

Abstract
Antibiotic resistance in bacteria is ever changing and adapting, as once-novel β-lactam antibiotics are losing their efficacy, primarily due to the production of β-lactamases. Metallo-β-lactamases (MBLs) efficiently inactivate a broad range of β-lactam antibiotics, including carbapenems, and are often coexpressed with other antibacterial resistance factors. The rapid dissemination of MBLs and lack of novel antibacterials pose an imminent threat to global health. In an effort to better counter these resistance-conferring β-lactamases, an investigation of their natural evolution and resulting substrate specificity was employed. In this study, we elucidated the effects of different amino acid substitutions at position 67 in IMP-type MBLs on the ability to hydrolyze and confer resistance to a range of β-lactam antibiotics. Wild-type β-lactamases IMP-1 and IMP-10 and mutants IMP-1-V67A and IMP-1-V67I were characterized biophysically and biochemically, and MICs for Escherichia coli cells expressing these enzymes were determined. We found that all variants exhibited catalytic efficiencies (kcat/Km) equal to or higher than that of IMP-1 against all tested β-lactams except penicillins, against which IMP-1 and IMP-1-V67I showed the highest kcat/Km values. The substrate-specific effects of the different amino acid substitutions at position 67 are discussed in light of their side chain structures and possible interactions with the substrates. Docking calculations were employed to investigate interactions between different side chains and an inhibitor used as a β-lactam surrogate. The differences in binding affinities determined experimentally and computationally seem to be governed by hydrophobic interactions between residue 67 and the inhibitor and, by inference, the β-lactam substrates.
INTRODUCTION
The use of β-lactam antibiotics by humans creates selective pressure that drives the rapid evolution of antibacterial resistance mechanisms in bacteria. The most important of these mechanisms is the production of β-lactamases. These enzymes inactivate β-lactams via hydrolysis of the β-lactam ring and are divided into four different classes (A, B, C, and D) based on sequence similarity and structural homology (1). Classes A, C, and D utilize an active site serine to facilitate hydrolysis, whereas class B or metallo-β-lactamases (MBLs) utilize one or two active site Zn(II) ions. MBLs are of particular clinical concern because of their broad substrate capacity (penicillins, cephalosporins, cephamycins, and carbapenems), insensitivity to commercially available β-lactamase inhibitors, and rapid dissemination via horizontal gene transfer (2–5). MBLs are divided into three subclasses (B1, B2, and B3) based on structure and Zn(II)-coordinating residues. The general class B β-lactamase (BBL) amino acid numbering scheme (6, 7) is utilized here. Among the most widespread and clinically relevant MBLs are the imipenemase (IMP)-type enzymes of subclass B1, which contain two Zn(II) cofactors (8–11). Reoccurring outbreaks of IMP-producing pathogens have been reported throughout Europe and the Asian-Pacific region (12). At present, 52 distinct IMP-type enzymes have been reported (13).
IMP-1 β-lactamase was originally isolated from Serratia marcescens in Japan in 1991 (14), but it has also been found in Pseudomonas aeruginosa (15), Klebsiella pneumoniae (16), Acinetobacter baumannii (17), and Alcaligenes xylosoxidans (18). The IMP-10 variant was first isolated from P. aeruginosa in Japan in 1997 and subsequently characterized (19). The encoding gene, blaIMP-10, differs from blaIMP-1 by a point mutation at nucleotide 145 from G to T, which translates into a V67F amino acid change in the substrate binding site. F67 can also be found in the variants IMP-20 (GenBank accession code [GBAC] AB196988), IMP-26 (20), IMP-40 (GBAC AB753457), IMP-41 (GBAC AB753458), IMP-43, and IMP-44 (21). Based on currently published biochemical data, enzymes with F67 show relatively high activity toward cephalosporins and carbapenems (kcat/Km values similar to [19] or up to an order of magnitude higher than [21] those of the corresponding enzymes with V67) but low activity toward penicillins except carbenicillin (kcat/Km values about 1/10 [21] or less [19] of those of the corresponding enzymes with V67).
Position 67 is located at the base of the flexible active site loop L3 (22), which is composed of two antiparallel β strands connected by a β turn (β hairpin loop) and includes residues 58 to 67 (23) (Fig. 1). Due to L3's flexibility and topology (forming one side of and covering the active site), it is believed to be involved in substrate binding, as it acts to accommodate molecules of various widths and depths (24). Other residues in L3, such as W64 (25) and residue 59 (26), were shown to have a significant impact on substrate specificity. In a codon randomization study of IMP-1 (27), it was shown that position 67 tolerates a variety of mutations depending on the antibacterial used for selection. In the same study, the residue at position 67 was shown to contribute to the hydrophobic pocket formed at the base of L3 and is hypothesized to interact with R1 groups of β-lactam substrates (27). Furthermore, position 67 is considered to be under positive selective pressure, which emphasizes its significance in the molecular evolution of IMP enzymes (28). Interestingly, the importance of residue 67 in substrate binding was also demonstrated for VIM-type MBLs (29), in which it is invariably tyrosine among 46 variants reported to date (13). Among the 16 NDM-type MBLs reported so far (13), this residue is consistently valine.
FIG 1.
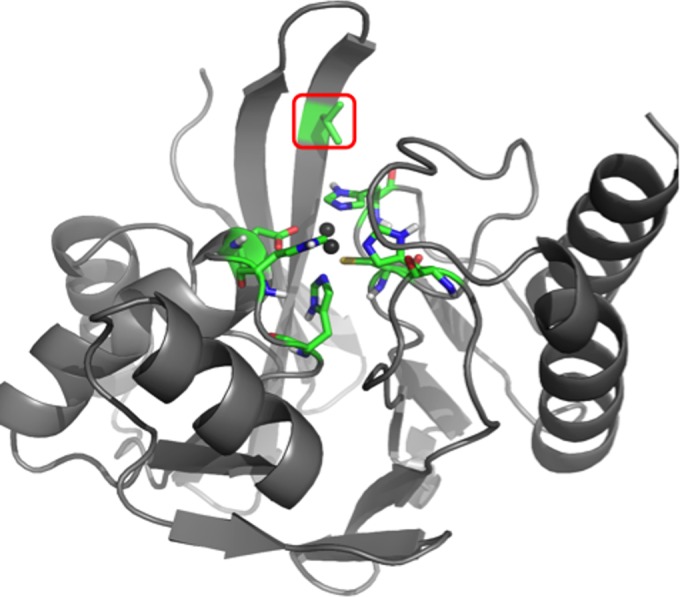
3D model of IMP-1 metallo-β-lactamase, generated with PyMOL (www.pymol.com) by using the crystal structure 1DD6 (23). The enzyme backbone is represented as a gray ribbon, and atoms of key active site residues are shown as sticks colored in green (carbon), red (oxygen), blue (nitrogen), yellow (sulfur), and white (hydrogen). The Zn(II) ions are represented by dark gray spheres. The residue under investigation at position 67 is indicated by a red box.
Based on this apparent significance of residue 67 and because a systematic biochemical analysis has not been reported to date, the aim of the present study was to elucidate the effects of different amino acid substitutions found naturally at position 67 in IMP-type MBLs on the β-lactamases' ability to hydrolyze and confer resistance to β-lactam antibiotics. Other amino acids found at position 67 are isoleucine and alanine. I67 is found in the naturally occurring MBLs IMP-9 (30), IMP-31 (GBAC KF148593), IMP-35 (31), and IMP-45 (32), and A67 is found in the IMP-21 variant (GBAC AB204557), while all other variants harbor V67, like IMP-1. To our knowledge, biochemical data are not available for the variants harboring I67 and A67. We investigated the impacts of these residues independently of the other sequence changes in these variants and therefore created the artificial variants IMP-1-V67I and IMP-1-V67A and compared them to IMP-1 and IMP-10 with respect to their biochemical and biophysical properties as well as their potential to confer resistance on Escherichia coli.
MATERIALS AND METHODS
Antibacterials.
Ampicillin, benzylpenicillin, cephalothin, chloramphenicol, imipenem, and aztreonam were purchased from MP Biomedicals, LLC (Illkirch, France). Ceftazidime and meropenem were purchased from USP (Rockville, MD). Cefotaxime was purchased from Tokyo Chemical Industry Co. Ltd. (Tokyo, Japan), cefoxitin from Biosynth (Itaska, IL), chromacef from Sopharmia (St. Joseph, MO), and kanamycin from Fisher Scientific (Fair Lawn, NJ).
Site-directed mutagenesis.
Plasmids pET26b and pBC SK(+), each individually carrying blaIMP-10, blaIMP-1-V67I, and blaIMP-1-V67A, were created by PCR-based site-directed mutagenesis using the previously described vectors pET26b-blaIMP-1 (kindly provided by James Spencer) and pBC SK(+)-blaIMP-1 (33) as templates. Primers V67A ForL/RevL and V67I ForL/RevL (see Table S1 in the supplemental material) were used to create pET26b-blaIMP-1-V67A and pET26b-blaIMP-1-V67I, respectively. Primers V67A For/Rev and V67I For/Rev were used to create plasmids pBC SK(+)-blaIMP-1-V67A and pBC SK(+)-blaIMP-1-V67I, respectively. Both pET26b and pBC SK(+) plasmids carrying the blaIMP-10 gene were produced using the V67F For/Rev primers. All primers were purchased from Integrated DNA Technologies, Inc. (San Diego, CA).
After detection of a PCR product via agarose gel electrophoresis, DpnI (Promega, Madison, WI) digestion, and deionization using steps 6 to 10 of a QIAprep Spin miniprep kit (Qiagen, Valencia, CA), E. coli TG1 electrocompetent cells (Lucigen, Middleton, WI) were transformed with the PCR product via electroporation. Transformants were incubated at 37°C overnight on lysogeny broth (LB) agar plates with either 50 μg/ml kanamycin (cells containing pET26b) or 34 μg/ml chloramphenicol [cells containing pBC SK(+)]. Resulting bacterial colonies were cultivated in LB with the appropriate antibiotic overnight at 37°C, plasmids were isolated (QIAprep Spin miniprep kit; Qiagen), and the blaIMP genes were sequenced at Eurofins MWG Operon (Huntsville, AL).
Expression and purification.
E. coli OverExpress C43 (DE3) cells (Lucigen, Middleton, WI) were transformed with the pET26b plasmids carrying blaIMP-1-V67A, blaIMP-1-V67I, and blaIMP-10 via electroporation. The enzymes were overexpressed and purified as described previously (3, 33). Finally, the enzymes were concentrated using 15-ml Amicon Ultra centrifugal filter units with a molecular mass cutoff of 10 kDa (Millipore, Billerica, MA), and the purities of the concentrated proteins were determined via sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE). A Coomassie (Bradford) protein assay (Fisher, Pittsburgh, PA) with bovine serum albumin (BSA) as a standard was used to determine the concentrations of the purified proteins.
Biophysical characterization.
The molecular weights of purified IMP-1-V67A, IMP-1-V67I, and IMP-10 were determined by electrospray ionization mass spectrometry (ESI-MS) using an Applied Biosystems QSTAR-XL tandem MS (MS/MS) system with an EKsigient nano-liquid chromatography two-dimensional system at the Protein/Peptide Microanalytical Laboratory (PPMAL) at the California Institute of Technology (Pasadena, CA). The purified proteins were dialyzed against zinc-free 50 mM morpholinepropanesulfonic acid (MOPS), pH 7.0, and the zinc content of each was determined using the 4-(2-pyridylazo)resorcinol (PAR) assay (34) as previously described (3, 35). The concentrations of the dialyzed proteins were determined as described above. Circular dichroism (CD) analysis was used to analyze the secondary structure and thermal stability of the four enzymes. Details can be found in the supplemental material.
Steady-state kinetic assay.
Steady-state kinetic experiments were conducted using an SQ-2802 UV-visible (UV-Vis) spectrophotometer with a Peltier element for temperature control (Unico, Dayton, NJ) controlled by UV-Vis Analyst software (MacroEasy Technologies, Ltd., Shanghai, China). Purified enzyme was prepared in 50 mM MOPS, pH 7.0, 100 μM ZnSO4, and 10 μg/ml BSA and preincubated at 30°C. Following the addition of preincubated (30°C) substrate at different concentrations, the initial velocities were determined by measuring the degradation of the substrate. The wavelengths and molar extinction coefficients used were described previously for doripenem (36) and all other substrates (2). The enzyme concentrations were adjusted to obtain <15% substrate hydrolysis within the first minute. Enzyme samples were prepared three times per substrate concentration, resulting in three sets of data per antibacterial. The kinetic constants were calculated for each data set by fitting them to the Michaelis-Menten equation, using Prism 6 (GraphPad Software, Inc., La Jolla, CA).
MIC assay.
E. coli ElectroMAX DH10B cells (Invitrogen, Grand Island, NY) transformed with the pBC SK(+) phagemid harboring the blaIMP-1-V67A, blaIMP-1-V67I, or blaIMP-10 gene were used for susceptibility assays. MIC assays with DH10B cells producing the various MBLs in the presence of 10 antibacterials were performed by a broth microdilution method following the guidelines of the Clinical and Laboratory Standards Institute (37), using antibiotic concentrations in the range of 0.03125 μg/ml to 1,024 μg/ml. An inoculum of 104 E. coli DH10B cells containing the corresponding blaIMP-carrying phagemid was grown in Mueller-Hinton II broth at 37°C for 20 h in the presence of one of the antibacterials, supplied in serial dilutions in a 96-well plate assay format. DH10B cells transformed with the pBC SK(+) phagemid lacking the MBL gene and cells lacking any phagemid served as negative controls, while DH10B cells with no antibacterials served as a positive control. Six independent measurements were carried out, and the median was designated the MIC.
Immunoblotting.
Immunoblotting was used to evaluate the normal expression levels of wild-type and artificial variants of IMP-1. This was done under conditions comparable to those in the susceptibility assays. Five-milliliter cultures of E. coli DH10B cells containing pBC SK(+) phagemids harboring the various blaIMP genes in Mueller-Hinton II broth containing 20 μg/ml chloramphenicol were grown at 37°C to an optical density at 600 nm (OD600) of 0.8. Fifty-microliter aliquots of whole cells from these cultures were pelleted and frozen overnight. Both whole cells and crude cell extracts were tested. For whole-cell analysis, pellets were resuspended in 20 μl of loading buffer. Crude cell extracts were prepared by resuspending pellets in 50 μl of 50 mM Tris-HCl (pH 7.4) containing 0.04 mg/ml lysozyme, 1 mM MgSO4, and 5 U/ml benzonuclease (Millipore Corp., Billerica, MA), incubating them at room temperature for 30 min, and removing cell debris by centrifugation at 10,000 × g for 5 min. Whole cells or crude cell extracts were then separated via SDS-PAGE and subsequently transferred to a polyvinylidene difluoride (PVDF) membrane (Novex, Life Technologies, Carlsbad, CA) by electroblotting. Immunostaining was carried out as described previously (38).
Inhibition experiments.
eMolecules compound 6142342 was purchased from ChemDiv (San Diego, CA), initially dissolved in dimethyl sulfoxide, and then further diluted in assay buffer. For kinetic assays carried out as described for the steady-state kinetic assays described above, the final solution contained 320 pM purified MBL, 10 μg/ml BSA, and either no or 40 μM compound 6142342 in the kinetic assay buffer described above plus 2 mM 3-[(3-cholamidopropyl)-dimethylammonio]-1-propanesulfonate (CHAPS) detergent. The percent residual activity was defined as the initial velocity for the conversion of 6 μM chromacef in the presence of compound 6142342 divided by the initial velocity without the compound. Percent inhibition was defined as 100% minus the residual activity. The experiments were carried out in triplicate.
Molecular docking.
The three-dimensional (3D) structure of an IMP-1 ligand, eMolecules compound 6142342 (Fig. 2), identified using the Shape Signatures algorithm (39), was constructed using ArgusLab (www.arguslab.com). The inhibitor was built with a deprotonated hydroxyl group and an overall charge of −1e at pH 7.0, as depicted at the ZINC docking database (www.zinc.docking.org). The appropriate hybridizations were then assigned to the molecule, followed by minimization in fewer than 100 steps, using the semi-empirical method Austin Model 1 (AM1) (40). AutoDock Tools (41) was used to prepare the docking files. The IMP-1 crystal structure (PDB entry 1DD6) (23) was used as the receptor file. The protonation states of the Zn(II)-coordinating histidine residues (H116, H118, H196, and H263) were modified to ε-protonated tautomers with the deprotonated N-ζ pointing toward the Zn(II) ions. Furthermore, charge (+1.4e) (42) and van der Waals parameters (σ = 1.95 Å; ε = 0.25 kcal/mol) (43) for the Zn(II) ions were used as described in a previous study (26). To balance the molecular system, the Zn(II) ligands (3H for Zn1 and DCH for Zn2) were given an additional charge of +0.2e each. The IMP-10, IMP-1-V67A, and IMP-1-V67I enzymes were created from PDB entry 1DD6 by using the “mutagenesis” function in PyMOL (www.pymol.com) before being prepared as described above.
FIG 2.

Chemical structures of representative β-lactam substrates used in this study, as well as compound 6142342, used as an IMP inhibitor. For cefotaxime, the positions of the R1 and R2 groups are indicated exemplarily.
The maximum numbers of energy evaluations and generations were set to 2,500,000 and 27,000, respectively. The mutation and crossover rates were set to 0.02 and 0.8, respectively. The rest of the parameters were kept at their default values, and all docking calculations were performed without constraints. Conformations that constitute each cluster were defined by a root mean square deviation tolerance of 2.0 Å. The docking simulations were run on Windows, using the AutoGrid 4.0 and AutoDock 4.2 programs (41). The highest-ranking conformation (lowest binding energy) for each enzyme inhibitor complex was analyzed visually using PyMOL.
RESULTS AND DISCUSSION
Expression, purification, and biophysical characterization of MBL variants.
All four β-lactamases were expressed well, and between 5 and 15 mg per liter of culture could be purified to homogeneity (>95% purity as judged by SDS-PAGE) from the soluble fraction after cell lysis (Table 1). Mass spectrometry gave molecular masses close to the theoretical values. Approximately two Zn(II) ions were determined per protein molecule for all enzymes by using the PAR assay (3, 34) (Table 1).
TABLE 1.
Biophysical characteristics of the purified enzymes
| Enzymea | % soluble | Yield (mg/liter) | Mol wt from ESI-MS (theoretical mol wt) | Tm (°C) | No. of Zn(II) ions/enzyme moleculeb |
|---|---|---|---|---|---|
| IMP-1-V67A | 25 | 7.4 | 25,088 (25,085) | 71 | 1.8 ± 0.1 |
| IMP-1 (V67) | 80 | 5.1 | 25,112 (25,113) | 70 | 1.8 ± 0.1 |
| IMP-1-V67I | 25 | 15.1 | 25,129 (25,127) | 72 | 1.6 ± 0.1 |
| IMP-10 (F67) | 50 | 5.6 | 25,160 (25,161) | 67 | 2.3 ± 0.2 |
The enzymes are presented by increasing size of residue 67, with the smallest being alanine (IMP-1-V67A) and the largest being phenylalanine (IMP-10). The amino acid identities are shown in bold. Data for IMP-1 are from reference 33.
Data are average values for three measurements ± standard deviations.
Circular dichroism (CD) spectra of IMP-1, IMP-10, and IMP-1-V67I are superimposable and are characteristic of the typical αββα fold (44) common among all MBLs and comparable to those of other IMP variants (3, 38, 45) (see Fig. S1 in the supplemental material). The spectrum of IMP-1-V67A is slightly different in shape, consistent with a relatively higher α-helical portion in this variant. This observation was further validated by quantitative secondary structure analysis of the spectra by using DichroWeb (46, 47) with the CDSSTR method (48) and reference set 7 (49) (see Fig. S2 and Table S2). It is conceivable that alanine, which has a high helix propensity (50, 51), disrupts the β hairpin loop structure and induces formation of a small α helix in L3. Thermal denaturation was monitored by CD analysis (see Fig. S3) and revealed very similar melting temperatures (Tm) for all four enzymes, ranging from 67 to 72°C (Table 1).
In summary, all enzymes behaved very similarly during expression and purification, bound two Zn(II) ions, maintained the overall MBL fold, with a small change in secondary structure for IMP-1-V67A, and were thermally stable under physiological as well as kinetic assay (30°C) conditions.
Kinetic constants.
The β-lactamase activities of purified IMP-10, IMP-1-V67I, and IMP-1-V67A were determined with the following 10 substrates: cephalothin, cefotaxime, ceftazidime, cefoxitin, benzylpenicillin, ampicillin, imipenem, meropenem, doripenem, and aztreonam (Fig. 2). The resulting kinetic constants (kcat, Km, and kcat/Km) are shown in Table 2, together with those of IMP-1 reported previously (33). The activity profiles of the four recombinant enzymes indicate that they hydrolyzed all tested β-lactams except for aztreonam. IMP-10 and the two mutant enzymes (IMP-1-V67A and IMP-1-V67I) exhibited significantly higher catalytic efficiencies (kcat/Km) than those of IMP-1 toward the cephalosporins with neutral R groups (cephalothin and cefotaxime) and the more recently introduced carbapenems (meropenem and doripenem), which likely also have neutral R groups in the MBL active site (33). IMP-1 and the two mutants (IMP-1-V67A and IMP-1-V67I) showed comparable catalytic efficiencies toward the β-lactams from these groups that have positively charged R2 groups (the cephalosporin ceftazidime and the carbapenem imipenem), while those of IMP-10 were 2 to 6 times higher. All enzymes showed comparable kcat/Km values toward the cephamycin cefoxitin. Interestingly, the situation was opposite toward the penicillins: IMP-1 was the most efficient enzyme, followed closely by IMP-1-V67I, while IMP-1-V67A and IMP-10 were significantly less efficient, in agreement with a previous report on IMP-10 (19). Increased kcat/Km values compared to those of other enzymes were almost exclusively due to increased kcat values, and only in the case of doripenem were they also due to decreased Km values.
TABLE 2.
Kinetic constants of the four enzymes studied against 10 β-lactam antibacterialsa
| β-Lactam |
kcat (s−1) |
Km (μM) |
kcat/Km (μM−1 s−1) |
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| IMP-1-V67A | IMP-1 (V67) | IMP-1-V67I | IMP-10 (F67) | IMP-1-V67A | IMP-1 (V67) | IMP-1-V67I | IMP-10 (F67) | IMP-1-V67A | IMP-1 (V67) | IMP-1-V67I | IMP-10 (F67) | |
| Cephalothin | 500 ± 20 | 50 ± 1 | 470 ± 20 | 450 ± 40 | 9.2 ± 0.6 | 4.5 ± 0.2 | 8.4 ± 0.5 | 8 ± 1 | 55 ± 2 | 11.1 ± 0.2 | 55 ± 1 | 56 ± 2 |
| Cefotaxime | 230 ± 10 | 16.4 ± 0.4 | 46 ± 2 | 140 ± 10 | 14 ± 1 | 5.1 ± 0.8 | 5.8 ± 0.5 | 11 ± 1 | 17 ± 1 | 3.2 ± 0.5 | 8.0 ± 0.3 | 13 ± 1 |
| Ceftazidime | 8 ± 1 | 14 ± 1 | 29 ± 6 | 66 ± 8 | 25 ± 6 | 55 ± 7 | 50 ± 10 | 49 ± 10 | 0.32 ± 0.05 | 0.25 ± 0.02 | 0.59 ± 0.03 | 1.4 ± 0.1 |
| Cefoxitin | 27 ± 1 | 16.2 ± 0.1 | 22 ± 1 | 22.5 ± 0.5 | 4.1 ± 0.4 | 4.0 ± 0.1 | 6.7 ± 0.5 | 3.5 ± 0.5 | 6.6 ± 0.5 | 4.0 ± 0.1 | 3.2 ± 0.1 | 6.4 ± 0.6 |
| Benzylpenicillin | 41 ± 1 | 2,000 ± 100 | 860 ± 70 | 110 ± 20 | 98 ± 15 | 530 ± 50 | 350 ± 60 | 300 ± 90 | 0.42 ± 0.05 | 3.8 ± 0.1 | 2.5 ± 0.2 | 0.37 ± 0.07 |
| Ampicillin | 38 ± 2 | 260 ± 30 | 310 ± 30 | 126 ± 12 | 85 ± 9 | 210 ± 30 | 360 ± 60 | 660 ± 80 | 0.45 ± 0.02 | 1.2 ± 0.1 | 0.9 ± 0.1 | 0.19 ± 0.01 |
| Imipenem | 114 ± 5 | 105 ± 5 | 160 ± 20 | 780 ± 90 | 31 ± 3 | 35 ± 4 | 36 ± 7 | 60 ± 10 | 3.6 ± 0.1 | 3.0 ± 0.2 | 4.6 ± 0.4 | 13 ± 1 |
| Meropenem | 390 ± 30 | 22 ± 1 | 53 ± 1 | 114 ± 3 | 19 ± 2 | 2.9 ± 0.2 | 2.3 ± 0.1 | 6.3 ± 0.2 | 21 ± 1 | 7.7 ± 0.4 | 23 ± 1 | 18.2 ± 0.2 |
| Doripenem | 140 ± 10 | 37 ± 8 | 134 ± 6 | 52 ± 1 | 21 ± 2 | 35 ± 6 | 18 ± 2 | 6.7 ± 0.7 | 7.0 ± 0.2 | 1.1 ± 0.1 | 7.7 ± 0.6 | 7.8 ± 0.6 |
| Aztreonam | ND | ND | ND | ND | ||||||||
Amino acid identities are shown in bold. Data for IMP-1 are from reference 33. ND, not detectable.
Antibacterial susceptibility.
The antibacterial resistance profiles conferred to E. coli DH10B cells by expression of the investigated MBLs were established by determining the MICs of the same 10 β-lactams. All wild-type and artificial mutant MBLs conferred clinically relevant levels of resistance to the tested β-lactams, except aztreonam, while cells not expressing any MBL (negative controls) were susceptible (Table 3). Different levels of penetration into the bacterial periplasm may explain why MICs of different antibiotics do not necessarily correlate with their kinetic constants. For instance, the MICs of cephalothin and cefoxitin are similar, although both the kcat and kcat/Km values are an order of magnitude higher for cephalothin. The kcat/Km values of cefoxitin and imipenem are similar, but the MICs of cefoxitin are about 2 orders of magnitude higher. For the cephalosporins, the penicillins, and imipenem, the MICs of a given antibiotic are for the most part consistent with the kinetic constants kcat and kcat/Km exhibited by different enzymes, give or take one serial dilution. For the neutral carbapenems, meropenem and doripenem, IMP-10 especially shows higher MICs than those with IMP-1 (8-fold), while those with the other enzymes are comparable. The high MICs of these antibiotics are not easily explained by kcat/Km values, because IMP-1-V67A and IMP-1-V67I exhibit similarly high kcat/Km values but lower MICs. It is possible that both kcat and Km need to be in the right ranges to result in a high MIC. We previously made a similar observation with IMP-25, which exhibited a lower kcat/Km value toward meropenem than IMP-1 and IMP-6, but its MIC was 8-fold and 2-fold higher, respectively (33). For both IMP-25 and IMP-10, the kcat value is on the order of 100 s−1. Perhaps this is an ideal turnover rate under periplasmic conditions. Clearly, more research is needed to resolve this issue.
TABLE 3.
MIC assay of E. coli DH10B cells with pBC SK(+) phagemid harboring blaIMP variant genes as well as controls (phagemid without a bla gene and no phagemid)a
| β-Lactam | MIC (μg/ml) for E. coli DH10B |
|||||
|---|---|---|---|---|---|---|
| pBC SK(+)-blaIMP-1-V67A | pBC SK(+)-blaIMP-1 (V67) | pBC SK(+)-blaIMP-1-V67I | pBC SK(+)-blaIMP-10 (F67) | pBC SK(+) | No phagemid | |
| Cephalothin | 1,024 | 512 | 1,024 | 1,024 | 16 | 16 |
| Cefotaxime | 256 | 256 | 128 | 512 | 0.125 | 0.25 |
| Ceftazidime | 128 | 256 | 512 | 1,024 | 0.5 | 0.5 |
| Cefoxitin | 1,024 | 1,024 | 1,024 | 512 | 8 | 16 |
| Benzylpenicillin | 32 | 256 | 128 | 64 | 32 | 32 |
| Ampicillin | 16 | 256 | 256 | 32 | 4 | 8 |
| Imipenem | 8 | 4 | 16 | 16 | 0.25 | 0.25 |
| Meropenem | 32 | 16 | 16 | 128 | <0.0625 | 0.0625 |
| Doripenem | 8 | 16 | 8 | 128 | 0.0625 | 0.125 |
| Aztreonam | 0.125 | 0.25 | 0.125 | 0.25 | 0.25 | 0.25 |
Amino acid identities are shown in bold. Data for blaIMP-1, pBC SK(+), and no phagemid are from reference 33.
Immunoblotting.
To test whether different expression levels of functional enzyme could have affected the MICs, immunoblot analysis with a previously reported IMP-1-specific antibody (38) was employed. The amounts of enzyme detected by the antibody in crude cell lysates and using whole cells were comparable (Fig. 3). One interesting feature observed was that IMP-1-V67A migrated slightly less than the other enzymes in the SDS-PAGE gel. Even though denaturing conditions were used, this could possibly be related to the slightly different secondary structure observed in its CD spectrum (see Fig. S1 and S2 and Table S2 in the supplemental material) and to its slightly different thermal denaturation kinetics (see Fig. S3).
FIG 3.
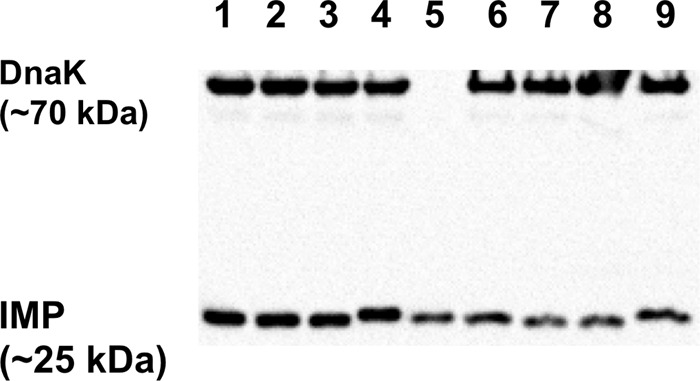
Immunoblot of the four IMP enzymes investigated. Lanes 1 to 4, crude cell lysates; lane 5, 50 ng purified IMP-1; lanes 6 to 9, whole cells. Lanes 1 and 6, IMP-1 (V67); lanes 2 and 7, IMP-10 (F67); lanes 3 and 8, IMP-1-V67I; lanes 4 and 9, IMP-1-V67A. DnaK was used as a constitutively expressed control.
Experimental and computational inhibition study.
In order to obtain a better understanding of the binding determinants between β-lactam substrates and residue 67 (and the L3 loop as a whole), an inhibitor that structurally resembles typical β-lactams was tested on the four β-lactamases both experimentally and computationally by docking. Using a substrate for such a study would not necessarily yield conclusive results, because Km correlates with Ks only under certain conditions. Therefore, binding of compound 6142342 (eMolecules) (Fig. 2), which was identified as part of another study with the Shape Signatures algorithm (39) as a modest inhibitor of IMP-1, was investigated. This compound was tested on the four enzymes and exhibited between 33% (IMP-1-V67A) and 85% (IMP-1) inhibition at 40 μM (Table 4).
TABLE 4.
Experimental and computational results of inhibition study with compound 6142342
| Enzyme | Experimental % inhibition with 40 μM inhibitora | Computational result |
|||||
|---|---|---|---|---|---|---|---|
| Binding energy (kcal/mol)b | Inhibitor-enzyme distance (Å)c |
||||||
| O-1–Zn1 | O-1–Zn2 | O-3–Zn1 | O-2–K224 N-ζ | N-3–K150a N-ζ | |||
| IMP-1-V67Ad | 33 ± 10 | −10.7 ± 0.5 | 2.3 | 1.9 | 2.1 | 2.9 | 3.4 |
| IMP-1 (V67) | 85 ± 5 | −11.8 ± 0.8 | 2.1 | 1.9 | 2.0 | 3.1 | 3.2 |
| IMP-1-V67I | 58 ± 11 | −11.4 ± 0.4 | 2.3 | 1.8 | 1.9 | 2.7 | 3.6 |
| IMP-10 (F67) | 49 ± 5 | −11.0 ± 0.6 | 2.1 | 1.9 | 2.2 | 3.0 | 3.3 |
Percent inhibition values are the averages for three experiments ± standard deviations.
The binding energies reported are the average binding energies of the highest-ranked clusters of docking conformations ± standard deviations.
The distances reported are distances between inhibitor atoms and enzyme atoms found in the highest-ranked docking conformation (see Fig. 4B). O-1 is the nitro group oxygen coordinating the two Zn(II) ions, O-2 is the nitro group oxygen hydrogen bonding with K224, O-3 is the deprotonated phenol oxygen coordinating Zn1, and N-3 is the benzothiazole nitrogen that interacts electrostatically with K150a.
Amino acid identities are shown in bold.
In order to elucidate binding interactions and to compare potential differences in substrate binding among the four β-lactamases, docking simulations were carried out with AutoDock 4.2 (41). Out of 50 docking conformations for each complex, between 4 and 31 were found in the highest-ranked cluster, with mean binding energies of these clusters falling between −11.8 and −10.7 kcal/mol. Interestingly, there is a clear correlation between the experimental percent inhibition values and these binding energies (R2 = 0.96) (Fig. 4A). The lowest-binding-energy conformations of the four complexes were nearly identical. Figure 4B exemplarily shows the complex with IMP-1, and characteristic interactions between the enzymes and the inhibitor are summarized in Table 4. The aromatic nitro group coordinated both Zn(II) ions and formed a hydrogen bond with K224 N-ζ. The hydroxyl oxygen on the nitrophenol ring (O-3), which was assumed to be deprotonated under physiological conditions, also acted as a Zn1 ligand. The nitrogen (N-3) in the benzothiazole ring system, which corresponds to R1 of β-lactams, formed an electrostatic interaction with K150a N-ζ. Lastly, the benzyl substituent of compound 6142342, which corresponds to R2 of β-lactams, formed hydrophobic interactions with residue 67 mutated in this study, as well as with V61 and W64. Since the docked conformations are so similar between the four enzymes, it is likely that interaction between the benzyl substituent or R2 of substrates and these residues plays a major role in the observed inhibitor binding and substrate specificities. Interestingly, inhibitor binding decreases with an increasing size of residue 67 (V → I → F), with the exception of A, which may be due to alanine forming an unfavorable void or an altered secondary structure of L3 creating clashes in IMP-1-V67A.
FIG 4.
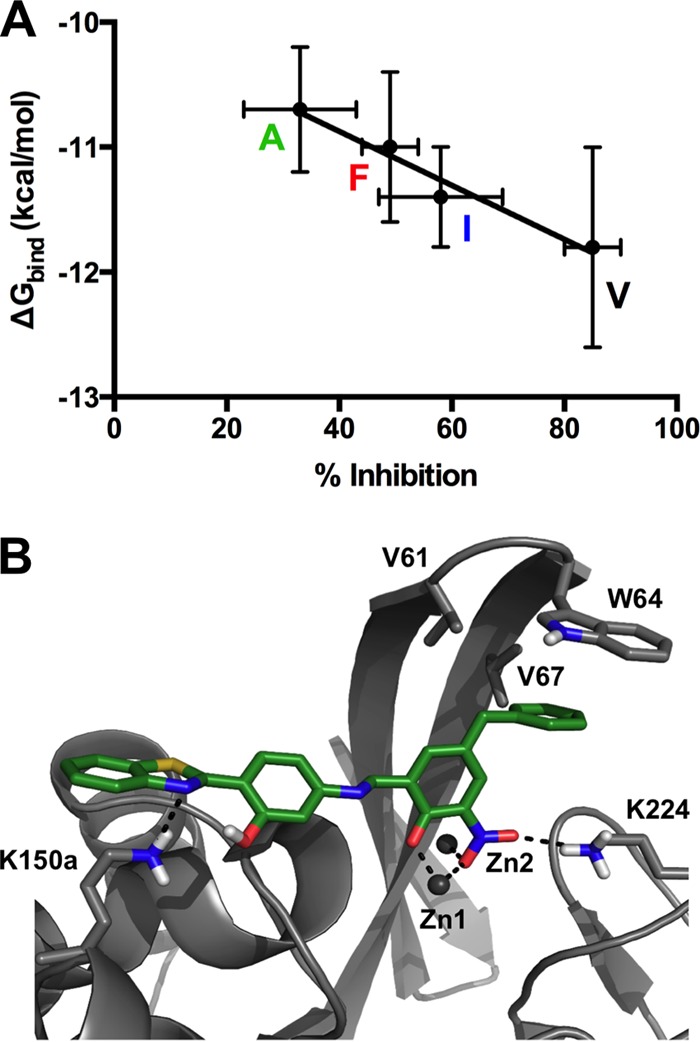
Docking study of molecule 6142342 with different IMP variants. (A) Graphical representation of docking results and experimentally determined percent inhibition data. Colored letters indicate the identities of residue 67. The R2 value of the linear regression is 0.96. (B) Graphical representation of molecule 6142342 docked into the active site of IMP-1 in its lowest-binding-energy conformation. The color code is the same as in Fig. 1, except that protein residue carbons are displayed in gray and carbons of molecule 6142342 are shown in green.
Interpretation of results with respect to MBL evolution.
Investigation of the role of position 67 in IMP-type MBLs is warranted, since it has been shown to be under positive selection pressure (28) and both its mutation frequency (number of variants with substitutions at this position) and variability (number of different amino acid identities at this position) are high (52). At the time of the latter publication (2012), those numbers were 5 and 4, respectively; now they are 12 and 4. Interestingly, the mutation frequency at position 67 relative to IMP-1 has been increasing exponentially, while the variability has remained constant after the first 20 variants (see Fig. S4 in the supplemental material), suggesting that having one of three mutations (V67A, V67I, or V67F) may provide an evolutionary advantage but that other amino acids may not be easily accessible or beneficial. Therefore, it was expected that studying the effects of the four amino acids found naturally at position 67 would provide valuable insights into MBL evolution.
Consistent with residue 67's role in forming a hydrophobic pocket together with V61 and W64 (23, 26, 27) (Fig. 4B), all amino acids found at position 67 are hydrophobic. They increase in size and bulkiness from alanine over valine (IMP-1) and isoleucine to phenylalanine (IMP-10). Phenylalanine is the only residue that could possibly interact with a substrate electrostatically, through the π electrons in its aromatic ring. Thus, substrate preference may be dominated primarily based on fit and, in the case of F67, electrostatic interactions. In the following, we discuss the observed activities toward the different substrate groups and attempt to rationalize them based on the residue 67 side chain structures.
Penicillins.
A consistent observation in the report of Iyobe and coworkers (19) and the present study is that IMP-10 is much less capable of inactivating penicillins than IMP-1, as observed by kcat/Km measurements as well as MICs. This can be extended to IMP-1-V67I, which is almost as efficient as IMP-1, and IMP-1-V67A, which is less efficient, like IMP-10. It appears that penicillins are optimally inactivated when residue 67 is either valine or isoleucine, both of which are branched hydrophobic residues that could pack nicely with the axial methyl groups on the dihydrothiazine ring of penicillins, which has been highlighted in numerous studies (21, 25, 53). Crystal structures of IMP-1 in complex with a substrate, intermediate, or product are not available; however, in a crystal structure of the MBL NDM-1 in complex with hydrolyzed benzylpenicillin (PDB code 4EYF) (54), the axial methyl group pointing toward loop L3 is very close to V73, the residue that corresponds to position 67 in the BBL numbering scheme (closest C C distance of 4.3 Å in chain A and 4.4 Å in chain B). The distance between this residue and the phenyl ring in R1 is >5 Å, arguing for a larger significance of the axial methyl groups in interacting with residue 67. It is conceivable that replacing the branched side chains of valine and isoleucine with just a methyl group in alanine would create an unfavorable void, whereas replacing them with a benzyl moiety in phenylalanine would create unfavorable clashes. If A67 did indeed induce an α helix, as mentioned above, that could also generate unfavorable clashes. The increased inactivation of and resistance against ampicillin conferred by IMP-1 and IMP-1-V67I are in agreement with a random active site residue selection study by Materon and coworkers, which showed that in the presence of ampicillin IMP-1 can only tolerate the V67I mutation, whereas a variety of other amino acids were selected with other antibacterials (cefotaxime, cefaloridine, and imipenem) (27).
C distance of 4.3 Å in chain A and 4.4 Å in chain B). The distance between this residue and the phenyl ring in R1 is >5 Å, arguing for a larger significance of the axial methyl groups in interacting with residue 67. It is conceivable that replacing the branched side chains of valine and isoleucine with just a methyl group in alanine would create an unfavorable void, whereas replacing them with a benzyl moiety in phenylalanine would create unfavorable clashes. If A67 did indeed induce an α helix, as mentioned above, that could also generate unfavorable clashes. The increased inactivation of and resistance against ampicillin conferred by IMP-1 and IMP-1-V67I are in agreement with a random active site residue selection study by Materon and coworkers, which showed that in the presence of ampicillin IMP-1 can only tolerate the V67I mutation, whereas a variety of other amino acids were selected with other antibacterials (cefotaxime, cefaloridine, and imipenem) (27).
Cephalosporins and cephamycin.
Catalytic efficiencies against the neutral cephalosporins cephalothin and cefotaxime were increased for all variants relative to that of IMP-1; however, this was not apparent in the MICs, which, as mentioned above, may be due to the fact that these catalytic efficiencies are so high that other factors, such as diffusion and membrane permeation, may become limiting factors for MICs. At the molecular level, the increased efficiency may be explained by the fact that the neutral but polar acetoxymethyl R2 group of cephalothin and cefotaxime (Fig. 2) can interact favorably with a range of residues. In the work of Materon et al. (27), several amino acids were tolerated when cefotaxime was used for selection, including the hydrophobic isoleucine and phenylalanine residues studied here, but also the neutral and polar amino acids serine, tyrosine, and glutamine. In a recently reported crystal structure of NDM-1 in complex with a cefuroxime intermediate, which also has a neutral but polar R2 group (carbamoyloxymethyl), the closest heavy atom-heavy atom distance between V73 (V67 in BBL numbering) and R2 is 4.2 Å in both chains A and B (55). For the cephamycin cefoxitin, which has an identical R2 group, there is no significant difference in kinetic constants or MICs between the IMP variants studied. This could be due to the additional 7-methoxy group keeping loop L3 at a distance and keeping residue 67 from interacting with the β-lactam. For ceftazidime, which has a positively charged pyridinium methyl R2 group (Fig. 2), IMP-1-V67I and IMP-10 (F67) exhibit 2-fold- and 4-fold-increased kcat, kcat/Km, and MIC values compared to those of IMP-1, suggesting that the R2 group favorably interacts with the I67 and F67 side chains, possibly through π stacking of the aromatic rings in the latter case. In Materon et al.'s study (27), isoleucine and phenylalanine were selected at position 67 with cephaloridine, which has an R2 group identical to that of ceftazidime. Recent studies in which a ceftazidime anionic intermediate was modeled in the IMP-1 active site by molecular dynamic simulations (26, 56) and the observation that the structure of compound 6142342 docked into the IMP-1 active site (Fig. 4B) may provide some insight. In both cases, the aromatic ring of R2 is located in the hydrophobic pocket formed by W64 and residue 67, supporting the scenario proposed above.
Carbapenems.
The V67F mutation (IMP-10) has a significant effect on MICs of carbapenems (8-fold higher for meropenem and doripenem and 4-fold higher for imipenem), which correlates well with the catalytic efficiencies (2- to 4-fold higher for meropenem and doripenem and 1.6-fold higher for imipenem). The V67F mutation in the IMP-type MBLs IMP-43 and IMP-44 has been shown to have impacts on carbapenem susceptibility and activity similar to those of IMP-7 and IMP-11, respectively, which have valine at position 67 (21). Mutation of V67 to alanine or isoleucine also has a positive effect on catalytic efficiencies against meropenem and doripenem but not imipenem. Thus, it seems that A67, I67, and F67 can better accommodate meropenem and doripenem than V67 can, while only F67 can better accommodate imipenem. Similar to the cephalosporins with neutral R2 groups, the carbapenems with neutral R2 groups (meropenem and doripenem) could interact more favorably with the larger isoleucine and phenylalanine residues, while with A67 other factors might be at play, such as a change in the secondary structure of L3. In a crystal structure of NDM-1 with hydrolyzed meropenem (PDB code 4EYL) (54), the pyrrolidine ring in R2 of meropenem (Fig. 2) is closest to V73 (V67 in BBL numbering) (CC distance of 4.2 Å in chain A and 3.7 Å in chain B), supporting this scenario. It would make sense that imipenem, which has a positively charged (iminomethyl)amino group at that position, would interact less favorably with hydrophobic residues, and indeed, catalytic efficiencies and MICs are smaller for imipenem than for the other carbapenems. With phenylalanine, there could be a more favorable interaction between the π electrons of the phenyl ring and the (iminomethyl)amino group, offering a rationale for why IMP-10 is the most efficient enzyme against imipenem. Interestingly, Materon et al. did not select phenylalanine at position 67 with imipenem, but instead found tyrosine, cysteine, serine, and threonine (27), all of which would be capable of engaging in electrostatic interactions with a positively charged imipenem R2 group.
Conclusions.
This study of IMP variants with all naturally observed amino acids at position 67 yielded interesting insights into MBL evolution. First, as evident from the expression levels of functional enzyme, metal content, secondary structure, and thermal stability, none of the substitutions investigated had any detrimental effect on the fitness of these enzymes with regard to these biophysical properties. Second, the activity profiles of the enzymes against a range of β-lactams allow for an interesting hypothesis: that the original enzyme IMP-1 that appeared in the early 1990s was a broad-spectrum enzyme that efficiently inactivated all older β-lactam antibiotics, i.e., penicillins, the first-generation cephalosporin cephalothin, the neutral third-generation cephalosporin cefotaxime, the cephamycin cefoxitin, and, moderately, the early carbapenem imipenem (hence its name imipenemase). Over time, possibly selected by exposure to newer β-lactams, variants with mutations at position 67 appeared, including IMP-9 with I67 in 2000 (57) and IMP-10 with F67, isolated from different strains from 1997 to 2000 (19). Both residues increased resistance to imipenem, the charged third-generation cephalosporin ceftazidime, and, especially for F67, the more recently introduced carbapenems meropenem and doripenem, which were FDA approved in 1996 and 2007, respectively. On the flip side, these enzymes lost their effectiveness against penicillins, which are probably rarely used against the bacteria from which they were isolated. In other words, these enzymes had become “specialists” for the inactivation of newer, last-resort antibacterials, and over time, residues other than valine at position 67 have increasingly accumulated in the pool of IMP variants (see Fig. S4 in the supplemental material). Alanine at position 67 has been explored in only one variant so far, namely, IMP-21, which was published in GenBank in 2004 (GBAC AB204557) but never published in the form of an article. The A67 variant in isolation as studied here (IMP-21 has many other mutations relative to IMP-1) provides little to no advantage over IMP-1 against the newer drugs in terms of MIC, consistent with the fact that this amino acid has not been found in other variants to date.
Interestingly, only four amino acids have been realized in IMP enzymes (see Fig. S4 in the supplemental material), and many of the other amino acids at position 67 that yielded viable β-lactamases in Materon et al.'s study (27) were not found naturally. There are two likely reasons for this: (i) mutations yielding these substitutions may require more than one nucleotide change and therefore be very unlikely; and (ii) they might not provide any significant advantage over the already existing variants, because they do not increase resistance to a particular antibacterial or do so only at the expense of decreased resistance to other important antibacterials.
Understanding the impacts of mutations such as those studied here is an important step toward deciphering the mechanisms of antibacterial resistance and its evolution. Such knowledge may allow for educated adjustments in our current antibacterial use (whether in medicine or animal husbandry [58, 59]), with the goal of alleviating selective pressure, as well as aiding in the design of novel antibacterials and MBL inhibitors.
Supplementary Material
ACKNOWLEDGMENTS
Research reported in this publication was supported in part by funds from the National Institute of Allergy and Infectious Diseases of the National Institutes of Health, under award numbers R01AI063517 and R01AI100560 to R.A.B., and from the National Institute of Environmental Health Sciences of the National Institutes of Health, under award number P30 ES005022 to W.J.W. This study was also supported by funds and/or facilities provided by the Cleveland Department of Veterans Affairs, Veterans Affairs Merit Review Program award 1I01BX001974, and Geriatric Research Education and Clinical Center award VISN 10 to R.A.B.
We thank James Spencer, University of Bristol, for providing the pET26b-blaIMP-1 vector.
The content is solely the responsibility of the authors and does not represent the official views of the National Institutes of Health or the Department of Veterans Affairs.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AAC.01651-15.
REFERENCES
- 1.Ambler RP. 1980. The structure of β-lactamases. Philos Trans R Soc Lond B Biol Sci 289:321–331. doi: 10.1098/rstb.1980.0049. [DOI] [PubMed] [Google Scholar]
- 2.Laraki N, Franceschini N, Rossolini GM, Santucci P, Meunier C, de Pauw E, Amicosante G, Frere JM, Galleni M. 1999. Biochemical characterization of the Pseudomonas aeruginosa 101/1477 metallo-β-lactamase IMP-1 produced by Escherichia coli. Antimicrob Agents Chemother 43:902–906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Oelschlaeger P, Mayo SL, Pleiss J. 2005. Impact of remote mutations on metallo-β-lactamase substrate specificity: implications for the evolution of antibiotic resistance. Protein Sci 14:765–774. doi: 10.1110/ps.041093405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhao WH, Hu ZQ. 2011. IMP-type metallo-β-lactamases in Gram-negative bacilli: distribution, phylogeny, and association with integrons. Crit Rev Microbiol 37:214–226. doi: 10.3109/1040841X.2011.559944. [DOI] [PubMed] [Google Scholar]
- 5.Chouchani C, Marrakchi R, Henriques I, Correia A. 2013. Occurrence of IMP-8, IMP-10, and IMP-13 metallo-β-lactamases located on class 1 integrons and other extended-spectrum β-lactamases in bacterial isolates from Tunisian rivers. Scand J Infect Dis 45:95–103. doi: 10.3109/00365548.2012.717712. [DOI] [PubMed] [Google Scholar]
- 6.Galleni M, Lamotte-Brasseur J, Rossolini GM, Spencer J, Dideberg O, Frere JM, Metallo-β-Lactamases Working Group. 2001. Standard numbering scheme for class B β-lactamases. Antimicrob Agents Chemother 45:660–663. doi: 10.1128/AAC.45.3.660-663.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Garau G, Garcia-Saez I, Bebrone C, Anne C, Mercuri P, Galleni M, Frere JM, Dideberg O. 2004. Update of the standard numbering scheme for class B β-lactamases. Antimicrob Agents Chemother 48:2347–2349. doi: 10.1128/AAC.48.7.2347-2349.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bush K. 2010. Alarming β-lactamase-mediated resistance in multidrug-resistant Enterobacteriaceae. Curr Opin Microbiol 13:558–564. doi: 10.1016/j.mib.2010.09.006. [DOI] [PubMed] [Google Scholar]
- 9.Cornaglia G, Giamarellou H, Rossolini GM. 2011. Metallo-β-lactamases: a last frontier for β-lactams? Lancet Infect Dis 11:381–393. doi: 10.1016/S1473-3099(11)70056-1. [DOI] [PubMed] [Google Scholar]
- 10.Miriagou V, Cornaglia G, Edelstein M, Galani I, Giske CG, Gniadkowski M, Malamou-Lada E, Martinez-Martinez L, Navarro F, Nordmann P, Peixe L, Pournaras S, Rossolini GM, Tsakris A, Vatopoulos A, Canton R. 2010. Acquired carbapenemases in Gram-negative bacterial pathogens: detection and surveillance issues. Clin Microbiol Infect 16:112–122. doi: 10.1111/j.1469-0691.2009.03116.x. [DOI] [PubMed] [Google Scholar]
- 11.Oelschlaeger P, Ai N, Duprez KT, Welsh WJ, Toney JH. 2010. Evolving carbapenemases: can medicinal chemists advance one step ahead of the coming storm? J Med Chem 53:3013–3027. doi: 10.1021/jm9012938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bush K, Fisher JF. 2011. Epidemiological expansion, structural studies, and clinical challenges of new β-lactamases from gram-negative bacteria. Annu Rev Microbiol 65:455–478. doi: 10.1146/annurev-micro-090110-102911. [DOI] [PubMed] [Google Scholar]
- 13.Bush K, Palzkill TG, Jacoby GA. 2015. β-Lactamase classification and amino acid sequences for TEM, SHV and OXA extended-spectrum and inhibitor resistant enzymes. Lahey Clinic, Burlington, MA: http://www.lahey.org/studies/other.asp-table1 Accessed 6 July 2015. [Google Scholar]
- 14.Osano E, Arakawa Y, Wacharotayankun R, Ohta M, Horii T, Ito H, Yoshimura F, Kato N. 1994. Molecular characterization of an enterobacterial metallo-β-lactamase found in a clinical isolate of Serratia marcescens that shows imipenem resistance. Antimicrob Agents Chemother 38:71–78. doi: 10.1128/AAC.38.1.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Watanabe M, Iyobe S, Inoue M, Mitsuhashi S. 1991. Transferable imipenem resistance in Pseudomonas aeruginosa. Antimicrob Agents Chemother 35:147–151. doi: 10.1128/AAC.35.1.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Koh TH, Babini GS, Woodford N, Sng LH, Hall LM, Livermore DM. 1999. Carbapenem-hydrolysing IMP-1 β-lactamase in Klebsiella pneumoniae from Singapore. Lancet 353:2162. doi: 10.1016/S0140-6736(05)75604-X. [DOI] [PubMed] [Google Scholar]
- 17.Jones RN, Deshpande LM, Bell JM, Turnidge JD, Kohno S, Hirakata Y, Ono Y, Miyazawa Y, Kawakama S, Inoue M, Hirata Y, Toleman MA. 2004. Evaluation of the contemporary occurrence rates of metallo-β-lactamases in multidrug-resistant Gram-negative bacilli in Japan: report from the SENTRY Antimicrobial Surveillance Program (1998–2002). Diagn Microbiol Infect Dis 49:289–294. doi: 10.1016/j.diagmicrobio.2004.04.007. [DOI] [PubMed] [Google Scholar]
- 18.Shibata N, Doi Y, Yamane K, Yagi T, Kurokawa H, Shibayama K, Kato H, Kai K, Arakawa Y. 2003. PCR typing of genetic determinants for metallo-β-lactamases and integrases carried by gram-negative bacteria isolated in Japan, with focus on the class 3 integron. J Clin Microbiol 41:5407–5413. doi: 10.1128/JCM.41.12.5407-5413.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Iyobe S, Kusadokoro H, Takahashi A, Yomoda S, Okubo T, Nakamura A, O'Hara K. 2002. Detection of a variant metallo-β-lactamase, IMP-10, from two unrelated strains of Pseudomonas aeruginosa and an Alcaligenes xylosoxidans strain. Antimicrob Agents Chemother 46:2014–2016. doi: 10.1128/AAC.46.6.2014-2016.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Koh TH, Khoo CT, Tan TT, Arshad MA, Ang LP, Lau LJ, Hsu LY, Ooi EE. 2010. Multilocus sequence types of carbapenem-resistant Pseudomonas aeruginosa in Singapore carrying metallo-β-lactamase genes, including the novel bla(IMP-26) gene. J Clin Microbiol 48:2563–2564. doi: 10.1128/JCM.01905-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tada T, Miyoshi-Akiyama T, Shimada K, Shimojima M, Kirikae T. 2013. IMP-43 and IMP-44 metallo-β-lactamases with increased carbapenemase activities in multidrug-resistant Pseudomonas aeruginosa. Antimicrob Agents Chemother 57:4427–4432. doi: 10.1128/AAC.00716-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Meini MR, Llarrull LI, Vila AJ. 2014. Evolution of metallo-β-lactamases: trends revealed by natural diversity and evolution. Antibiotics (Basel) 3:285–316. doi: 10.3390/antibiotics3030285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Concha NO, Janson CA, Rowling P, Pearson S, Cheever CA, Clarke BP, Lewis C, Galleni M, Frere JM, Payne DJ, Bateson JH, Abdel-Meguid SS. 2000. Crystal structure of the IMP-1 metallo β-lactamase from Pseudomonas aeruginosa and its complex with a mercaptocarboxylate inhibitor: binding determinants of a potent, broad-spectrum inhibitor. Biochemistry 39:4288–4298. doi: 10.1021/bi992569m. [DOI] [PubMed] [Google Scholar]
- 24.Scrofani SD, Chung J, Huntley JJ, Benkovic SJ, Wright PE, Dyson HJ. 1999. NMR characterization of the metallo-β-lactamase from Bacteroides fragilis and its interaction with a tight-binding inhibitor: role of an active-site loop. Biochemistry 38:14507–14514. doi: 10.1021/bi990986t. [DOI] [PubMed] [Google Scholar]
- 25.Moali C, Anne C, Lamotte-Brasseur J, Groslambert S, Devreese B, Van Beeumen J, Galleni M, Frere JM. 2003. Analysis of the importance of the metallo-β-lactamase active site loop in substrate binding and catalysis. Chem Biol 10:319–329. doi: 10.1016/S1074-5521(03)00070-X. [DOI] [PubMed] [Google Scholar]
- 26.Pegg KM, Liu EM, Lacuran AE, Oelschlaeger P. 2013. Biochemical characterization of IMP-30, a metallo-β-lactamase with enhanced activity toward ceftazidime. Antimicrob Agents Chemother 57:5122–5126. doi: 10.1128/AAC.02341-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Materon IC, Beharry Z, Huang W, Perez C, Palzkill T. 2004. Analysis of the context dependent sequence requirements of active site residues in the metallo-β-lactamase IMP-1. J Mol Biol 344:653–663. doi: 10.1016/j.jmb.2004.09.074. [DOI] [PubMed] [Google Scholar]
- 28.Pal A, Tripathi A. 2013. An in silico approach for understanding the molecular evolution of clinically important metallo-β-lactamases. Infect Genet Evol 20:39–47. doi: 10.1016/j.meegid.2013.07.028. [DOI] [PubMed] [Google Scholar]
- 29.Yamaguchi Y, Jin W, Matsunaga K, Ikemizu S, Yamagata Y, Wachino J, Shibata N, Arakawa Y, Kurosaki H. 2007. Crystallographic investigation of the inhibition mode of a VIM-2 metallo-β-lactamase from Pseudomonas aeruginosa by a mercaptocarboxylate inhibitor. J Med Chem 50:6647–6653. doi: 10.1021/jm701031n. [DOI] [PubMed] [Google Scholar]
- 30.Xiong J, Hynes MF, Ye H, Chen H, Yang Y, M'Zali F, Hawkey PM. 2006. bla(IMP-9) and its association with large plasmids carried by Pseudomonas aeruginosa isolates from the People's Republic of China. Antimicrob Agents Chemother 50:355–358. doi: 10.1128/AAC.50.1.355-358.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pournaras S, Kock R, Mossialos D, Mellmann A, Sakellaris V, Stathopoulos C, Friedrich AW, Tsakris A. 2013. Detection of a phylogenetically distinct IMP-type metallo-β-lactamase, IMP-35, in a CC235 Pseudomonas aeruginosa from the Dutch-German border region (Euregio). J Antimicrob Chemother 68:1271–1276. doi: 10.1093/jac/dkt004. [DOI] [PubMed] [Google Scholar]
- 32.Wang Y, Wang X, Schwarz S, Zhang R, Lei L, Liu X, Lin D, Shen J. 2014. IMP-45-producing multidrug-resistant Pseudomonas aeruginosa of canine origin. J Antimicrob Chemother 69:2579–2581. doi: 10.1093/jac/dku133. [DOI] [PubMed] [Google Scholar]
- 33.Liu EM, Pegg KM, Oelschlaeger P. 2012. The sequence-activity relationship between metallo-β-lactamases IMP-1, IMP-6, and IMP-25 suggests an evolutionary adaptation to meropenem exposure. Antimicrob Agents Chemother 56:6403–6406. doi: 10.1128/AAC.01440-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hunt JB, Neece SH, Ginsburg A. 1985. The use of 4-(2-pyridylazo)resorcinol in studies of zinc release from Escherichia coli aspartate transcarbamoylase. Anal Biochem 146:150–157. doi: 10.1016/0003-2697(85)90409-9. [DOI] [PubMed] [Google Scholar]
- 35.Fast W, Wang Z, Benkovic SJ. 2001. Familial mutations and zinc stoichiometry determine the rate-limiting step of nitrocefin hydrolysis by metallo-β-lactamase from Bacteroides fragilis. Biochemistry 40:1640–1650. doi: 10.1021/bi001860v. [DOI] [PubMed] [Google Scholar]
- 36.Tremblay LW, Fan F, Blanchard JS. 2010. Biochemical and structural characterization of Mycobacterium tuberculosis β-lactamase with the carbapenems ertapenem and doripenem. Biochemistry 49:3766–3773. doi: 10.1021/bi100232q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Clinical and Laboratory Standards Institute. 2012. Methods for dilution antimicrobial susceptibility tests for bacteria that grow aerobically; approved standard, ninth edition. M07-A9. Clinical and Laboratory Standards Institute, Wayne, PA. [Google Scholar]
- 38.Pegg KM, Liu EM, George AC, LaCuran AE, Bethel CR, Bonomo RA, Oelschlaeger P. 2014. Understanding the determinants of substrate specificity in IMP family metallo-β-lactamases: the importance of residue 262. Protein Sci 23:1451–1460. doi: 10.1002/pro.2530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zauhar RJ, Moyna G, Tian L, Li Z, Welsh WJ. 2003. Shape signatures: a new approach to computer-aided ligand- and receptor-based drug design. J Med Chem 46:5674–5690. doi: 10.1021/jm030242k. [DOI] [PubMed] [Google Scholar]
- 40.Dewar MJS, Zoebisch EG, Healy EF, Stewart JJP. 1985. The development and use of quantum-mechanical molecular models. 76. AM1: a new general-purpose quantum-mechanical molecular model. J Am Chem Soc 107:3902–3909. doi: 10.1021/Ja00299a024. [DOI] [Google Scholar]
- 41.Morris GM, Goodsell DS, Halliday RS, Huey R, Hart WE, Belew RK, Olson AJ. 1998. Automated docking using a Lamarckian genetic algorithm and an empirical binding free energy function. J Comput Chem 19:1639–1662. doi:. [DOI] [Google Scholar]
- 42.Irwin JJ, Raushel FM, Shoichet BK. 2005. Virtual screening against metalloenzymes for inhibitors and substrates. Biochemistry 44:12316–12328. doi: 10.1021/bi050801k. [DOI] [PubMed] [Google Scholar]
- 43.Stote RH, Karplus M. 1995. Zinc binding in proteins and solution: a simple but accurate nonbonded representation. Proteins 23:12–31. doi: 10.1002/prot.340230104. [DOI] [PubMed] [Google Scholar]
- 44.Carfi A, Pares S, Duee E, Galleni M, Duez C, Frere JM, Dideberg O. 1995. The 3-D structure of a zinc metallo-β-lactamase from Bacillus cereus reveals a new type of protein fold. EMBO J 14:4914–4921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Oelschlaeger P, Mayo SL. 2005. Hydroxyl groups in the ββ sandwich of metallo-β-lactamases favor enzyme activity: a computational protein design study. J Mol Biol 350:395–401. doi: 10.1016/j.jmb.2005.04.044. [DOI] [PubMed] [Google Scholar]
- 46.Whitmore L, Wallace BA. 2004. DICHROWEB, an online server for protein secondary structure analyses from circular dichroism spectroscopic data. Nucleic Acids Res 32:W668–W673. doi: 10.1093/nar/gkh371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Whitmore L, Wallace BA. 2008. Protein secondary structure analyses from circular dichroism spectroscopy: methods and reference databases. Biopolymers 89:392–400. doi: 10.1002/bip.20853. [DOI] [PubMed] [Google Scholar]
- 48.Compton LA, Johnson WC Jr. 1986. Analysis of protein circular dichroism spectra for secondary structure using a simple matrix multiplication. Anal Biochem 155:155–167. doi: 10.1016/0003-2697(86)90241-1. [DOI] [PubMed] [Google Scholar]
- 49.Sreerama N, Woody RW. 2000. Estimation of protein secondary structure from circular dichroism spectra: comparison of CONTIN, SELCON, and CDSSTR methods with an expanded reference set. Anal Biochem 287:252–260. doi: 10.1006/abio.2000.4880. [DOI] [PubMed] [Google Scholar]
- 50.Bryson JW, Betz SF, Lu HS, Suich DJ, Zhou HX, O'Neil KT, DeGrado WF. 1995. Protein design: a hierarchic approach. Science 270:935–941. doi: 10.1126/science.270.5238.935. [DOI] [PubMed] [Google Scholar]
- 51.Meiler J, Mueller M, Zeidler A, Schmaeschke F. 2001. Generation and evaluation of dimension-reduced amino acid parameter representations by artificial neural networks. J Mol Model 7:360–369. doi: 10.1007/s008940100038. [DOI] [Google Scholar]
- 52.Widmann M, Pleiss J, Oelschlaeger P. 2012. Systematic analysis of metallo-β-lactamases using an automated database. Antimicrob Agents Chemother 56:3481–3491. doi: 10.1128/AAC.00255-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bebrone C, Anne C, Kerff F, Garau G, De Vriendt K, Lantin R, Devreese B, Van Beeumen J, Dideberg O, Frere JM, Galleni M. 2008. Mutational analysis of the zinc- and substrate-binding sites in the CphA metallo-β-lactamase from Aeromonas hydrophila. Biochem J 414:151–159. doi: 10.1042/BJ20080375. [DOI] [PubMed] [Google Scholar]
- 54.King DT, Worrall LJ, Gruninger R, Strynadka NC. 2012. New Delhi metallo-β-lactamase: structural insights into β-lactam recognition and inhibition. J Am Chem Soc 134:11362–11365. doi: 10.1021/ja303579d. [DOI] [PubMed] [Google Scholar]
- 55.Feng H, Ding J, Zhu D, Liu X, Xu X, Zhang Y, Zang S, Wang DC, Liu W. 2014. Structural and mechanistic insights into NDM-1 catalyzed hydrolysis of cephalosporins. J Am Chem Soc 136:14694–14697. doi: 10.1021/ja508388e. [DOI] [PubMed] [Google Scholar]
- 56.Oelschlaeger P, Schmid RD, Pleiss J. 2003. Modeling domino effects in enzymes: molecular basis of the substrate specificity of the bacterial metallo-β-lactamases IMP-1 and IMP-6. Biochemistry 42:8945–8956. doi: 10.1021/bi0300332. [DOI] [PubMed] [Google Scholar]
- 57.Xiong J, Alexander DC, Ma JH, Deraspe M, Low DE, Jamieson FB, Roy PH. 2013. Complete sequence of pOZ176, a 500-kilobase IncP-2 plasmid encoding IMP-9-mediated carbapenem resistance, from outbreak isolate Pseudomonas aeruginosa 96. Antimicrob Agents Chemother 57:3775–3782. doi: 10.1128/AAC.00423-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Oelschlaeger P, Toney JH. 13 October 2009. Are human microflora influenced by environmental exposure to antibiotics? Science http://www.sciencemag.org/content/325/5944/1128/reply Accessed 8 July 2015.
- 59.Kuemmerer K. 2008. Substance flow associated with medical care—significance of different sources, p 43–59. In Pharmaceuticals in the environment: sources, fate, effects and risks. Springer, Berlin, Germany. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.