

Abstract
Clostridium difficile is a leading cause of antibiotic-associated diarrhea and the etiologic agent responsible for C. difficile infection. Toxin A (TcdA) and toxin B (TcdB) are nearly indispensable virulence factors for Clostridium difficile pathogenesis. Given the toxin-centric mechanism by which C. difficile pathogenesis occurs, the selective sequestration with neutralization of TcdA and TcdB by nonantibiotic agents represents a novel mode of action to prevent or treat C. difficile-associated disease. In this preclinical study, we used quantitative enzyme immunoassays to determine the extent by which a novel drug, calcium aluminosilicate uniform particle size nonswelling M-1 (CAS UPSN M-1), is capable of sequestering TcdA and TcdB in vitro. The following major findings were derived from the present study. First, we show that CAS UPSN M-1 efficiently sequestered both TcdA and TcdB to undetectable levels. Second, we show that CAS UPSN M-1's affinity for TcdA is greater than its affinity for TcdB. Last, we show that CAS UPSN M-1 exhibited limited binding affinity for nontarget proteins. Taken together, these results suggest that ingestion of calcium aluminosilicate might protect gastrointestinal tissues from antibiotic- or chemotherapy-induced C. difficile infection by neutralizing the cytotoxic and proinflammatory effects of luminal TcdA and TcdB.
INTRODUCTION
Clostridium difficile is a leading cause of antibiotic-associated diarrhea (AAD) and is the etiologic agent responsible for C. difficile-associated infection (CDI). CDI typically starts as a mild diarrhea but rapidly degenerates into a variety of potentially life-threatening conditions, including sepsis syndrome and pseudomembranous colitis (1). In the United States, approximately 330,000 cases of CDI are estimated to occur each year (2); however, the incidence of C. difficile infection continues to increase (2, 3). The increasing CDI rates highlight the fact that the current infection control procedures and treatment options are insufficient.
In the health care setting, C. difficile endospores are transmitted to patients via the fecal-oral route (4). Following exposure, the host's gastrointestinal microbiota typically either quells a nascent C. difficile infection or suppresses it to subclinical levels (5). As a result of the latter, approximately 20% of hospitalized adults become asymptomatic C. difficile carriers, and the carriage rates approach 50% for patients in long-term care (6–9). The likelihood of development of CDI increases in patients with dysbiotic gastrointestinal microbiota, since C. difficile can thrive in the dysbiotic niche (5, 10). This dysbiosis is often the result of nonspecific chemotherapies that are used to treat conditions unrelated to C. difficile infection (e.g., antibacterial agents or antineoplastic drugs).
The antibiotics metronidazole and vancomycin are currently used to treat CDI (11). Unfortunately, given the conflicting roles of antibiotics in the establishment and resolution of CDI, C. difficile AAD recurs in up to 1 in 5 patients (12). These already high reoccurrence rates are expected to increase if C. difficile strains with intermediate and complete resistance to metronidazole and vancomycin emerge (13). Taken together, these alarming trends illustrate an urgent need for the development of novel and efficacious therapies to treat CDI, including nontraditional therapeutic agents.
C. difficile is an extracellular pathogen, and it typically does not invade host tissues. While a number of C. difficile-encoded virulence factors are responsible for C. difficile carriage and pathogenesis, toxin A (TcdA) and toxin B (TcdB) are among the best studied (14). Once secreted into the colon, these cytotoxic protein-based enzymes are translocated across the membrane bilayer and into the cytosol by receptor-mediated endocytosis (15). Once inside the cell, these glycosyltransferases trigger altered cellular transcription, which results in significant cellular apoptosis and tissue remodeling (16–18). TcdA and TcdB are also strongly proinflammatory, which exacerbates their effects on structural and functional changes in tissue integrity (19, 20). Together, these inflammation-related activities contribute to the progressive ablation of gastrointestinal function that is characteristic of CDI. In animal models, the administration of purified C. difficile TcdA induces the hallmark symptoms of an acute, pseudomembranous colitis-like condition: edema, gastrointestinal inflammation, cellular necrosis, and gastroenteritis in the absence of the bacterium (19, 21–23). The administration of TcdB elicits similar effects, albeit to a lesser degree (22, 24). As a result, these protein-based enzymes have been ascribed as nearly indispensable determinants for C. difficile pathogenesis.
Given the toxin-centric mechanism by which C. difficile pathogenesis occurs, the selective sequestration with neutralization of TcdA and TcdB by nonantibiotic agents represents a novel mode of action to prevent or treat C. difficile-associated diseases (25, 26). To date, four C. difficile toxin-binding agents (i.e., cholestyramine, colestipol, Synsorb 90, and tolevamer) have been examined in preclinical studies (25). Of these toxin-binding agents, only three have been tested in clinical studies. Unfortunately, none of these agents has proven to be as efficacious as traditional antibiotic therapies. Nevertheless, it is important to continue to develop new candidate therapies. In this article, we describe the characterization of calcium aluminosilicate uniform particle size nonswelling M-1 (CAS UPSN M-1), a novel calcium aluminosilicate agent that has been developed to selectively bind to and neutralize large clostridial protein toxins. Calcium aluminosilicate is recognized by the Food and Drug Administration (FDA) as a generally regarded as safe (GRAS) additive, which can be used as a supplement to foods at levels up to 2% (wt/wt) (27).
MATERIALS AND METHODS
Protein-based cytotoxic enzymes and reagents.
Lyophilized C. difficile TcdA and C. difficile TcdB were stored according to the manufacturer's specifications (Calbiochem, Gibbstown, NJ). TcdA and TcdB were resuspended in 10 mM 2,2-bis(hydroxymethyl)-2,2′,2-nitrilotriethanol (bis-Tris) buffer (Sigma-Aldrich, St. Louis, MO) and maintained on ice prior to being assayed. All other chemicals were molecular biology grade and stored as recommended by the manufacturer. A SevenMulti conductivity meter (Mettler Toledo, Columbus, OH) was used for pH measurements.
Putative toxin-binding agent.
Calcium aluminosilicate uniform particle size nonswelling M-1 (CAS UPSN M-1), the novel sequestering agent used in this study, was provided by Salient Pharmaceuticals Incorporated (Houston, TX).
Quantitative enzyme immunoassay for toxin quantification.
The concentration of C. difficile TcdA or TcdB was measured using the Premier Toxins A&B enzyme immunoassay (EIA) according to the manufacturer's instructions (Meridian Bioscience, Inc., Cincinnati, OH), except that a series of assay positive-control samples (i.e., TcdA and TcdB reference standards at a range of known concentrations) was incorporated into each repeated measurement; the concentrations of these reference standards typically ranged from 5 to 20 ng/ml. In brief, the resultant quantitative enzyme immunoassay (qEIA) uses C. difficile TcdA- and TcdB-specific polyclonal antibodies to capture TcdA and TcdB and to noncovalently anchor them to the solid-phase EIA support matrix. The matrix-bound toxins were subsequently complexed with horseradish peroxidase (HRP)-conjugated mouse anti-toxin A (monoclonal) or goat anti-toxin B (polyclonal) antibodies, respectively. After the removal of the unbound HRP-antibody conjugates, the degradation of urea hydrogen peroxide by toxin-bound horseradish peroxidase was assayed in the presence of the reducing cosubstrate 3,3′,5,5′-tetramethyl-(1,1′-biphenyl)-4,4′-diamine (TMB). Phosphoric acid (1 M) was used to arrest the reaction. The terminal chromophore benzidine-4,4′-diimine (BZDI), an oxidized derivative of TMB, was measured in arbitrary units (AU) at an optical density of 450 nm (OD450) using an Infinite M200 microplate reader (Tecan US, Inc., Durham, NC). Infinite M200 i-Control software was used to generate custom EIA microplate templates to speed data acquisition and to ensure accurate sample assignation.
Toxin sequestration assays.
This qEIA was used to assess the sequestration (an aggregate of adsorption and absorption) of large clostridial protein toxins by calcium aluminosilicate. Unless otherwise indicated, calcium aluminosilicate was suspended up to a final concentration of 0.5 mg/ml in 10 mM bis-Tris (pH 6.5) and preequilibrated to 37°C in a Thermomixer R shaking incubator (Eppendorf, Hauppauge, NY) equipped with a 1.5-ml block with constant agitation at 1,200 rpm. The individual sequestration reactions were started by the addition of the toxin to a final concentration of 10 ng/ml (TcdA) or 15 ng/ml (TcdB), unless otherwise indicated. Sequestration reaction mixtures were then incubated for 10 min at 37°C with constant agitation at 1,200 rpm in a Thermomixer R. The sequestration reaction was stopped when the residual calcium aluminosilicate and calcium aluminosilicate-bound toxin complexes were pelleted by centrifugation (2 min at 21,130 × g) in a 5424 benchtop centrifuge (Eppendorf). Following centrifugation, the clarified supernatant was carefully removed by aspiration, transferred to a 1.5-ml tube, and chilled on ice prior to toxin quantification using the qEIA described above.
Heterologous, nontarget protein binding and SDS-PAGE.
Calcium aluminosilicate (5 mg/ml) was coincubated with the SeeBlue Plus2 protein ladder (750 μg/ml; Invitrogen, San Diego, CA), which contains a number of nontarget proteins. Samples were incubated at 37°C for 20 min with constant agitation (1,200 rpm). The reaction was stopped when the residual calcium aluminosilicate and calcium aluminosilicate-bound protein complexes were pelleted by centrifugation (2 min at 21,130 × g) in an Eppendorf 5424 benchtop centrifuge. The supernatant was carefully transferred to a fresh tube and set on ice. The pellet containing the residual calcium aluminosilicate and calcium aluminosilicate-bound protein complexes was resuspended in 1 volume of buffer and agitated for 20 min (1,200 rpm). The unbound calcium aluminosilicate and calcium aluminosilicate-bound protein complexes were again pelleted by centrifugation (2 min at 21,130 × g). The resultant pellet eluate, including any proteins eluted from the calcium aluminosilicate, was carefully transferred to a fresh tube. Negative-control samples (i.e., SeeBlue Plus2 devoid of calcium aluminosilicate) were otherwise treated identically to the experimental samples.
The original supernatant and the pellet eluate were subjected to PAGE. Samples (15 μl) were loaded into the 1-mm wells of a NuPAGE Novex Tris-acetate gel (Invitrogen) using 1× lithium dodecyl sulfate (LDS) sample buffer (Invitrogen). The protein electrophoresis was carried out (150 V for 1 h) in an Xcell SureLock mini cell (Invitrogen) and 1× NuPAGE Tris-acetate SDS running buffer (Invitrogen). Proteins were stained using the SimplyBlue SafeStain (Invitrogen), according to the manufacturer's instruction. The results were captured using the FluorChem HD2 documentation system with a 5-MHz cooled digital charge-coupled-device camera (Alpha Innotech).
Biostatistics.
Raw qEIA data were captured using Infinite M200 i-Control software, exported to Excel (Microsoft Corporation, Redmond, WA), and analyzed using Prism (GraphPad Software, Inc., La Jolla, CA). Unless otherwise indicated, the data represent at least three repeated measures. The data are expressed as the mean (x̄) ± either the standard error of the mean (SEM) or the standard deviation (SD), as noted. Calibration curves were generated by least-squares regression. Analysis of variance (ANOVA) was used to determine the statistical significance of the measured differences between treatments. When significant differences were detected by ANOVA, Bonferroni tests were performed post hoc in order to explore these differences. An associated P value of <0.05 was considered statistically significant. The base 10 logarithm (log10) of each data point was calculated, and a calibration curve for the interpolation of unknowns was generated using least-squares linear regression.
RESULTS
Optimization of a qEIA to detect TcdA and TcdB.
Samples containing known concentrations of TcdA or TcdB were used to generate standard curves. The standard curves from each of six randomly selected vials of TcdB are plotted in Fig. 1. A one-factor ANOVA found that toxin vial-specific effects were statistically significant (P < 0.0051); however, post hoc comparisons using the Bonferroni test indicated that, with the exception of one obvious outlier (P < 0.05), the differences between the remaining five curves were statistically nonsignificant (P > 0.05). For this experimental subset (n = 5), the Pearson product-moment correlation coefficient (r) indicated a strong, colinear relationship between the toxin concentration and optical density that was statistically significant between interexperimental EIA replicates (r = 0.9078, P = 0.0047). Similar results were seen for TcdA (n = 5), although the differences between the individual vials of TcdA were statistically nonsignificant (P = 0.8707). As seen with TcdB, a strong colinear relationship between the toxin concentration and optical density was observed. This colinear relationship was statistically significant between interexperimental qEIA replicates (r = 0.9973, P = 0.0027). In separate experiments that examined the effect of toxin thermostability, the differences in the qEIA reactivity following short-term (e.g., 8 h) incubation on ice were found to be statistically nonsignificant (P > 0.05). As a result, individual toxin vials were used to conduct multiple sequestration assays within a single workday and were then discarded.
FIG 1.
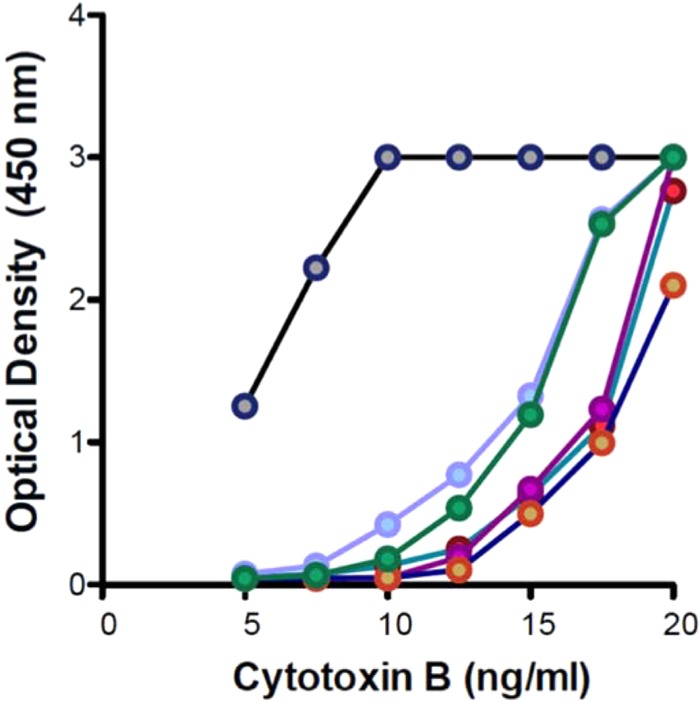
Untransformed EIA calibration curves. The mean optical density (y axis) for each of six randomly selected cytotoxin B EIA calibration curves is plotted as a function of the cytotoxin B concentration (x axis). The data are expressed as means ± SEM.
Data distribution and transformation.
The descriptive statistic skewness (g1) and the Kolmogorov-Smirnov (K-S) normality test were used to examine the data distribution within the TcdA and TcdB data sets. While both data sets skewed right (TcdA, g1 = 0.6626; TcdB, g1 = 1.331), the K-S test indicated that neither exhibited significant differences from the Gaussian distribution (P > 0.10). Nevertheless, the log10 of each data point was calculated to normalize the data. The linear regressions of the mean TcdA (n = 4; R2 = 0.968) and TcdB (n = 5; R2 = 0.966) reference curves are plotted in Fig. 2. Signal response plateaus were observed at very high toxin concentrations and very low toxin concentrations (data not shown).
FIG 2.
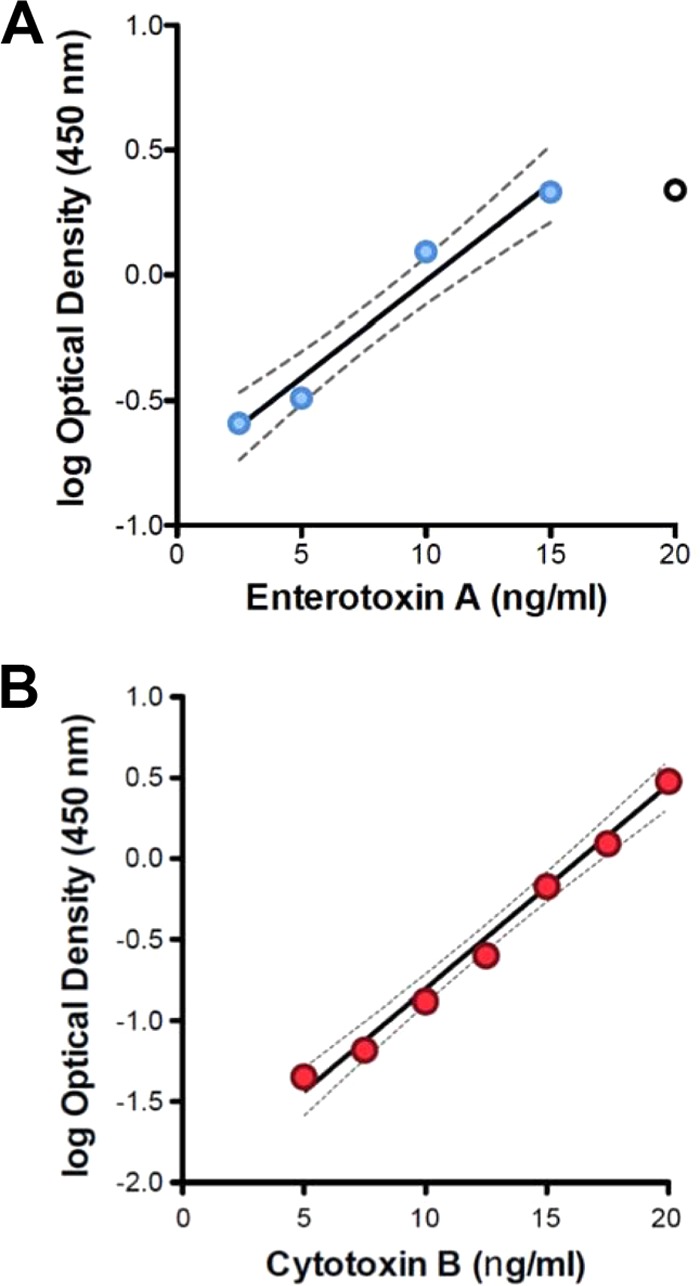
Linear regression of representative logarithm-transformed qEIA calibration data. The logarithm (base 10) of the optical density of each reference standard (y axis) was determined by EIA and plotted against the toxin concentration (x axis). Circles denote the median calibration data from n repeated replicates. The solid line denotes the linear regression with 95% confidence limits, and the dashed lines denote the boundaries of the calculated 95% confidence interval. (A) Linear regression of enterotoxin A data (n = 5, R2 = 0.968). (B) Linear regression of cytotoxin B calibration data (n = 4, R2 = 0.966).
Calcium aluminosilicate does not affect the pH of the qEIA system.
In order to determine if calcium aluminosilicate might artificially diminish the sensitivity of the qEIA by affecting the pH of the buffer system, calcium aluminosilicate was supplemented to a final working concentration of 0.5 mg/ml in 100 mM Tris (pH 6.5), and the pH was measured (n = 3) once the mixture was equilibrated to 37°C. The incorporation of calcium aluminosilicate up to 0.5 mg/ml did not affect the pH of the buffer system (x̄, 6.52; SD, 0.01) compared to that of a buffer control that was devoid of calcium aluminosilicate (x̄, 6.50; SD, 0.001), as expected.
Effective, dose-dependent sequestration of large C. difficile cytotoxic enzymes by calcium aluminosilicate.
Calcium aluminosilicate was assessed for its ability to reduce the concentration of large clostridial protein-based cytotoxic enzymes in vitro. During these dose-response experiments, the concentration of TcdA or TcdB (Fig. 3) was fixed at 10 ng/ml, while the calcium aluminosilicate concentration was varied 100-fold (i.e., 0.05 mg/ml, 0.075 mg/ml, 0.1 mg/ml, 0.3 mg/ml, 0.5 mg/ml, 0.75 mg/ml, 1 mg/ml, 2 mg/ml, 3 mg/ml, and 5 mg/ml). Calcium aluminosilicate efficiently sequestered TcdA and TcdB in vitro. The differences in the mean endpoint OD450 values between the assay negative-control (i.e., vehicle devoid of TcdA) samples (x̄, 0.044 AU; SD, 0.001 AU) and the experimental samples supplemented with calcium aluminosilicate up to 2 mg/ml (x̄, 0.049 AU; SD, 0.004 AU), 3 mg/ml (x̄, 0.049 AU; SD, 0.005 AU), and 5 mg/ml (x̄, 0.048 AU; SD, 0.004 AU) were statistically nonsignificant (P > 0.05).
FIG 3.

Selective and dose-dependent adsorption of C. difficile toxins by CAS UPSN M-1 in vitro. CAS UPSN M-1 was supplemented to a final concentration between 0.05 and 5 mg/ml (100-fold range) (x axis), while the toxin concentration was fixed to 10 μg/ml (dashed line). After a 10-min coincubation, the mineral-toxin complexes were removed by centrifugation, and the residual concentrations of enterotoxin A (blue) and cytotoxin B (red) were determined by quantitative EIA (y axis). The data are expressed as the means ± SD. The statistical significance of the differences in the toxin-specific binding ability is reported for each concentration. ns, statistically nonsignificant (P > 0.05); **, statistically significant (P < 0.01); ***, statistically significant (P < 0.001).
The raw qEIA measurements obtained from the experimental samples were converted to residual toxin concentrations using the intraexperimental EIA calibration curves. The differences in the residual TcdA concentrations between the assay positive-control (i.e., TcdA-containing samples devoid of calcium aluminosilicate) samples and the experimental samples containing calcium aluminosilicate supplemented up to 0.05 mg/ml (x̄, 11.11 ng/ml; SD, 1.82 ng/ml), 0.075 mg/ml (x̄, 11.35 ng/ml; SD, 0.22 ng/ml), and 0.1 mg/ml (x̄, 10.93 ng/ml; SD, 0.06 ng/ml) were statistically nonsignificant (Fig. 3). In contrast, statistically significant differences in the residual TcdA concentrations were measured between the assay positive-control and experimental samples containing calcium aluminosilicate supplemented to 0.3 mg/ml (x̄, 8.54 ng/ml; SD, 1.19 ng/ml; P < 0.05), 0.5 mg/ml (x̄, 4.88 ng/ml; SD, 0.80 ng/ml; P < 0.001), 0.75 mg/ml (x̄, 3.20 ng/ml; SD, 0.65 ng/ml; P < 0.001), 1 mg/ml (x̄, 1.67 ng/ml; SD, 0.71 ng/ml), 2 mg/ml (x̄, below the lower limit of detection [bLLD]; P < 0.001), 3 mg/ml (x̄, bLLD; P < 0.001), and 5 mg/ml (x̄, bLLD; P < 0.001). The lower limits of detection for this qEIA are approximately 1.4 ng/ml for TcdA and 2.4 ng/ml for TcdB.
The efficiency by which the calcium aluminosilicate sequestered TcdB was also explored (Fig. 3). The differences in endpoint OD450 measurements between the assay negative-control (i.e., vehicle devoid of TcdB) samples (x̄, 0.047 AU; SD, 0.004 AU) and the experimental samples supplemented with calcium aluminosilicate up to 3 mg/ml (x̄, 0.043 AU; SD, 0.001 AU) and 5 mg/ml (x̄, 0.041 AU; SD, 0.002 AU) were statistically nonsignificant (P > 0.05). As performed with TcdA, the raw qEIA measurements were converted to residual toxin concentrations. The differences in the residual TcdB concentrations between the assay positive-control (i.e., TcdB-containing samples devoid of calcium aluminosilicate) samples and the experimental samples containing calcium aluminosilicate supplemented up to 0.05 mg/ml (x̄, 9.07 ng/ml; SD, 0.06 ng/ml), 0.075 mg/ml (x̄, 8.75 ng/ml; SD, 0.05 ng/ml), 0.1 mg/ml (x̄, 9.02 ng/ml; SD, 1.38 ng/ml), and 0.3 mg/ml (x̄, 8.44 ng/ml; SD, 0.8 ng/ml) were statistically nonsignificant (P > 0.05). In contrast, statistically significant differences in residual TcdB concentrations were measured between the assay positive-control and the experimental samples containing calcium aluminosilicate supplemented up to 0.5 mg/ml (x̄, 6.27 ng/ml; SD, 0.36 ng/ml; P < 0.001), 0.75 mg/ml (x̄, 5.47 ng/ml; SD, 0.64 ng/ml; P < 0.001), 1 mg/ml (x̄, 4.99 ng/ml; SD, 1.23 ng/ml; P < 0.001), 2 mg/ml (x̄, 3.75 ng/ml; P < 0.001), 3 mg/ml (x̄, bLLD; P < 0.001), and 5 mg/ml (x̄, bLLD; P < 0.001).
Protein-binding activity of calcium aluminosilicate.
The selectivity of calcium aluminosilicate's protein-binding activity was explored further using a variation of the C. difficile toxin-binding assay described above. During these experiments, calcium aluminosilicate (5 mg/ml) was challenged with a commercial protein cocktail that contained a number of nontarget proteins, including myosin, bovine serum albumin, and glutamate dehydrogenase. Representative SDS-PAGE gels of the supernatant and the pellet eluate can be found in Fig. 4. CAS UPSN M-1 bound nontarget proteins but did so with various efficiencies. Indeed, CAS UPSN M-1 bound myosin and bovine serum albumin inefficiently, while glutamate dehydrogenase was bound efficiently. Attempts to elute proteins bound to CAS UPSN M-1 were unsuccessful (Fig. 4, lanes 4 and 5), which suggests that proteins bound to CAS UPSN M-1 are bound tightly.
FIG 4.
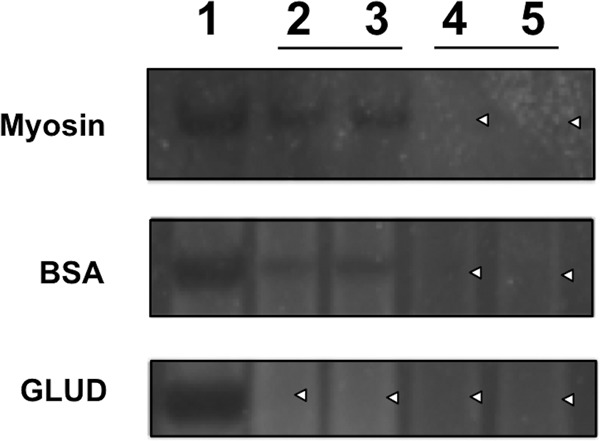
Nontherapeutic protein-binding assay. SDS-PAGE gels illustrating differential binding of myosin (top panel), bovine serum albumin (BSA) (middle panel), and glutamate dehydrogenase (GLUD) (bottom panel). Lane 1, untreated reference protein (negative control); lanes 2 and 3, CAS UPSN M-1-treated protein samples (duplicates); lanes 4 and 5, eluates of protein-bound-CAS UPSN M-1 complexes (duplicates). White carets mark the lanes that are devoid of a protein band.
DISCUSSION
The initial toxin concentrations used in this study were selected because they approximated the median concentration of TcdA (4.3 ng/ml) that is typically found in the stools of patients with C. difficile-associated diarrhea (range, 0.6 ng/ml to 19 μg/ml) (28). As such, the qEIA protocol developed in this study enabled C. difficile TcdA and TcdB quantification at clinically relevant concentrations. No hook effect was obvious for either toxin at toxin concentrations between 5 and 15 ng/ml, which constituted the linear range for this qEIA. The high-dose hook effect occurs when the antigen negatively affects the binding capacity of the reporter antibody or when it is added in excess of the reporter antibody (29). The intraexperimental EIA calibration curves generated using this qEIA protocol enabled toxin quantification via interpolation and, thus, facilitated the conversion of optical density measurements to residual toxin concentrations. Furthermore, given the high degree of reproducibility, this qEIA supported interexperimental comparisons between repeated measurements (e.g., randomized block experiments).
This assay revealed that calcium aluminosilicate efficiently removed both TcdA and TcdB at physiologically relevant concentrations. Indeed, calcium aluminosilicate neutralized TcdA to subclinical levels in vitro. As for tolevamer, protein binding by calcium aluminosilicate does not occur in a generalized or otherwise indiscriminate fashion and, thus, displays a degree of target specificity. Tolevamer is an anionic, high-molecular-weight polymer (>400 kDa) that was developed to neutralize TcdA and TcdB. Tolevamer has been shown to ameliorate CDI-like symptoms in hamsters (30). In addition, tolevamer has demonstrated therapeutic efficacy in a number of phase II and phase III clinical studies (25, 26). While effective, tolevamer's cure rate was found to be inferior to those of vancomycin and metronidazole (26, 31). Surprisingly, however, the rate of CDI reoccurrence was generally lower with tolevamer than with either vancomycin or metronidazole (26).
TcdA and TcdB are postulated to be paralogs (32, 33). As a result, these proteins share significant amino acid sequence similarity to one another, especially at their amino- and carboxy-terminal regions. The two toxins share approximately 47% identity to each other and approximately 68% sequence similarity (data not shown). Both proteins are composed of three well-characterized functional domains (15). The amino terminus of the protein encodes a peptidase C80-type glycosyltransferase domain and a proximal substrate recognition domain. The hydrophobic middle region is putatively involved in membrane translocation. The carboxy terminus of the protein encodes the clostridial repetitive oligopeptides (CROPS) (also known as cell wall-binding [CWB] domains). The carboxy-terminal CROPS facilitate calcium-dependent host cell recognition (33) and may also play a role in the sequestration of TcdA and TcdB by calcium aluminosilicate. Proteins that are evolutionarily and/or structurally related to TcdA and TcdB might also be viable therapeutic targets for calcium aluminosilicate; however, additional research is required to test this hypothesis.
In addition to the similarities noted above, a number of toxin-specific differences were also observed. For example, the lowest experimental concentration of calcium aluminosilicate for which there was no observable effect was 0.1 mg/ml for TcdA but 0.3 mg/ml for TcdB. The minimum effective concentration (i.e., the threshold dose) for calcium aluminosilicate was 0.3 mg/ml for TcdA but 0.5 mg/ml for TcdB. Under these conditions, the calcium aluminosilicate concentration that achieved the maximum efficacy (EC100) was 2 mg/ml for TcdA, but the concentration of calcium aluminosilicate that provided approximately 50% of the maximum effect (EC50) for TcdA was 0.5 mg/ml. In contrast, the EC100 and EC50 for TcdB were 3 mg/ml and 1 mg/ml, respectively. While calcium aluminosilicate sequesters both protein-based cytotoxic enzymes, these results suggest that its affinity for TcdA is greater than its affinity for TcdB.
As antibiotic-resistant pathogens continue to emerge, the development of nonantibiotic treatment options represents a timely therapeutic approach to CDI management. Calcium aluminosilicate exhibited potent C. difficile TcdA- and TcdB-neutralizing activity and selective protein binding in vitro. Given the well-documented safety profile of calcium aluminosilicate (34), these studies provide in vitro evidentiary support of our hypothesis that ingestion of calcium aluminosilicate might protect gastrointestinal tissues and accelerate a patient's recovery from antibiotic- or chemotherapy-induced C. difficile-associated diarrhea by neutralizing the cytotoxic effects of luminal TcdA and TcdB. Depending on its relative effectiveness and tolerability during downstream clinical studies, CAS UPSN M-1, the novel sequestration agent described in this study, may be used to complement or, possibly, replace existing antibiotic therapies for the treatment of CDI. However, it is beyond the scope of this current study to examine the biological effects of calcium aluminosilicate in vivo.
ACKNOWLEDGMENTS
Salient Pharmaceuticals Incorporated (Houston, TX) provided the calcium aluminosilicate (CAS UPSN M-1) used in this study without cost. Salient Pharmaceuticals, Incorporated provided funding for this study to Texas A&M AgriLife Research (J.M.S.). Additional support was provided by the U.S. Department of Agriculture, Cooperative State Research, Education and Extension Service Hatch project TEX 09436 and Texas A&M AgriLife Research. The sponsors had no role in the data collection or analysis, the production of the submitted manuscript, or the decision to submit the manuscript for publication.
We thank Trung Nguyen and Lynn Jones for technical support. In addition, we thank Richard Scruggs at Salient Pharmaceuticals Incorporated for insightful conversations. Last, we thank the anonymous reviewers for their valuable comments.
REFERENCES
- 1.Eaton SR, Mazuski JE. 2013. Overview of severe Clostridium difficile infection. Crit Care Clin 29:827–839. doi: 10.1016/j.ccc.2013.06.004. [DOI] [PubMed] [Google Scholar]
- 2.Lessa FC, Gould CV, McDonald LC. 2012. Current status of Clostridium difficile infection epidemiology. Clin Infect Dis 55(Suppl 2):S65–S70. doi: 10.1093/cid/cis319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gerding DN. 2010. Global epidemiology of Clostridium difficile infection in 2010. Infect Control Hosp Epidemiol 31(Suppl 1):S32–S34. doi: 10.1086/655998. [DOI] [PubMed] [Google Scholar]
- 4.Sunenshine RH, McDonald LC. 2006. Clostridium difficile-associated disease: new challenges from an established pathogen. Cleve Clin J Med 73:187–197. doi: 10.3949/ccjm.73.2.187. [DOI] [PubMed] [Google Scholar]
- 5.Britton RA, Young VB. 2014. Role of the intestinal microbiota in resistance to colonization by Clostridium difficile. Gastroenterology 146:1547–1553. doi: 10.1053/j.gastro.2014.01.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Riggs MM, Sethi AK, Zabarsky TF, Eckstein EC, Jump RL, Donskey CJ. 2007. Asymptomatic carriers are a potential source for transmission of epidemic and nonepidemic Clostridium difficile strains among long-term care facility residents. Clin Infect Dis 45:992–998. doi: 10.1086/521854. [DOI] [PubMed] [Google Scholar]
- 7.Ryan J, Murphy C, Twomey C, Paul Ross R, Rea MC, MacSharry J, Sheil B, Shanahan F. 2010. Asymptomatic carriage of Clostridium difficile in an Irish continuing care institution for the elderly: prevalence and characteristics. Ir J Med Sci 179:245–250. doi: 10.1007/s11845-009-0361-1. [DOI] [PubMed] [Google Scholar]
- 8.Rea MC, O'Sullivan O, Shanahan F, O'Toole PW, Stanton C, Ross RP, Hill C. 2012. Clostridium difficile carriage in elderly subjects and associated changes in the intestinal microbiota. J Clin Microbiol 50:867–875. doi: 10.1128/JCM.05176-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hung YP, Lee JC, Lin HJ, Liu HC, Wu YH, Tsai PJ, Ko WC. 2015. Clinical impact of Clostridium difficile colonization. J Microbiol Immunol Infect 48:241−248. doi: 10.1016/j.jmii.2014.04.011. [DOI] [PubMed] [Google Scholar]
- 10.Pérez-Cobas AE, Artacho A, Ott SJ, Moya A, Gosalbes MJ, Latorre A. 2014. Structural and functional changes in the gut microbiota associated to Clostridium difficile infection. Front Microbiol 5:335. doi: 10.3389/fmicb.2014.00335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Luciano JA, Zuckerbraun BS. 2014. Clostridium difficile infection: prevention, treatment, and surgical management. Surg Clin North Am 94:1335–1349. doi: 10.1016/j.suc.2014.08.006. [DOI] [PubMed] [Google Scholar]
- 12.Fekety R, McFarland LV, Surawicz CM, Greenberg RN, Elmer GW, Mulligan ME. 1997. Recurrent Clostridium difficile diarrhea: characteristics of and risk factors for patients enrolled in a prospective, randomized, double-blinded trial. Clin Infect Dis 24:324–333. doi: 10.1093/clinids/24.3.324. [DOI] [PubMed] [Google Scholar]
- 13.Shah D, Dang MD, Hasbun R, Koo HL, Jiang ZD, DuPont HL, Garey KW. 2010. Clostridium difficile infection: update on emerging antibiotic treatment options and antibiotic resistance. Expert Rev Anti Infect Ther 8:555–564. doi: 10.1586/eri.10.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Awad MM, Johanesen PA, Carter GP, Rose E, Lyras D. 2014. Clostridium difficile virulence factors: insights into an anaerobic spore-forming pathogen. Gut Microbes 5:579−593. doi: 10.4161/19490976.2014.969632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Voth DE, Ballard JD. 2005. Clostridium difficile toxins: mechanism of action and role in disease. Clin Microbiol Rev 18:247–263. doi: 10.1128/CMR.18.2.247-263.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Just I, Gerhard R. 2004. Large clostridial cytotoxins. Rev Physiol Biochem Pharmacol 152:23–47. doi: 10.1007/s10254-004-0033-5. [DOI] [PubMed] [Google Scholar]
- 17.Feltis BA, Wiesner SM, Kim AS, Erlandsen SL, Lyerly DL, Wilkins TD, Wells CL. 2000. Clostridium difficile toxins A and B can alter epithelial permeability and promote bacterial paracellular migration through HT-29 enterocytes. Shock 14:629–634. doi: 10.1097/00024382-200014060-00010. [DOI] [PubMed] [Google Scholar]
- 18.Farrow MA, Chumbler NM, Lapierre LA, Franklin JL, Rutherford SA, Goldenring JR, Lacy DB. 2013. Clostridium difficile toxin B-induced necrosis is mediated by the host epithelial cell NADPH oxidase complex. Proc Natl Acad Sci U S A 110:18674–18679. doi: 10.1073/pnas.1313658110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hirota SA, Iablokov V, Tulk SE, Schenck LP, Becker H, Nguyen J, Al Bashir S, Dingle TC, Laing A, Liu J, Li Y, Bolstad J, Mulvey GL, Armstrong GD, MacNaughton WK, Muruve DA, MacDonald JA, Beck PL. 2012. Intrarectal instillation of Clostridium difficile toxin A triggers colonic inflammation and tissue damage: development of a novel and efficient mouse model of Clostridium difficile toxin exposure. Infect Immun 80:4474–4484. doi: 10.1128/IAI.00933-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Vohra P, Poxton IR. 2012. Induction of cytokines in a macrophage cell line by proteins of Clostridium difficile. FEMS Immunol Med Microbiol 65:96–104. doi: 10.1111/j.1574-695X.2012.00952.x. [DOI] [PubMed] [Google Scholar]
- 21.Lyerly DM, Saum KE, MacDonald DK, Wilkins TD. 1985. Effects of Clostridium difficile toxins given intragastrically to animals. Infect Immun 47:349–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lima AA, Lyerly DM, Wilkins TD, Innes DJ, Guerrant RL. 1988. Effects of Clostridium difficile toxins A and B in rabbit small and large intestine in vivo and on cultured cells in vitro. Infect Immun 56:582–588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sun X, Savidge T, Feng H. 2010. The enterotoxicity of Clostridium difficile toxins. Toxins (Basel) 2:1848–1880. doi: 10.3390/toxins2071848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Savidge TC, Pan WH, Newman P, O'Brien M, Anton PM, Pothoulakis C. 2003. Clostridium difficile toxin B is an inflammatory enterotoxin in human intestine. Gastroenterology 125:413–420. doi: 10.1016/S0016-5085(03)00902-8. [DOI] [PubMed] [Google Scholar]
- 25.Weiss K. 2009. Toxin-binding treatment for Clostridium difficile: a review including reports of studies with tolevamer. Int J Antimicrob Agents 33:4–7. doi: 10.1016/j.ijantimicag.2008.07.011. [DOI] [PubMed] [Google Scholar]
- 26.Bauer MP, van Dissel JT. 2009. Alternative strategies for Clostridium difficile infection. Int J Antimicrob Agents 33(Suppl 1):S51–S56. doi: 10.1016/S0924-8579(09)70018-4. [DOI] [PubMed] [Google Scholar]
- 27.Electronic Code of Federal Regulations. Title 21. Food and drugs. Chapter I. Food and Drug Administration. Subchapter B. Part 182. Substances generally recognized as safe. Subpart C. Anticaking agents. 21 CFR 182. 2729. http://www.gpo.gov/fdsys/pkg/CFR-2011-title21-vol3/pdf/CFR-2011-title21-vol3-sec182-2729.pdf.
- 28.Solomon K, Webb J, Ali N, Robins RA, Mahida YR. 2005. Monocytes are highly sensitive to Clostridium difficile toxin A-induced apoptotic and nonapoptotic cell death. Infect Immun 73:1625–1634. doi: 10.1128/IAI.73.3.1625-1634.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fernando SA, Wilson GS. 1992. Studies of the ‘hook’ effect in the one-step sandwich immunoassay. J Immunol Methods 151:47–66. doi: 10.1016/0022-1759(92)90104-2. [DOI] [PubMed] [Google Scholar]
- 30.Barker RH Jr, Dagher R, Davidson DM, Marquis JK. 2006. Review article: tolevamer, a novel toxin-binding polymer: overview of preclinical pharmacology and physicochemical properties. Aliment Pharmacol Ther 24:1525–1534. doi: 10.1111/j.1365-2036.2006.03157.x. [DOI] [PubMed] [Google Scholar]
- 31.Louie TJ, Peppe J, Watt CK, Johnson D, Mohammed R, Dow G, Weiss K, Simon S, John JF Jr, Garber G, Chasan-Taber S, Davidson DM, Tolevamer Study Investigator Group . 2006. Tolevamer, a novel nonantibiotic polymer, compared with vancomycin in the treatment of mild to moderately severe Clostridium difficile-associated diarrhea. Clin Infect Dis 43:411–420. doi: 10.1086/506349. [DOI] [PubMed] [Google Scholar]
- 32.Hofmann F, Herrmann A, Habermann E, von Eichel-Streiber C. 1995. Sequencing and analysis of the gene encoding the alpha-toxin of Clostridium novyi proves its homology to toxins A and B of Clostridium difficile. Mol Gen Genet 247:670–679. doi: 10.1007/BF00290398. [DOI] [PubMed] [Google Scholar]
- 33.Demarest SJ, Salbato J, Elia M, Zhong J, Morrow T, Holland T, Kline K, Woodnutt G, Kimmel BE, Hansen G. 2005. Structural characterization of the cell wall binding domains of Clostridium difficile toxins A and B; evidence that Ca2+ plays a role in toxin A cell surface association. J Mol Biol 346:1197–1206. doi: 10.1016/j.jmb.2004.12.059. [DOI] [PubMed] [Google Scholar]
- 34.Mitchell NJ, Kumi J, Aleser M, Elmore SE, Rychlik KA, Zychowski KE, Romoser AA, Phillips TD, Ankrah NA. 2014. Short-term safety and efficacy of calcium montmorillonite clay (UPSN) in children. Am J Trop Med Hyg 91:777–785. doi: 10.4269/ajtmh.14-0093. [DOI] [PMC free article] [PubMed] [Google Scholar]