

Abstract
Therapies that are safe, effective, and not vulnerable to developing resistance are highly desirable to counteract bacterial infections. Host-directed therapeutics is an antimicrobial approach alternative to conventional antibiotics based on perturbing host pathways subverted by pathogens during their life cycle by using host-directed drugs. In this study, we identified and evaluated the efficacy of a panel of host-directed drugs against respiratory infection by nontypeable Haemophilus influenzae (NTHi). NTHi is an opportunistic pathogen that is an important cause of exacerbation of chronic obstructive pulmonary disease (COPD). We screened for host genes differentially expressed upon infection by the clinical isolate NTHi375 by analyzing cell whole-genome expression profiling and identified a repertoire of host target candidates that were pharmacologically modulated. Based on the proposed relationship between NTHi intracellular location and persistence, we hypothesized that drugs perturbing host pathways used by NTHi to enter epithelial cells could have antimicrobial potential against NTHi infection. Interfering drugs were tested for their effects on bacterial and cellular viability, on NTHi-epithelial cell interplay, and on mouse pulmonary infection. Glucocorticoids and statins lacked in vitro and/or in vivo efficacy. Conversely, the sirtuin-1 activator resveratrol showed a bactericidal effect against NTHi, and the PDE4 inhibitor rolipram showed therapeutic efficacy by lowering NTHi375 counts intracellularly and in the lungs of infected mice. PDE4 inhibition is currently prescribed in COPD, and resveratrol is an attractive geroprotector for COPD treatment. Together, these results expand our knowledge of NTHi-triggered host subversion and frame the antimicrobial potential of rolipram and resveratrol against NTHi respiratory infection.
INTRODUCTION
Strategies for managing infectious diseases have mainly focused on targeting enzymes of pathogens, with antibiotics being the most notable example of this approach. However, among the serious disadvantages of this pathogen-directed strategy are the development of microbial drug resistance (1) and the difficulty in treating intracellular pathogens (2). Despite the growing need for new antimicrobials, the rates of discovery for novel antibiotics are declining (3). Therefore, new broad-spectrum therapeutics that are safe, effective, and not vulnerable to the development of bacterial resistance are needed (4).
Pathogens exploit and subvert various host cell factors for survival and growth in an otherwise hostile environment. An alternative antimicrobial approach is to perturb host cell pathways used by bacteria at various stages of their life cycle (adhesion, entry, growth, etc.). This strategy, termed host-directed therapeutics, promotes the use of host-directed antimicrobial drugs (5). Identification of host targets requires a detailed understanding of host-pathogen interactions. In the present study, we used global expression profiling to elucidate cellular pathways exploited by nontypeable Haemophilus influenzae (NTHi) to infect airway epithelia and evaluated drugs that, by perturbing these host cell targets, may limit infection by this opportunistic pathogen.
Although typically a commensal of the nasopharynx, the Gram-negative bacterium H. influenzae, especially in its unencapsulated or nontypeable form, is also an opportunistic pathogen causing middle-ear infections, conjunctivitis, community-acquired pneumonia, exacerbations of chronic obstructive pulmonary disease (COPD), and occasionally, invasive disease (6). Infections frequently persist and recur despite the host's development of bactericidal antibodies and the use of antibiotics. Our current understanding of the molecular mechanisms involved in NTHi infection remains limited, but identical strains have been repeatedly isolated from the lungs of COPD patients in serial clinic visits, suggesting that NTHi presents features that promote chronic infection (7, 8). NTHi is a facultative intracellular pathogen (9–16), and epithelial invasion may allow bacterial cells to temporarily evade the immune system and/or therapeutic interventions. Indeed, a correlation between the occurrence of intracellular NTHi and persistent infection has been proposed (17, 18). Based on these observations, a comprehensive understanding of host factors hijacked by NTHi to invade airways epithelia may lead to the identification of targets for host-directed drugs with antimicrobial potential.
NTHi infection is also an inflammatory process (19). Chronic respiratory patients are prescribed anti-inflammatory therapies, which may influence NTHi infection. In fact, glucocorticoids modify NTHi gene expression (20), suppress host inflammation via the upregulation of IRAK-M, enhance NTHi-induced Toll-like receptor 2 (TLR2), and inhibit NTHi-induced MUC5A expression via mitogen-activated protein kinase (MAPK) phosphatase MKP-1-dependent inhibition of p38 MAPK (21–24). Moreover, although glucocorticoids attenuate NTHi-triggered inflammation in vivo, they also compromise bacterial clearance (25). The β2 receptor agonist salmeterol seems to protect the respiratory epithelium against H. influenzae-induced damage, but inhalation of this bronchodilator may negatively influence NTHi clearance from the murine airways (26, 27). The phosphodiesterase 4 (PDE4) inhibitor roflumilast dampens NTHi-triggered inflammation in the lung and middle ear by upregulation of the deubiquitinase CYLD (28), but it may also synergize with NTHi to upregulate PDE4B2 expression via a cross talk between PKA-Cβ and p65, therefore contributing to chemokine induction (29).
Altogether, the proposed relationship between NTHi intracellular location and persistence, as well as the observed modulation of the NTHi-host cell interplay by anti-inflammatory therapies, prompted us to hypothesize that drugs perturbing host pathways used by NTHi to enter airway cells could have antimicrobial potential against this pathogen. Based on this notion, we screened for host cell pathways affected upon infection by a NTHi clinical isolate by analyzing whole-genome expression profiling in order to identify potential host target candidates that can be inhibited by commercially available drugs, including some U.S. Food and Drug Administration (FDA)-approved compounds. Such inhibitors were tested for their bactericidal effect, host cell toxicity, effect on NTHi-host cell interplay, and effect on NTHi respiratory infection in vivo. By focusing on drugs that were not cytotoxic, that impair NTHi invasion of epithelial cells, and that lower bacterial counts in vivo, we identified the PDE4 inhibitor rolipram as a nonbactericidal host-directed drug able to dampen NTHi epithelial invasion and to enhance mice lung clearance. Moreover, our analysis revealed a bactericidal effect for the sirtuin-1 activator resveratrol (RESV) against NTHi. We present here evidence for the antimicrobial potential of rolipram and resveratrol against NTHi respiratory infection.
MATERIALS AND METHODS
Bacterial strains, media, and growth conditions.
NTHi strains were grown at 37°C and 5% CO2 on chocolate-agar (bioMérieux) or on brain heart infusion (BHI) agar supplemented with 10 μg of hemin/ml and 10 μg of NAD/ml, referred to here as sBHI. NTHi liquid cultures were grown in sBHI. NTHi375 is a genome-sequenced clinical isolate previously used in host-pathogen interplay studies (13, 14, 30).
Interfering drugs and molecules.
Caffeic acid phenethyl ester (CAPE), BAY11-7083, SB202190, SP600125, dexamethasone (DEX), fluticasone propionate (FP), human epidermal growth factor (EGF), resveratrol (RESV), theophylline (TEOPH), TAPI-2 acetate salt, salmeterol (SALM), formoterol fumarate dehydrate (FORM), (S)-(−)-propranolol hydrochloride (PROP), N6,2′-O-dibutyryladenosine 3′,5′-cyclic monophosphate sodium salt (db-cAMP), and 3-isobutyl-1-methylxanthine (IBMX) were purchased from Sigma-Aldrich. Ro31-8220, roflumilast 3-(cyclopropylmethoxy)-N-(3,5-dichloro-4-pyridinyl)-4-(difluoromethoxy)benzamide (ROFLUM), rolipram (R,S)-4-(3-(cyclopentyloxy)-4-methoxyphenyl)pyrrolidin-2-one (ROLIP), and 8-(4-chlorophenylthio)-2′-O-methyladenosine 3′,5′-cyclic monophosphate monosodium (8-pCPT-2′-O-Me-cAMP) were purchased from Santa Cruz Biotechnology. PD98059, GM6001, fluvastatin (FLUV), and protein kinase A (PKA) inhibitor 14-22 amide (PKI) were purchased from Calbiochem. Stock solutions for CAPE (17.6 mM), BAY11-7083 (6 mM), SB202190 (15.1 mM), SP600125 (22.7 mM), PD98059 (18.7 mM), FP (1 mM), RESV (20 mM), GM6001 (10 mM), FORM (10 mM), IBMX (560 mM), and ROFLUM (12.4 mM) were prepared in dimethyl sulfoxide (DMSO). Stock solutions for Ro31-8220 (1.8 mM), DEX (1 mM), TAPI-2 (4.8 mM), FLUV (5 mM), PROP (2.6 mM), db-cAMP (50.8 mM), 8-pCPT-2′-O-Me-cAMP (2 mM), and PKI (0.4 mM) were prepared in distilled water. EGF (165 nM) was dissolved in 10 mM acetic acid, TEOPH (2.7 mM) was dissolved in 0.1 M NaOH, SALM (0.2 mM) was dissolved in phosphate-buffered saline (PBS), and ROLIP (18.2 mM) was dissolved in ethanol. Drugs were diluted to the required working concentrations in Earle's balanced salt solution (EBSS).
To test bacterial viability in the presence of the interfering agents, a bacterial suspension recovered with 1 ml of PBS from a freshly grown chocolate-agar plate, was adjusted to an optical density at 600 nm (OD600) of 1 (∼109 CFU/ml), and 100 μl of this suspension was incubated for 2 h with the working concentration of each drug (or an equivalent volume of vehicle solution used as a control [CON]) in 1 ml of EBSS. Serial dilutions were plated on sBHI agar. Volumes used for each vehicle solution did not reduce bacterial viability (data not shown). The data are expressed as the percentage of viability compared to CON bacteria, considered to be 100% (see Fig. S1 in the supplemental material). Viability assays were carried out in duplicate on at least two separate occasions (n ≥ 4).
Resveratrol susceptibility assay.
A bacterial suspension recovered with 1 ml of PBS from a freshly grown chocolate-agar plate was adjusted to an OD600 of 0.5 (∼5 × 108 CFU/ml). Resveratrol was serially diluted in sBHI (225, 175, 112.5, 56.25, 28.125, 14, 7, 3.5, 1.75, and 0.88 μg/ml). Portions (80 μl) of each resveratrol dilution were transferred to individual wells in 96-well microtiter plates (Iwaki); 20 μl of the previously prepared bacterial suspension was added to each well, followed by incubation for 40 min at 37°C. In parallel, 20 μl of the bacterial suspension was added to 80 μl of sBHI containing DMSO volumes identical to those used for each resveratrol dilution. Bacteria were serially diluted in PBS and plated on sBHI agar. The results are expressed as a percentage of the colony count of bacteria not exposed to resveratrol, considered to be 100%. Experiments were performed in duplicate on at least four independent occasions (n ≥ 8).
Cell culture and bacterial infection.
Carcinomic human alveolar basal epithelial cells (A549, ATCC CCL-185) were maintained and seeded in 24-well tissue culture plates as described previously (14). Adhesion and invasion assays were performed and processed as previously described (13, 14). The results are expressed as CFU per well. When indicated, the cells were pretreated in EBSS with the panel of interfering molecules as follows: (i) 20 min with 20 μM TEOPH; (ii) 30 min with 1 mM IBMX; (iii) 1 h with 52.8 μM CAPE, 5 μM BAY11-7083, 50 μM SP600125, 200 μM GM6001, 100 μM TAPI-2, 50 to 200 μM FLUV, 1 mM db-cAMP, 1 μM ROFLUM, 10 μM ROLIP, or 1 μM PKI; (iv) 90 min with 50 μM PD98059; (v) 2 h with 1 μM Ro31-8220, 30 μM SB202190, 1 μM DEX, 10 μM FORM, or 100 μM 8-pCPT-2′-O-Me-cAMP; (vi) 3 h with 20 μM PROP; (vii) 4 h with 1 μM FP or 20 μM RESV; and (viii) 16 h with 82.5 to 165 nM EGF or 1 μM SALM. When necessary, A549 cells were incubated at 33 or 41°C 24 h before and during NTHi infection. When no bactericidal effect was observed, drug exposure was maintained during bacterium-cell contact. When a bactericidal effect was observed (see Fig. S1 in the supplemental material), cells were pretreated as indicated above, and the drugs were removed prior to infection. In all cases, the cytotoxicity on A549 cells at each drug working concentration was tested by measuring the release of lactate dehydrogenase with a CytoTox-96 nonradioactive cytotoxicity assay (Promega) and by microscopy (data not shown). Experiments were performed in triplicate on at least three independent occasions (n ≥ 9).
Immunofluorescence microscopy.
A549 cells were seeded on 13-mm circular coverslips, infections were performed as described previously (13, 14), and infected cells were incubated in RPMI 1640 containing 10% fetal calf serum (FCS), HEPES 10 mM, and gentamicin at 200 μg/ml for 60 min. The cells were then washed, fixed, and stained as previously described (13, 14). NTHi375 was stained with rabbit anti-NTHi serum diluted 1:600 (14). GM1 ganglioside was stained with Vibrio cholerae toxin subunit B conjugated to Texas Red (Molecular Probes) at 5 μg/ml. Donkey anti-rabbit conjugated to Cy2 secondary antibody (Jackson) was diluted 1:100. Samples were analyzed with a Leica TCS SP5 confocal microscope.
RNA extraction, microarray hybridization, and data analysis.
Total RNA was isolated using a NucleoSpin RNAII kit (Macherey-Nagel), including an on-column DNase treatment step. Total RNA quality was evaluated using RNA 6000 Nano LabChips (Agilent 2100 Bioanalyzer). All samples had intact 18S and 28S rRNA bands with RNA integrity numbers (RIN) between 9.3 and 9.7 and an RNA A260/A280 ratio of 2.1. For microarray hybridization, samples (n = 4) were processed using manufacturer protocols and hybridized to the Agilent Human 1A Microarray V2 with a single labeling. Data normalization was performed by using a quantile algorithm. After quality assessment, a filtering process was performed to eliminate low-expression probes. Applying the criterion of an expression value >64 in three samples for each experimental condition, 18,235 probes were selected for statistical analysis. R and Bioconductor (31) were used for preprocessing and statistical analysis. LIMMA (Linear Models for Microarray Data) (32) was used to find out the probes that showed significant differential expression between experimental conditions. Genes were selected as significant using a B statistic cutoff of B > 1. Functional enrichment analysis of Gene Ontology (GO) categories was carried out using a standard hypergeometric test (33). The biological knowledge extraction was complemented through the use of Ingenuity Pathway Analysis (Ingenuity Systems).
Real-time quantitative PCR (RT-qPCR).
cDNA was synthesized from total RNA (1 μg) using SuperScript II reverse transcriptase reagents (Invitrogen). PCR amplification was performed by using Thermo Scientific Luminaris HiGreen qPCR master mix (Thermo Scientific). Fluorescence data were analyzed with ABI 7900HT Prism sequence detector software (Applied Biosystems). The primer pairs, designed with Primer-BLAST software (NCBI), are shown in Table S1 in the supplemental material. Relative mRNA levels of each gene were normalized by using GAPDH (glyceraldehyde-3-phosphate dehydrogenase) as an internal control. The relative quantities of mRNAs were obtained by using the comparative threshold cycle (CT) method. Data are expressed as the relative expression on infected cells compared to uninfected cells (considered to be 1; see Fig. S3 in the supplemental material). All measures were carried out in duplicate and at least three times (n ≥ 6).
Transient transfections.
A549 cells were seeded, transfected, and infected as previously described (13). Two small interfering RNAs (siRNAs) for each gene (a and b) and a scramble nonsilencing RNA were synthesized by Qiagen (see Table S2 in the supplemental material). siRNA (40 nM) was used for transfection. The cells were infected at 48 h posttransfection. RNA interference (RNAi) efficiency was assessed at the mRNA level by RT-qPCR on cell extracts at 48 h posttransfection, as detailed above. Validation of interference is expressed as the percentage of mRNA expression on siRNA transfected compared to AS-CON-transfected cells, which was considered to be 100% (see Fig. S4 in the supplemental material). Knockdown and subsequent infection assays were carried out in duplicate and on at least in three separate occasions (n ≥ 6).
Secretion of IL-8.
A549 cells were maintained, seeded, and infected for 2 h, as described previously (10, 34). Cells were washed three times with PBS and incubated for 6 h in RPMI 1640 medium containing 10% FCS, 10 mM HEPES, and gentamicin (100 μg/ml). Supernatants were collected from the wells, cell debris was removed by centrifugation, and samples were frozen at −80°C. Interleukin-8 (IL-8) levels in the supernatants were measured by enzyme-linked immunosorbent assay (ELISA; Abnova, catalog no. KA0115) with a sensitivity of <2 pg/ml. When necessary, cells were pretreated with each drug or with vehicle solution, which was kept during infection, including the gentamicin incubation period. For fluvastatin, the cells were treated in EBSS prior and during infection; during the gentamicin incubation period, this drug was added to RPMI 1640 with 10 mM HEPES and 100 μg of gentamicin/ml, in the absence or presence of FCS. In all cases, infections were performed in duplicate on at least two separate occasions (n ≥ 4). The results are expressed as IL-8 pg/ml.
NTHi mouse lung infection.
A CD1 mouse model of NTHi respiratory infection was used as described previously (7, 10, 34, 35). NTHi375 was used for lung infection, and mice were randomly divided into the following six groups (n = 5): (i) control A (intraperitoneally [i.p.] administered vehicle solution), (ii) control B (oroesophageal gavage administered vehicle solution), (iii) a group treated daily with dexamethasone 10 days before infection, (iv) a group treated daily with fluvastatin 10 days before infection, (v) a group treated with fluvastatin 24, 12, and 1 h before infection and 6 h postinfection (hpi), and (vi) a group treated with rolipram 24, 12, and 1 h prior to infection and 6 hpi. Dexamethasone treatment was performed at a dose of 2.5 mg/kg of body weight in 0.1 ml of water and administered i.p. (36); fluvastatin treatments were performed at doses of 1 or 30 mg/kg of body weight in 0.1 ml of water and administered i.p. (37). Rolipram treatment was performed at a dose of 10 mg/kg of body weight in 0.1 ml of PBS-ethanol (2:1) and administered by oroesophageal gavage (Popper & Sons, Inc.) (38). NTHi intranasal infection was performed, and the lungs were processed as previously described (7, 10, 34, 35) at 12 or 24 hpi. Each lung homogenate was serially 10-fold diluted in PBS, and 100 μl of each dilution was spread in triplicate on sBHI-agar (detection limit, <10 CFU/lung). The results are expressed as means ± the standard deviations (SD) of individual log10 CFU/lung.
Statistical analysis.
For gene expression, bacterial viability, cell infection, IL-8 secretion, and bacterial loads in lungs, the means ± the SD were calculated, and statistical comparisons of means were performed using a two-tailed Student t test. In all cases, a P value of <0.05 was considered statistically significant. Analyses were performed using the Prism software, version 4, for PC statistical package (GraphPad Software).
Database accession number.
Microarray data files were submitted to GEO (Gene Expression Omnibus) database and are available under accession number GSE69134.
RESULTS
Gene expression profiling identifies host functions differentially expressed in NTHi-infected A549 human type II pneumocytes.
To examine the human airway epithelial transcriptional response to NTHi infection, we performed expression profiling of A549 cells infected with the clinical isolate NTHi strain 375 (here, NTHi375) (I) or in cells left uninfected (NI). The samples were collected at 2 h postinfection. This infection condition was used to mimic early stages of the infectious process, when the pathogen may modulate the expression of host functions to favor its entry into the cells. Transcriptomic analysis using microarrays revealed 402 probes corresponding to 352 genes differentially expressed (B > 1) in A549-infected cells compared to control NI cells. A heat map of highly induced or repressed genes (B > 1 and logFC > 1.5), clustered genes in two groups: those that were upregulated and those that were downregulated in infected cells (Fig. 1A). Some of the most representative canonical pathways enriched with a Fisher exact test (P < 0.05) using Ingenuity Pathways Analysis (IPA) software included inflammatory responses (i.e., IL-17, IL-6, and IL-8) and cell signaling (i.e., glucocorticoid receptor, protein kinase A [PKA], and phosphatidylinositol 3-kinase [PI3K]/AKT signaling) (Fig. 1B). Moreover, 25-gene clustering networks were created by the IPA software. The network with the highest score, together with networks 13, 14, and 19 (data not shown) were related to ubiquitin C (see Fig. S2A in the supplemental material). We also focused on interactive networks 3 and 21, where we found SIRT1, encoding the NAD-dependent deacetylase sirtuin-1 (see Fig. S2B in the supplemental material), and HMGCR, encoding the 3-hydroxy-3-methyl-glutaryl-CoA reductase (HMGCR; see Fig. S2D in the supplemental material), respectively. In agreement with the enriched canonical pathways, we found several phosphodiesterases (PDEs) in interactive network 8 (see Fig. S2C in the supplemental material). Supporting the observed transcriptional changes, RT-qPCR showed significantly enhanced expression for IL-8, AREG, CXCL-1, and DUSP1 genes in A549-infected cells versus control uninfected cells (see Fig. S3 in the supplemental material).
FIG 1.

NTHi alters the human airways epithelial transcriptomic response. (A) Intensity heat map and clustering of differentially expressed genes (B > 1, logFC > 2) during infection (normalized expression values are shown). A549 cells were either not infected (NI) or infected (I) with NTHi375 for 2 h. Each row represents one gene that was either upregulated (red), downregulated (green), or not affected (black) under each of the conditions relative to uninfected cells. (B) Selection of enriched functions associated with differentially expressed genes with a Fisher exact test P value threshold set at 0.01 (red line) determined using IPA software.
Drug repurposing against NTHi respiratory infection.
Genome expression profiling revealed drug targets within the NTHi infection transcriptional signature whose pharmacological interference could impair NTHi cell invasion and, potentially, respiratory infection. For example, PI3K/AKT signaling showed differential expression (Fig. 1B), and its involvement in NTHi epithelial invasion has been reported (13, 14). Moreover, based on the occurrence of genes that are differentially expressed in NTHi-infected cells and that encode known drug targets, we observed several repurposing opportunities for launched drugs which are not currently used in anti-NTHi therapy. These genes included SIRT1 (a target of resveratrol), HMGCR (a target of statins), and PDE4B (the major PDE isoform expressed in lung and a target of roflumilast N-oxide) (see Fig. S2 in the supplemental material). Our analysis also suggested an antimicrobial potential for the pharmacological interference of glucocorticoid receptor signaling and inflammation (Fig. 1B). We selected a panel of host target candidates and assessed the effect of their pharmacological interference on NTHi respiratory infection.
Glucocorticoid receptor signaling and NTHi respiratory infection.
IPA-based analysis showed that the highest-rated function associated with relative expression of NTHi-infected/noninfected cells was glucocorticoid receptor signaling (Fig. 1B). Glucocorticoids enhance NTHi-induced TLR2 upregulation via MKP1 phosphatase-dependent inhibition of p38 MAPK (21, 24). MKP1 also seems to lead to reduced activation of the extracellular signal-regulated kinase (ERK) MAPK (39). Genome profiling revealed that the MKP1 encoding gene DUSP1 and p38 MAPK signaling are overexpressed in NTHi-infected cells. To determine whether DUSP1 and p38 MAPK play a role in NTHi cell infection, bacterial invasion was tested in the presence or absence of Ro31-8220 or SB202190 inhibitors, respectively, which reduced NTHi375 epithelial invasion (Ro31-8220, P = 0.01; SB202190, P = 0.05) (Fig. 2B). Cell pretreatment with the ERK inhibitor PD98059 did not alter NTHi375 cell invasion (Fig. 2B). Given that NTHi-induced activation of JNK2 kinase has been reported (28), we also tested the effect of cell exposure to the JNK inhibitor SP600125 and found a significant reduction of NTHi375 invasion (P < 0.005). None of these drugs was bactericidal (see Fig. S1 in the supplemental material), or modified bacterial epithelial adhesion (Fig. 2A). Despite these observations, cell treatment with two clinically used glucocorticoids, dexamethasone and fluticasone propionate (the dose range tested for both drugs was 0.01 to 1 μM), reduced IL-8 secretion by NTHi-infected cells (DEX, P < 0.0005; FP, P < 0.0001) but did not modify NTHi epithelial adhesion or invasion (Fig. 2C to E). We also assessed the effect of dexamethasone administration in vivo by using a mouse model of NTHi respiratory infection and a regimen consisting of DEX given in 2.5-mg/kg daily administrations for 10 days prior to infection. At 24 hpi, the NTHi burden in the lungs of mice treated with dexamethasone was similar to that found in the lungs of control A (CON) untreated mice (Fig. 2F). These results showed that, although glucocorticoids reduce NTHi-triggered IL-8 secretion and although the inhibition of some glucocorticoid signaling elements impairs NTHi375 cell invasion, glucocorticoids do not modulate NTHi epithelial invasion. Neither do they display an NTHi clearing effect in vivo.
FIG 2.
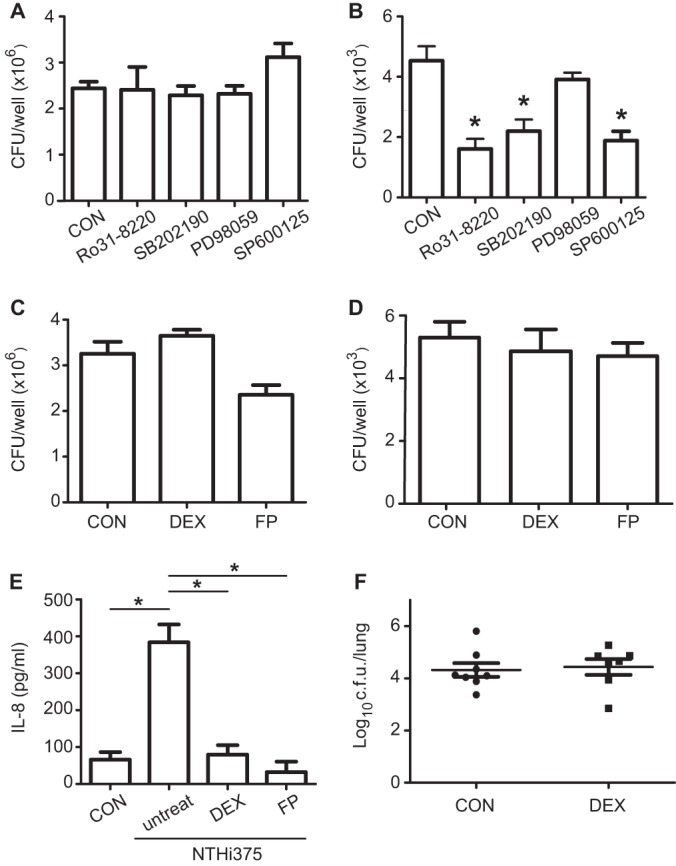
Effect of glucocorticoid receptor/MAK signaling interference on NTHi respiratory infection. (A and C) A549 cells were treated with Ro31-8220, SB202190, PD98059, or SP600125 (A) or with dexamethasone (DEX) or fluticasone propionate (FP) (C) and then infected with NTHi375 to determine adhesion compared to control (CON) untreated cells. (B and D) Cells were treated with Ro31-8220, SB202190, PD98059, or SP600125 (B) or with DEX or FP (D) and then infected with NTHi375 to determine invasion compared to control (CON) untreated cells. The mean numbers for NTHi375 invasion into Ro31-8220-, SB202190-, and SP600125-treated cells were lower than those obtained for control cells (Ro31-8220, P = 0.01; SB202190, P = 0.05; SP600125, P < 0.005). (E) Effect of DEX and FP on NTHi375-mediated inflammatory response, as measured by IL-8 secreted by A549 cells. CON, cells uninfected that did not receive any agent; untreat, cells infected with NTHi375 that did not receive any agent. NTHi375-infected cells triggered IL-8 secretion (P > 0.0001); IL-8 release was lower in DEX (P < 0.0005)- and FP (P < 0.0001)-treated NTHi375-infected cells than in untreated infected A549 cells. (F) Effect of DEX on lung bacterial loads in mice infected by NTHi375. Mice were infected intranasally with ∼108 bacteria/mouse. DEX (2.5 mg/kg) was administered i.p. daily 10 days prior to infection. Bacterial counts in lungs were determined at 24 hpi (log10 CFU/lung).
Inflammation signaling and epithelial infection by NTHi.
IPA-based analysis also showed that high-rated functions associated with the relative expression of infected versus noninfected (I/NI) cells included inflammation signaling pathways (Fig. 1B). These observations agreed with previous reports showing that NTHi infection is an inflammatory process to be induced in a TRAF6/IKK/NF-κB-dependent manner (19). To determine whether NF-κB plays a role in NTHi location upon epithelial infection, we tested the effect of the NF-κB inhibitors CAPE and BAY11-7083 on bacterial cell invasion. Given that both inhibitors were bactericidal for NTHi375 (see Fig. S1 in the supplemental material), the cells were pretreated prior but not during infection. CAPE (P < 0.05), but not BAY11-7083, reduced NTHi375 cell invasion (Fig. 3B). These inhibitors did not modify NTHi375 epithelial adhesion (Fig. 3A). BAY11-7083 is an irreversible inhibitor of IKKα and IκBα phosphorylation. Although CAPE inhibits NF-κB (40), it may have additional properties, such as inhibition of PI3K/AKT signaling (41). AKT phosphorylation implication in NTHi cell invasion (14) could explain the different results obtained for CAPE and BAY11-7083.
FIG 3.
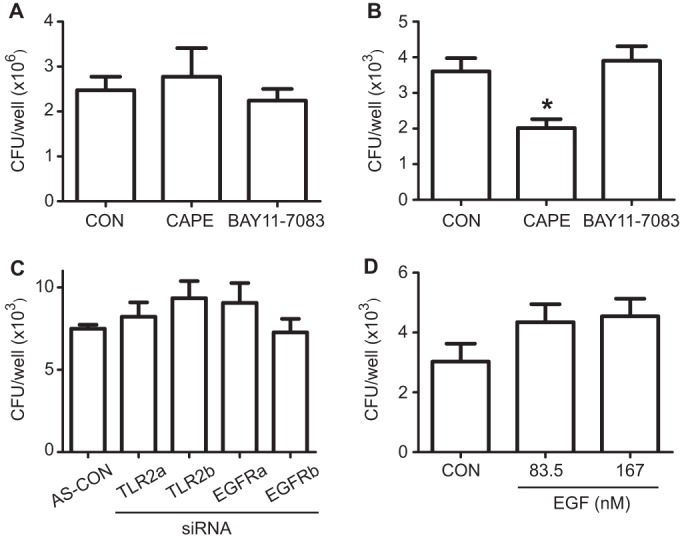
The interference of inflammation signaling has a limited effect on NTHi cell infection. Cells were left untreated or pretreated with CAPE or BAY11-7083 and then infected with NTHi375 to determine epithelial adhesion (A) or invasion (B). Comparable NTHi375 adhesion to A549 pretreated and control (CON) untreated cells was observed. The amount of intracellular NTHi375 decreased significantly upon cell pretreatment with CAPE (P < 0.05), but not with BAY11-7083. (C) Cells were treated with two siRNAs, a and b, to TLR2 and EGFR and then infected with NTHi375 to determine invasion. Comparable NTHi375 invasion of A549 siRNA-treated and control (AS-CON) cells was observed. (D) Cells were exposed to increasing concentrations of EGF and infected with NTHi375 to determine invasion, which remained unchanged.
Given that NTHi-triggered inflammation seems to be mediated by TLR2-dependent and epidermal growth factor receptor (EGFR)-dependent signaling (19, 42), we also sought to determine whether these molecules play a role in NTHi epithelial infection. NTHi375 invasion of cells transfected with two siRNAs (a and b) to TLR2 and two siRNAs (a and b) to EGFR (see Fig. S4 in the supplemental material) was similar to that observed for cells transfected with AllStars control siRNA (Fig. 3C). Similarly, exogenous addition of the natural EFGR ligand EGF did not modify NTHi375 cell invasion (Fig. 3D). In summary, the interference of a number of host elements known to be involved in NTHi-triggered inflammation did not modify bacterial entry into airway epithelial cells.
Histone/protein deacetylation and NTHi epithelial infection.
Gene clustering network created by the IPA software showed intermolecular connections for SIRT1, encoding the deacetylase sirtuin-1 (see Fig. S2B in the supplemental material). SIRT1 is a negative regulator of matrix metalloproteinase-9 (MMP9) (43) and protects against emphysema (44). Given that COPD relates to an inappropriate elevation of MMP9 (45) and to lung reduction of SIRT1 (43), the activation of SIRT1 could be an attractive therapeutic approach (46). To determine whether SIRT1 plays a role in NTHi epithelial invasion, cells were transfected with two siRNAs to SIRT1, a and b (see Fig. S4 in the supplemental material). NTHi375 invasion into cells transfected with SIRT1 siRNAs was similar to that observed for cells transfected with control AllStars siRNA (Fig. 4A). Given that the observed SIRT1 relative expression of I/NI cells could negatively regulate MMPs, we hypothesized that MMP blockage should not impair NTHi cell invasion. This was the case when NTHi375 cell invasion was tested in the presence of the MMP inhibitors GM6001 and TAPI-2 (Fig. 4B). Similarly, these drugs did not modulate bacterial cell adhesion (see Fig. S5A in the supplemental material).
FIG 4.

Effect of sirtuin-1-targeting drugs on NTHi viability and infection of A549 cells. (A) Cells were treated with two siRNAs, a and b, to SIRT1, and infected with NTHi375. Comparable NTHi375 invasion of A549 siRNA-treated and control (AS-CON) cells was observed. (B) Effect of GM6001 and TAPI-2 on NTHi375 invasion of A549 cells. Comparable NTHi375 invasion of A549-treated and control untreated cells was observed. (C) Effect of resveratrol (RESV) on NTHi375 viability as determined by comparing bacterial counts in sBHI containing RESV (□) to those obtained in sBHI containing equivalent volumes of DMSO (○). RESV displayed a dose-dependent bactericidal effect; NTHi375 survival was <50% at an RESV concentration of >175 μg/ml (P < 0.0001). (D) Effect of RESV and theophylline (TEOPH) on NTHi375 cell infection. Cells were left untreated, pretreated with RESV, or treated with TEOPH and then infected with NTHi375. Comparable NTHi375 invasion was observed for RESV/THEOPH-treated and control untreated cells.
Therapeutic activation of SIRT1 could synergize with NTHi infection, which may have unknown consequences in the lungs of the chronically infected patients. Based on this notion, we assessed the effect of resveratrol, a plant polyphenol that activates SIRT1 (47), on NTHi infection. Of note, resveratrol is bactericidal against some bacteria (48). We tested resveratrol microbicidal activity against NTHi and found that it decreased NTHi375 viability in a dose-dependent manner and that bacterial survival was less than 50% at a resveratrol concentration of >175 μg/ml (P < 0.0001) (Fig. 4C). Differently, cell pretreatment with resveratrol did not alter NTHi375 cell adhesion or invasion (Fig. 4D and see Fig. S5B in the supplemental material). As observed for resveratrol, the nonbactericidal histone deacetylase inducer theophylline (49) did not alter NTHi375 epithelial adhesion or invasion (Fig. 4D and see Fig. S5B in the supplemental material).
These results indicate that modulation of histone/protein deacetylases did not interfere with NTHi entry into airways epithelial cells. Importantly, resveratrol showed antimicrobial potential as a natural bactericidal against NTHi.
Effect of cholesterol modulation on NTHi respiratory infection.
Gene clustering network created by the IPA software showed intermolecular connections for HMGCR (see Fig. S2D in the supplemental material), the rate-controlling enzyme of the mevalonate pathway, which produces cholesterol and other isoprenoids (50). We showed previously that cholesterol is a component of the host membrane lipid rafts important for NTHi epithelial invasion (14). Supporting this notion, a temperature increase that could affect host plasma membrane fluidity (51), enhanced NTHi375 cell invasion (P < 0.0001) without modifying adhesion (Fig. 5A and B), and a recruitment of the glycosphingolipid GM1 ganglioside to the NTHi infection site could be observed (Fig. 5C). Importantly, HMGCR is the therapeutic target of statins, which are hypolipidemic drugs used to lower cholesterol in people at risk of cardiovascular disease (52). Targeting cholesterol has also become an attractive antimicrobial approach because many pathogens exploit cholesterol or lipid rafts to establish infection (53). We next sought to determine whether targeting cholesterol by a statin could modulate NTHi infection. Cell treatment with fluvastatin reduced NTHi375 cell invasion in a dose-dependent manner (FLUV, 200 μM; P < 0.05), without altering adhesion (Fig. 5D and E). Statin effect was restored by cholesterol replenishment with FCS (Fig. 5F). In the absence of FCS, fluvastatin also reduced IL-8 secretion in NTHi375-infected cells (P < 0.0001) (Fig. 5G). We next determined the effect of fluvastatin in vivo by using a mouse model of NTHi pulmonary infection and two statin regimens consisting of (i) FLUV (1 mg/kg) administration daily for 10 days prior to infection and mouse sacrifice at 24 hpi and (ii) FLUV (30 mg/kg) administration 24, 12, and 1 h prior to infection and 6 hpi and mouse sacrifice at 12 hpi. In both cases, NTHi burden in the lungs of mice treated with fluvastatin was similar to that found in the lungs of control A (CON) untreated mice (Fig. 5H). In summary, fluvastatin administration reduced NTHi invasion and IL-8 secretion by epithelial cells but did not enhance bacterial clearance in vivo.
FIG 5.

Effect of fluvastatin on NTHi respiratory infection. A549 cells were incubated at 37°C (CON), at 33°C, or at 41°C for 24 h and then infected with NTHi375 to determine epithelial adhesion (A) or invasion (B). Cell incubation at 41°C increased intracellular bacterial numbers (P < 0.0001) compared to control cells. (C) Vibrio cholerae toxin subunit B conjugated to Texas Red (red) was used to stain GM1 ganglioside. Cells were infected with NTHi375 (green). GM1 recruitment around infecting bacteria could be observed. (D to F) A549 cells were treated with various fluvastatin (FLUV) concentrations and infected with NTHi375 to determine adhesion (D) or invasion (E and F). NTHi375 adhesion to A549-treated and control (CON) untreated cells was comparable. The amount of intracellular NTHi375 decreased in a dose-dependent manner upon FLUV treatment (FLUV, 200 μM; P < 0.05). FLUV-mediated reduced NTHi375 cell invasion was restored by replenishing cholesterol with FCS (P < 0.0001). (G) Effect of FLUV on NTHi375-mediated inflammation. CON, cells uninfected that did not receive FLUV; untreat, cells infected with NTHi375 that did not receive FLUV; FLUV−FCS, cells infected with NTHi375 treated with FLUV in the absence of FCS; FLUV+FCS, cells infected with NTHi375 treated with FLUV in the presence of FCS. IL-8 was quantified by ELISA at 8 hpi. IL-8 release was lower in FLUV (P < 0.05)-treated infected A549 cells on RPMI 1640 medium without FCS during the gentamicin incubation period. (H) Effect of FLUV in the lungs of mice infected by NTHi. Mice were infected intranasally with ∼108 bacteria/mouse. FLUV (1 mg/kg) was administered i.p. daily 10 days prior to infection (left, black symbols); FLUV (30 mg/kg) was administered i.p. 24, 12, and 1 h before infection and at 6 hpi (right, open symbols). Control A untreated mice (CON) are represented by circles; FLUV-treated mice are represented by squares. Bacterial counts in lungs were determined at 24 hpi (left) or 12 hpi (right), respectively (log10 CFU/lung).
Modulation of PKA signaling and respiratory infection by NTHi.
We reported previously that intracellular cyclic AMP (cAMP) increase by activation of adenylate cyclases reduces NTHi cell invasion (13). Intracellular cAMP formation is initiated by stimulation of Gs-protein-coupled 7-transmembrane receptors, such as the β2-adrenergic receptors (54), and its levels are tightly controlled by PDEs (55). The downstream cAMP effectors are PKA and the cAMP-regulated guanine nucleotide exchange factors Epac1 and Epac2 (54). The PKA signaling axis contains several drug targets, including β2 adrenoreceptors and PDEs, targeted by β2 agonists and PDE inhibitors, respectively (56, 57). Given that our IPA analysis showed a high-rated function for PKA signaling in NTHi375-infected cells (Fig. 1B) and that gene clustering network 8 showed intermolecular connections for several PDEs (see Fig. S2C in the supplemental material), we assessed the effect of interfering PKA signaling on NTHi infection. First, we tested the effect of the β2 agonists salmeterol and formoterol. Cell treatment with formoterol, but not with salmeterol, reduced (P = 0.0005) NTHi375 invasion (Fig. 6A). These β2 agonists did not alter NTHi375 epithelial adhesion (see Fig. S5C in the supplemental material). Formoterol effect was restored by the adrenoceptor blocker propranolol (P < 0.005) (Fig. 6A). Second, intracellular cAMP levels were increased by cell exposure to (i) db-cAMP, a mimic of endogenous cAMP, (ii) IBMX, a nonselective PDE inhibitor, and (iii) rolipram and roflumilast, two PDE4 inhibitors. Cell exposure to these agents reduced NTHi375 invasion (db-cAMP, P < 0.05; ROFLUM, P < 0.05; ROLIP, P < 0.005) (Fig. 6B). Third, we tested the effect of cAMP effector modulation. In agreement with previous data obtained by using H-89 (13), cell exposure to the PKA inhibitor PKI increased significantly (P < 0.0001) NTHi375 invasion; conversely, the selective EPAC activator 8-pCPT-2′-O-Me-cAMP did not modify bacterial invasion (Fig. 6C). None of these drugs altered NTHi epithelial adhesion (see Fig. S5D in the supplemental material). We next determined the effect of rolipram administration in vivo by using a mouse model of NTHi lung infection and a regimen consisting of rolipram (10 mg/kg) oral administration at 24, 12, and 1 h prior to infection and 6 hpi. At 12 hpi, bacterial burden in the lungs of mice treated with rolipram was lower (P < 0.05) than that found in the lungs of control B untreated mice (Fig. 6D).
FIG 6.

PDE inhibitors have therapeutic potential against respiratory infection by NTHi. Effect of β2 agonists on NTHi375 infection of A549 type II pneumocytes. Cells received various treatments—salmeterol (SALM), formoterol (FORM), or propranolol (PROP) (A), db-cAMP, IBMX, roflumilast (ROFLUM), or rolipram (ROLIP) (B), or 8-pCPT-2′-O-Me-cAMP or PKI (C)—and bacterial invasion was assessed. The mean numbers for NTHi375 invasion into FORM-treated cells were lower than those obtained for control (CON) untreated cells (P = 0.0005). The FORM-mediated effect was restored by the addition of PROP (P < 0.005). The mean numbers for NTHi375 invasion into db-cAMP-, ROFLUM-, and ROLIP-treated cells were lower than those obtained for CON cells (db-cAMP, P < 0.05; ROFLUM, P < 0.05; ROLIP, P < 0.01). The mean numbers for NTHi375 invasion into PKI-treated cells were higher than those obtained for CON cells (P < 0.0001). (D) Effect of ROLIP on bacterial counts in lungs. Mice were infected intranasally with ∼108 bacteria/mouse. ROLIP (10 mg/kg) was administered orally 24, 12, and 1 h prior to infection and at 6 hpi. Bacterial counts were determined at 12 hpi (log10 CFU/lung). The NTHi375 counts were significantly lower in the lungs of mice treated with ROLIP than in those of control B (CON) untreated mice (P < 0.05).
These results indicate that a therapeutic increase in intracellular cAMP through β2 agonists or PDE inhibitors reduced NTHi cell invasion. Moreover, rolipram reduced lung bacterial counts in vivo, supporting its potential as a host-directed therapy against NTHi respiratory infection.
DISCUSSION
This study investigated the efficacy of host-directed antimicrobial therapies against respiratory infection by NTHi. Our approach relied on the identification of host cell pathways subverted by this pathogen for intracellular invasion. Therapeutic modulation of such pathogen-hijacked host functions may have an antimicrobial effect and, ultimately, impair the progression of the infection. We used cell genome profiling and unraveled a NTHi-triggered cell transcriptional signature, which revealed a panel of host-directed target candidates whose therapeutic potential was further explored by analyzing the effect of their interference by specific drugs. Our work on cultured cells and murine model systems of NTHi infection narrowed the microarray-generated panel of host target and drug candidates to resveratrol, a bactericidal natural polyphenol which also activates sirtuin-1, and to the nonbactericidal inhibition of PDE4. We acknowledge that genome expression profiling was performed on one cultured cell line with one NTHi clinical isolate, which may limit the results obtained. However, to our knowledge, this is the first study based on host-directed therapeutics devoted to identify novel treatment opportunities against NTHi and also the first report on the antimicrobial potential of resveratrol and rolipram against NTHi respiratory infection.
Given that epithelial invasion may allow NTHi to temporarily evade the host immune system (17, 18), we took intracellular invasion as an assay to test the microarray-generated panel of host target and drug candidates. This type of assay has been used to perform a chemical screening of drugs blocking the entry and/or the intracellular growth of Coxiella burnetii, Legionella pneumophila, Brucella abortus, Rickettsia conorii, or Mycobacterium tuberculosis (58, 59). Of note, some of the drugs tested here did not alter NTHi adhesion to and invasion of A549 epithelial cells or, if they were found to impair NTHi cell invasion, they did not favor bacterial clearance in vivo. Our results highlight the advantages and limitations of genome expression profiling, demonstrating that this screening method generates an expression signature contributing to our global understanding of host functions subverted by the pathogen and also that, as a discovery method for host therapeutics, this approach needs to be further sustained by experimental models.
Several inflammation-related genes and cell functions were differentially expressed in NTHi-infected cells. This observation, in agreement with previous studies (19), attracted our interest because (i) persistent NTHi respiratory infection is linked to COPD exacerbation and irreversible progression, (ii) inflammation is a hallmark of COPD, and (iii) COPD patients are administered anti-inflammatory drugs. Moreover, glucocorticoids modulate NTHi-triggered inflammation signaling (21, 24). Here, we observed that the inhibition of MKP1, p38 MAPK and JNK, but not of ERK MAPK, NF-κB, TLR2, or EGFR, reduced NTHi invasion. Conversely, although dexamethasone and fluticasone propionate reduced the epithelial inflammatory response elicited by NTHi, it neither affected NTHi invasion nor had a beneficial effect on bacterial clearance from the lungs of infected mice. Our results support a previous report showing that dexamethasone treatment on cigarette-smoke-exposed mice challenged with NTHi compromised the clearance of the bacteria (25). Treatment of respiratory patients with inhaled glucocorticoids has also been associated with an excess risk of pneumonia hospitalization, followed by death (60). These evidences suggest that corticosteroids may facilitate infections despite their anti-inflammatory efficacy.
Cardiovascular comorbidity generates a frequent use of statins in COPD patients whose effect on preventing exacerbations in moderate-to-severe patients is under debate (61, 62). Previous studies have suggested the involvement of cholesterol-rich lipid rafts in NTHi intracellular invasion (13, 14), and expression profiling network clustering highlighted HMGCR in NTHi-infected cells. Here, we observed that fluvastatin reduced NTHi invasion and the inflammatory response elicited by NTHi on epithelial cells. However, it did not have a beneficial effect on bacterial clearance from the lungs of infected mice. These results highlight the need of using multiple experimental models, especially in vivo model systems. We acknowledge that statins are a group of cholesterol-lowering drugs with different properties, such as different antimicrobial effects (63). Here, we examined fluvastatin administered i.p. to mice. Other statins, together with alternative routes of administration in vivo, will be the subject of further studies.
Modulation of intracellular cAMP is another therapeutic approach used to treat COPD, achieved by using β2 agonists and PDE4 inhibitors. Given that PKA signaling was differentially expressed in NTHi-infected cells and considering previous reports on PDE4B overexpression upon NTHi infection (28, 29), we used a panel of drugs to interfere PKA signaling at different levels. A549 cells have been shown to express β-adrenoreceptors (64), and we used two β2 agonists that yielded different results in terms of NTHi intracellular invasion. The structure, affinity, efficacy, kinetics, and ability to elevate intracellular cAMP vary for salmeterol and formoterol (57, 65), which could explain the differences observed. We also assessed the effect of increasing intracellular cAMP by inhibiting PDEs. Both nonselective and PDE4 selective inhibition reduced NTHi epithelial invasion and, importantly, in vivo treatment with rolipram showed a bacterial clearing effect. Supporting the therapeutic use of targeting PDE4, the ototopical postinoculation administration of roflumilast seems to suppress NTHi-induced inflammation in vivo (28). Roflumilast is currently administered to COPD patients (66). Despite its anti-inflammatory effect (28), it seems to also synergize with NTHi to induce PDE4B, which may contribute to developing tolerance (29). Future work will further analyze the effect of roflumilast on NTHi respiratory infection.
Although we sought here to identify antimicrobial drugs targeting host targets, we also observed a bactericidal effect for resveratrol against NTHi, an effect similar to that shown before for other bacteria (48). Given that resveratrol is both bactericidal against NTHi and an activator of sirtuin-1 and that it has been proposed to have beneficial health effects due to its antioxidant and anti-inflammatory properties (67), we speculate that resveratrol could be a useful therapy against respiratory NTHi infection associated with COPD. Further studies will tackle the resveratrol bactericidal effect and mechanisms on collections of NTHi clinical isolates of different pathological origins and its efficacy in vivo.
In sum, our genome expression profiling of host target candidates for intervention against NTHi respiratory infection revealed a whole range of therapeutic opportunities, and our further assessment showed a therapeutic potential for resveratrol and rolipram. Host modulation is likely to avoid selective pressure for the evolution of microbial resistance and to afford broad-spectrum protection, but it must not deleteriously affect helpful elements of the immune response (68). This tight balance should be necessarily taken into consideration by researchers and clinicians interested in exploring the antimicrobial benefits of host-directed therapeutics.
Supplementary Material
ACKNOWLEDGMENTS
We thank Antonio López-Gómez and Javier Margareto for technical work.
J.M. is funded by Ph.D. studentship BES-2013-062644 from the Ministerio Economía y Competitividad-MINECO, Spain. This study was funded by grants from ISCIII (PS09/00130), the Ministerio Economía y Competitividad (MINECO SAF2012-31166), and the Departamento de Salud Gobierno Navarra (359/2012) to J.G. CIBERES is an initiative from ISCIII, Spain.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AAC.01278-15.
REFERENCES
- 1.Martinez JL. 2014. General principles of antibiotic resistance in bacteria. Drug Discov Today Technol 11:33–39. doi: 10.1016/j.ddtec.2014.02.001. [DOI] [PubMed] [Google Scholar]
- 2.Ranjan A, Pothayee N, Seleem MN, Boyle SM, Kasimanickam R, Riffle JS, Sriranganathan N. 2012. Nanomedicine for intracellular therapy. FEMS Microbiol Lett 332:1–9. doi: 10.1111/j.1574-6968.2012.02566.x. [DOI] [PubMed] [Google Scholar]
- 3.Stanton TB. 2013. A call for antibiotic alternatives research. Trends Microbiol 21:111–113. doi: 10.1016/j.tim.2012.11.002. [DOI] [PubMed] [Google Scholar]
- 4.Frank CG, Bengoechea JA. 2010. Functional genomics to identify therapeutic prophylactic targets. Environ Microbiol Rep 2:219–227. doi: 10.1111/j.1758-2229.2009.00068.x. [DOI] [PubMed] [Google Scholar]
- 5.Schwegmann A, Brombacher F. 2008. Host-directed drug targeting of factors hijacked by pathogens. Sci Signal 1:re8. doi: 10.1126/scisignal.129re8. [DOI] [PubMed] [Google Scholar]
- 6.Agrawal A, Murphy TF. 2011. Haemophilus influenzae infections in the H. influenzae type b conjugate vaccine era. J Clin Microbiol 49:3728–3732. doi: 10.1128/JCM.05476-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Garmendia J, Viadas C, Calatayud L, Mell JC, Marti-Lliteras P, Euba B, Llobet E, Gil C, Bengoechea JA, Redfield RJ, Liñares J. 2014. Characterization of nontypable Haemophilus influenzae isolates recovered from adult patients with underlying chronic lung disease reveals genotypic and phenotypic traits associated with persistent infection. PLoS One 9:e97020. doi: 10.1371/journal.pone.0097020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Murphy TF, Brauer AL, Schiffmacher AT, Sethi S. 2004. Persistent colonization by Haemophilus influenzae in chronic obstructive pulmonary disease. Am J Respir Crit Care Med 170:266–272. doi: 10.1164/rccm.200403-354OC. [DOI] [PubMed] [Google Scholar]
- 9.Clementi CF, Hakansson AP, Murphy TF. 2014. Internalization and trafficking of nontypeable Haemophilus influenzae in human respiratory epithelial cells and roles of IgA1 proteases for optimal invasion and persistence. Infect Immun 82:433–444. doi: 10.1128/IAI.00864-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Euba B, Moleres J, Viadas C, Ruiz de los Mozos I, Valle J, Bengoechea JA, Garmendia J. 2015. Relative contribution of P5 and Hap surface proteins to nontypable Haemophilus influenzae interplay with the host upper and lower airways. PLoS One 10:e0123154. doi: 10.1371/journal.pone.0123154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Forsgren J, Samuelson A, Ahlin A, Jonasson J, Rynnel-Dagoo B, Lindberg A. 1994. Haemophilus influenzae resides and multiplies intracellularly in human adenoid tissue as demonstrated by in situ hybridization and bacterial viability assay. Infect Immun 62:673–679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ketterer MR, Shao JQ, Hornick DB, Buscher B, Bandi VK, Apicella MA. 1999. Infection of primary human bronchial epithelial cells by Haemophilus influenzae: macropinocytosis as a mechanism of airway epithelial cell entry. Infect Immun 67:4161–4170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.López-Gomez A, Cano V, Moranta D, Morey P, García del Portillo F, Bengoechea JA, Garmendia J. 2012. Host cell kinases, α5 and β1 integrins, and Rac1 signalling on the microtubule cytoskeleton are important for non-typable Haemophilus influenzae invasion of respiratory epithelial cells. Microbiology 158:2384–2398. doi: 10.1099/mic.0.059972-0. [DOI] [PubMed] [Google Scholar]
- 14.Morey P, Cano V, Martí-Lliteras P, López-Gomez A, Regueiro V, Saus C, Bengoechea JA, Garmendia J. 2011. Evidence for a non-replicative intracellular stage of nontypable Haemophilus influenzae in epithelial cells. Microbiology 157:234–250. doi: 10.1099/mic.0.040451-0. [DOI] [PubMed] [Google Scholar]
- 15.St Geme JW III, Falkow S. 1990. Haemophilus influenzae adheres to and enters cultured human epithelial cells. Infect Immun 58:4036–4044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Swords WE, Buscher BA, Ver Steeg Ii K, Preston A, Nichols WA, Weiser JN, Gibson BW, Apicella MA. 2000. Non-typeable Haemophilus influenzae adhere to and invade human bronchial epithelial cells via an interaction of lipooligosaccharide with the PAF receptor. Mol Microbiol 37:13–27. doi: 10.1046/j.1365-2958.2000.01952.x. [DOI] [PubMed] [Google Scholar]
- 17.Clementi CF, Murphy TF. 2011. Non-typeable Haemophilus influenzae invasion and persistence in the human respiratory tract. Front Cell Infect Microbiol 1:1. doi: 10.3389/fcimb.2011.00001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Garmendia J, Martí-Lliteras P, Moleres J, Puig C, Bengoechea JA. 2012. Genotypic and phenotypic diversity of the noncapsulated Haemophilus influenzae: adaptation and pathogenesis in the human airways. Int Microbiol 15:159–172. [DOI] [PubMed] [Google Scholar]
- 19.Wang WY, Lim JH, Li JD. 2012. Synergistic and feedback signaling mechanisms in the regulation of inflammation in respiratory infections. Cell Mol Immunol 9:131–135. doi: 10.1038/cmi.2011.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Earl CS, Keong TW, An SQ, Murdoch S, McCarthy Y, Garmendia J, Ward J, Dow JM, Yang L, O'Toole GA, Ryan RP. 2015. Haemophilus influenzae responds to glucocorticoids used in asthma therapy by modulation of biofilm formation and antibiotic resistance. EMBO Mol Med 7:1018–1033. doi: 10.15252/emmm.201505088. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 21.Imasato A, Desbois-Mouthon C, Han J, Kai H, Cato AC, Akira S, Li JD. 2002. Inhibition of p38 MAPK by glucocorticoids via induction of MAPK phosphatase-1 enhances nontypeable Haemophilus influenzae-induced expression of Toll-like receptor 2. J Biol Chem 277:47444–47450. doi: 10.1074/jbc.M208140200. [DOI] [PubMed] [Google Scholar]
- 22.Komatsu K, Jono H, Lim JH, Imasato A, Xu H, Kai H, Yan C, Li J-D. 2008. Glucocorticoids inhibit nontypeable Haemophilus influenzae-induced MUC5AC mucin expression via MAPK phosphatase-1-dependent inhibition of p38 MAPK. Biochem Biophys Res Commun 377:763–768. doi: 10.1016/j.bbrc.2008.10.091. [DOI] [PubMed] [Google Scholar]
- 23.Miyata M, Lee JY, Susuki-Miyata S, Wang WY, Xu H, Kai H, Kobayashi KS, Flavell RA, Li JD. 2015. Glucocorticoids suppress inflammation via the upregulation of negative regulator IRAK-M. Nat Commun 6:6062. doi: 10.1038/ncomms7062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shuto T, Imasato A, Jono H, Sakai A, Xu H, Watanabe T, Rixter D, Kai H, Andalibi A, Linthicum F, Guan Y, Han J, Cato A, Lim D, Akira S, Li J. 2002. Glucocorticoids synergistically enhance nontypeable Haemophilus influenzae-induced Toll-like receptor 2 expression via a negative crosstalk with p38 MAP kinase. J Biol Chem 277:17263–17270. doi: 10.1074/jbc.M112190200. [DOI] [PubMed] [Google Scholar]
- 25.Gaschler GJ, Skrtic M, Zavitz CC, Lindahl M, Onnervik PO, Murphy TF, Sethi S, Stampfli MR. 2009. Bacteria challenge in smoke-exposed mice exacerbates inflammation and skews the inflammatory profile. Am J Respir Crit Care Med 179:666–675. doi: 10.1164/rccm.200808-1306OC. [DOI] [PubMed] [Google Scholar]
- 26.Dowling RB, Johnson M, Cole PJ, Wilson R. 1998. Effect of salmeterol on Haemophilus influenzae infection of respiratory mucosa in vitro. Eur Respir J 11:86–90. doi: 10.1183/09031936.98.11010086. [DOI] [PubMed] [Google Scholar]
- 27.Maris N, Florquin S, van't Veer C, de Vos A, Buurman W, Jansen H, van der Poll T. 2006. Inhalation of β2 agonists impairs the clearance of nontypable Haemophilus influenzae from the murine respiratory tract. Respir Res 7:57. doi: 10.1186/1465-9921-7-57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Komatsu K, Lee JY, Miyata M, Hyang Lim J, Jono H, Koga T, Xu H, Yan C, Kai H, Li JD. 2013. Inhibition of PDE4B suppresses inflammation by increasing expression of the deubiquitinase CYLD. Nat Commun 4:1684. doi: 10.1038/ncomms2674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Susuki-Miyata S, Miyata M, Lee BC, Xu H, Kai H, Yan C, Li JD. 2015. Cross-talk between PKA-Cβ and p65 mediates synergistic induction of PDE4B by roflumilast and NTHi. Proc Natl Acad Sci U S A 112:E1800–E1809. doi: 10.1073/pnas.1418716112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mell JC, Sinha S, Balashov S, Viadas C, Grassa CJ, Ehrlich GD, Nislow C, Redfield RJ, Garmendia J. 2014. Complete genome sequence of Haemophilus influenzae strain 375 from the middle ear of a pediatric patient with otitis media. Genome Announc 2:e01245-14. doi: 10.1128/genomeA.01245-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gentleman R, Carey V, Huber W, Irizarry R, Dudoit S. 2005. Bioinformatics and computational biology solutions using R and Bioconductor. Springer, New York, NY. [Google Scholar]
- 32.Smyth GK. 2004. Linear models and empirical bayes methods for assessing differential expression in microarray experiments. Stat Appl Gen Mol Biol 3:Article3. [DOI] [PubMed] [Google Scholar]
- 33.Draghici S. 2003. Data analysis tools for DNA microarrays. Chapman & Hall/CRC, London, England. [Google Scholar]
- 34.Euba B, Moleres J, Viadas C, Barberan M, Caballero L, Grillo MJ, Bengoechea JA, de-Torres JP, Linares J, Leiva J, Garmendia J. 2015. Relationship between azithromycin susceptibility and administration efficacy for nontypeable Haemophilus influenzae respiratory infection. Antimicrob Agents Chem 59:2700–2712. doi: 10.1128/AAC.04447-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Morey P, Viadas C, Euba B, Hood DW, Barberan M, Gil C, Grillo MJ, Bengoechea JA, Garmendia J. 2013. Relative contributions of lipooligosaccharide inner and outer core modifications to nontypeable Haemophilus influenzae pathogenesis. Infect Immun 81:4100–4111. doi: 10.1128/IAI.00492-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Krakauer T, Buckley M. 2006. Dexamethasone attenuates staphylococcal enterotoxin B-induced hypothermic response and protects mice from superantigen-induced toxic shock. Antimicrob Agents Chemother 50:391–395. doi: 10.1128/AAC.50.1.391-395.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jin Y, Tachibana I, Takeda Y, He P, Kang S, Suzuki M, Kuhara H, Tetsumoto S, Tsujino K, Minami T, Iwasaki T, Nakanishi K, Kohmo S, Hirata H, Takahashi R, Inoue K, Nagatomo I, Kida H, Kijima T, Ito M, Saya H, Kumanogoh A. 2013. Statins decrease lung inflammation in mice by upregulating tetraspanin CD9 in macrophages. PLoS One 8:e73706. doi: 10.1371/journal.pone.0073706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Soares AC, Souza DG, Pinho V, Vieira AT, Barsante MM, Nicoli JR, Teixeira M. 2003. Impaired host defense to Klebsiella pneumoniae infection in mice treated with the PDE4 inhibitor rolipram. Br J Pharmacol 140:855–862. doi: 10.1038/sj.bjp.0705517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang WY, Komatsu K, Huang Y, Wu J, Zhang W, Lee JY, Miyata M, Xu H, Li JD. 2014. CYLD negatively regulates nontypeable Haemophilus influenzae-induced IL-8 expression via phosphatase MKP-1-dependent inhibition of ERK. PLoS One 9:e112516. doi: 10.1371/journal.pone.0112516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Natarajan K, Singh S, Burke TR, Grunberger D, Aggarwal BB. 1996. Caffeic acid phenethyl ester is a potent and specific inhibitor of activation of nuclear transcription factor NF-κB. Proc Natl Acad Sci U S A 93:9090–9095. doi: 10.1073/pnas.93.17.9090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chuu C-P, Lin H-P, Ciaccio MF, Kokontis JM, Hause RJ, Hiipakka RA, Liao S, Jones RB. 2012. Caffeic acid phenethyl ester suppresses the proliferation of human prostate cancer cells through inhibition of p70S6K and Akt signaling networks. Cancer Prevent Res 5:788–797. doi: 10.1158/1940-6207.CAPR-12-0004-T. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Xu X, Steere RR, Fedorchuk CA, Pang J, Lee J-Y, Lim JH, Xu H, Pan ZK, Maggirwar SB, Li J-D. 2011. Activation of epidermal growth factor receptor is required for NTHi-induced NF-κB-dependent inflammation. PLoS One 6:e28216. doi: 10.1371/journal.pone.0028216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nakamaru Y, Vuppusetty C, Wada H, Milne JC, Ito M, Rossios C, Elliot M, Hogg J, Kharitonov S, Goto H, Bemis JE, Elliott P, Barnes PJ, Ito K. 2009. A protein deacetylase SIRT1 is a negative regulator of metalloproteinase-9. FASEB J 23:2810–2819. doi: 10.1096/fj.08-125468. [DOI] [PubMed] [Google Scholar]
- 44.Yao H, Chung S, Hwang J-W, Rajendrasozhan S, Sundar IK, Dean DA, McBurney MW, Guarente L, Gu W, Rönty M, Kinnula VL, Rahman I. 2012. SIRT1 protects against emphysema via FOXO3-mediated reduction of premature senescence in mice. J Clin Invest 122:2032–2045. doi: 10.1172/JCI60132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Russell RE, Culpitt SV, DeMatos C, Donnelly L, Smith M, Wiggins J, Barnes PJ. 2002. Release and activity of matrix metalloproteinase-9 and tissue inhibitor of metalloproteinase-1 by alveolar macrophages from patients with chronic obstructive pulmonary disease. Am J Respir Cell Mol Biol 26:602–609. doi: 10.1165/ajrcmb.26.5.4685. [DOI] [PubMed] [Google Scholar]
- 46.Ito K, Colley T, Mercado N. 2012. Geroprotectors as a novel therapeutic strategy for COPD, an accelerating aging disease. Int J Chron Obstruct Pulmon Dis 7:641–652. doi: 10.2147/COPD.S28250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Knutson MD, Leeuwenburgh C. 2008. Resveratrol and novel potent activators of SIRT1: effects on aging and age-related diseases. Nutr Rev 66:591–596. doi: 10.1111/j.1753-4887.2008.00109.x. [DOI] [PubMed] [Google Scholar]
- 48.Nawrocki EM, Bedell HW, Humphreys TL. 2013. Resveratrol is cidal to both classes of Haemophilus ducreyi. Int J Antimicrob Agents 41:477–479. doi: 10.1016/j.ijantimicag.2013.02.008. [DOI] [PubMed] [Google Scholar]
- 49.Cosio BG, Soriano JB. 2009. Theophylline again? Reasons for believing. Eur Respir J 34:5–6. doi: 10.1183/09031936.00011309. [DOI] [PubMed] [Google Scholar]
- 50.Singh RP, Kumar R, Kapur N. 2003. Molecular regulation of cholesterol biosynthesis: implications in carcinogenesis. J Environ Pathol Toxicol Oncol 22:75–92. [DOI] [PubMed] [Google Scholar]
- 51.Hoffmann C, Berking A, Agerer F, Buntru A, Neske F, Chhatwal GS, Ohlsen K, Hauck CR. 2010. Caveolin limits membrane microdomain mobility and integrin-mediated uptake of fibronectin-binding pathogens. J Cell Sci 123:4280–4291. doi: 10.1242/jcs.064006. [DOI] [PubMed] [Google Scholar]
- 52.Opie LH. 2015. Present status of statin therapy. Trends Cardiovasc Med 25:216–225. doi: 10.1016/j.tcm.2014.10.002. [DOI] [PubMed] [Google Scholar]
- 53.Lafont F, van der Goot FG. 2005. Bacterial invasion via lipid rafts. Cell Microbiol 7:613–620. doi: 10.1111/j.1462-5822.2005.00515.x. [DOI] [PubMed] [Google Scholar]
- 54.Dekkers BG, Racke K, Schmidt M. 2013. Distinct PKA and Epac compartmentalization in airway function and plasticity. Pharmacol Ther 137:248–265. doi: 10.1016/j.pharmthera.2012.10.006. [DOI] [PubMed] [Google Scholar]
- 55.Houslay MD. 2010. Underpinning compartmentalized cAMP signalling through targeted cAMP breakdown. Trends Biochem Sci 35:91–100. doi: 10.1016/j.tibs.2009.09.007. [DOI] [PubMed] [Google Scholar]
- 56.Barnes PJ. 2006. Novel signal transduction modulators for the treatment of airway diseases. Pharmacol Ther 109:238–245. doi: 10.1016/j.pharmthera.2005.08.001. [DOI] [PubMed] [Google Scholar]
- 57.Johnson M. 2001. β2-adrenoceptors: mechanisms of action of β2-agonists. Paediatr Respir Rev 2:57–62. [DOI] [PubMed] [Google Scholar]
- 58.Czyz DM, Potluri LP, Jain-Gupta N, Riley SP, Martinez JJ, Steck TL, Crosson S, Shuman HA, Gabay JE. 2014. Host-directed antimicrobial drugs with broad-spectrum efficacy against intracellular bacterial pathogens. mBio 5:e01534-14. doi: 10.1128/mBio.01534-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Stanley SA, Barczak AK, Silvis MR, Luo SS, Sogi K, Vokes M, Bray M-A, Carpenter AE, Moore CB, Siddiqi N, Rubin EJ, Hung DT. 2014. Identification of host-targeted small molecules that restrict intracellular Mycobacterium tuberculosis growth. PLoS Pathog 10:e1003946. doi: 10.1371/journal.ppat.1003946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ernst P, Gonzalez AV, Brassard P, Suissa S. 2007. Inhaled corticosteroid use in chronic obstructive pulmonary disease and the risk of hospitalization for pneumonia. Am J Respir Crit Care Med 176:162–166. doi: 10.1164/rccm.200611-1630OC. [DOI] [PubMed] [Google Scholar]
- 61.Criner GJ, Connett JE, Aaron SD, Albert RK, Bailey WC, Casaburi R, Cooper JAD, Curtis JL, Dransfield MT, Han MK, Make B, Marchetti N, Martinez FJ, Niewoehner DE, Scanlon PD, Sciurba FC, Scharf SM, Sin DD, Voelker H, Washko GR, Woodruff PG, Lazarus SC. 2014. Simvastatin for the prevention of exacerbations in moderate-to-severe COPD. N Engl J Med 370:2201–2210. doi: 10.1056/NEJMoa1403086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Young RP, Hopkins RJ, Agusti A. 2014. Statins as adjunct therapy in COPD: how do we cope after STATCOPE? Thorax 69:891–894. doi: 10.1136/thoraxjnl-2014-205814. [DOI] [PubMed] [Google Scholar]
- 63.Jerwood S, Cohen J. 2008. Unexpected antimicrobial effect of statins. J Antimicrob Chemother 61:362–364. [DOI] [PubMed] [Google Scholar]
- 64.Abraham G, Kneuer C, Ehrhardt C, Honscha W, Ungemach FR. 2004. Expression of functional β2-adrenergic receptors in the lung epithelial cell lines 16HBE14o(−), Calu-3 and A549. Biochem Biophys Acta 1691:169–179. doi: 10.1016/j.bbamcr.2004.02.002. [DOI] [PubMed] [Google Scholar]
- 65.Donnelly LE, Tudhope SJ, Fenwick PS, Barnes PJ. 2010. Effects of formoterol and salmeterol on cytokine release from monocyte-derived macrophages. Eur Respir J 36:178–186. doi: 10.1183/09031936.00158008. [DOI] [PubMed] [Google Scholar]
- 66.Lipari M, Benipal H, Kale-Pradhan P. 2013. Roflumilast in the management of chronic obstructive pulmonary disease. Am J Health System Pharm 70:2087–2095. doi: 10.2146/ajhp130114. [DOI] [PubMed] [Google Scholar]
- 67.Juhasz B, Varga B, Gesztelyi R, Kemeny-Beke A, Zsuga J, Tosaki A. 2010. Resveratrol: a multifunctional cytoprotective molecule. Curr Pharm Biotechnol 11:810–818. doi: 10.2174/138920110793262079. [DOI] [PubMed] [Google Scholar]
- 68.Hancock RE, Nijnik A, Philpott DJ. 2012. Modulating immunity as a therapy for bacterial infections. Nat Rev Microbiol 10:243–254. doi: 10.1038/nrmicro2745. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.