzed and graphically reported by using GraphPad Prism (v.4) software, and statistical significance was examined by the Student t test.
Spontaneous rifampin-resistant mutant generation assay.
Rifampin-susceptible strain 93/4286 of N. meningitidis was plated onto GC agar supplemented with 36 μg/ml of rifampin. The plates were incubated at 37°C with 5% CO2 for 24 h. Single colonies were replated on GC agar containing rifampin at 36 μg/ml. The MICs of rifampin for the randomly selected mutants were determined by the broth dilution method (27). The strains were grown in GC broth containing concentrations of antibiotic ranging from 0 to 3 μg/ml for the wild-type strain and from 0 to 800 μg/ml for the rifampin-resistant mutants (Table 1). In both cases, the MIC is denoted by the concentration interval that encompassed the upper limit of growth and the first concentration that could not support visible bacterial growth after 24 h.
TABLE 1.
Features of N. meningitidis rifampin-resistant mutants investigated in this study
| Strain | rpoB nucleotide substitution | Deduced RpoB amino acid substitution | Rifampin MICa (μg/ml) | Rifampin MBCb (μg/ml) | Rifampin cluster |
|---|---|---|---|---|---|
| 93/4286 | None (wild type) | None | 0.5 | 0.8 | |
| H553Y mutant | C1663T | H553Y | 250–280 | 300 | I |
| H553R mutant | A1664G | H553R | 250–280 | 350 | I |
| S549F mutant | C1651T | S549F | 100 | 200 | I |
The concentration interval indicated for MIC denotes the range within which the true MIC for rifampin exists.
MBC, minimal bactericidal concentration.
DNA procedures and transformant resistant strains.
High-molecular-weight genomic DNA from the N. meningitidis strains was prepared as previously reported (6). DNA fragments were isolated by using acrylamide slab gels and recovered by electroelution as described before (28). The whole rpoB gene (4,185 bp) was amplified from the genomic DNA of wild-type and selected mutant strains by PCR. For this purpose, different sets of primers were designed according to the sequence of the N. meningitidis FAM18 strain (GenBank accession number NC_008767.1) and are reported in Table 2. Amplification reactions were as follows: 1 min of denaturation at 94°C, 1 min of annealing at 60°C, and 2 min of extension at 72°C for a total of 30 cycles. Reactions were carried out in a MyCycler thermal cycler (Bio-Rad). The amplicons were sequenced by Sanger methods. Primer synthesis and DNA sequencing were performed by Ceinge Biotecnologie Avanzate s.c.ar.l., Naples, Italy. DNA sequence analysis was carried out by using GeneJockey sequence processor software (Biosoft). Homology sequence analysis was carried out by using the multiple-sequence alignment tool Clustal W (http://www.ebi.ac.uk/Tools/msa/clustalw2/). The rpoB gene sequences of rifampin-resistant strains were compared with the whole rpoB gene sequence of strain 93/4286 and strain FAM18 provided by the database, in order to identify point mutations in clusters I, II, and III (15).
TABLE 2.
Oligonucleotides used in PCR assays and real-time RT-PCR analyses
| Primer name | Sequence |
|---|---|
| rpoB1 | 5′-GATCTGATTGATTCGGAAACCGGC-3′ |
| rpoB2 | 5′-CATCTGAATCCAAATCGGCATTCGCC-3′ |
| rpoBint1 | 5′-ATGGGTTACTTCAGAAGGGGTTGG-3′ |
| rpoBint2 | 5′-GAGCTGACTGAAAACCAATTCCGC-3′ |
| rpoB 3L | 5′-TTGTCTGCCATCGAAGAAGGC-3′ |
| rpoB 3R | 5′-AAGTGTAACGATCATCCGCAGCC-3′ |
| rpoB 4L | 5′-ACCGATTTGGGCGAATTGGCTTTGG-3′ |
| rpoB 4R | 5′-GTAATTTCGCTGCCTTTAGCCAGC-3′ |
| rpoB 5L | 5′-CCGTATCGAACGTATGATTGTGG-3′ |
| rpoB 5R | 5′-TCTGTTCAAGAACTCGCGCAACTCG-3′ |
| rpoB 6L | 5′-AAAAGGTATCGGCGAGCCTATCG-3′ |
| rpoB 6R | 5′-AAGGCCGTCTGAAAACTTCTGC-3′ |
| rpoB 7L | 5′-ATGCGTGCCGTTGCTTTCAAACG-3′ |
| rpoB 7R | 5′-AACCACGGTAGGGAATGATGC-3′ |
| rpoB 8L | 5′-AAGGTAAGACGCACTCTTCCG-3′ |
| rpoB 8R | 5′-CTCATTGATGTAAAGGGTCG-3′ |
| katA-L | 5′-AATTCGCCCACTTCAATCAG-3′ |
| katA-R | 5′-CATCAGCGCGACTTATACGA-3′ |
To generate strain 93/4286 with the H553Y, H553R, and S549F substitutions in RpoB (93/4286RpoBH553Y, 93/4286RpoBH553R, and 93/4286RpoBS549F), the chromosomal DNA from each rifampin-resistant mutant was used to transform the 93/4286 wild-type strain as previously described (29). Transformants were selected on GC agar medium supplemented with rifampin (36 μg/ml). The rpoB gene was sequenced as described above to confirm the allelic replacement by the transforming DNA in the recipient strain.
Competition assay.
Pairwise competition experiments were used to estimate the in vitro fitness of each rifampin-resistant mutant relative to that of the susceptible strain. Equal numbers of CFU of the rifampin-susceptible and rifampin-resistant bacteria were mixed together (1:1), and the bacteria were allowed to grow together competitively in antibiotic-free GC broth at 37°C. The initial and final number of parental and mutated bacterial cells was determined by standard plate counting on antibiotic-free GC agar followed by pick-and-patch testing, in which a portion of each colony was streaked onto GC agar medium with or without rifampin (36 μg/ml). Competition assays were carried out across a small number of generations starting with an initial inoculum of about 106 bacteria to reach a final number of about 109 bacteria. Indeed, with fastidious bacteria such as pathogenic Neisseria spp., growth from a lower initial inoculum may be difficult (generally due to susceptibility to pH and divalent cation concentrations and a tendency toward autolysis) and/or affected by considerable variability that may mask eventual differences between competing strains (30). The experiments were performed five times with five independent cultures, and the statistical significance of the results was examined by the Student t test. The number of generations (g) of the isogenic mutant and parental strain that occurred in the mixed broth was calculated as described by Billington et al. (31): g = (log B − log A)/log2, where A is the number of CFU per milliliter at time zero and B is the number of CFU per milliliter at an optical density at 600 nm (OD600) of 1.0. The difference in fitness between two competing strains was calculated using the function described by Sander et al. (32). Hence, D0 − 1.0OD can be interpreted as the natural logarithm of the quotient of the growth rates of the competing strains. D0 − 1.0OD is equal to 0 if there is no difference in fitness between the competing strains, D0 − 1.0OD is negative if rifampin resistance reduces bacterial fitness, and D0 − 1.0OD is positive if resistance increases bacterial fitness relative to that of the rifampin-susceptible competitor strain. The cost per generation (cpg) was calculated as cpg = 1 − eD0-1.0OD (32) (see Table 3).
TABLE 3.
Fitness cost of investigated Rifr mutations
| Mutant strain | No. of generationsa |
Cost per generationb | D0 − 1.0ODb,c | Relative fitness (gR/gS)b | |
|---|---|---|---|---|---|
| gS | gR | ||||
| S549F | 9.97 | 4.20 | 0.34 ± 0.12 | −0.40 ± 0.17 | 0.42 ± 0.24 |
| H553R | 8.99 | 7.48 | 0.11 ± 0.02 | −0.12 ± 0.03 | 0.83 ± 0.04 |
| H553Y | 9.02 | 9.1 | −0.018 ± 0.04 | 0.018 ± 0.04 | 0.99 ± 0.006 |
The number of generations of the mutant strain (gR) and of the wild-type strain (gS) was calculated as described by Billington and coworkers (31). The values are the means from five independent experiments.
The values are presented as the means ± SDs from five independent experiments.
The difference in fitness (D0 − 1.0OD) was estimated by direct competition against an equal number of CFU of the rifampin-susceptible wild-type strain.
Determination of meningococcal survival/growth within THP-1 cells.
Phagocytosis assays were performed as previously described (33, 34). THP-1 cells were used. These human monocyte cells were grown in RPMI 1640 medium (Lonza) supplemented with 10% heat-inactivated fetal bovine serum and 2 mM l-glutamine and were differentiated to macrophages using 0.8 nM phorbol myristic acid for 4 to 5 days. THP-1 cells were incubated with the wild-type strain, the rifampin-resistant mutants, and transformant resistant strains at a multiplicity of infection (MOI) of 10 for 1 h in a humidified atmosphere at 37°C with 5% CO2, washed twice with 1× phosphate-buffered saline (PBS) to eliminate the majority of extracellular bacteria, and then exposed to gentamicin (Biolife) to kill the remaining extracellular bacteria. Cells were washed extensively with 1× PBS to remove gentamicin and dead extracellular bacteria and subsequently destroyed using saponin (0.5%; Sigma-Aldrich) to release intracellular bacteria. Gentamicin treatment was performed at 40 μg/ml, a concentration 10-fold above the MIC, for 30 min at 37°C with 5% CO2; the MICs of gentamicin for all strains used in this study were comparable, and the strains were equally insensitive to saponin. For quantification of the intracellular bacteria released from THP-1 cells, the lysed cell suspension was plated and the numbers of CFU were counted on the next day. When required, cells were reincubated in culture medium for various times after gentamicin treatment and treated as mentioned above. In all experiments, the bacteria were centrifuged (60 × g) onto cells to start the assay. The experiments were performed in quadruplicate, and statistical significance was examined by the Student t test.
SNP sensitivity assay.
The 93/4286 strain was grown on GC broth, and the H553Y mutant and the corresponding 93/4286RpoBH553Y transformant were grown on GC broth supplemented with rifampin (36 μg/ml) at 37°C with shaking until the OD600 was 1.0. When bacteria reached an OD600 of 1.0, 10 μl of a scalar dilution was spotted on GC agar plates with increasing concentrations (0, 0.1, 0.2, and 0.3 mM) of sodium nitroprusside (SNP; Sigma-Aldrich), a nitric oxide (NO) generator, and then the plates were incubated overnight at 37°C with 5% CO2. In parallel, each dilution was also plated onto GC agar and GC agar with rifampin for determination of the viable counts and incubated overnight under the same conditions described above (35). The experiments were performed in triplicate, and statistical significance was examined by the Student t test.
Hydrogen peroxide sensitivity assays.
The wild-type strain was grown on GC agar, and the H553Y mutant and the corresponding 93/4286RpoBH553Y transformant were grown on GC agar supplemented with rifampin overnight at 37°C with 5% CO2. Some colonies of each strain were resuspended in MCDA-1, and the cells were harvested by centrifugation at 2,000 rpm for 10 min at room temperature. The pellets were then resuspended in 15 μl of MCDA-1, spotted onto MCDA-2 agar, and incubated overnight under the same conditions described above. After 24 h, the bacteria were grown in MCDA-2 until the OD600 reached 0.6. The bacterial suspensions were centrifuged at 2,000 rpm for 10 min. The cells were resuspended in 30 ml MCDA-2 and were finally incubated at 37°C with vigorous shaking for 30 min. Then, the cultures were split into three aliquots, increasing concentrations (0, 0.1, and 0.2 mM) of stabilized H2O2 (Sigma-Aldrich) were added to each sample, and the reaction mixtures were incubated for 20 min with shaking at 37°C. After incubation, in order to remove the excess H2O2, the cells were harvested by centrifugation at 2,000 rpm. Each sample was suspended in 300 μl of MCDA-2. The cells were collected by centrifugation at 13,000 rpm and were suspended in 1 ml of MCDA with an [Na+] of 121.6 mM. Viable counts were evaluated by the CFU method on GC agar (for the susceptible strain) and GC agar supplemented with rifampin (for the resistant strains). The experiments were performed in triplicate. The results obtained were analyzed and graphically reported by using GraphPad Prism (v.4) software.
RNA-seq and bioinformatics analysis.
Wild-type strain 93/4286 and the H553Y mutant were grown to middle logarithmic phase (OD600, 0.6) in the chemically defined medium MCDA with an [Na+] of 61.6 mM. Total bacterial RNAs were then extracted by use of an RNeasy minikit (Qiagen) according to the manufacturer's instructions. Before extraction, samples were treated with 2 volumes of RNA Protect Bacteria reagent (Qiagen). DNA contamination was avoided by on-column treatment with an RNase-free DNase set (Qiagen) according to the manufacturer's instructions. This procedure was performed in triplicate for each strain. The concentration and integrity of the RNA samples were assessed by measurement of the A260/A280 and A260/A230 ratios and were verified using a NanoDrop Lite spectrophotometer (Thermo Scientific).
Next-generation sequencing experiments with quality control samples and bioinformatics analysis were performed as a service by Genomix4life S.R.L. (Baronissi, Salerno, Italy). Indexed libraries were prepared from 1 μg of each purified RNA with a TruSeq stranded total RNA sample preparation kit (Illumina) according to the manufacturer's instructions. Libraries were quantified using an Agilent 2100 bioanalyzer (Agilent Technologies) and pooled such that each index-tagged sample was present in equimolar amounts, with the final concentration of the pooled samples being 2 nM. The pooled samples were subject to cluster generation and sequencing using an Illumina HiSeq 2500 system (Illumina) in a 2 × 100 paired-end format at a final concentration of 8 pmol.
The raw sequence (.fastq) files generated underwent quality control analysis using the FastQC program (v.0.11.2; http://www.bioinformatics.babraham.ac.uk/projects/fastqc/), and the quality-checked reads were then aligned to the reference genome (N. meningitidis serogroup C strain FAM18, NCBI reference sequence with GenBank accession number NC_008767.1, version GI:121633901) using bowtie2 software (v.2.2.4) (36) with standard parameters.
Using the reads mapped to the genome, the HTSeq-count program (37) was used to calculate the expression values of each transcript. A given gene was considered expressed when at least ≥10 reads were detected. Differentially expressed transcripts were identified using the DESeq2 program (v.1.6.3) (38). The raw read counts were used as input to DESeq2 for calculation of the normalized signal for each transcript in the samples, and differential expression was reported as the fold change (FC) along with the associated adjusted P values (computed according to the Benjamini-Hochberg method).
Real-time RT-PCR experiments.
Semiquantitative analysis of the katA transcript normalized to the level of expression of the 16S rRNA gene was performed by real-time reverse transcriptase (RT) PCR. Total RNA samples were treated with RNase-free recombinant DNase I (10 U/μl; Roche) at 37°C for 45 min, and finally, the RNA was dissolved in RNase-free water. Then, for each sample, total RNAs (2 μg) were reverse transcribed into cDNA by using a random hexamer (2.5 μM) with SuperScript RT (5 U/μl; Invitrogen) in the presence of 0.01 M dithiothreitol and 2 U/μl RNase inhibitor at 37°C for 50 min. The reactions were stopped by heat inactivation at 70°C for 15 min. For semiquantitative analysis, about 0.05 to 0.1% of each reverse transcription reaction mixture was used to run a real-time PCR on an iCycler iQ apparatus (Bio-Rad) with SYBR Green JumpStart Taq ReadyMix (Sigma-Aldrich) and katA-specific primer pair katA-L/katA-R, which generated a PCR product 167 bp long. Samples were run in the real-time PCR in triplicate, and statistical significance was examined by the Student t test. To quantify gene expression, we expressed the data obtained using the 2−ΔΔCT method, where CT is threshold cycle. The real-time PCR conditions were 30 s at 94°C, 30 s at 60°C, and 30 s at 72°C for 40 cycles, and the melting curve was performed at 60°C to 95°C with a ramping of 0.5°C/10 s.
Measurement of intracellular glutathione pool.
To analyze the intracellular glutathione pool, each strain was initially resuspended in MCDA-1 and spotted onto MCDA-2 agar as described above for the hydrogen peroxide sensitivity assays. Bacteria were grown in MCDA-2 to an OD600 of 1.0, and then about 109 cells were washed twice in H2O and centrifuged at 4,000 rpm for 10 min. Next, 3 volumes of 5% sulfosalicylic acid was added to the cells, which were treated with lysozyme (1 mg/ml; Sigma-Aldrich), and the cells were freeze-thawed twice in liquid nitrogen and then left for 5 min at 4°C. Finally, samples were centrifuged at 13,000 rpm for 10 min, and the glutathione concentrations in the supernatants were measured with a glutathione assay kit (Sigma-Aldrich) according to the manufacturer's instructions. The experiments were performed in triplicate, and statistical significance was examined by the Student t test.
Quantitative determination of meningococcal capsular polysaccharide.
The relative amount of meningococcal capsular polysaccharide was determined by using two methods: (i) immunoblotting and (ii) the resorcinol method for quantitative determination of sialic acid (39).
For the immunoblot assay, cultures of meningococcal wild-type strain 93/4286 or the H553Y mutant grown in MCDA-2 medium to late logarithmic phase were resuspended in 1× PBS and the growth was normalized to an optical density (A600) of 1. Dilutions of the bacterial suspensions were transferred onto polyvinylidene difluoride (PVDF) membranes from Millipore (Milan, Italy) under vacuum by using a slot blot manifold (Bethesda Research Laboratories). The PVDF membranes were preventatively immersed in 100% methanol for a few seconds, transferred into distilled water for 2 to 3 min, and then incubated in a transfer buffer (25 mM Tris, 192 mM glycine, 20% methanol, pH 8.3). After blotting, the membranes were air dried, blocked in 5% milk in 1× PBS for 30 min at room temperature, incubated for 2 h with a primary antibody against serogroup C meningococci (R30166901; Remel Europe Ltd.) at a 1:500 dilution, washed in Tween 0.1% in 1× PBS, and then incubated for 1 h with a secondary antibody conjugated with horseradish peroxidase at a 1:5,000 dilution. The bands were visualized using enhanced chemiluminescence (GE, Milan, Italy). All of the incubations and washings were carried out at room temperature with gentle shaking.
For the resorcinol assay, extracts enriched in capsular polysaccharide were prepared from wild-type strain 93/4286 or the H553Y mutant grown to late logarithmic phase in GC or MCDA-2 broth as described by Gotschlich (40) and modified as described previously (41), and the sialic acid content was determined by the resorcinol reaction (39) with N-acetylneuraminic acid (Sigma-Aldrich) as a standard. The sialic acid content was normalized to the optical density at 600 nm of the bacterial cultures.
RESULTS AND DISCUSSION
Selection of spontaneous rifampin-resistant rpoB mutants from N. meningitidis serogroup C strain 93/4286.
Thirty-eight spontaneous rifampin-resistant mutants were isolated from serogroup C strain 93/4286 on GC agar medium supplemented with 36 μg/ml rifampin. Literature data report that in bacteria, resistance to rifampin is mostly due to missense mutations mapping within an evolutionarily conserved region of the rpoB gene encoding the RNA polymerase β chain (15). The whole rpoB gene of the rifampin-resistant mutants and the wild-type strain was therefore amplified and subjected to nucleotide sequencing. The homology analysis demonstrated three types of mutations mapping in cluster I (21): (i) a C-to-T transition at position 1663 of the nucleotide sequence of the coding region of N. meningitidis rpoB, (ii) an A-to-G transition at position 1664, and (iii) a C-to-T transition at position 1651 (Table 1; see also File S1 in the supplemental material). These mutations resulted, respectively, in the following missense substitutions: H553Y, H553R, and S549F. The results of sequence analysis through the Neisseria multilocus sequence typing website (http://pubmlst.org/neisseria) showed that the rpoB alleles correspond to rpoB4 (wild-type strain, ET-37 ST-11 complex), rpoB16 (H553Y mutant), rpoB10 (H553R mutant), and rpoB23 (S549F mutant).
In order to establish the degree of Rifr of the selected mutants, MICs were determined by the broth dilution method (27). The assay showed that the amino acid substitutions H553Y and H553R resulted in a more than 500-fold increase in antibiotic resistance, while the S549F substitution resulted in an increase of about 200-fold compared to that for the wild-type strain (Table 1).
Evaluation of fitness of the rifampin-resistant mutants under in vitro conditions.
In order to evaluate the fitness costs of rifampin resistance-conferring mutations, the growth and stationary-phase survival of wild-type strain 93/4286 and its derivative H553Y, H553R, and S549F mutants were preliminarily analyzed in GC broth. Bacteria were cultivated at 37°C or 32°C. The H553Y and H553R mutants exhibited growth curves comparable to those of the wild-type strain at both temperatures (Fig. 1B). In contrast, the S549F mutant exhibited a partial temperature-sensitive (Ts) growth phenotype. The rate of growth of this mutant was slightly lower than that of the wild-type strain at 32°C, but the growth of the mutant was almost completely abolished at 37°C.
This result is consistent with literature data demonstrating the Ts phenotype of E. coli with the corresponding S522F substitution (42). It has been known for a long time that in E. coli some Rifr-conferring mutations at the rpoB locus, including the corresponding S522F substitution, have a Ts effect on growth, because RNA polymerase containing the mutated subunit is either inactive or unable to assemble properly at the nonpermissive temperature (43). Moreover, there is evidence that although the RNA polymerase is a metabolically stable component in exponentially growing bacteria, degradation of the full-sized subunits has been found in two cases, i.e., several temperature-sensitive mutants with a defect in the assembly of RNA polymerase and the stationary-phase cells of a wild-type bacterium (44). A defect in subunit assembly and the metabolic instability of mutated RNA polymerase may therefore account for the phenotype of the rpoB S549F mutant in rich GC medium. The Ts phenotype may explain why the S549F substitution has rarely been observed in clinical isolates of N. meningitidis (8). On the other hand, one may expect the occurrence of the S549F mutant in the nasopharynx of healthy carriers, as the nasopharyngeal temperature, approximately 34°C, is lower than 37°C (45).
As there is evidence that several mutations conferring Rifr in E. coli confer a growth advantage in aging colonies (46), survival during stationary phase was also evaluated. Bacteria were grown in GC broth to late logarithmic phase (OD600, about 1.5) at 37°C or 32°C, and then bacterial viability was determined during the stationary phase by the CFU method (Fig. 1C). The results did not show any significant difference between any of the strains except the S549F mutant, which could not be cultivated at 37°C, as mentioned above.
Growth rate and generation time are accepted measures of a fitness deficit associated with antibiotic resistance. A fitness deficit can be determined by using resistant and susceptible strains in competition assays with pairs of strains, where two strains of interest are mixed together in equal proportions and left to compete head-to-head in a common environment (47). To evaluate the fitness deficit, equal numbers of CFU of rifampin-susceptible and rifampin-resistant bacteria were mixed together (1:1), and the bacteria were allowed to grow competitively in antibiotic-free GC broth at 37°C. The in vitro competition assay showed a reduction in the growth rate of the H553R mutant during the logarithmic phase of growth compared to that of the wild-type strain, providing a relative fitness of the mutant of 83% compared with that of the wild type; in contrast, the H553Y substitution did not seem to affect significantly bacterial fitness in vitro (Table 3; see also Fig. S1 in the supplemental material). The different fitness costs associated with the H553Y and the H553R substitutions in the meningococcal RNA polymerase β chain gene were consistent with the findings of a previous study addressing the physiological cost of rifampin resistance induced in vitro in M. tuberculosis (31). No relationship between the cost of the rpoB mutation and the level of resistance to rifampin (the MIC) was detected (Table 1). Interestingly, the growth defect of the Ts S549F mutant at 37°C was partially rescued when it was cocultivated with the wild-type strain, with the relative fitness being 42% (Table 3).
The growth and stationary-phase survival of the Rifr mutants were then assayed in MCDA chemically defined medium with different sodium concentrations (Fig. 2A to E). Indeed, during infection of the human host, N. meningitidis colonizes microenvironments with very different sodium concentrations, and there is evidence that environmental sodium levels affect meningococcal resistance to oxidative stress (48). In the human naso-oropharynx, the sodium concentrations in nasal secretions typically range from 90 to 148 meq/liter (49), while salivary sodium levels fluctuate widely and range from 4 and 40 meq/liter, depending on the flow rate, but average about 26 meq/liter (50). In intracellular host microenvironments, which meningococci may face during traversal of epithelial and endothelial cell barriers, sodium levels fluctuate between 10 and 15 meq/liter, with some degree of variation occurring depending on the tissue (51, 52). In human plasma and interstitial fluids, the normal concentration range of sodium is 136 to 145 meq/liter (53). Slightly higher values are generally observed in the cerebrospinal fluid (CSF) of healthy subjects, with the CSF sodium concentration/plasma sodium concentration ratio being 1.05, on average (54). CSF sodium levels, however, undergo circadian fluctuation (55), and both serum and CSF sodium concentrations may be significantly lower during acute bacterial meningitis with a syndrome of the inappropriate secretion of the antidiuretic hormone (56).
FIG 2.

Growth and stationary-phase survival of rifampin-resistant strains in minimal medium. Growth (A to E, left) and stationary-phase survival (A to E, right) curves for mutants and the wild-type strain in MCDA medium with increasing sodium concentrations ranging from 21.6 mM to 121.6 mM. The experiments were performed in triplicate with three independent cultures for each medium, and statistical significance was examined by the Student t test. Results are indicated as means ± SDs. Asterisks indicate statistical significance (*, P < 0.05; **, P < 0.01; ***, P < 0.001).
In our in vitro experiments, we used the chemically defined medium MCDA with sodium concentrations ranging from 21.6 to 121.6 mM. This range of values is permissive for optimal meningococcal growth (26, 48) and extensively overlaps the slightly higher range encountered under in vivo conditions. The growth of the mutants was similar to that of the wild-type strain under all conditions tested. Notably, the S549F mutant was able to grow in the chemically defined medium at 37°C (Fig. 2A to E, left). The stationary-phase survival of this strain was, however, significantly reduced under all conditions examined (Fig. 2A to E, right). The survival of the H553R mutant was reduced as a function of the sodium concentration, while the survival of the H553Y mutant was not significantly affected, consistent with the results of the competition assay (Table 3).
Evaluation of survival and growth of the rifampin-resistant mutants in a differentiated THP-1 human monocyte line.
We then evaluated the survival and growth of the wild-type 93/4286 strain and the rifampin-resistant mutants after entry into differentiated cells of the THP-1 human monocyte cell line. Differentiated monocytes and bacteria were challenged at an MOI of 1:10 (Fig. 3A). After 1 h of incubation followed by gentamicin treatment to eliminate extracellular and/or adherent bacteria, the number of viable intracellular bacteria was determined by the CFU method. The results demonstrated a reduced level of entry of the H553R and S549F mutants into the differentiated monocytes (Fig. 3A) and decreased intracellular survival of all rifampin-resistant mutants during the first hour of incubation compared to the findings for the wild-type strain. After this time, the number of viable intracellular bacteria started to increase, consistent with a previous report (33). The increase was slower for the H553R and S549F mutants and occurred with delayed kinetics for the H553Y mutant compared to the times for the wild-type strain. This result, on the one hand, confirmed the reduced fitness of the H553R and S549F mutants observed in the competition experiments (Table 3), and, on the other hand, it brought to light a reduced capacity of the H553Y mutant to grow/survive in differentiated monocytes.
FIG 3.

Survival and growth of wild-type strain 93/4286, the H553Y mutant, and the 93/4286RpoBH553Y transformant in differentiated human THP-1 monocytes and evaluation of sensitivity to nitrosative and oxidative injury. (A and B) Survival and growth of wild-type strain 93/4286 and derivative rifampin-resistant mutants (A) and transformants (B) in THP-1 cells. Differentiated THP-1 cells (105 cells/well) were infected with meningococci at an MOI of 10, treated with gentamicin, and reincubated in RPMI 1640 medium for the indicated times. After saponin lysis, the numbers of CFU from intracellular bacteria were scored. Values are means from at least four independent experiments. Results are shown as the relative number of CFU per well ± SD. (C) Effect of the H553Y substitution on sensitivity to nitrosative injury. The H553Y mutant, transformant 93/4286RpoBH553Y, and the wild-type strain were grown to OD600 of 1.0 (corresponding to about 1 ×109 CFU for each strain) and then exposed to increasing concentrations of the nitric oxide generator SNP, and the viable bacteria were evaluated by the CFU method. Values represent the means from three independent experiments ± SDs. (D) Effect of the H553Y substitution on sensitivity to oxidative killing. Bacteria were incubated with H2O2 over a range of concentrations (0 to 0.2 mM) for 20 min in MCDA-2, and the number of surviving bacteria was determined by the CFU method. Data are shown as the means ± SDs from three independent experiments, each with triplicate samples. (E) Semiquantitative analysis of transcriptional changes in the katA gene by real-time RT-PCR. Transcription was performed with retrotranscribed total RNA isolated from the wild-type strain and H553Y mutant that had been grown in MCDA-2 and exposed to increasing concentrations of H2O2. Results are reported as the fold change in the level of expression of the katA transcript, normalized to the 16S mRNA level, compared to the level of expression in the wild-type strain grown with 0 mM H2O2. (F) Intracellular glutathione levels. The levels of the intracellular glutathione pool in bacterial strains were determined in MCDA-2. The H553Y mutant had a significantly reduced amount of intracellular glutathione after exposure to 0.2 mM H2O2. Values represent the means ± SDs from three independent experiments. The statistical significance of all above-mentioned experiments was determined by Student's t test. Asterisks indicate statistical significance (*, P < 0.05; **, P < 0.01; ***, P < 0.001).
To rule out the possibility that mutations in genes other than rpoB could be responsible for the phenotype observed in THP-1 cells, transformation experiments were performed. The entire chromosomal DNA from each rifampin-resistant mutant was used to transform wild-type strain 93/4286. Sequence analysis of the whole rpoB gene demonstrated correct allelic replacement in the corresponding transformants 93/4286RpoBH553Y, 93/4286RpoBH553R, and 93/4286RpoBS549F. Differentiated THP-1 cells were then challenged with the three transformants. The result of this experiment demonstrated a reduced capacity of all transformants to survive/grow inside these cells, confirming the causative role of the mutations in the survival/growth defect (Fig. 3B).
Evaluation of sensitivity of the H553Y mutant to oxidative injury.
In the evaluation of sensitivity of the mutants to oxidative injury, we focused on the H553Y mutant. This mutant exhibited a clear growth/survival defect in differentiated THP-1 cells (Fig. 3A) but no significant reduction in fitness in vitro (Table 3; see also Fig. S1 in the supplemental material). We evaluated the ability of this mutant to counteract the oxidative damage. Defenses against oxidative stress are, indeed, crucial for the survival of N. meningitidis in phagocytic cells (48). Reactive oxygen species (ROS), including superoxide (O2·−) and hydrogen peroxide (H2O2), are encountered by bacteria during aerobic respiration and interactions with phagocytic cells. In addition, nitric oxide (NO) and reactive nitrogen species (RNS) generated via the NO synthase pathway are important effectors during the innate immune response.
Preliminarily, sodium nitroprusside (SNP) was used as an NO donor to analyze the resistance to oxidative burst of the N. meningitidis Rifr mutants (35). Assessment of viability after exposure to SNP demonstrated that the H553Y mutant and the corresponding 93/4286RpoBH553Y transformant were moderately more resistant to nitrosative injuries than parental strain 93/4286 (Fig. 3C). To assay for resistance against oxidative killing, the meningococcal strains were grown in MCDA medium with a 61.6 mM sodium concentration (MCDA-2) and exposed to increasing concentrations of hydrogen peroxide (Fig. 3D). The assay showed a slightly reduced ability of the mutant and transformant to counteract the oxidative injury. Compared to the survival of the wild-type strain, the survival of the H553Y mutant was reduced about 10-fold upon exposure to 0.2 mM hydrogen peroxide. A similar reduction was observed with the 93/4286RpoBH553Y transformant.
Transcriptomic analysis of the H553Y mutant and wild-type strain.
To investigate all the transcriptional changes that could be associated with the phenotype of the H553Y mutant, comparative RNA-seq analysis was carried out. For this purpose, the H553Y mutant and parental strain 93/4286 were grown to middle logarithmic phase in MCDA-2 medium before total RNA extraction. High-throughput RNA sequencing was carried out by using the Illumina technology, and differentially expressed transcripts were identified using DESeq2, as detailed in Materials and Methods. Differential expression is reported as the FC along with the associated adjusted P values in Tables S1 and S2 in the supplemental material, showing, respectively, the genes up- and downregulated genes in the H553Y mutant compared to their expression in the wild-type strain. By filtering data with P values of ≤0.05 and an FC cutoff of ≥1.5, a total of 628 differentially expressed genes (DEGs; 280 upregulated and 348 downregulated genes in the H553Y mutant) were identified. Data mapping to the reference genome of reference strain FAM18 revealed that most of the DEGs were clustered in putative operons.
A large proportion of DEGs with known functions coded for proteins involved in bacterial metabolism, genetic information processing, and the host-pathogen interaction. In particular, many genes coding for enzymes catalyzing key steps of amino acid biosynthesis (histidine, phenylalanine, tyrosine, tryptophan, arginine, lysine, valine, isoleucine) and uptake (glutamic acid, cysteine, aromatic amino acids, methionine) and several tRNA synthetases were upregulated in the H553Y mutant (see Table S1 in the supplemental material), while most of genes coding for ribosomal proteins and tRNA were downregulated (see Table S2 in the supplemental material). Many genes coding for proteins involved in protein folding, turnover, and degradation were also upregulated (see Table S1 in the supplemental material). This pattern is consistent with the finding that strains with certain rpoB mutations causing Rifr, including the equivalent H481Y substitution in S. aureus (57) and the H437Y substitution in Streptomyces (58, 59), exhibit a stringent-like phenotype. The downregulation of dksA (see Table S2 in the supplemental material), coding for an RNA polymerase binding protein that plays a crucial role in the stringent response and whose promoter is regulated by DksA and GDP 3′-diphosphate (ppGpp) by a negative-feedback mechanism in E. coli (60), is also consistent with the stringent-like phenotype of the H553Y mutant. A stringent behavior might also account for the upregulation of ppk, coding for polyphosphate kinase, which is subject to stringent control (61). The downregulation of both chpA, coding for the ChpA/MazF mRNA interferase (which cleaves translated RNAs) of the MazE/MazF toxin/antitoxin system, and trmA, coding for an enzyme that modifies the transfer-messenger RNA involved in MazE/MazF control (62), further supports this view (see Table S2 in the supplemental material). In fact, it was reported that artificial overproduction of ppGpp inhibited transcription of the chpA (mazEF) locus in E. coli (63).
In addition to genes involved in amino acid biosynthesis and uptake, ribosome biogenesis and translation, protein folding, turnover, and degradation, a number of genes involved in carbon and energy metabolism were differentially expressed in the wild-type strain and its derivative H553Y mutant. The upregulation of most of genes involved in the tricarboxylic acid (TCA) cycle and acetyl-phosphate pathway in the H553Y mutant (see Table S1 in the supplemental material) is consistent with previously published proteomic data (64). Several genes involved in key steps of the Entner-Doudoroff pathway were, in contrast, downregulated, possibly to channel more glucose toward aromatic amino acids (through the pentose phosphate pathway) and histidine biosynthesis. The downregulation of genes of the de novo purine and pyrimidine biosynthetic pathways (and the upregulation of genes of the salvage pathways) may contribute to metabolic adjustment in the H553Y mutant because these pathways share with the histidine and tryptophan biosynthetic pathways the common precursor 5′-phosphoribosyl-1′-pyrophosphate (PRPP) (see Table S2 in the supplemental material). Also noteworthy was the upregulation of nearly all genes involved in peptidoglycan (PG) and lipooligosaccharide (LOS) biosynthesis (see Table S1 in the supplemental material); the exceptions were rfaC, rfaD, and lst, which were, in contrast, downregulated (see Table S2 in the supplemental material). The lst gene is responsible for LOS sialylation, which plays an important role in meningococcal virulence (65).
Another interesting result of the RNA-seq analysis was the downregulation of gpxA, coding for glutathione peroxidase, and sodB and sodC, encoding two meningococcal superoxide dismutases in the H553Y mutant (see Table S2 in the supplemental material). This finding is consistent with the enhanced sensitivity of this mutant to oxidative injury (Fig. 3D). Such a phenotype could, however, also be associated with both reduced transcriptional activation of the catalase gene katA upon exposure to hydrogen peroxide (determined by real-time RT-PCR) (Fig. 3E) and the reduced pool of intracellular glutathione in the hydrogen peroxide-exposed H553Y mutant (Fig. 3F) compared to the level of transcriptional activation and the size of the intracellular glutathione pool in parental strain 93/4286. In particular, the glutathione pool was decreased to less than 50% of that for the parental strain in the mutant exposed to 0.2 mM hydrogen peroxide.
A number of genes coding for proteins of the transcription machinery, RNA processing, and degradation were upregulated in the H553Y mutant (see Table S1 in the supplemental material). Upregulation of greA may be associated with the stringent phenotype of the H553Y mutant, as there is evidence that GreA plays a major role in the ppGpp-DksA regulatory network (66). nusA upregulation is also expected because equivalent mutations in E. coli (RpoB H526Y) and Bacillus subtilis (H482Y) are responsible for severe defects in transcription termination, and the E. coli RpoB H526Y substitution suppresses the antitermination defect of the nusA1 allele (67–69).
In the H553Y mutant, the upregulation of a considerable number of genes involved in DNA transformation and DNA recombination and repair (see Table S1 in the supplemental material) and the downregulation of many genes coding for DNA restriction and modification enzymes (see Table S2 in the supplemental material) are also noteworthy because they suggest the existence of functional links between rifampin resistance, a stringent-like phenotype, and genome plasticity. However, the most important trait of the transcriptional profile of the H553Y mutant is represented by the downregulation of most of the genes that play key roles in the host-pathogen interaction and virulence (see Table S2 in the supplemental material), including those coding for meningococcal type IV pili (pilE, pilX, comP, pilM, pilP, pilZ), porins (porA), adhesins/invasins (opa-1700, nadA, mafA, NMC2075), IgA protease (iga), IgG-binding proteins involved in biofilm formation (tspB), and a meningococcus-specific two-partner secretion system (hrpA-hrpB) that has recently been shown to be involved in adhesion (70), biofilm formation (71), and intracellular survival and vacuole escape (34).
Interestingly, most of genes involved in capsular polysaccharide biosynthesis (siaA, siaB, siaC, siaDC, ctrC, and lipB) were downregulated in the H553Y mutant compared to their level of regulation in the wild-type strain (see Table S2 in the supplemental material). To validate this result, the relative amount of capsular polysaccharide in wild-type strain 93/4286 and its H553Y derivative mutant was determined by using immunoblotting (Fig. 4A) and the resorcinol method for quantitative determination of sialic acid (Fig. 4B). The results demonstrated a reduction in the amount of capsular polysaccharide of about 2.5-fold in the H553Y mutant grown in MCDA-2 medium to late logarithmic phase compared with that in 93/4286. The reduction was even more pronounced (about 4-fold) when the strains were grown in rich GC medium. This result is of great interest due to the essential role of the meningococcal capsule in meningococcal virulence. The antiphagocytic properties of the disease-associated capsules are essential for bacterial growth in the bloodstream, a prerequisite for septicemia and meningitis (72). In addition, there is evidence that the meningococcal capsule is fundamental for the intracellular survival of this microorganism in both human phagocytic and nonphagocytic cells (33). Moreover, it has recently been shown that the serogroup B meningococcal capsule directly mediates the interaction between bacteria and microtubules and inhibits tubulin polymerization in vitro (41).
FIG 4.
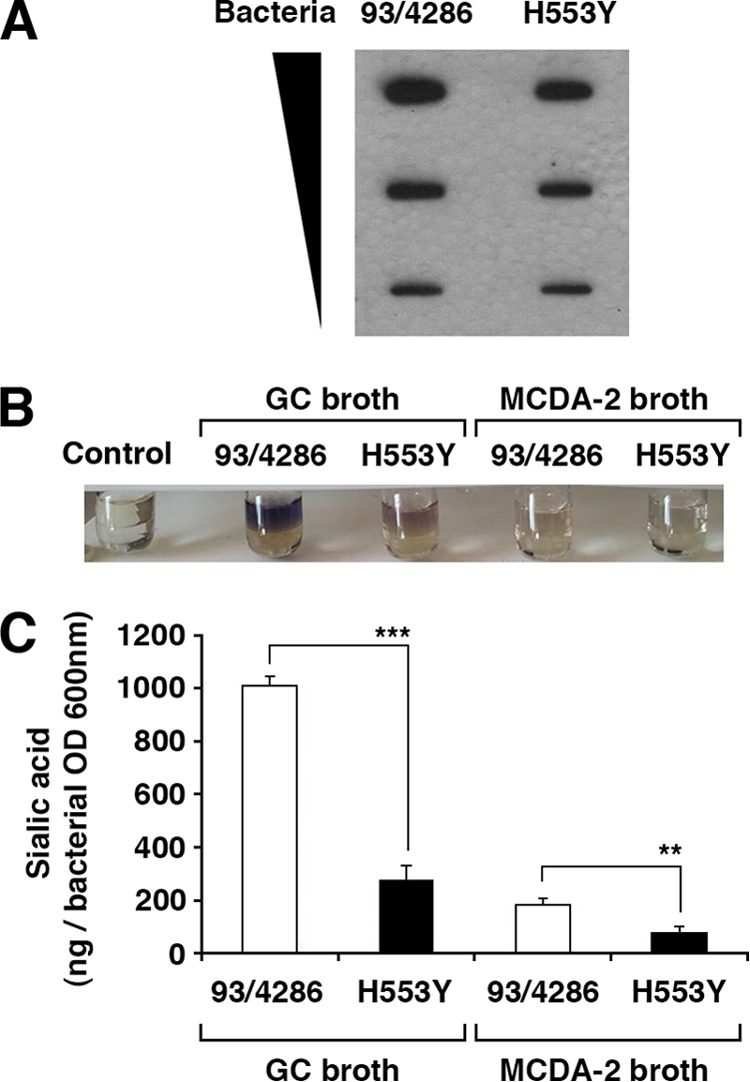
Quantitative determination of meningococcal capsular polysaccharide. (A) Immunoblot assay. Wild-type strain 93/4286 or the H553Y mutant grown in MCDA-2 medium to late logarithmic phase was resuspended in 1× PBS, and growth was normalized to an OD600 of 1. Dilutions of the bacterial suspensions (0.09 to 0.01 as shown) were transferred onto PVDF membranes by slot blotting. After blotting, the membranes were air dried, blocked in 5% milk in 1× PBS, incubated for 2 h with a primary antibody against serogroup C meningococci, washed in Tween 0.1% in 1× PBS, and then incubated for 1 h with a secondary antibody conjugated with horseradish peroxidase at a 1:5,000 dilution. (B and C) Resorcinol assay for quantitative determination of sialic acid. Wild-type strain 93/4286 or the H553Y mutant was grown to late logarithmic phase either in MCDA-2 medium or in GC medium, as indicated. Extracts enriched in capsular polysaccharide were then prepared, and the sialic acid content was determined by the chromogenic resorcinol reaction with N-acetylneuraminic acid as a standard. (B) The chromogenic reaction in a representative set of samples is shown. (C) The sialic acid content was normalized to the optical density at 600 nm of the bacterial cultures. Asterisks indicate statistical significance (**, P < 0.05; ***, P < 0.005).
Altogether these findings suggest that the reduced growth/survival of the H553Y mutant in differentiated THP-1 cells (Fig. 3A) not only may be due to reduced resistance to oxidative injury (Fig. 3D) but also may be due to downregulation of a number of bacterial determinants involved in intracellular survival. These findings also reveal the existence in N. meningitidis of a peculiar link between the stringent phenotype (associated with the H553Y substitution) and virulence very different from that found in other pathogenic bacteria, including broad-niche/host-range enteric bacteria and pseudomonads that rely on a stringent response for full virulence (73–75). In contrast, in narrow-niche/host-range N. meningitidis strains, most of the genes that play key roles in the host-pathogen interaction and virulence seem to be repressed in a stringent background. Although we are aware of the inherent limitations of in vitro systems in their ability to model processes that occur in vivo, we propose that this mechanism may account for the rarity of Rifr isolates in medical practice. This hypothesis could be tested in future experiments by using other rpoB mutants, relA-defective meningococcal mutants unable to trigger the stringent response, and animal models to better reproduce certain human host environments.
Supplementary Material
ACKNOWLEDGMENTS
This research was supported in part by POR Campania FSE 2007-2013, Project CRÈME, Project CCM 2011 (Prevenzione Universale, Malattie Infettive, Analisi di malattie emergenti e riemergenti in relazione ai flussi migratory), and PRIN 2012 (grant number 2012WJSX8K, Host-microbe interaction models in mucosal infections: development of novel therapeutic strategies).
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AAC.01746-15.
REFERENCES
- 1.Cuevas LE, Kazembe P, Mughogho GK, Tillotson GS, Hart CA. 1995. Eradication of nasopharyngeal carriage of Neisseria meningitidis in children and adults in rural Africa: a comparison of ciprofloxacin and rifampicin. J Infect Dis 171:728–731. doi: 10.1093/infdis/171.3.728. [DOI] [PubMed] [Google Scholar]
- 2.Deal WB, Sanders E. 1969. Efficacy of rifampin in treatment of meningococcal carriers. N Engl J Med 281:641–645. doi: 10.1056/NEJM196909182811203. [DOI] [PubMed] [Google Scholar]
- 3.Kaiser AB, Hennekens CH, Saslaw MS, Hayes PS, Bennett JV. 1974. Seroepidemiology and chemoprophylaxis disease due to sulfonamide-resistant Neisseria meningitidis in a civilian population. J Infect Dis 130:217–224. doi: 10.1093/infdis/130.3.217. [DOI] [PubMed] [Google Scholar]
- 4.Blakebrough IS, Gilles HM. 1980. The effect of rifampicin on meningococcal carriage in family contacts in northern Nigeria. J Infect 2:137–143. doi: 10.1016/S0163-4453(80)91159-7. [DOI] [PubMed] [Google Scholar]
- 5.Guttler RB, Counts GW, Avent CK, Beaty HN. 1971. Effect of rifampin and minocycline on meningococcal carrier rates. J Infect Dis 124:199–205. doi: 10.1093/infdis/124.2.199. [DOI] [PubMed] [Google Scholar]
- 6.Bucci C, Lavitola A, Salvatore P, Del Giudice L, Massardo DR, Bruni CB, Alifano P. 1999. Hypermutation in pathogenic bacteria: frequent phase variation in meningococci is a phenotypic trait of a specialized mutator biotype. Mol Cell 3:435–445. doi: 10.1016/S1097-2765(00)80471-2. [DOI] [PubMed] [Google Scholar]
- 7.Colicchio R, Pagliarulo C, Lamberti F, Vigliotta G, Bruni CB, Alifano P, Salvatore P. 2006. RecB-dependent mutator phenotype in Neisseria meningitidis strains naturally defective in mismatch repair. DNA Repair 5:1428–1438. doi: 10.1016/j.dnarep.2006.07.001. [DOI] [PubMed] [Google Scholar]
- 8.Skoczynska A, Ruckly C, Hong E, Taha MK. 2009. Molecular characterization of resistance to rifampicin in clinical isolates of Neisseria meningitidis. Clin Microbiol Infect 15:1178–1181. doi: 10.1111/j.1469-0691.2009.02783.x. [DOI] [PubMed] [Google Scholar]
- 9.Stefanelli P, Fazio C, La Rosa G, Marianelli C, Muscillo M, Mastrantonio P. 2001. Rifampicin-resistant meningococci causing invasive disease: detection of point mutations in the rpoB gene and molecular characterization of the strains. J Antimicrob Chemother 47:219–222. doi: 10.1093/jac/47.2.219. [DOI] [PubMed] [Google Scholar]
- 10.Ercis S, Köseoğlu O, Salmanzadeh-Ahrabi S, Ercis M, Akin L, Hasçelik C. 2005. The prevalence of nasopharyngeal Neisseria meningitidis carriage, serogroup distribution, and antibiotic resistance among healthy children in Cankaya municipality schools of Ankara Province. Mikrobiyol Bul 39:411–420. [PubMed] [Google Scholar]
- 11.Kremastinou J, Tzanakaki G, Levidiotou S, Markou F, Themeli E, Voyiatzi A, Psoma E, Theodoridou M, Blackwell CC. 2003. Carriage of Neisseria meningitidis and Neisseria lactamica in northern Greece. FEMS Immunol Med Microbiol 39:23–29. doi: 10.1016/S0928-8244(03)00174-3. [DOI] [PubMed] [Google Scholar]
- 12.Rohani MY, Ahmad Afkhar F, Amir MA, Muhd Amir K, Sahura H, Fairuz A, Norazah A, Monalisa AR, Tay AW, Azizah M, Norzarila Z, Lim KE. 2007. Serogroups and antibiotic susceptibility patterns of Neisseria meningitidis isolated from army recruits in a training camp. Malays J Pathol 29:91–94. [PubMed] [Google Scholar]
- 13.Yagupsky P, Ashkenazi S, Block C. 1993. Rifampicin-resistant meningococci causing invasive disease and failure of chemoprophylaxis. Lancet 341:1152–1153. doi: 10.1016/0140-6736(93)93171-V. [DOI] [PubMed] [Google Scholar]
- 14.Mounchetrou Njoya I, Deghmane A, Taha M, Isnard H, Parent du Chatelet I. 2012. A cluster of meningococcal disease caused by rifampicin-resistant C meningococci in France, April 2012. Euro Surveill 17(34):pii=20254 http://www.eurosurveillance.org/ViewArticle.aspx?ArticleId=20254. [PubMed] [Google Scholar]
- 15.Jin DJ, Gross CA. 1988. Mapping and sequencing of mutations in the Escherichia coli rpoB gene that lead to rifampicin resistance. J Mol Biol 202:45–58. doi: 10.1016/0022-2836(88)90517-7. [DOI] [PubMed] [Google Scholar]
- 16.Brandis G, Pietsch F, Alemayehu R, Hughes D. 2015. Comprehensive phenotypic characterization of rifampicin resistance mutations in Salmonella provides insight into the evolution of resistance in Mycobacterium tuberculosis. J Antimicrob Chemother 70:680–685. doi: 10.1093/jac/dku434. [DOI] [PubMed] [Google Scholar]
- 17.O'Neill AJ, Huovinen T, Fishwick CW, Chopra I. 2006. Molecular genetic and structural modeling studies of Staphylococcus aureus RNA polymerase and the fitness of rifampin resistance genotypes in relation to clinical prevalence. Antimicrob Agents Chemother 50:298–309. doi: 10.1128/AAC.50.1.298-309.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wichelhaus TA, Böddinghaus B, Besier S, Schäfer V, Brade V, Ludwig A. 2002. Biological cost of rifampin resistance from the perspective of Staphylococcus aureus. Antimicrob Agents Chemother 46:3381–3385. doi: 10.1128/AAC.46.11.3381-3385.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gagneux S, Long CD, Small PM, Van T, Schoolnik GK, Bohannan BJ. 2006. The competitive cost of antibiotic resistance in Mycobacterium tuberculosis. Science 312:1944–1946. doi: 10.1126/science.1124410. [DOI] [PubMed] [Google Scholar]
- 20.Brandis G, Hughes D. 2013. Genetic characterization of compensatory evolution in strains carrying rpoB Ser531Leu, the rifampicin resistance mutation most frequently found in clinical isolates. J Antimicrob Chemother 68:2493–2497. doi: 10.1093/jac/dkt224. [DOI] [PubMed] [Google Scholar]
- 21.Carter PE, Abadi FJ, Yakubu DE, Pennington TH. 1994. Molecular characterization of rifampin-resistant Neisseria meningitidis. Antimicrob Agents Chemother 38:1256–1261. doi: 10.1128/AAC.38.6.1256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nolte O, Müller M, Reitz S, Ledig S, Ehrhard I, Sonntag HG. 2003. Description of new mutations in the rpoB gene in rifampicin-resistant Neisseria meningitidis selected in vitro in a stepwise manner. J Med Microbiol 52:1077–1081. doi: 10.1099/jmm.0.05371-0. [DOI] [PubMed] [Google Scholar]
- 23.Taha MK, Zarantonelli ML, Ruckly C, Giorgini D, Alonso JM. 2006. Rifampin-resistant Neisseria meningitidis. Emerg Infect Dis 12:859–860. doi: 10.3201/eid1205.051296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Catlin BW. 1973. Nutritional profiles of Neisseria gonorrhoeae, Neisseria meningitidis, and Neisseria lactamica in chemically defined media and the use of growth requirements for gonococcal typing. J Infect Dis 128:178–194. doi: 10.1093/infdis/128.2.178. [DOI] [PubMed] [Google Scholar]
- 25.Pagliarulo C, Salvatore P, De Vitis LR, Colicchio R, Monaco C, Tredici M, Talà A, Bardaro M, Lavitola A, Bruni CB, Alifano P. 2004. Regulation and differential expression of gdhA encoding NADP-specific glutamate dehydrogenase in Neisseria meningitidis clinical isolates. Mol Microbiol 51:1757–1772. doi: 10.1111/j.1365-2958.2003.03947.x. [DOI] [PubMed] [Google Scholar]
- 26.Monaco C, Talà A, Spinosa MR, Progida C, De Nitto E, Gaballo A, Bruni CB, Bucci C, Alifano P. 2006. Identification of a meningococcal l-glutamate ABC transporter operon essential for growth in low-sodium environments. Infect Immun 74:1725–1740. doi: 10.1128/IAI.74.3.1725-1740.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Varaldo PE. 2002. Antimicrobial resistance and susceptibility testing: an evergreen topic. J Antimicrob Chemother 50:1–4. [DOI] [PubMed] [Google Scholar]
- 28.Sambrook J, Russell DW. 2001. Molecular cloning: a laboratory manual, 3rd ed Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. [Google Scholar]
- 29.Frosch M, Schultz E, Glenn-Calvo E, Meyer TF. 1990. Generation of capsule-deficient Neisseria meningitidis strains by homologous recombination. Mol Microbiol 4:1215–1218. doi: 10.1111/j.1365-2958.1990.tb00697.x. [DOI] [PubMed] [Google Scholar]
- 30.Annear DI, Wild B. 1982. Growth of Neisseria gonorrhoeae in brain heart infusion. J Clin Pathol 35:118–119. doi: 10.1136/jcp.35.1.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Billington OJ, McHugh TD, Gillespie SH. 1999. Physiological cost of rifampin resistance induced in vitro in Mycobacterium tuberculosis. Antimicrob Agents Chemother 43:1866–1869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sander P, Springer B, Prammananan T, Sturmfels A, Kappler M, Pletschette M, Böttger EC. 2002. Fitness cost of chromosomal drug resistance-conferring mutations. Antimicrob Agents Chemother 46:1204–1211. doi: 10.1128/AAC.46.5.1204-1211.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Spinosa MR, Progida C, Talà A, Cogli L, Alifano P, Bucci C. 2007. The Neisseria meningitidis capsule is important for intracellular survival in human cells. Infect Immun 75:3594–3603. doi: 10.1128/IAI.01945-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Talà A, Progida C, De Stefano M, Cogli L, Spinosa MR, Bucci C, Alifano P. 2008. The HrpB-HrpA two-partner secretion system is essential for intracellular survival of Neisseria meningitidis. Cell Microbiol 10:2461–2482. doi: 10.1111/j.1462-5822.2008.01222.x. [DOI] [PubMed] [Google Scholar]
- 35.Talà A, De Stefano M, Bucci C, Alifano P. 2008. Reverse transcriptase-PCR differential display analysis of meningococcal transcripts during infection of human cells: up-regulation of priA and its role in intracellular replication. BMC Microbiol 8:131. doi: 10.1186/1471-2180-8-131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Langmead B, Salzberg SL. 2012. Fast gapped-read alignment with Bowtie 2. Nat Methods 9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Anders S, Pyl PT, Huber W. 2015. HTSeq—a Python framework to work with high-throughput sequencing data. Bioinformatics 31:166–169. doi: 10.1093/bioinformatics/btu638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Love MI, Huber W, Anders S. 2014. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Miettinen TA, Takki-Luukkainen IT. 1959. Use of butyl acetate in determination of sialic acid. Acta Chem Scand 13:856–858. doi: 10.3891/acta.chem.scand.13-0856. [DOI] [Google Scholar]
- 40.Gotschlich EC. 1975. Development of polysaccharide vaccines for the prevention of meningococcal disease. Monogr Allergy 9:245–251. [PubMed] [Google Scholar]
- 41.Talà A, Cogli L, De Stefano M, Cammarota M, Spinosa MR, Bucci C, Alifano P. 2014. Serogroup-specific interaction of Neisseria meningitidis capsular polysaccharide with host cell microtubules and effects on tubulin polymerization. Infect Immun 82:265–274. doi: 10.1128/IAI.00501-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jin DJ, Gross CA. 1989. Characterization of the pleiotropic phenotypes of rifampin-resistant rpoB mutants of Escherichia coli. J Bacteriol 171:5229–5231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kirschbaum JB, Claeys IV, Nasi S, Molholt B, Miller JH. 1975. Temperature-sensitive RNA polymerase mutants with altered subunit synthesis and degradation. Proc Natl Acad Sci U S A 72:2375–2379. doi: 10.1073/pnas.72.6.2375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ishihama A, Fujita N, Glass RE. 1987. Subunit assembly and metabolic stability of E. coli RNA polymerase. Proteins 2:42–53. doi: 10.1002/prot.340020106. [DOI] [PubMed] [Google Scholar]
- 45.Keck T, Leiacker R, Riechelmann H, Rettinger G. 2000. Temperature profile in the nasal cavity. Laryngoscope 110:651–654. doi: 10.1097/00005537-200004000-00021. [DOI] [PubMed] [Google Scholar]
- 46.Wrande M, Roth JR, Hughes D. 2008. Accumulation of mutants in “aging” bacterial colonies is due to growth under selection, not stress-induced mutagenesis. Proc Natl Acad Sci U S A 105:11863–11868. doi: 10.1073/pnas.0804739105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Pope CF, McHugh TD, Gillespie SH. 2010. Methods to determine fitness in bacteria. Methods Mol Biol 642:113–121. doi: 10.1007/978-1-60327-279-7_9. [DOI] [PubMed] [Google Scholar]
- 48.Talà A, Monaco C, Nagorska K, Exley RM, Corbett A, Zychlinsky A, Alifano P, Tang CM. 2011. Glutamate utilization promotes meningococcal survival in vivo through avoidance of the neutrophil oxidative burst. Mol Microbiol 81:1330–1342. doi: 10.1111/j.1365-2958.2011.07766.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Eichner H, Behbehani AA, Hochstrasser K. 1983. Diagnostic value of nasal secretions, current state: normal values. 1. Laryngol Rhinol Otol (Stuttg) 62:561–565. doi: 10.1055/s-2007-1008498. [DOI] [PubMed] [Google Scholar]
- 50.White AG, Entmacher PS, Rubin G, Leiter L. 1955. Physiological and pharmacological regulation of human salivary electrolyte concentrations; with a discussion of electrolyte concentrations of some other exocrine secretions. J Clin Invest 34:246–255. doi: 10.1172/JCI103077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Madelin G, Kline R, Walvick R, Regatte RR. 2014. A method for estimating intracellular sodium concentration and extracellular volume fraction in brain in vivo using sodium magnetic resonance imaging. Sci Rep 4:4763. doi: 10.1038/srep04763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Rose AM, Valdes R Jr. 1994. Understanding the sodium pump and its relevance to disease. Clin Chem 40:1674–1685. [PubMed] [Google Scholar]
- 53.Bhave G, Neilson EG. 2011. Body fluid dynamics: back to the future. J Am Soc Nephrol 22:2166–2181. doi: 10.1681/ASN.2011080865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Schain RJ. 1964. Cerebrospinal fluid and serum cation levels. Arch Neurol 11:330–333. doi: 10.1001/archneur.1964.00460210108012. [DOI] [PubMed] [Google Scholar]
- 55.Harrington MG, Salomon RM, Pogoda JM, Oborina E, Okey N, Johnson B, Schmidt D, Fonteh AN, Dalleska NF. 2010. Cerebrospinal fluid sodium rhythms. Cerebrospinal Fluid Res 7:3. doi: 10.1186/1743-8454-7-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sambrook MA. 1974. The relationship between cerebrospinal fluid and plasma electrolytes in patients with meningitis. J Neurol Sci 23:265–273. doi: 10.1016/0022-510X(74)90230-5. [DOI] [PubMed] [Google Scholar]
- 57.Gao W, Cameron DR, Davies JK, Kostoulias X, Stepnell J, Tuck KL, Yeaman MR, Peleg AY, Stinear TP, Howden BP. 2013. The RpoB H481Y rifampicin resistance mutation and an active stringent response reduce virulence and increase resistance to innate immune responses in Staphylococcus aureus. J Infect Dis 207:929–939. doi: 10.1093/infdis/jis772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ochi K, Tanaka Y, Tojo S. 2014. Activating the expression of bacterial cryptic genes by rpoB mutations in RNA polymerase or by rare earth elements. J Ind Microbiol Biotechnol 41:403–414. doi: 10.1007/s10295-013-1349-4. [DOI] [PubMed] [Google Scholar]
- 59.Talà A, Wang G, Zemanova M, Okamoto S, Ochi K, Alifano P. 2009. Activation of dormant bacterial genes by Nonomuraea sp. strain ATCC 39727 mutant-type RNA polymerase. J Bacteriol 191:805–814. doi: 10.1128/JB.01311-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Chandrangsu P, Lemke JJ, Gourse RL. 2011. The dksA promoter is negatively feedback regulated by DksA and ppGpp. Mol Microbiol 80:1337–1348. doi: 10.1111/j.1365-2958.2011.07649.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Rao NR, Liu S, Kornberg A. 1998. Inorganic polyphosphate in Escherichia coli: the phosphate regulon and the stringent response. J Bacteriol 180:2186–2193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Christensen SK, Pedersen K, Hansen FG, Gerdes K. 2003. Toxin-antitoxin loci as stress-response-elements: ChpAK/MazF and ChpBK cleave translated RNAs and are counteracted by tmRNA. J Mol Biol 332:809–819. doi: 10.1016/S0022-2836(03)00922-7. [DOI] [PubMed] [Google Scholar]
- 63.Aizenman E, Engelberg-Kulka H, Glaser G. 1996. An Escherichia coli chromosomal “addiction module” regulated by guanosine 3′,5′-bispyrophosphate: a model for programmed bacterial cell death. Proc Natl Acad Sci U S A 93:6059–6063. doi: 10.1073/pnas.93.12.6059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Neri A, Mignogna G, Fazio C, Giorgi A, Schininà ME, Stefanelli P. 2010. Neisseria meningitidis rifampicin resistant strains: analysis of protein differentially expressed. BMC Microbiol 24:246. doi: 10.1186/1471-2180-10-246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Jones DM, Borrow R, Fox AJ, Gray S, Cartwright KA, Poolman JT. 1992. The lipooligosaccharide immunotype as a virulence determinant in Neisseria meningitidis. Microb Pathog 13:219–224. doi: 10.1016/0882-4010(92)90022-G. [DOI] [PubMed] [Google Scholar]
- 66.Vinella D, Potrykus K, Murphy H, Cashel M. 2012. Effects on growth by changes of the balance between GreA, GreB, and DksA suggest mutual competition and functional redundancy in Escherichia coli. J Bacteriol 194:261–273. doi: 10.1128/JB.06238-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Jin DJ, Cashel M, Friedman DI, Nakamaru Y, Walter WA, Gross CA. 1988. Effects of rifampicin resistant rpoB mutations on antitermination and interaction with nusA in Escherichia coli. J Mol Biol 204:247–261. doi: 10.1016/0022-2836(88)90573-6. [DOI] [PubMed] [Google Scholar]
- 68.Heisler LM, Feng G, Jin DJ, Gross CA, Landick R. 1996. Amino acid substitutions in the two largest subunits of Escherichia coli RNA polymerase that suppress a defective Rho termination factor affect different parts of the transcription complex. J Biol Chem 271:14572–14583. doi: 10.1074/jbc.271.24.14572. [DOI] [PubMed] [Google Scholar]
- 69.Ingham CJ, Furneaux PA. 2000. Mutations in the subunit of the Bacillus subtilis RNA polymerase that confer both rifampicin resistance and hypersensitivity to NusG. Microbiology 146:3041–3049. doi: 10.1099/00221287-146-12-3041. [DOI] [PubMed] [Google Scholar]
- 70.Schmitt C, Turner D, Boesl M, Abele M, Frosh M, Kurzai O. 2007. A functional two-partner secretion system contributes to adhesion of Neisseria meningitidis to epithelial cells. J Bacteriol 189:7968–7976. doi: 10.1128/JB.00851-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Neil RB, Apicella MA. 2009. Role of HrpA in biofilm formation of Neisseria meningitidis and regulation of the hrpBAS transcripts. Infect Immun 77:2285–2293. doi: 10.1128/IAI.01502-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Jarvis G, Vedros N. 1987. Sialic acid of group B Neisseria meningitidis regulates alternative complement pathway activation. Infect Immun 55:174–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Alifano P, Palumbo C, Pasanisi D, Talà A. 2015. Rifampicin-resistance, rpoB polymorphism and RNA polymerase genetic engineering. J Biotechnol 202:60–77. doi: 10.1016/j.jbiotec.2014.11.024. [DOI] [PubMed] [Google Scholar]
- 74.Dalebroux ZD, Svensson SL, Gaynor EC, Swanson MS. 2010. ppGpp conjures bacterial virulence. Microbiol Mol Biol Rev 74:171–199. doi: 10.1128/MMBR.00046-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Potrykus K, Cashel M. 2008. (p)ppGpp: still magical? Annu Rev Microbiol 62:35–51. doi: 10.1146/annurev.micro.62.081307.162903. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.