

Abstract
Various protease inhibitors (PIs) currently are becoming available for treatment of hepatitis C virus (HCV). For genotype 1, substitutions at NS3 protease positions 155, 156, and 168 are the main determinants of PI resistance. For other genotypes, similar substitutions were selected during PI treatment but were not characterized systematically. To elucidate the impact of key PI resistance substitutions on genotypes 2 to 6, we engineered the substitutions R155A/E/G/H/K/Q/T, A156G/S/T/V, and D/Q168A/E/G/H/N/V into HCV recombinants expressing genotype 2 to 6 proteases. We evaluated viral fitness and sensitivity to nine PIs (telaprevir, boceprevir, simeprevir, asunaprevir, vaniprevir, faldaprevir, paritaprevir, deldeprevir, and grazoprevir) in Huh7.5 cells. We found that most variants showed decreased fitness compared to that of the original viruses. Overall, R155K, A156G/S, and D/Q168A/E/H/N/V variants showed the highest fitness; however, genotype 4 position 168 variants showed strong fitness impairment. Most variants tested were resistant to several PIs. Resistance levels varied significantly depending on the specific substitution, genotype, and PI. For telaprevir and boceprevir, specific 155 and 156, but not 168, variants proved resistant. For the remaining PIs, most genotype 2, 4, 5, and 6, but not genotype 3, variants showed various resistance levels. Overall, grazoprevir (MK-5172) had the highest efficacy against original viruses and variants. This is the first comprehensive study revealing the impact of described key PI resistance substitutions on fitness and PI resistance of HCV genotypes 2 to 6. In conclusion, the studied substitutions induced resistance to a panel of clinically relevant PIs, including the newer PIs paritaprevir, deldeprevir, and grazoprevir. We discovered complex patterns of resistance, with the impact of substitutions varying from increased sensitivity to high resistance.
INTRODUCTION
Hepatitis C virus (HCV) chronically infects ∼150 million people worldwide. Interferon-free treatment regimens based on direct-acting antivirals (DAAs) are currently being defined (1). Despite their high efficacy, DAA-resistant variants are expected to develop in 5 to 15% of treated patients and might be spreading in populations (1, 2). The HCV NS3 protease (NS3P) is, in association with cofactor NS4A, essential for HCV replication; furthermore, it inactivates cellular proteins mediating innate antiviral responses (3). NS3P inhibitors (PIs) are expected to be an important component of interferon-free treatment regimens (1). Telaprevir, boceprevir, simeprevir, and paritaprevir have been licensed, while several additional PIs are in the final stages of clinical development (1). For HCV, six epidemiologically important genotypes have been described, differing in ∼30% of their sequence and in their sensitivity to antivirals (1, 4–7). In Europe and the Americas, genotype 1 is most common, followed by genotypes 2 and 3. However, worldwide, genotypes 4, 5, and 6 cause >20% of all infections and are spreading beyond their primary geographical localizations in Africa and Asia (8).
Initial studies of DAAs have focused on genotype 1. For this genotype, substitutions at NS3P amino acid (aa) positions 155, 156, and 168 are selected during PI treatment in the clinic and in in vitro studies and have been demonstrated to confer high-level PI resistance in vitro (9, 10). The likelihood with which a resistant variant is selected during treatment and persists following the end of treatment is determined by the degree of resistance and by the fitness of the specific variant (9). Recent reports suggest that genotypes 2 to 6 also acquire substitutions at NS3P positions 155, 156, and 168 during PI treatment; however, their impact on resistance to different PIs and on viral fitness has not been characterized systematically (11–26). In addition, substitutions at these positions are found at low frequency in treatment-naive HCV-infected patients (9, 10, 20, 27–29). Thus, substitutions at NS3P aa 155, 156, and 168 appear to be key determinants of PI resistance.
Using cell culture infectious HCV recombinants with genotype 2 to 6-specific NS3P and NS4A (5, 7, 30, 31), we have characterized the impact of 17 previously described PI resistance substitutions at NS3P positions 155, 156, and 168 on viral fitness and resistance to nine clinically relevant PIs, including PIs developed for improved efficacy against various genotypes and resistant variants, such as paritaprevir, deldeprevir, and grazoprevir. While most variants showed decreased fitness and resistance to several PIs, this comprehensive study revealed complex patterns depending on the specific substitution, the genotype, and the PI.
MATERIALS AND METHODS
HCV recombinants.
Molecular clones with NS3P/NS4A specific for genotype(isolate) 1a(TN) (31, 32), 2a(J6) (5, 33), 2a(JFH1) (30, 34), 3a(452) (5, 35), 3a(S52) (5, 36), 4a(ED43) (7, 36), 5a(SA13) (5), and 6a(HK6a) (5) were previously developed (see Fig. S1 in the supplemental material). Variants with nucleotide changes specified in Table S1, encoding amino acid substitutions at NS3P position 155, 156, or 168 (relative H77 [GenBank accession number AF009606] amino acid reference numbers are used throughout) were generated using restriction enzyme-based cloning of PCR amplicons (Pfu DNA polymerase; Stratagene) or of chemically synthesized DNA fragments (GenScript). HCV sequences of final DNA preparations were confirmed (Macrogen).
Transfection, viral passage, and evaluation of Huh7.5 cell cultures.
Huh7.5 cells were transfected with HCV RNA transcripts using Lipofectamine and a standardized protocol (37); in each experiment transcripts from control viruses were included to ensure interassay reproducibility. For viral passage, naive cells were infected with culture supernatant (37). Cells were split three times weekly; HCV-specific immunostaining was done using primary antibody anti-NS5A-9E10 for 2a(JFH1), 2a(J6), 3a(S52), 3a(452), 5a(SA13), and 6a(HK6a) recombinants (5) or a combination of anti-NS5A-9E10 and anti-Core-C7-50 (Enzo Life Sciences) for 1a(TN) and 4a(ED43) recombinants (7, 31); the percentage of HCV antigen-positive cells was estimated by fluorescence microscopy (37). Culture supernatant infectivity titers were determined as focus-forming units (FFU) per milliliter following the infection of triplicate cultures on poly-d-lysine-coated 96-well plates (Nunc) with serially diluted supernatants, immunostaining with the primary antibodies indicated above, and automated FFU counting (5, 6, 36).
Direct sequence analysis of NS3P/NS4A of cell culture-produced HCV.
Methods for RNA extraction from culture supernatant, reverse transcription-PCR, and direct sequence analysis have been described (37). The complete NS3P/NS4A sequence was analyzed (5). Primers are specified in prior publications for 1a(TN) (31); 2a(J6) (5); 2a(JFH1) (37); 3a(452), 3a(S52), 5a(SA13), and 6a(HK6a) (5); and 4a(ED43) (7).
HCV concentration-response assays and statistical analysis.
Overall, experiments were conducted as described previously (5). Briefly, 5 × 103 cells per well, plated the previous day on poly-d-lysine-coated 96-well plates (Nunc), were infected with supernatants from first- or second-viral-passage cultures; low-titer supernatants were concentrated using Amicon Ultra-15 centrifugal filter units (Millipore). The PIs used, telaprevir (VX-950), boceprevir (SCH 503034), simeprevir (TMC435350), asunaprevir (BMS-650032), vaniprevir (MK-7009), faldaprevir (BI 201335), paritaprevir (ABT-450), deldeprevir (ACH-2684), and grazoprevir (MK-5172), all were purchased from Acme Bioscience and were dissolved in dimethyl sulfoxide. Cells were treated 24 h postinfection with a dilution series of each of these inhibitors; in general, each concentration was tested in triplicate. Seventy-two hours postinfection, immunostaining was conducted as described for infectivity titration (5). Single HCV antigen-positive cells were counted automatically, counts from treated wells were related to means of counts from infected, nontreated wells, and sigmoidal concentration-response curves were fitted [Y = Top/(1 + 10[log10(EC50) − X] × Hill slope)] to obtained data following transformation of X values. Log10 median effective concentration (EC50) and standard errors (SE) of log10(EC50) from replicate experiments were used to calculate inverse-variance weighted means of log10(EC50) with SE and 95% confidence intervals (CI). Mean differences between log10(EC50) of variants versus original recombinants with SE and 95% CI were calculated from the inverse-variance weighted-mean log10(EC50) values. Inverse logarithmic transformation rendered median EC50 with 95% CI and median fold differences with 95% CI. P values were determined by Z test. Cell viability was monitored using the CellTiter 96 AQueous one solution cell proliferation assay (Promega) (5).
RESULTS
We engineered the PI resistance-associated substitutions R155A/E/G/H/K/Q/T, A156G/S/T/V, and D/Q168A/E/G/H/N/V (9–26) into HCV recombinants with genotype(isolate) 2a(JFH1)-, 3a(S52)-, 4a(ED43)-, 5a(SA13)-, and 6a(HK6a)-specific NS3P/NS4A (see Fig. S1 and Table S1 in the supplemental material) (5, 7, 30). To evaluate whether in vitro assays reflected findings previously reported for genotype 1, we also engineered six selected substitutions into a 1a(TN) recombinant (31): R155K/Q, A156G/S, and D168A/H. After transfection of Huh7.5 hepatoma cells with RNA transcripts of a total of 91 variants, we evaluated viral fitness compared to that of the original recombinants by (i) determination of peak supernatant infectivity titers, defined as the highest representative titer at the peak of infection; (ii) evaluation of viral spread kinetics by determination of comparative infectivity titers, defined as the titer of the NS3P variant on the day at which the original recombinant achieved peak titer; (iii) evaluation of viral spread kinetics by determination of the percentage of HCV antigen-expressing cells; and (iv) evaluation of genetic stability of the developed recombinants by direct sequence analysis of NS3P/NS4A of viruses passaged to naive cells (Fig. 1; also see Fig. S2 and Table S1). For viruses that had maintained the engineered amino acid substitution, concentration-response profiles and median EC50s were determined for nine PIs (see Table 2; also see Fig. S3 to Fig. S8).
FIG 1.

Fitness characteristics of HCV genotype 1 to 6 recombinants with substitutions at NS3P positions 155, 156, or 168. For HCV recombinants with NS3P/NS4A of genotype(isolate) 1a(TN) (A), 2a(JFH1) (B), 3a(S52) (C), 4a(ED43) (D), 5a(SA13) (E), and 6a(HK6a) (F), HCV RNA transcripts of original recombinants and variants with the indicated substitutions were transfected into Huh7.5 cells. Numbers above bars indicate the day posttransfection at which HCV infection had spread to ≥80% of culture cells, as estimated by immunostaining. Titers are means from triplicates ± standard errors of the means (SEM) and were calculated as described in Materials and Methods; the lower cutoff, indicated by the y axis break, was up to 2.2 log10 FFU/ml. Peak supernatant infectivity titers were defined as the highest representative titer at the peak of infection. Comparative infectivity titers were defined as the titer of the variant on the day at which the original recombinant achieved peak titer. In the few instances where the variant achieved a peak infectivity titer prior to the original recombinant, the peak infectivity titer of the variant is given. Transfections for recombinants in each panel were not necessarily done in the same experiment. However, in each transfection experiment the original virus was included and showed similar spread kinetics. Comparative titers of variants were always obtained on the day at which the original recombinant in the respective transfection experiment reached peak titer. #, Comparative titer was not determined; na, not applicable; nv, nonviable (recombinants for which transfection cultures did not show any HCV antigen-positive cells during at least 3 weeks of follow-up were defined as nonviable, and no titer was determined); ns, no viral spread despite the presence of HCV antigen-positive cells (virus did not infect ≥80% of culture cells); the peak infectivity titer was not determined. Supernatants derived from transfection cultures at the peak of infection were used to inoculate first viral passage cultures. The results of the direct sequencing of NS3P/NS4A, indicated by color shading, revealed whether (i) substitutions were genetically stable, meaning that the engineered NS3P substitution was maintained and no additional substitutions were acquired in NS3P/NS4A (green shading), (ii) the engineered NS3P substitution was maintained, but one or more additional substitutions were found in NS3P/NS4A (for details, see Table S1 in the supplemental material) (yellow shading), or (iii) the engineered NS3P substitution had reverted to the original amino acid but not to the original nucleotide sequence or had changed to another residue than the original amino acid or the engineered substitution (orange shading); in some instances additional substitutions were acquired. No shading indicates that recombinants with the respective substitutions were nonviable or viral spread was not observed, resulting in first viral passage attempts remaining unsuccessful. Footnote a, data on genetic stability were obtained from the first passage of replicate transfections, which showed similar kinetics as the transfections shown; footnote b, in a replicate experiment, 6a(HK6a)R155G maintained the engineered substitution and acquired the additional NS3P substitution D168V. In addition to variants described in Results, we studied the impact of R155K/T and A156S/T on 2a(J6) and 3a(452) (see Table S1).
TABLE 2.
Effect of substitutions at NS3P position 155, 156, or 168 on sensitivity of HCV genotypes 1 to 6 to nine PIsa
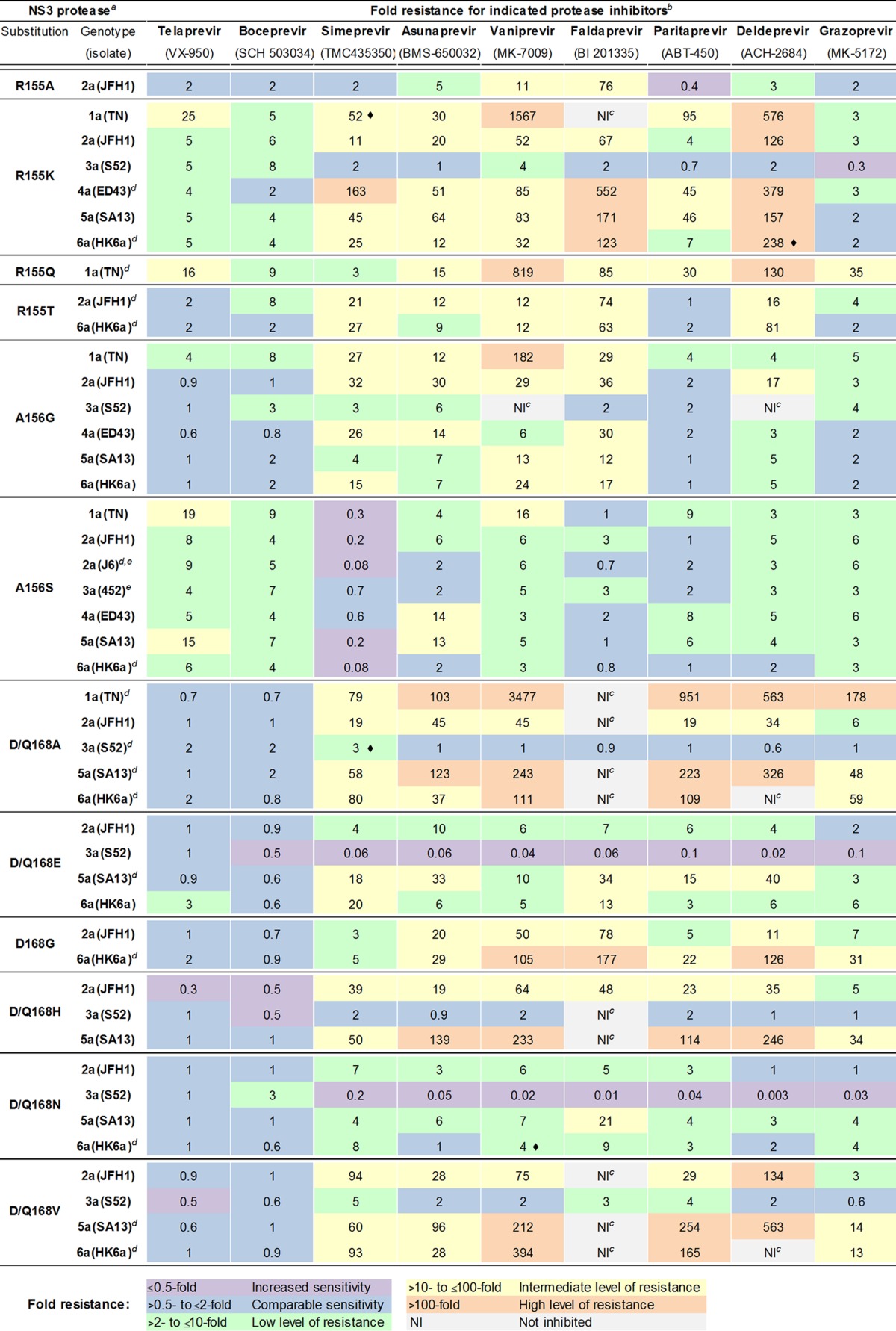
Viruses were from passage experiments; the NS3P/NS4A sequence was confirmed by direct sequencing.
Original recombinants and variants with indicated NS3P substitutions were tested in concentration-response assays against the indicated PIs; representative experiments are shown in Fig. S3 to S10 in the supplemental material. Mean EC50s and median fold EC50 differences for variants versus original recombinants were calculated as described in Materials and Methods. Shown are fold difference values. Color shadings indicate different levels of sensitivity/resistance. Differences were statistically significant (P < 0.05) unless indicated by a diamond (◆). Statistical analysis is not included for treatments of variants with comparable sensitivity to original viruses and of variants for which 50% inhibition was not achieved.
NI, not inhibited; for these variants, 50% inhibition was not achieved by the highest applied concentration of vaniprevir (15,000 nM), faldaprevir (5,000 nM), or deldeprevir (5,000 nM). For faldaprevir and deldeprevir, cytotoxicity was clearly observed at 15,000 nM.
For the following variants, additional substitution(s) had been acquired in NS3P/NS4A of first- or second-passage virus stocks used for treatment: 4a(ED43)R155K, 113V/i in NS3P; 1a(TN)R155Q, V6A in NS4A; 2a(JFH1)R155T, D168D/A in NS3P; 6a(HK6a)R155T, D168A in NS3P; 2a(J6)A156S, T72T/a and P86L in NS3P; 6a(HK6a)A156S stock used for asunaprevir treatment only, C72S in NS3P; 1a(TN)D168A, Y134C in NS3P; 3a(S52)Q168A, K62R in NS3P; 5a(SA13)D168A, L25V in NS4A; 6a(HK6a)D168A, K62R in NS3P; 5a(SA13)D168E, V6V/L in NS4A; 6a(HK6a)D168G, K62R in NS3P; 6a(HK6a)D168N, K62R in NS3P; 5a(SA13)D168V, I17 M in NS3P; 6a(HK6a)D168V, K62R in NS3P. For 6a(HK6a)R155K, direct sequencing indicated that a minor virus population had reverted to the original amino acid.
In addition to variants described in Results, we tested 2a(J6)A156S and 3a(452)A156S (see Table S1 in the supplemental material).
Effect of NS3P substitutions previously described to confer genotype 1 PI resistance on fitness and resistance of the 1a(TN) recombinant.
Of the two recombinants with substitutions at position 155 (R155K and R155Q), 1a(TN)R155K showed high fitness with relatively fast viral spread kinetics, resulting in infection of most culture cells on day 15 posttransfection (Fig. 1; also see Fig. S2 and Table S1 in the supplemental material). Further, 1a(TN)R155K yielded a high comparative and peak infectivity titer. Finally, 1a(TN)R155K was genetically stable, meaning that it maintained the engineered substitution without the acquisition of additional substitutions in NS3P/NS4A following viral passage to naive cells. In contrast, 1a(TN)R155Q showed delayed spread kinetics. While the peak infectivity titer was similar to that of 1a(TN), passaged 1a(TN)R155Q had acquired an additional substitution in NS4A (see Table S1). 1a(TN)A156G and 1a(TN)A156S showed relatively high fitness and were genetically stable. In contrast, 1a(TN)D168A and 1a(TN)D168H showed delayed spread kinetics. Following viral passage, 1a(TN)D168A acquired an additional substitution in NS3P, while D168H reverted to the original amino acid but not to the original nucleotide sequence.
In accordance with previous data (7, 31), the tested PIs showed differential efficacy against the original 1a(TN). Telaprevir and boceprevir were least efficient. Increased efficacy was found for asunaprevir, simeprevir, faldaprevir, and especially for vaniprevir (Table 1; also see Fig. S3 in the supplemental material). Grazoprevir showed exceptional efficacy. We also tested paritaprevir and deldeprevir for the first time in our infectious systems, which proved as efficient as vaniprevir and nearly as efficient as grazoprevir against 1a(TN).
TABLE 1.
Efficacy of PIs against HCV recombinants with genotype 1- to 6-specific NS3P/NS4A
| Genotypea (isolate) | Mean EC50 for indicated protease inhibitorb (nM) (95% CI) |
||||||||
|---|---|---|---|---|---|---|---|---|---|
| Telaprevir (VX-950) | Boceprevir (SCH 503034) | Simeprevir (TMC435350) | Asunaprevir (BMS-650032) | Vaniprevir (MK-7009) | Faldaprevir (BI 201335) | Paritaprevir (ABT-450) | Deldeprevir (ACH-2684) | Grazoprevir (MK-5172) | |
| 1a(TN) | 133 (120–149) | 82 (73–92) | 38 (36–41) | 54 (49–60) | 1.7 (1.5–2.0) | 14 (12–16) | 1.9 (1.7–2.0) | 1.8 (1.7–2.0) | 0.7 (0.7–0.8) |
| 2a(JFH1) | 514 (482–548) | 623 (586–662) | 87 (83–91) | 190 (149–243) | 66 (60–73) | 48 (41–57) | 38 (32–45) | 15 (13–18) | 9.2 (8.2–10.4) |
| 2a(J6) | 805 (736–880) | 1,178 (1,058–1,311) | 310 (274–352) | 785 (647–952) | 39 (33–48) | 125 (93–169) | 33 (24–46) | 31 (23–43) | 7.1 (6.4–8.0) |
| 3a(S52) | 3,368 (3,063–3,705) | 1,219 (1,118–1,329) | 1,956 (1,727–2,214) | 3,715 (2,684–5,143) | 2,030 (1,865–2,209) | 1,480 (1,120–1,956) | 677 (497–922) | 1,572 (1,380–1,790) | 78 (71–86) |
| 3a(452) | 4,227 (3,891–4,591) | 1,175 (1,004–1,375) | 3,350 (2,337–4,802) | 3,839 (3,019–4,883) | 2,158 (1,744–2,670) | 1,968 (1,502–2,578) | 839 (602–1,171) | 956 (717–1,276) | 75 (64–89) |
| 4a(ED43) | 2,159 (2,018–2,309) | 1,387 (1,194–1,611)c | 4.4 (4.1–4.7) | 38 (35–42) | 7.4 (6.7–8.2) | 2.3 (2.0–2.7)c | 1.8 (1.0–3.3) | 0.5 (0.4–0.5) | 1.3 (1.3–1.4) |
| 5a(SA13) | 823 (730–928) | 794 (734–858) | 150 (138–164) | 83 (68–100) | 27 (25–29) | 14 (13–15)c | 8.8 (7.7–10.0) | 7.9 (7.1–8.7) | 5.3 (5.2–5.5)c |
| 6a(HK6a) | 447 (418–477) | 628 (589–669) | 87 (81–92) | 330 (267–409) | 31 (29–33) | 19 (17–21) | 22 (18–27) | 18 (16–22) | 4.9 (4.5–5.2) |
The indicated original recombinants without putative resistance substitutions were tested in concentration-response assays against the nine indicated PIs.
Mean EC50s and 95% CI were calculated as described in Materials and Methods.
Value was reported previously (7).
For 1a(TN), 155 and 156 variants showed low-to-intermediate resistance to telaprevir and boceprevir, while D168A did not confer resistance to these PIs (Table 2; also see Fig. S3 in the supplemental material). For simeprevir, R155K, A156G, and D168A caused intermediate resistance, whereas R155Q caused low resistance and A156S caused increased sensitivity. For asunaprevir, D168A caused high resistance, while the other tested substitutions caused low-to-intermediate resistance. For vaniprevir, R155K, R155Q, A156G, and D168A caused high resistance, while A156S conferred intermediate resistance. Also for faldaprevir, very high resistance was observed for the R155K and D168A variants, which were not inhibited by noncytotoxic faldaprevir concentrations. Conversely, R155Q and A156G conferred only intermediate resistance, while A156S did not confer resistance. For paritaprevir, R155K and R155Q caused intermediate resistance, while A156G and A156S caused low resistance; in contrast, D168A caused high resistance. For deldeprevir, R155K, R155Q, and D168A conferred high resistance, while A156G and A156S conferred low resistance. Finally, for grazoprevir, D168A conferred high resistance, while R155Q conferred intermediate resistance and R155K, A156G, and A156S caused low resistance.
In conclusion, previously described clinically relevant genotype 1 PI resistance substitutions at NS3P positions 155, 156, and 168 had differential effects on the viral fitness of the 1a(TN) virus. Further, the nine PIs investigated in this study showed differential efficacy against the original 1a(TN) virus. Finally, the tested substitutions conferred various levels of resistance to nine PIs. Of note, in our 1a(TN) infectious system, substitutions identified in genotype 1-infected patients following treatment with specific PIs conferred relatively high resistance to these PIs, confirming the clinical relevance of our in vitro findings (38).
Effect of putative resistance substitutions on the fitness of genotype 2 to 6 recombinants.
Most substitutions at position 155 were not tolerated in genotype 2 to 6 recombinants. Thus, recombinants with R155E/G/H/Q were either nonviable, as defined by the absence of HCV antigen-positive cells during 3 weeks following transfection, or the introduced substitution had reverted or changed following viral passage (Fig. 1; also see Fig. S2 and Table S1 in the supplemental material). Among R155A/T variants, only 2a(JFH1) and 6a(HK6a) variants spread in transfection cultures, but they showed decreased fitness. These four variants spread following viral passage and maintained the introduced substitutions. While 2a(JFH1)R155A was genetically stable, 6a(HK6a)R155A, 2a(JFH1)R155T, and 6a(HK6a)R155T acquired the additional NS3P substitution D168A (Fig. 1; also see Table S1). In reverse genetic experiments we showed that D168A compensated for fitness impairment induced by R155T (Fig. 2). Thus, following transfection, 2a(JFH1)R155T,D168A and 6a(HK6a)R155T,D168A were comparable to the respective original viruses regarding spread kinetics and infectivity titers. Further, passaged R155T,D168A variants were genetically stable. In contrast to most other 155 variants, all R155K variants spread in transfection cultures, showing various fitness levels. With the exception of 4a(ED43)R155K, these variants were genetically stable.
FIG 2.

Identification of additional NS3P substitutions compensating for decreased fitness induced by PI resistance substitutions. HCV RNA transcripts of 2a(JFH1) and 6a(HK6a) recombinants with R155T and D168A singly and in combination (A), as well as 6a(HK6a) recombinants with K62R and D168A/G/N/V singly and in combination (B), were transfected into Huh7.5 cells. Infectivity titers were determined at the indicated days posttransfection. Titers are means from triplicates ± SEM; the lower cutoff, indicated by the y axis break, was up to 2.3 log10 FFU/ml. The day at which viral infection had spread to ≥80% of culture cells, as estimated by immunostaining, is indicated. Supernatants from the peak of infection were used to inoculate first viral passage cultures. Results from direct sequencing of NS3P are indicated. One representative experiment is shown. In a replicate experiment (data not shown), including all recombinants shown in panels A and B, viral spread kinetics in transfection were comparable to those of the experiment shown. Direct sequencing of NS3P of first-passage viruses showed results identical to those of the experiment shown, except for 2a(JFH1)R155T, which did not acquire D168A in the first but only in the second viral passage, and 6a(HK6a)D168V, which also acquired K62R but showed an additional partial reversion of D168V. Footnote a, data for the indicated recombinants are identical.
All variants with A156G and A156S were viable. Except for 3a(S52)A156S, these variants spread in transfection cultures, mostly showing minor fitness impairment, and were genetically stable. In contrast, A156T and A156V were not tolerated in any of the developed recombinants; all A156T/V variants reverted following passage.
Substitutions at position 168 had differential effects on the fitness of variants of different genotypes. For 2a(JFH1), most 168 variants showed relatively high fitness, and all were genetically stable. 3a(S52), 5a(SA13), and 6a(HK6a) 168 variants were viable but showed various degrees of fitness impairment; of the 18 variants, only 5a(SA13)D168E and 6a(HK6a)D168E/H showed spread kinetics comparable to those of the respective original recombinants. We were able to generate first-passage virus stocks for 17 of these variants, while 3a(S52)Q168G infected few cells in transfection cultures and could not be passaged. 3a(S52)Q168E/H/N/V, 5a(SA13)D168H/N, and 6a(HK6a)D168E/H were genetically stable. In contrast, 5a(SA13)D168G reverted and 5a(SA13)D168A/E/V acquired additional substitutions (see Table S1 in the supplemental material). 3a(S52)Q168A and 6a(HK6a)D168A/G/N/V all acquired the additional NS3P substitution K62R. For 6a(HK6a), using reverse genetic studies, we showed that K62R compensated for fitness impairment induced by D168A/G/N/V (Fig. 2). For 4a(ED43), none of the substitutions at position 168 were tolerated. Of the six developed variants, only 4a(ED43)D168E/H/N/V were viable, showing impaired fitness. Of these, only 4a(ED43)D168E could be passaged, but it had reverted following passage.
In conclusion, the engineered substitutions had differential impacts on the fitness of genotype 2 to 6 recombinants. Thus, most substitutions at position 155 were not tolerated and only R155K variants showed relatively high fitness for most genotypes. Substitutions at position 156 induced minor (A156G/S) versus major (A156T/V) fitness impairment. Most substitutions at position 168 did not induce major fitness impairment for genotypes 2, 3, 5, and 6; however, none of these substitutions were tolerated for genotype 4. In addition, we found that D168A compensated for fitness impairment induced by R155T and that K62R compensated for fitness impairment induced by D168A/G/N/V for certain recombinants. Overall, we were able to generate virus stocks of 40 genotype 2 to 6 NS3P variants for use in PI treatment studies (Table 2).
Effect of putative resistance substitutions on PI sensitivity of genotypes 2 to 6.
All of the 40 NS3P variants showed resistance to at least a subset of the PIs tested and had different levels of resistance depending on the PI, the genotype, and the specific substitution. An exception was genotype 3, for which substitutions at position 168 had a limited effect on PI sensitivity. While results and representative examples are shown in Table 2 and Fig. S4 to S10 in the supplemental material, observed patterns are highlighted below.
(i) Level of PI sensitivity and resistance of original and variant genotype 2 to 6 recombinants depended on the PI.
In accordance with previous studies (5, 7), PIs had differential efficacy against the original genotype 2 to 6 recombinants. As observed for genotype 1, overall, telaprevir and boceprevir showed the lowest efficacy, being least efficient against genotypes 3 and 4 (Table 1). However, most of the developed genotype 2 to 6 NS3P variants showed only low to no resistance to these PIs (Table 2). Simeprevir and asunaprevir were more efficacious than telaprevir and boceprevir against original recombinants of genotypes 2, 5, and 6 and had greatly improved efficacy against genotype 4. Variants mainly showed low to intermediate resistance to these PIs, but instances of increased sensitivity also were observed. Compared to that of simeprevir and asunaprevir, vaniprevir and faldaprevir showed increased efficacy against original recombinants of most genotypes; however, several variants showed high resistance to these two compounds. Paritaprevir and deldeprevir showed further increased efficacy against original recombinants of most genotypes. Many variants showed low or no resistance to paritaprevir, but there were also instances of intermediate to high resistance. Compared to paritaprevir, more variants showed high resistance to deldeprevir. Overall, grazoprevir showed the highest efficacy against original recombinants and variants. Thus, none of the 40 genotype 2 to 6 variants showed high resistance and only 6 showed intermediate resistance to grazoprevir.
(ii) Level of PI resistance depended on the NS3P substitution.
R155K, which was tolerated in all genotypes, and A156S were the main determinants of resistance to telaprevir and boceprevir (Table 2). In addition, for genotypes 2, 4, 5, and 6, R155K caused intermediate to high resistance to simeprevir, asunaprevir, vaniprevir, faldaprevir, and deldeprevir. Paritaprevir showed increased efficacy against specific R155K variants, while grazoprevir showed high efficacy against most R155K variants. R155A, tested for 2a(JFH1), did not confer resistance to telaprevir, boceprevir, simeprevir, paritaprevir, or grazoprevir but conferred low to intermediate resistance to remaining PIs. 2a(JFH1)R155T and 6a(HK6a)R155T were sensitive or showed only low resistance to telaprevir, boceprevir, paritaprevir, and grazoprevir, but they showed mostly intermediate resistance to the remaining PIs.
At NS3P position 156, A156G and A156S could be tested for representative isolates of all genotypes. A156G did not confer resistance to telaprevir and boceprevir, except for 3a(S52)A156G, which showed low resistance to boceprevir. A156G caused mostly low to intermediate resistance to simeprevir, asunaprevir, vaniprevir, faldaprevir, and deldeprevir; however, 3a(S52)A156G was not inhibited by the highest applied concentration of vaniprevir and deldeprevir. The A156G variants showed no resistance to paritaprevir and low to no resistance to grazoprevir. In contrast to A156G, for genotypes 2 to 6, A156S conferred mostly low resistance to telaprevir and boceprevir. Also, A156S conferred mostly low resistance to vaniprevir, deldeprevir, and grazoprevir. A156S variants showed no, low, or intermediate resistance to asunaprevir. The majority of A156S variants showed no resistance to faldaprevir and pariteprevir, while low resistance was observed for the remainder of these variants. Interestingly, for all genotypes, A156S either had no effect or caused increased sensitivity to simeprevir.
Most substitutions at position 168 were tolerated in genotypes 2, 3, 5, and 6. Except for 6a(HK6a)D168E and 3a(S52)Q168N, none of the resulting variants showed >2-fold resistance to telaprevir or boceprevir. NS3P variants showed variable sensitivity to the remaining PIs, with the level of resistance depending on the specific substitution. Thus, for genotypes 2, 5, and 6, tested D168A/G/H/V variants mostly caused intermediate to high resistance; D168E/N mostly caused low to intermediate resistance for these genotypes. For genotype 3, NS3P 168 variants mostly showed increased sensitivity or low to no resistance.
(iii) Level of PI sensitivity and resistance depended on genotype.
As previously observed, PI efficacy was influenced by the genotype (Table 1) (5, 7). Thus, all PIs showed comparatively low efficacy against original genotype 3 recombinants. While most substitutions at position 168 did not confer additional resistance to genotype 3, most substitutions at positions 155 and 156 conferred resistance to several PIs, albeit to lower levels than to other genotypes. For telaprevir and boceprevir, however, substitutions at positions 155 and 156 conferred levels of resistance to genotype 3 similar to those for other genotypes (Table 2). Genotype 4 also showed natural resistance to telaprevir and boceprevir, which was further increased by R155K and A156S. The remaining PIs, especially faldaprevir, paritaprevir, deldeprevir, and grazoprevir, had high efficacy against genotype 4. Of these PIs, grazoprevir showed the highest efficacy against the three genotype 4 variants that could be tested. Finally, in most instances, substitutions at position 168 conferred greater resistance to genotypes 5 and 6 than to genotype 2 for most PIs except telaprevir and boceprevir.
DISCUSSION
We systematically studied the impact of previously described key PI resistance substitutions at NS3P positions 155, 156, and 168 on viral fitness and PI resistance for HCV genotypes 2 to 6. While most substitutions decreased viral fitness, the level of fitness impairment depended on the specific substitution and genotype. For genotypes 2 and 6, we identified NS3P substitutions compensating for fitness impairment induced by specific resistance substitutions at positions 155 and 168. While most substitutions conferred resistance to several PIs, including newer PIs developed for increased efficacy against resistant variants, their impact varied from increased sensitivity to high resistance. Thus, we identified complex patterns of resistance, determined by the specific PI, the substitution, and the genotype.
Clinical trials allow for the identification of putative PI resistance substitutions (9, 10). However, characterization of their impact on viral fitness and resistance requires in vitro studies. Enzymatic assays and replicon systems only allow studying NS3P activity and viral replication (39). In contrast, HCV infectious culture systems allow studies in the context of the complete viral life cycle. Results in these systems reflect in vivo findings regarding PI sensitivity and resistance (Table 2; also see Fig. S3 in the supplemental material) (5–7, 31). Nevertheless, it should be noted that results obtained in these systems might be influenced by the presence of cell culture-adaptive substitutions. In general, except for certain JFH1-based chimeric genotype 2a or 2b recombinants (5, 17, 30, 35, 40–42), HCV infectious culture systems depend on cell culture-adaptive substitutions (39). Such adaptive substitutions might facilitate the interaction of HCV proteins with host factors or of HCV proteins of different genotypes in the case of chimeric recombinants. It might be preferable to carry out studies in full-length recombinants (43). However, efficient full-length recombinants currently are only available for genotypes 1a, 2a, and 2b (31, 44–49). Recent reports indicate that HCV genotypes 2 to 6 acquire substitutions examined in this study de novo when subjected to PI treatment in vivo and in vitro (11–26). However, only a few studies have investigated the impact of such substitutions on PI resistance. In line with our findings, in an enzymatic assay, R155K and A156S conferred telaprevir resistance for genotype 2 and R155K conferred resistance for genotype 3 (16). Further, certain substitutions of position 168 conferred ciluprevir and/or danoprevir resistance to Jc-1-based constructs with genotype 1 to 6-specific NS3P/NS4A (11, 15). In addition, A156T/V mediated ciluprevir resistance for genotype 4a (15). Finally, in a cell culture-infectious genotype 2a recombinant, A156S conferred resistance to telaprevir, and A156G and D168A/V conferred resistance to ciluprevir; cross-resistance was not observed (13). Compared to previous studies, we used a broad and systematic approach for a head-to-head comparison of the effect of various key resistance substitutions at NS3P positions 155, 156, and 168 on fitness and resistance of HCV genotypes 2 to 6 in the context of the full viral life cycle. Further, our study is unique, since a broad panel of PIs, including newly developed PIs, was studied. In future studies it might be of interest to refine the resistance profiles of individual PIs by inducing viral escape under treatment. Such studies might reveal which substitutions engineered in the present study are preferably selected under treatment. Further, they might reveal additional resistance substitutions and the major determinants of resistance for different genotypes and PIs.
Previous studies suggested that most PI resistance substitutions resulted in fitness impairment and that variants with low fitness were unlikely to persist long term in vivo; however, impaired fitness could be rescued by the acquisition of compensating substitutions (9, 10). In this study, the developed variants showed great variation of fitness depending on the specific substitution and the genotype. Certain variants acquired additional substitutions in NS3P or NS4A, possibly compensating for fitness impairment induced by the engineered resistance substitution. For other variants, the engineered substitution reverted to the original amino acid but not to the original nucleotide sequence or had changed to another residue than the original amino acid or the engineered substitution. These substitutions might have been maintained under treatment pressure, driving coselection of compensating substitutions. Compared to genotype 3, 4, and 5 variants, more genotype 2 and 6 variants were viable and were able to spread in cell culture following transfection (Fig. 1). However, several of these variants were not genetically stable, especially for genotype 6. Compared to JFH1-based recombinants with genotype-specific NS3P/NS4A, the original 2a(JFH1) virus is characterized by favorable replication/growth characteristics, which might facilitate reversion of the introduced substitution or selection of compensating substitutions (5, 7). In addition, the genetic context is likely to influence the effect of the introduced resistance substitutions on viral fitness.
Most substitutions of the highly conserved R155 resulted in strong fitness impairment and many R155 variants reverted, possibly due to the fact that arginine is encoded by six different codons. An exception was the R155K variants, which showed relatively high fitness, possibly due to similar physicochemical properties of lysine and arginine. Certain substitutions at position 155 apparently were permissible given that an additional substitution at position 168 was acquired (Fig. 1; also see Table S1 in the supplemental material). In reverse genetic studies we showed that R155T was permissible in 2a(JFH1) and 6a(HK6a) when combined with D168A (Fig. 2), possibly restoring the described interactions of position 155 and 168 (50, 51). In line with our findings, a previous study suggested relatively high fitness of genotype 1a R155K/T versus R155G/Q variants in vitro (52). In addition, the fact that R155K was frequently found in genotype 1 and, more recently, also in genotype 2, 3, and 5 PI-treated patients indicates a relatively high fitness (9, 10, 14, 16, 18, 21, 26, 53). Among substitutions at the highly conserved A156, A156G/S generally resulted in minor fitness impairment, whereas A156T/V resulted in strong fitness impairment. This is in line with previous studies, suggesting relatively high fitness of genotype 1a, 2a, and 6 A156S versus A156T/V variants in vitro and in vivo (13, 52–54), and the fact that A156G/S variants were frequently found in genotype 1 and, more recently, also in genotype 2 and 3 PI-treated patients (9, 10, 14, 16, 21, 26). Changes at position 168, harboring a highly conserved glutamine in genotype 3 and aspartic acid in other genotypes, did not result in major fitness impairment for most genotypes, as was previously suggested for genotype 1 (52). However, all genotype 4 168 variants showed strongly impaired fitness. Further, 3a(S52)Q168A and 6a(HK6a)D168A/G/N/V acquired the additional NS3P substitution K62R, which restored the fitness of the 6a(HK6a) variants (Fig. 2).
In future studies it might be of interest to investigate if specific substitutions have a similar effect on the fitness of different isolates of the same genotype. In an initial approach, we engineered R155K/T and A156S/T into 2a(J6) and 3a(452) recombinants (5). Fitness characteristics of the resulting variants were relatively different from those observed for the respective 2a(JFH1) and 3a(S52) variants (see Table S1 in the supplemental material). In general, 2a(JFH1) variants had greater fitness than 2a(J6) variants, suggesting a positive correlation between the fitness of the original recombinant and fitness of derived variants (5).
We confirmed previously observed differences in the potency of the tested PIs against genotypes 1 to 6 (Table 1) (5, 7). Grazoprevir showed exceptional in vitro potency, followed by deldeprevir and paritaprevir, which we tested here for the first time in our infectious cell culture systems. Compared to other original recombinants, 1a(TN) showed high sensitivity to all PIs and 4a(ED43) showed high sensitivity to all PIs except telaprevir and boceprevir. This lack of sensitivity to telaprevir and boceprevir might be explained by V170, found for the majority of genotype 4a isolates and associated with PI resistance (alignment of 17 genotype 4a NS3P sequences deposited in the European HCV database, generated July 2015 [55], and the 2008 Web alignment from the Los Alamos HCV database [56]) (5, 9). The comparatively low sensitivity of original genotype 3a recombinants to all PIs might be explained by Q168, conserved for genotype 3a, and V170, found in 3a(S52) (alignment of 28 genotype 3a NS3P sequences deposited in the European HCV database, generated July 2015 [55], and the 2008 Web alignment from the Los Alamos HCV database [56]) (5, 9, 10). The testing of additional genotype 2a and 3a isolates suggested that there were no major EC50 differences for different isolates of the same genotype (Table 1; also see Fig. S4, S5, S9, and S10 in the supplemental material) (5, 7). Given the limited efficacy of PIs as well as of some NS5A- and nonnucleotidic polymerase inhibitors against genotype 3, this epidemiologically important genotype might emerge as relatively difficult to treat with interferon-free DAA-based combination treatment regimens (1, 57). For genotype 3, future treatment regimens might be based on nucleotidic polymerase inhibitors with high efficacy against different genotypes. Combinations with newer PIs, such as grazoprevir and NS5A inhibitors, showing improved efficacy are being evaluated (ClinicalTrials.gov NCT02133131 and NCT02332720).
We found that most described PI resistance substitutions at NS3P positions 155, 156, and 168 were able to confer resistance to simeprevir, asunaprevir, vaniprevir, faldaprevir, paritaprevir, deldeprevir, and grazoprevir for genotypes 2, 4, 5, and 6; natural resistance of genotype 3 was further increased by selected substitutions. An exception was the relatively high sensitivity of most R155K, R155T, and A156G variants to grazoprevir; the high efficacy of this PI against genotype 1 R155K variants in enzymatic assays and replicons has been reported previously (58). Further exceptions were the relatively high sensitivity of most R155T, A156G, and A156S variants to paritaprevir and of most A156S variants to faldaprevir; A156S variants were highly sensitive to simeprevir. As reported for genotype 1, substitutions at position 168 did not confer resistance to telaprevir and boceprevir (9, 10, 50), revealing a pangenotypic pattern. R155K and A156S mainly conferred resistance of genotypes 2 to 6 to telaprevir and boceprevir. Thus, we observed complex resistance patterns with levels of resistance depending on the specific PI, the substitution, and the genotype.
NS3P resistance substitutions are thought to influence PI binding. A given substitution might have a differential effect on PI binding for different genotypes. Further, the structure of a given PI might affect its ability to bind despite the presence of a substitution. In future studies, molecular modeling might be used to explain differences in resistance levels observed for different PIs (50). Testing an additional genotype 2a isolate, we found that A156S conferred similar levels of resistance to most PIs for 2a(JFH1) and 2a(J6) (Table 2; also see Fig. S4 and S9 in the supplemental material). It is possible that NS3P substitutions acquired in first-passage viruses in addition to the engineered substitution influenced PI sensitivity (45). In contrast, it is less likely that additional substitutions in NS4A influenced PI sensitivity (42).
Relatively low levels of in vitro resistance can translate into reduced clinical response rates. For instance, Q80K, reported to induce low-level (∼5- to 10-fold) resistance in vitro, can result in significantly reduced response rates to simeprevir-based regimens in the clinic (1, 10). In addition, EC50 values of many resistant variants obtained in this study exceeded reported PI minimum plasma concentrations. Thus, variants showing resistance in this study might confer PI resistance in vivo.
In conclusion, this study systematically characterizes the impact of a great number of key PI resistance substitutions at NS3P positions 155, 156, and 168 on fitness and resistance of HCV genotypes 2 to 6. The impact on viral fitness depended on the specific substitution and the genotype, with most variants showing various levels of fitness impairment. The impact on resistance to nine clinically relevant PIs depended on the specific substitution, the genotype, and the PI, with most variants showing various levels of resistance to several PIs; however, increased sensitivity also was observed. This study significantly increases the knowledge on determinants of genotype 2 to 6 resistance to a large panel of clinically relevant PIs.
Supplementary Material
ACKNOWLEDGMENTS
We thank Lubna Ghanem, Anna-Louise Sørensen, and Lotte Mikkelsen for laboratory assistance; Steen Ladelund for statistical advice; Charles Rice (Rockefeller University) for reagents; and Bjarne Ø. Lindhardt, Ove Andersen, Jens Ole Nielsen (Copenhagen University Hospital, Hvidovre), and Carsten Geisler (University of Copenhagen) for support.
This work was supported by grants from Copenhagen University Hospital, Hvidovre (S.B.J., S.B.N.S., and J.M.G.), Region H Foundation (J.B. and J.M.G.), The Lundbeck Foundation (S.R., J.B., and J.M.G.), The Novo Nordisk Foundation (Y.-P.L., J.B., and J.M.G.), The Danish Council for Independent Research, Medical Sciences (J.B. and J.G.), The A. P. Møller and Chastine McKinney Møller Foundation (J.B. and J.M.G.), The Danish Cancer Society (J.B. and J.M.G.), Ph.D. stipends from the Faculty of Health Sciences, University of Copenhagen (S.B.J. and S.B.N.S.), and Individual Postdoctoral Stipends from the Danish Council for Independent Research, Medical Sciences (D.G.H. and S.R.). J.B. is the 2014 recipient of an advanced top researcher grant from the Danish Council for Independent Research and the 2015 recipient of the Novo Nordisk Prize.
We have no conflicts of interest to report.
Authors made the following contributions: study concept and design (J.B. and J.M.G.), acquisition of data (S.B.J., S.B.N.S, D.G.H., S.R., and Y.-P.L.), analysis and interpretation of data (S.B.J., S.B.N.S, J.B., and J.M.G.), drafting of the manuscript (S.B.J. and J.M.G.), revision of the manuscript (S.B.J., S.B.N.S, J.B., and J.M.G.), statistical analysis (S.B.J., S.B.N.S, and J.M.G.), obtaining funding (S.B.J., S.B.N.S, D.G.H., S.R., Y.-P.L., J.B., and J.M.G.), and study supervision (J.B. and J.M.G.).
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AAC.01953-15.
REFERENCES
- 1.Pawlotsky JM. 2014. New hepatitis C therapies: the toolbox, strategies, and challenges. Gastroenterology 146:1176–1192. doi: 10.1053/j.gastro.2014.03.003. [DOI] [PubMed] [Google Scholar]
- 2.Franco S, Tural C, Nevot M, Molto J, Rockstroh JK, Clotet B, Martinez MA. 2014. Detection of a sexually transmitted hepatitis C virus protease inhibitor-resistance variant in a human immunodeficiency virus-infected homosexual man. Gastroenterology 147:599–601. doi: 10.1053/j.gastro.2014.05.010. [DOI] [PubMed] [Google Scholar]
- 3.Moradpour D, Penin F. 2013. Hepatitis C virus proteins: from structure to function. Curr Top Microbiol Immunol 369:113–142. doi: 10.1007/978-3-642-27340-7_5. [DOI] [PubMed] [Google Scholar]
- 4.Smith DB, Bukh J, Kuiken C, Muerhoff AS, Rice CM, Stapleton JT, Simmonds P. 2014. Expanded classification of hepatitis C virus into 7 genotypes and 67 subtypes: updated criteria and genotype assignment web resource. Hepatology 59:318–327. doi: 10.1002/hep.26744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gottwein JM, Scheel TK, Jensen TB, Ghanem L, Bukh J. 2011. Differential efficacy of protease inhibitors against HCV genotypes 2a, 3a, 5a, and 6a NS3/4A protease recombinant viruses. Gastroenterology 141:1067–1079. doi: 10.1053/j.gastro.2011.06.004. [DOI] [PubMed] [Google Scholar]
- 6.Scheel TK, Gottwein JM, Mikkelsen LS, Jensen TB, Bukh J. 2011. Recombinant HCV variants with NS5A from genotypes 1-7 have different sensitivities to an NS5A inhibitor but not interferon-alpha. Gastroenterology 140:1032–1042. doi: 10.1053/j.gastro.2010.11.036. [DOI] [PubMed] [Google Scholar]
- 7.Li YP, Ramirez S, Humes D, Jensen SB, Gottwein JM, Bukh J. 2014. Differential sensitivity of 5′UTR-NS5A recombinants of hepatitis C virus genotypes 1-6 to protease and NS5A inhibitors. Gastroenterology 146:812–821. doi: 10.1053/j.gastro.2013.11.009. [DOI] [PubMed] [Google Scholar]
- 8.Antaki N, Craxi A, Kamal S, Moucari R, Van der Merwe S, Haffar S, Gadano A, Zein N, Lai CL, Pawlotsky JM, Heathcote EJ, Dusheiko G, Marcellin P. 2010. The neglected hepatitis C virus genotypes 4, 5 and 6: an international consensus report. Liver Int 30:342–355. doi: 10.1111/j.1478-3231.2009.02188.x. [DOI] [PubMed] [Google Scholar]
- 9.Vermehren J, Sarrazin C. 2012. The role of resistance in HCV treatment. Best Pract Res Clin Gastroenterol 26:487–503. doi: 10.1016/j.bpg.2012.09.011. [DOI] [PubMed] [Google Scholar]
- 10.Schneider MD, Sarrazin C. 2014. Antiviral therapy of hepatitis C in 2014: do we need resistance testing? Antiviral Res 105:64–71. doi: 10.1016/j.antiviral.2014.02.011. [DOI] [PubMed] [Google Scholar]
- 11.Imhof I, Simmonds P. 2010. Development of an intergenotypic hepatitis C virus (HCV) cell culture method to assess antiviral susceptibilities and resistance development of HCV NS3 protease genes from HCV genotypes 1 to 6. J Virol 84:4597–4610. doi: 10.1128/JVI.02698-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lenz O, Verbinnen T, Lin TI, Vijgen L, Cummings MD, Lindberg J, Berke JM, Dehertogh P, Fransen E, Scholliers A, Vermeiren K, Ivens T, Raboisson P, Edlund M, Storm S, Vrang L, de Kock H, Fanning GC, Simmen KA. 2010. In vitro resistance profile of the hepatitis C virus NS3/4A protease inhibitor TMC435. Antimicrob Agents Chemother 54:1878–1887. doi: 10.1128/AAC.01452-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cheng G, Chan K, Yang H, Corsa A, Pokrovskii M, Paulson M, Bahador G, Zhong W, Delaney W. 2011. Selection of clinically relevant protease inhibitor-resistant viruses using the genotype 2a hepatitis C virus infection system. Antimicrob Agents Chemother 55:2197–2205. doi: 10.1128/AAC.01382-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Foster GR, Hezode C, Bronowicki JP, Carosi G, Weiland O, Verlinden L, van Heeswijk R, van Baelen B, Picchio G, Beumont M. 2011. Telaprevir alone or with peginterferon and ribavirin reduces HCV RNA in patients with chronic genotype 2 but not genotype 3 infections. Gastroenterology 141:881–889. doi: 10.1053/j.gastro.2011.05.046. [DOI] [PubMed] [Google Scholar]
- 15.Imhof I, Simmonds P. 2011. Genotype differences in susceptibility and resistance development of hepatitis C virus to protease inhibitors telaprevir (VX-950) and danoprevir (ITMN-191). Hepatology 53:1090–1099. doi: 10.1002/hep.24172. [DOI] [PubMed] [Google Scholar]
- 16.De Meyer S, Ghys A, Foster GR, Beumont M, Van BB, Lin TI, Dierynck I, Ceulemans H, Picchio G. 2013. Analysis of genotype 2 and 3 hepatitis C virus variants in patients treated with telaprevir demonstrates a consistent resistance profile across genotypes. J Viral Hepat 20:395–403. doi: 10.1111/jvh.12046. [DOI] [PubMed] [Google Scholar]
- 17.Gottwein JM, Jensen SB, Li YP, Ghanem L, Scheel TK, Serre SB, Mikkelsen L, Bukh J. 2013. Combination treatment with hepatitis C virus protease and NS5A inhibitors is effective against recombinant genotype 1a, 2a, and 3a viruses. Antimicrob Agents Chemother 57:1291–1303. doi: 10.1128/AAC.02164-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lenz O, Vijgen L, Berke JM, Cummings MD, Fevery B, Peeters M, De Smedt G, Moreno C, Picchio G. 2013. Virologic response and characterisation of HCV genotype 2-6 in patients receiving TMC435 monotherapy (study TMC435-C202). J Hepatol 58:445–451. doi: 10.1016/j.jhep.2012.10.028. [DOI] [PubMed] [Google Scholar]
- 19.Silva MO, Treitel M, Graham DJ, Curry S, Frontera MJ, McMonagle P, Gupta S, Hughes E, Chase R, Lahser F, Barnard RJ, Howe AY, Howe JA. 2013. Antiviral activity of boceprevir monotherapy in treatment-naive subjects with chronic hepatitis C genotype 2/3. J Hepatol 59:31–37. doi: 10.1016/j.jhep.2013.02.018. [DOI] [PubMed] [Google Scholar]
- 20.Bronowicki JP, Ratziu V, Gadano A, Thuluvath PJ, Bessone F, Martorell CT, Pol S, Terg R, Younes Z, He B, Eley T, Cohen D, Yu F, Hernandez D, McPhee F, Mendez P, Hughes E. 2014. Randomized trial of asunaprevir plus peginterferon alfa and ribavirin for previously untreated genotype 1 or 4 chronic hepatitis C. J Hepatol 61:1220–1227. doi: 10.1016/j.jhep.2014.07.011. [DOI] [PubMed] [Google Scholar]
- 21.Lawitz E, Sullivan G, Rodriguez-Torres M, Bennett M, Poordad F, Kapoor M, Badri P, Campbell A, Rodrigues L Jr, Hu Y, Pilot-Matias T, Vilchez RA. 2015. Exploratory trial of ombitasvir and ABT-450/r with or without ribavirin for HCV genotype 1, 2, and 3 infection. J Infect 70:197–205. doi: 10.1016/j.jinf.2014.09.008. [DOI] [PubMed] [Google Scholar]
- 22.Chayama K, Notsumata K, Kurosaki M, Sato K, Rodrigues L Jr, Setze C, Badri P, Pilot-Matias T, Vilchez RA, Kumada H. 2015. Randomized trial of interferon- and ribavirin-free ombitasvir/paritaprevir/ritonavir in treatment-experienced hepatitis C virus-infected patients. Hepatology 61:1523–1532. doi: 10.1002/hep.27705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Moreno C, Hezode C, Marcellin P, Bourgeois S, Francque S, Samuel D, Zoulim F, Grange JD, Shukla U, Lenz O, Ouwerkerk-Mahadevan S, Fevery B, Peeters M, Beumont M, Jessner W. 2015. Efficacy and safety of simeprevir with PegIFN/ribavirin in naive or experienced patients infected with chronic HCV genotype 4. J Hepatol 62:1047–1055. doi: 10.1016/j.jhep.2014.12.031. [DOI] [PubMed] [Google Scholar]
- 24.Hezode C, Asselah T, Reddy KR, Hassanein T, Berenguer M, Fleischer-Stepniewska K, Marcellin P, Hall C, Schnell G, Pilot-Matias T, Mobashery N, Redman R, Vilchez RA, Pol S. 2015. Ombitasvir plus paritaprevir plus ritonavir with or without ribavirin in treatment-naive and treatment-experienced patients with genotype 4 chronic hepatitis C virus infection (PEARL-I): a randomised, open-label trial. Lancet 385:2502–2509. doi: 10.1016/S0140-6736(15)60159-3. [DOI] [PubMed] [Google Scholar]
- 25.Zeuzem S, Ghalib R, Reddy KR, Pockros PJ, Ari ZB, Zhao Y, Brown DD, Wan S, DiNubile MJ, Nguyen BY, Robertson MN, Wahl J, Barr E, Butterton JR. 2015. Grazoprevir-elbasvir combination therapy for treatment-naive cirrhotic and noncirrhotic patients with chronic hepatitis C virus genotype 1, 4, or 6 infection: a randomized trial. Ann Intern Med 163:1–13. doi: 10.7326/M15-0785. [DOI] [PubMed] [Google Scholar]
- 26.Kumada H, Sato K, Takehara T, Nakamuta M, Ishigami M, Chayama K, Toyota J, Suzuki F, Nakayasu Y, Ochi M, Yamada I, Okanoue T. 2015. Efficacy of telaprevir-based therapy for difficult-to-treat patients with genotype 2 chronic hepatitis C in Japan. Hepatol Res 45:745–754. doi: 10.1111/hepr.12416. [DOI] [PubMed] [Google Scholar]
- 27.Besse B, Coste-Burel M, Bourgeois N, Feray C, Imbert-Marcille BM, Andre-Garnier E. 2012. Genotyping and resistance profile of hepatitis C (HCV) genotypes 1-6 by sequencing the NS3 protease region using a single optimized sensitive method. J Virol Methods 185:94–100. doi: 10.1016/j.jviromet.2012.06.011. [DOI] [PubMed] [Google Scholar]
- 28.Paolucci S, Fiorina L, Piralla A, Gulminetti R, Novati S, Barbarini G, Sacchi P, Gatti M, Dossena L, Baldanti F. 2012. Naturally occurring mutations to HCV protease inhibitors in treatment-naive patients. Virol J 9:245. doi: 10.1186/1743-422X-9-245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Alves R, Queiroz AT, Pessoa MG, da Silva EF, Mazo DF, Carrilho FJ, Carvalho-Filho RJ, de Carvalho IM. 2013. The presence of resistance mutations to protease and polymerase inhibitors in hepatitis C virus sequences from the Los Alamos databank. J Viral Hepat 20:414–421. doi: 10.1111/jvh.12051. [DOI] [PubMed] [Google Scholar]
- 30.Lindenbach BD, Evans MJ, Syder AJ, Wolk B, Tellinghuisen TL, Liu CC, Maruyama T, Hynes RO, Burton DR, McKeating JA, Rice CM. 2005. Complete replication of hepatitis C virus in cell culture. Science 309:623–626. doi: 10.1126/science.1114016. [DOI] [PubMed] [Google Scholar]
- 31.Li YP, Ramirez S, Jensen SB, Purcell RH, Gottwein JM, Bukh J. 2012. Highly efficient full-length hepatitis C virus genotype 1 (strain TN) infectious culture system. Proc Natl Acad Sci U S A 109:19757–19762. doi: 10.1073/pnas.1218260109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sakai A, Takikawa S, Thimme R, Meunier JC, Spangenberg HC, Govindarajan S, Farci P, Emerson SU, Chisari FV, Purcell RH, Bukh J. 2007. In vivo study of the HC-TN strain of hepatitis C virus recovered from a patient with fulminant hepatitis: RNA transcripts of a molecular clone (pHC-TN) are infectious in chimpanzees but not in Huh7.5 cells. J Virol 81:7208–7219. doi: 10.1128/JVI.01774-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yanagi M, Purcell RH, Emerson SU, Bukh J. 1999. Hepatitis C virus: an infectious molecular clone of a second major genotype (2a) and lack of viability of intertypic 1a and 2a chimeras. Virology 262:250–263. doi: 10.1006/viro.1999.9889. [DOI] [PubMed] [Google Scholar]
- 34.Kato T, Furusaka A, Miyamoto M, Date T, Yasui K, Hiramoto J, Nagayama K, Tanaka T, Wakita T. 2001. Sequence analysis of hepatitis C virus isolated from a fulminant hepatitis patient. J Med Virol 64:334–339. doi: 10.1002/jmv.1055. [DOI] [PubMed] [Google Scholar]
- 35.Pietschmann T, Kaul A, Koutsoudakis G, Shavinskaya A, Kallis S, Steinmann E, Abid K, Negro F, Dreux M, Cosset FL, Bartenschlager R. 2006. Construction and characterization of infectious intragenotypic and intergenotypic hepatitis C virus chimeras. Proc Natl Acad Sci U S A 103:7408–7413. doi: 10.1073/pnas.0504877103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gottwein JM, Scheel TK, Callendret B, Li YP, Eccleston HB, Engle RE, Govindarajan S, Satterfield W, Purcell RH, Walker CM, Bukh J. 2010. Novel infectious cDNA clones of hepatitis C virus genotype 3a (strain S52) and 4a (strain ED43): genetic analyses and in vivo pathogenesis studies. J Virol 84:5277–5293. doi: 10.1128/JVI.02667-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gottwein JM, Scheel TK, Hoegh AM, Lademann JB, Eugen-Olsen J, Lisby G, Bukh J. 2007. Robust hepatitis C genotype 3a cell culture releasing adapted intergenotypic 3a/2a (S52/JFH1) viruses. Gastroenterology 133:1614–1626. doi: 10.1053/j.gastro.2007.08.005. [DOI] [PubMed] [Google Scholar]
- 38.Lontok E, Harrington P, Howe A, Kieffer T, Lennerstrand J, Lenz O, McPhee F, Mo H, Parkin N, Pilot-Matias T, Miller V. 10 June 2015. Hepatitis C virus drug resistance-associated substitutions: state of the art summary. Hepatology. doi: 10.1002/hep.27934. [DOI] [PubMed] [Google Scholar]
- 39.Lohmann V, Bartenschlager R. 2014. On the history of hepatitis C virus cell culture systems. J Med Chem 57:1627–1642. doi: 10.1021/jm401401n. [DOI] [PubMed] [Google Scholar]
- 40.Gottwein JM, Scheel TK, Jensen TB, Lademann JB, Prentoe JC, Knudsen ML, Hoegh AM, Bukh J. 2009. Development and characterization of hepatitis C virus genotype 1-7 cell culture systems: role of CD81 and scavenger receptor class B type I and effect of antiviral drugs. Hepatology 49:364–377. doi: 10.1002/hep.22673. [DOI] [PubMed] [Google Scholar]
- 41.Li YP, Gottwein JM, Scheel TK, Jensen TB, Bukh J. 2011. MicroRNA-122 antagonism against hepatitis C virus genotypes 1-6 and reduced efficacy by host RNA insertion or mutations in the HCV 5′ UTR. Proc Natl Acad Sci U S A 108:4991–4996. doi: 10.1073/pnas.1016606108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gottwein JM, Jensen SB, Serre SB, Ghanem L, Scheel TK, Jensen TB, Krarup H, Uzcategui N, Mikkelsen LS, Bukh J. 2013. Adapted J6/JFH1-based hepatitis C virus recombinants with genotype-specific NS4A show similar efficacies against lead protease inhibitors, alpha interferon, and a putative NS4A inhibitor. Antimicrob Agents Chemother 57:6034–6049. doi: 10.1128/AAC.01176-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wakita T, Pietschmann T, Kato T, Date T, Miyamoto M, Zhao Z, Murthy K, Habermann A, Krausslich HG, Mizokami M, Bartenschlager R, Liang TJ. 2005. Production of infectious hepatitis C virus in tissue culture from a cloned viral genome. Nat Med 11:791–796. doi: 10.1038/nm1268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Li YP, Ramirez S, Gottwein JM, Scheel TK, Mikkelsen L, Purcell RH, Bukh J. 2012. Robust full-length hepatitis C virus genotype 2a and 2b infectious cultures using mutations identified by a systematic approach applicable to patient strains. Proc Natl Acad Sci U S A 109:E1101–E1110. doi: 10.1073/pnas.1203829109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ramirez S, Li YP, Jensen SB, Pedersen J, Gottwein JM, Bukh J. 2014. Highly efficient infectious cell culture of three hepatitis C virus genotype 2b strains and sensitivity to lead protease, nonstructural protein 5A, and polymerase inhibitors. Hepatology 59:395–407. doi: 10.1002/hep.26660. [DOI] [PubMed] [Google Scholar]
- 46.Li YP, Ramirez S, Mikkelsen LS, Bukh J. 2015. Efficient infectious cell culture systems of the hepatitis C virus (HCV) prototype strains HCV-1 and H77. J Virol 89:811–823. doi: 10.1128/JVI.02877-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Russell RS, Meunier JC, Takikawa S, Faulk K, Engle RE, Bukh J, Purcell RH, Emerson SU. 2008. Advantages of a single-cycle production assay to study cell culture-adaptive mutations of hepatitis C virus. Proc Natl Acad Sci U S A 105:4370–4375. doi: 10.1073/pnas.0800422105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Date T, Kato T, Kato J, Takahashi H, Morikawa K, Akazawa D, Murayama A, Tanaka-Kaneko K, Sata T, Tanaka Y, Mizokami M, Wakita T. 2012. Novel cell culture-adapted genotype 2a hepatitis C virus infectious clone. J Virol 86:10805–10820. doi: 10.1128/JVI.07235-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yamane D, McGivern DR, Wauthier E, Yi M, Madden VJ, Welsch C, Antes I, Wen Y, Chugh PE, McGee CE, Widman DG, Misumi I, Bandyopadhyay S, Kim S, Shimakami T, Ikawa T, Whitmire JK, Heise MT, Dittmer DP, Kao CC, Pitson SM, Merrill AHJ, Reid LM, Lemon SM. 2014. Regulation of the hepatitis C virus RNA replicase by endogenous lipid peroxidation. Nat Med 20:927–935. doi: 10.1038/nm.3610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Romano KP, Ali A, Aydin C, Soumana D, Ozen A, Deveau LM, Silver C, Cao H, Newton A, Petropoulos CJ, Huang W, Schiffer CA. 2012. The molecular basis of drug resistance against hepatitis C virus NS3/4A protease inhibitors. PLoS Pathog 8:e1002832. doi: 10.1371/journal.ppat.1002832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lim SR, Qin X, Susser S, Nicholas JB, Lange C, Herrmann E, Hong J, Arfsten A, Hooi L, Bradford W, Najera I, Smith P, Zeuzem S, Kossen K, Sarrazin C, Seiwert SD. 2012. Virologic escape during danoprevir (ITMN-191/RG7227) monotherapy is hepatitis C virus subtype dependent and associated with R155K substitution. Antimicrob Agents Chemother 56:271–279. doi: 10.1128/AAC.05636-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Shimakami T, Welsch C, Yamane D, McGivern DR, Yi M, Zeuzem S, Lemon SM. 2011. Protease inhibitor-resistant hepatitis C virus mutants with reduced fitness from impaired production of infectious virus. Gastroenterology 140:667–675. doi: 10.1053/j.gastro.2010.10.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sarrazin C, Kieffer TL, Bartels D, Hanzelka B, Muh U, Welker M, Wincheringer D, Zhou Y, Chu HM, Lin C, Weegink C, Reesink H, Zeuzem S, Kwong AD. 2007. Dynamic hepatitis C virus genotypic and phenotypic changes in patients treated with the protease inhibitor telaprevir. Gastroenterology 132:1767–1777. doi: 10.1053/j.gastro.2007.02.037. [DOI] [PubMed] [Google Scholar]
- 54.Aloia AL, Eyre NS, Black S, Bent SJ, Gaeguta A, Guo Z, Narayana SK, Chase R, Locarnini S, Carr JM, Howe JA, Beard MR. 2015. Generation of a chimeric hepatitis C replicon encoding a genotype-6a NS3 protease and assessment of boceprevir (SCH503034) sensitivity and drug-associated mutations. Antivir Ther 20:271–280. doi: 10.3851/IMP2850. [DOI] [PubMed] [Google Scholar]
- 55.Combet C, Garnier N, Charavay C, Grando D, Crisan D, Lopez J, Dehne-Garcia A, Geourjon C, Bettler E, Hulo C, Le MP, Bartenschlager R, Diepolder H, Moradpour D, Pawlotsky JM, Rice CM, Trepo C, Penin F, Deleage G. 2007. euHCVdb: the European hepatitis C virus database. Nucleic Acids Res 35:D363–D366. doi: 10.1093/nar/gkl970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kuiken C, Hraber P, Thurmond J, Yusim K. 2008. The hepatitis C sequence database in Los Alamos. Nucleic Acids Res 36:D512–D516. doi: 10.1093/nar/gkm962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ampuero J, Romero-Gomez M, Reddy KR. 2014. Review article: HCV genotype 3–the new treatment challenge. Aliment Pharmacol Ther 39:686–698. doi: 10.1111/apt.12646. [DOI] [PubMed] [Google Scholar]
- 58.Summa V, Ludmerer SW, McCauley JA, Fandozzi C, Burlein C, Claudio G, Coleman PJ, DiMuzio JM, Ferrara M, Di FM, Gates AT, Graham DJ, Harper S, Hazuda DJ, McHale C, Monteagudo E, Pucci V, Rowley M, Rudd MT, Soriano A, Stahlhut MW, Vacca JP, Olsen DB, Liverton NJ, Carroll SS. 2012. MK-5172, a selective inhibitor of hepatitis C virus NS3/4a protease with broad activity across genotypes and resistant variants. Antimicrob Agents Chemother 56:4161–4167. doi: 10.1128/AAC.00324-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.