

Summary
Glioblastoma (GBM) is an aggressive brain tumor whose growth is driven by stem cell-like cells. BMP signaling triggers cell-cycle exit and differentiation of GBM stem cells (GSCs) and, therefore, might have therapeutic value. However, the epigenetic mechanisms that accompany differentiation remain poorly defined. It is also unclear whether cell-cycle arrest is terminal. Here we find only a subset of GSC cultures exhibit astrocyte differentiation in response to BMP. Although overtly differentiated non-cycling astrocytes are generated, they remain vulnerable to cell-cycle re-entry and fail to appropriately reconfigure DNA methylation patterns. Chromatin accessibility mapping identified loci that failed to alter in response to BMP and these were enriched in SOX transcription factor-binding motifs. SOX transcription factors, therefore, may limit differentiation commitment. A similar propensity for cell-cycle re-entry and de-differentiation was observed in GSC-derived oligodendrocyte-like cells. These findings highlight significant obstacles to BMP-induced differentiation as therapy for GBM.
Graphical Abstract
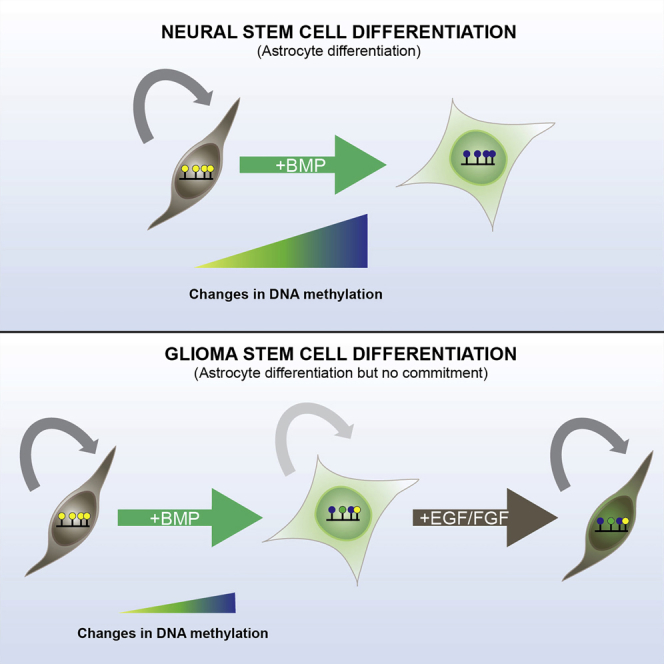
Highlights
-
•
Genome-wide profiling shows DNA methylation patterns during glioblastoma (GBM) differentiation
-
•
Delayed and incomplete epigenetic changes appear in GBM stem cells in response to BMP
-
•
SOX transcription factors may explain the lack of terminal differentiation
-
•
Lack of differentiation commitment limits the effectiveness of BMP-based therapies
BMP induces differentiation of glioblastoma stem cells (GSCs), but it remains unclear if differentiation commitment and permanent cell-cycle arrest occurs. Pollard, Beck, and colleagues report that differentiated progeny of GSCs fail to reconfigure DNA methylation patterns and are vulnerable to de-differentiation. Failure to suppress the activity of SOX transcription factors may explain this deficit.
Introduction
Many solid tumors display phenotypic and functional cellular heterogeneity reminiscent of normal tissues (Shackleton et al., 2009). An underlying developmental hierarchy therefore may exist, with a subset of malignant stem cell-like cells generating more differentiated non-malignant progeny. If malignant stem cells could be permanently forced into a non-proliferative and terminally differentiated state, then differentiation therapy might be highly effective.
Glioblastoma (GBM) is one of the most aggressive human cancers. GBMs contain distinct cellular subpopulations expressing neural stem (NS) and progenitor cell markers (e.g., NESTIN, SOX2, OLIG2, ASCL1, and FOXG1) (Dahlstrand et al., 1992, Singh et al., 2004, Suvà et al., 2014, Verginelli et al., 2013). Isolation and orthotopic xenotransplantation of GBM stem cells (GSCs) has confirmed their tumor-initiating potential (Singh et al., 2004). GBMs also harbor cells expressing markers associated with differentiated glial lineages (e.g., O4- and MBP-expressing oligodendrocytes or GFAP-expressing astrocytes). Astrocytes and oligodendrocytes are post-mitotic under normal homeostasis, but whether GSCs can generate terminally differentiated progeny remains a significant unresolved issue.
BMP signaling triggers cell-cycle exit and astrocyte differentiation of NS cells (Bonaguidi et al., 2005). Exposure of GBM-derived stem cells to BMP4 can drive astrocyte differentiation both in vitro and in vivo (Piccirillo et al., 2006), raising the prospect that this pathway could be exploited for differentiation therapy. It is assumed that GBM stem cell differentiation follows a unidirectional path; indeed, a gene regulatory network that defines the tumor-initiating state and distinguishes non-tumorigenic astrocytes has been defined recently (Suvà et al., 2014). There are, however, many key unresolved questions. Can BMP induce a consistent differentiation response widely across the spectrum of GBMs? Is astrocyte differentiation accompanied by extensive transcriptional and epigenetic modifications, and do these mirror normal NS cell differentiation? Are epigenetic gene regulatory mechanisms, such as DNA methylation or chromatin remodelling, able to stably suppress pro-tumorigenic pathways? Can differentiation cues trigger a permanent cell-cycle arrest?
Here we use primary human GBM-derived NS (GNS) cell cultures as a tractable and disease-relevant experimental model to determine the transcriptional and epigenetic changes accompanying differentiation. We assessed longitudinal genome-wide DNA methylation changes, chromatin accessibility, and the stability of the differentiated phenotype. Our findings highlight several important caveats that may undermine the utility of BMP signaling for the treatment of GBM.
Results
GNS Cell Lines Display Diverse Responses to Differentiation Cues
We first assessed differentiation responses in a panel of six previously reported GNS cell lines obtained from independent patient tumors (Pollard et al., 2009, Fael Al-Mayhani et al., 2009), which mirror the transcriptional subtypes of primary GBM (Figure S1A). Growth factor (GF) withdrawal from culture media typically results in spontaneous multi-lineage differentiation of GNS cells, with a subpopulation of cells undergoing cell-cycle exit (Pollard et al., 2009). However, a more pronounced and uniform astrocyte differentiation response is observed following 7 days of BMP4 treatment and GF withdrawal (no EGF or FGF-2).
Each of the six GNS cell lines tested displayed reduced proliferation in response to BMP to a greater extent than GF withdrawal alone, confirming the cytostatic effects of BMP signaling (Figure 1A). However, consistent with previous reports (Lee et al., 2008), we also observed clear variability between GNS cell lines in the degree of the BMP-induced cytostatic response and the proportion of cells that activated GFAP expression (Figure 1B). GNS cell lines G19 and G26 exhibited uniform upregulation of the astrocyte marker GFAP, whereas G144 and G166 failed to efficiently upregulate GFAP (Figure 1B). EdU incorporation assays and immunocytochemistry for the cell cycle marker MCM2 confirmed that >80% of cells were driven out of the cell cycle in G19 and G26 following 8 days of continuous BMP treatment (Figures 1B–1D). Thus, BMP does not effectively induce astrocyte differentiation across all patient-derived GNS cell lines. Only in G19 and G26 did we observe a robust cytostatic astrocyte differentiation response similar to genetically normal NS cell controls. Levels of BMPR1B expression may explain the differential responses observed in these two GSC lines, as reported previously (Lee et al., 2008); we found BMPR1B mRNA at >10-fold higher levels in G19 and G26 compared to other lines (Figure 1E). G19 and G26 therefore were used in subsequent experiments to explore transcriptional and epigenetic changes in differentiating astrocytes.
Figure 1.

BMP Treatment Reduces Proliferation of GNS and NS Cells
(A) Proliferation curves of seven GNS and two NS cells (NS-1 and NS-2). At day 7 all paired comparisons (GF versus BMP4) showed a significant difference in proliferation rate (p < 0.01).
(B) Cells were expanded in the GFs EGF and FGF-2 (GF) or exposed to BMP4 in the absence of GFs for 8 days (BMP). Proliferation was assessed by EdU (16 hr incorporation) and astrocyte differentiation using GFAP (red).
(C) Quantification of EdU-positive cells in proliferating conditions (GF), GF withdrawal (GF−), and BMP4 is shown.
(D) Immunostaining for cell-cycle marker MCM2 (red) and quantification (bottom) are shown.
(E) Relative mRNA expression levels of the BMPR1B in NS and GNS cell lines (fold change relative to normal brain). Error bars denote SD of two technical and two biological replicates for (C)–(E) (triplicates for immunostainings). Scale bars in (B) and (D), 100 μm.
BMP-Induced Transcriptional Changes Continue to Accrue over Many Weeks in Post-mitotic GBM-Derived Astrocytes
To first delineate the kinetics of transcriptional changes associated with the response to BMP4, we initially assessed mRNA expression of key markers over a time course of 8, 16, 32, and 48 days in G26. As anticipated, the NS cell-associated markers OLIG2 and EGFR genes were rapidly downregulated following 8 days of BMP-4 treatment; astrocyte markers GFAP and AQP4, as well as the BMP-signaling genes ID1–3, were upregulated (Figure 2A). However, we also noted continuous changes in levels of expression over the entire time course, up until 48 days. Similarly, slow kinetics of transcriptional change were observed by Taqman low-density array assays for a set of 72 genes that we had identified previously as candidate regulators of oncogenic pathways in GBM (Engström et al., 2012; Figure 2B). Thus, while cell-cycle exit and morphological changes occur rapidly in response to BMP treatment, there is also a slower accumulation of additional transcriptional changes that accrue over the course of weeks and months. These might be indicative of an epigenetic progression or epigenetic switching (i.e., a slow directional change until threshold or switch point is reached), which may be required for stabilization of the differentiated state. These later time points (32 and 48 days) therefore were included in the profiling of DNA methylation patterns described below.
Figure 2.

Kinetics of DNA Methylation Changes Induced by BMP in NS and GNS Cells
(A) The qRT-PCR analysis of G26 during a 48-day time course of BMP treatment. Error bars denote SD of four independent experiments (technical and biological duplicates).
(B) A panel of 72 genes, previously defined as differentially expressed between GNS and NS cells (Engström et al., 2012), was analyzed using Taqman low-density arrays on G26 cells treated for up to 48 days with BMP4.
(C) Total numbers of identified MVPs during BMP treatment for GNS cell lines (G19 and G26) and normal NS cells (NS-1) at each time point (BMP 8–48 days; B8, B16, B32, B48) are shown.
(D) Dendrogram shows the 450K methylation array data.
BMP-Induced Astrocyte Differentiation Is Accompanied by Delayed or Incomplete Reconfiguration of DNA Methylation Patterns in GNS Cells
Patterns of DNA methylation frequently are reconfigured during stem cell differentiation to stabilize and safeguard the differentiated state. Altered DNA methylation is, accordingly, a plausible molecular mechanism that could function in differentiating tumor stem cells to silence oncogenic pathways (Carén et al., 2013). We therefore first assessed whether G19 and G26 displayed methylation profiles in proliferating cultures prior to BMP treatment that were disease relevant. DNA methylation profiles across a panel of 13 different GNS cell lines were defined using the HumanMethylation450 BeadChips, and they were compared to data from primary tumors attained using the same platform as the Cancer Genome Atlas (TCGA). All GNS cells used in this study were found to display patterns of DNA methylation similar to primary GBM tumors (Figure S1B). None of the GNS cell lines displayed a glioma-CpG island hypermethylator phenotype (G-CIMP), which is a feature of secondary GBM. G19 and G26 cell cultures therefore harbor DNA methylation patterns similar to those of primary GBM tumors.
We next defined patterns of DNA methylation across a 48-day time course in post-mitotic BMP-induced differentiating astrocytes derived from G19, G26, plus normal reference NS cell controls. Surprisingly, few methylation variable positions (MVPs) accumulated in the first 8–16 days, and the vast majority emerged more slowly over a period of many weeks for each cell line, including normal NS cells (Figures 2C and 2D). Thus, we did not observe rapid widespread DNA methylation changes associated with astrocyte differentiation (within days), as has been described for the hematopoietic lineage (Ji et al., 2010).
MVPs were enriched at enhancers and gene bodies, consistent with a role in transcriptional regulation (Figure 3A), and we only rarely identified MVPs that reverted over the 48-day time course (<1% of probes in G26). There were no significant directional global changes in non-CpG methylation or hydroxymethylation, which is consistent with the observed gain of methylation (data not shown). Controls grown under normal self-renewal conditions (plus EGF and FGF-2; without BMP) for 48 days showed no significant alterations in methylation compared to day 0 samples.
Figure 3.

Analysis of Sites of DNA Methylation Alterations Imposed by BMP Treatment
(A) Percentage enrichment of epigenomic features (left), enhancers (middle), and PRC2 target genes (right) is shown (random resampling p value ≤ 0.001).
(B) Visualization of MVPs over the time course of BMP is shown for the following: the astrocyte marker S100A6, the neurotransmitter receptor GRIK2, and the WNT-signaling tumor suppressor gene SFRP2.
(C) GO analysis for the significantly altered MVPs with >30% change in methylation were analyzed using DAVID (GO BP_ALL).
(D) Publicly available methylation datasets for pilocytic astrocytoma and human brain orbitofrontal cortex (non-neuronal) were used to generate a set of GBM-specific MVPs (top 100 MVPs shown), and these sites are shown for the BMP-treated GNS cells. Each experiment represents biological and technical duplicates of each sample.
In G26, MVPs accumulated increasingly with the duration of BMP exposure at genes associated with neuronal and astrocyte differentiation, such as GRIK2 and S100A6, respectively (Figure 3B). Additionally, consistent with an appropriate differentiation response, gene ontology (GO) terms such as neurogenesis and cell differentiation emerged as statistically enriched in the set of 2,325 genes (with MVPs of more than 30% alteration in methylation frequency) (Figure 3C).
We also identified enrichment of polycomb repressor complex 2 (PRC2) target genes (Figure 4D), particularly CpG island/shore/shelf regions for G26 and NS-1 (p < 0.001) (Figure 3A). A set of GBM MVPs that are associated with primary GBM tumors, but not low-grade tumors or normal neural cell types, was compiled from published datasets (see Experimental Procedures). Approximately 12% of these sites, including those associated with SFRP2, a WNT-signaling pathway antagonist, were altered in response to BMP in G26 (Figure 3D). These data are consistent with G26 cell responses to BMP involving progression to a differentiated and less malignant state.
Figure 4.

RNA-Seq Analysis of BMP4-Treated G26 and NS-1 Cells
(A) Gene expression of the MVP-associated genes GRIK2, SFRP2, and S100A6 (left) and the two top downregulated genes OLIG1 and OLIG2 (right). Fold change of the average of the number of reads in the two passages is shown.
(B) Dendrogram of the RNA-seq data is shown.
(C) The mRNA expression levels for many PRC2 target genes are frequently altered during BMP treatment.
(D) The mRNA levels for FOXM1 and PLK1 are shown.
(E) Heatmap shows transcription factors associated with the tumor-propagating state that recently was defined (Suvà et al., 2014).
(F) Gene expression of DNA replication licensing proteins and cell-cycle regulators is shown relative to growth factors (GF) at day 0.
(G) Quantification of MCM2-positive cells from immunocytochemistry. Each experiment represents biological and technical duplicates of each sample. Error bars denote SD of replicates.
Despite the above observations, we noted that DNA methylation changes were delayed in G26 compared to normal NS cells (Figure 2C). Also, for G19 we identified only limited numbers of MVPs (<500 compared to >5,000 for NS and G26), even after 48 days of BMP treatment (Figure 2C). Thus, we observed an incomplete acquisition of altered DNA methylation patterns during the differentiation response for G19. Together these data indicate that, even within the subset of GNS cells that display strong cytostatic responses to BMP, there is a failure to rapidly and fully reconfigure differentiation-associated patterns of DNA methylation.
BMP Signaling Fails to Silence Transcription of Cell Cycle and DNA Replication Licensing Genes in GNS Cells
The large number of MVPs detected in G26 following BMP treatment prompted us to assess genome-wide transcriptional changes using RNA sequencing (RNA-seq). We confirmed that MVP-associated genes, S100A6, GRIK2, and SFRP2, displayed altered gene expression patterns in the expected direction (Figure 4A). As anticipated, astrocyte marker genes GFAP, AQP4, and ALDH1A1 (Adam et al., 2012) also were upregulated, as were components of the BMP-signaling pathway, such as SMADs and IDs.
Cluster analysis of mRNA expression also suggested two phases of transcriptional change, the first occurring as a rapid immediate response to BMP and a second occurring slowly over subsequent weeks, mirroring the time course of DNA methylation changes (Figures 4B and 4C). The most significantly downregulated genes in BMP-treated G26 cells were the well-studied NS cell regulators, OLIG1 (fold change 570, adjusted p value 6.6E−52) and OLIG2 (165-fold, adjusted p value 9.5E−42) (Figures 4A and 4E). Expression of many other PRC2 target genes also was altered, such as HES5, and components of the WNT-signaling pathway (WNT1, WNT5A, and FRZB) (Figure 4C). GO terms such as cell differentiation and nervous system development were identified in this set (data not shown). We also observed BMP-induced suppression of mRNA levels for many of the transcription factors that define the core gene regulatory network recently defined for GBM stem-like cells (Suvà et al., 2014; Figure 4E).
In G26, the majority of gene expression changes entailed an increase in expression level, whereas both upregulated and downregulated genes were detected in NS cells; this suggests a failure to appropriately silence genes in G26 (data not shown). This set of failed silencing genes included many DNA replication licensing proteins and cell-cycle regulators, including the mini-chromosome maintenance proteins (MCMs); CDT1; proliferating cell nuclear antigen (PCNA, a co-factor of DNA polymerase delta); MYBL2, involved in cell-cycle progression; and mitotic regulators FOXM1 and PLK1 (Figure 4F). We also confirmed this at the protein level using immunocytochemistry analysis of MCM2 (Figure 4G). Thus, while BMP can impose appropriate transcriptional changes associated with BMP-induced differentiation, there is incomplete silencing of expression of the genes involved in competence for cell-cycle re-entry.
GBM-Derived Astrocyte-like Cells Do Not Undergo Terminal Cell-Cycle Arrest
Stem cells within tissues that turn over rapidly, such as blood and skin, generate terminally differentiated progeny with a limited lifespan; differentiation therapy therefore can eradicate proliferating tumor cells (e.g., in acute promyelocytic leukemia [APL]; Sell, 2004). By contrast, in the nervous system astrocytes and oligodendrocytes are long-lived, and thus any differentiation therapy for GBM must ensure that differentiation is accompanied by robust suppression of proliferative potential.
While the majority of astrocytes in the adult brain are post-mitotic, the quiescent NS cell population in the adult subependymal zone displays astrocytic features, including GFAP expression (Doetsch et al., 1999). Additionally, under certain injury conditions, GFAP-expressing astrocytes can be proliferative (e.g., reactive gliosis). GFAP also is expressed by radial glia progenitors during fetal development. Hence, whether GFAP-expressing astrocytes induced following BMP treatment of GNS cells are irreversibly cell-cycle arrested or in a quiescent/G0 state has not yet been resolved. Failure to fully silence expression of DNA replication licensing components and incomplete reconfiguration of DNA methylation patterns prompted us to test if GNS cell-derived astrocytes are terminally cell-cycle arrested or instead driven to a transcriptional state with hallmarks of quiescent astrocyte stem cells.
Limited detection of MCM2 protein and EdU incorporation in the majority of the G26 cells in BMP-treated cultures, and failure of significant increases in cell numbers throughout the 48-day time course, suggested that BMP-treated G26 cells had withdrawn from cell cycle or were slow cycling (Figure 1A). To test whether proliferative potential was irrevocably lost, we tested the consequences of re-exposing non-cycling and overtly differentiated astrocytes to GFs (i.e., self-renewal conditions EGF and FGF-2, with no BMP). We found that, despite the extended time in a non-cycling state at both early and late stages of differentiation (7 or 54 days of BMP treatment), GNS cells immediately began proliferating rapidly again (Figures 5A and 5B). GNS cell-derived astrocytes are, therefore, not terminally cell-cycle arrested; they are able to readily re-enter the cell proliferation cycle upon withdrawal of differentiation stimulus.
Figure 5.

BMP-Treated Cells Do Not Undergo Terminal Differentiation
(A) G26 under self-renewal conditions (GF), treated with BMP4 for 7 (B7) and 54 days (B54), and when adding back GFs after the respective treatment times for 4 days (B7 + GF4 and B54 + GF4) is shown.
(B) Quantitation of EdU-positive cells from (A). Error bars denote SD of five technical replicates. Student’s two-sided t test was used for pairwise comparisons (∗p ≤ 0.05).
(C) The qPCR analysis of G26 to determine the effects of re-exposure to GFs on astrocyte markers (AQP4 and GFAP) and NS cell-associated markers (EGFR, SOX2, and OLIG2). Error bars denote SD of two technical replicates from two biological replicates relative to cultures in GFs at day 0.
(D) Dendrogram shows the 450K methylation data in G26 before and after the readdition of EGF and FGF-2 for 4 days (GF).
(E) Kaplan-Meier curve showing survival data of mice transplanted with G26 grown under self-renewal conditions (GF), treated with BMP4 for 7 (B7) and 54 days (B54), and when adding back GFs after the respective treatment times for 4 days (B7 + GF4 and B54 + GF4) (n = 4–7 mice in each group). Scale bars, 100 μm.
We next assessed DNA methylation patterns and differentiation markers in the astrocytes that re-entered cell cycle. GFAP and AQP4 mRNA were markedly downregulated, but not fully extinguished, within 4 days (Figure 5C), indicative of partial de-differentiation. This contrasts with the response in NS cells, where GFAP expression remained stable (and increased) after the 4 days (Figure S2). Transcription of OLIG2 and EGFR, two key regulators of the NS cell state, was increased, although it did not reach NS cell levels (Figure 5C). DNA methylation analysis indicated that, despite cell-cycle re-entry, the patterns of DNA methylation were not immediately reverted to the original stem cell state (Figure 5D). Together these data indicate that BMP-induced astrocytes remain capable of rapidly re-engaging in cell cycle, even after many weeks in a non-proliferative post-mitotic state, and they undergo a partial de-differentiation. Differentiated astrocytes therefore are not permanently driven to a terminally differentiated cell-cycle arrested state by BMP treatment, and they remain vulnerable to reacquisition of a proliferative stem cell-like state.
BMP4-Treated GNS Cells Remain Tumor Initiating
We next assessed whether BMP-treated cells remain tumorigenic. We performed orthotopic transplantation of the following: untreated G26 cells, BMP-treated G26 cells (either 7 or 54 days), and BMP-treated cells that were subsequently re-exposed to EGF and FGF-2 for 4 days (prior to transplantation). Cells were injected into the ventral forebrain of immunocompromised mice and we then monitored overall survival. BMP-treated cells did not show any significant increase in survival times (Figure 5E), suggesting that terminal differentiation of the cells is not achieved and the transplanted population is readily able to generate tumors.
Chromatin Accessibility Mapping Reveals SOX Motif-Enriched Regions that Fail to Close in BMP-Treated GNS Cells
We hypothesized that the failure of GNS cells to undergo differentiation commitment in response to BMP might result from an inappropriate chromatin remodelling at gene regulatory regions, such as enhancers. The assay for transposase-accessible chromatin using sequencing (ATAC-seq) provides a powerful method for defining chromatin compaction and nucleosome positioning genome-wide (Buenrostro et al., 2013). We used this technique to define in an unbiased manner whether genomic regions in GNS cells failed to be silenced relative to NS cells after 8 days of BMP treatment. Principal component analysis (PCA) confirmed significant BMP-induced chromatin remodelling in both GNS and NS cells (Figure 6A). To determine any regions of incomplete silencing specific to GNS cells, we identified differentially accessible regions that likely include enhancer elements. Those loci that failed to be silenced in G26 B8 were identified by the intersection of differentially more accessible loci in NS cells versus BMP-differentiated progeny shared with loci more accessible in the BMP-treated GNS cells versus BMP-treated NS cells. This analysis identified 268 loci (Figure 6B).
Figure 6.

ATAC-Seq Analysis of BMP4-Treated G26 and NS-1 Cells
(A) PCA plot shows ATAC-seq data.
(B) Heatmap generated for those loci that failed to be closed during BMP-induced differentiation in G26 compared to normal NS cells. KEGG glioma gene proximal loci (<10 kb) are labeled in red; cell-cycle genes in blue are the shared glioma cell-cycle loci in black.
(C) Genome browser view of the IGF1 glioma locus is shown.
(D) Motif enrichment analysis identified a high frequency of SOX motifs within those peaks identified in (B); two top motifs (lowest adjusted p values) for SOX2 are shown (see also Table S1 for full motif sets).
We used the Genomic Regions Enrichment of Annotations Tool (GREAT) to identify enriched genes associated with these regions. The top identified Kyoto Encyclopedia of Genes and Genomes (KEGG) category was glioma pathways (genes CAMK2B, CAMK2D, CALM1, IGF1, and PIK3R1). Several other cell-cycle regulators also were identified (Figure 6B). We determined which transcription factor-binding motifs were overrepresented in these loci and identified many binding motifs associated with SOX proteins (Table S1). Motifs for many developmental transcription factors, such as those of the SOX, GATA, POU, and FOX families, were enriched. These included motifs for SOXB1 family members, most notably the critical NS cell self-renewal factor SOX2 (Figure 6D). Together these data suggest that increased levels and/or activity of SOX proteins could undermine the ability of GNS cells to exit self-renewal.
GNS Cell-Derived Oligodendrocyte Cells Fail to Undergo Terminal Cell-Cycle Arrest and Are Able to De-differentiate
Oligodendrocyte differentiation of GNS cells might provide an alternative possibility for successful differentiation therapy for GBM. The cell surface marker O4 defines a pre-oligodendrocyte cell state, while MBP defines a more mature myelinating state (Zhang, 2001). Building on our previous studies (Pollard et al., 2009), we identified a subset of GNS cells (G144, G25, and G2) that could readily generate O4-expressing oligodendrocyte-like cells by the removal of GFs (Figures 7A–7C). O4-expressing cells emerge several days following GF withdrawal, acquire an elaborate oligodendrocyte-like branched morphology, and are non-proliferative when assessed by EdU incorporation (Figure 7A). Our qRT-PCR confirmed that MBP and MAG were activated (Figure 7D); however, by immunocytochemistry we found that MBP, a more mature oligodendrocyte marker, was expressed only in a handful of the O4-expressing cells, suggesting the majority of cells fail to fully mature (data not shown). Consistent with this, upon reintroduction of GFs for 4 days, it was clear that, similar to the BMP-induced astrocytes, the O4-positive cells remained competent to re-enter cell cycle (Figure 7E).
Figure 7.

Oligodendrocyte-like Cells Generated from GNS Cells Fail to Terminally Differentiate
(A) G144 cells stained with O4 (red) and EdU under self-renewal conditions (+GF) and under differentiating conditions (GF withdrawal, −GF) (left) and flow cytometry (right) are shown.
(B) Live O4 immunostaining in G19, G25, and G144 after GF withdrawal for 7 days is shown (green).
(C) Quantitation of flow analysis from (B). Error bars denote SD of biological replicates (n = 3–7).
(D) The qPCR analysis of the oligodendrocyte markers MBP and MAG. Error bars denote SD of biological and technical duplicates.
(E) EdU incorporation in proliferating G26 (left), after 10 days GF withdrawal (middle), and at re-exposure to EGF and FGF-2 (GF) (right). Quantification shows the same experiment and additional experiments at 20 days (six biological replicates).
(F) (Top) Experimental design for clonal analysis of differentiation commitment in O4-positive cells. (Bottom) Live immunostaining of O4 in the G144 culture after GF withdrawal (left), at single-cell deposition (middle), and after adding back GFs (right) is shown.
(G) Quantification of O4-positive cells after single-cell deposition indicates that O4-positive cells can re-enter cell cycle and proliferate. Total number of cells in each well was scored after 10 days of GF re-addition. Undifferentiated controls, UD; cell death, cross. Scale bars, 100 μm.
A significant reduction in the numbers of O4-positive cells was observed after only 48 hr of GF re-exposure, suggesting that the majority of O4 cells might be quickly reverting to the GNS state by de-differentiation. To test this directly, we made use of the fact that O4 is a live cell surface marker amenable to fluorescence-activated cell sorting (FACS), enabling single-cell isolation and clonal analysis (Figures 7F and 7G). We harvested the highest 1% O4-expressing subpopulation after 8 days of differentiation by FACS and deposited single cells in each well of a microplate. Surprisingly, we found that O4-expressing cells are able to readily re-enter cell cycle with similar frequency to O4-negative cells (both G144 and G25) (Figure 7G). Together these data demonstrate that, similar to astrocyte differentiation, oligodendrocyte-like cells generated from GNS cell differentiation do not readily undergo terminal differentiation and permanent cell-cycle arrest and remain vulnerable to de-differentiation.
Discussion
GBM-derived stem cells previously have been reported to differentiate in response to BMPs and to acquire a non-malignant astrocyte-like phenotype (Piccirillo et al., 2006). For differentiation therapy to be effective for GBM, it will be important to attain terminal cell-cycle arrest, both because of the nature of the disease (tumor cells widely disseminate through the brain by infiltration) and because of the nature of the differentiated cell types (neurons, oligodendrocytes, and astrocytes), which unlike tissues with a rapid cellular turnover are long-lived. Here we assessed the capacity of human GBM-derived stem cells to undergo appropriate terminal astrocyte and oligodendrocyte differentiation. Our findings suggest inter-tumoral heterogeneity in cytostatic differentiation responses, and a limited scope for proper epigenetic resetting could limit the effectiveness of differentiation therapy for GBM.
We first explored whether GBM stem cells derived from independent tumors would all respond similarly. Striking differences were observed among the distinct patient-derived GNS cell lines, with a spectrum of outcomes ranging from a full cytostatic response to lack of any phenotypic change following exposure to BMP4. It is likely that these differential responses in vitro would be mirrored by patient responses in a clinical setting and that only a subset of patients might respond to BMP4 differentiation therapy. Furthermore, BMP-unresponsive cells would be likely to become enriched following treatment and subsequent tumor growth due to selective pressures.
Second, we focused our attention on the subset of GNS cultures in which there was a strong cytostatic response (G26 and G19) and assessed whether differentiation was accompanied by reconfiguration of the epigenetic landscape (DNA methylation) in a manner similar to genetically normal NS cells. We found that one patient line (G26 cells) indeed responded with a significant number of reconfigured loci, including those of known importance to neuronal and glial differentiation. However, these responses were delayed, and RNA-seq analysis revealed that the expression of many genes controlling the cell cycle/DNA replication licensing apparatus was not extinguished. More significantly, for another cell line (G19), despite a uniform exit from cell cycle and upregulation of GFAP, we did not observe any striking changes in DNA methylation patterns. Thus, even in the two GNS cell lines that underwent BMP-induced cell-cycle exit, there was incomplete epigenetic progression associated with astrocyte differentiation.
Third, we tested the permanence of cell-cycle exit for both post-mitotic astrocytes and oligodendrocytes derived from the GNS cells. Although cells were initially overtly differentiated, they remained immediately responsive to re-exposure to EGF and FGF-2 and began proliferating. This rapid re-entry into cell cycle was unexpected, as these cells had striking morphological changes and molecular markers of differentiation, and they were kept in a non-cycling state for many weeks or months (for astrocytes). Clearly, a latent ability to re-enter a cycling state is not desirable for any prospective differentiation therapy. It is possible that the combination of BMP and increased cell-autonomous EGF/FGF signaling (due to genetic pathways) within GNS cells forces them into a quiescent astrocyte stem cell state, rather than triggering terminal astrocyte differentiation (Martynoga et al., 2013). Moreover, our GNS cell cultures may have been heterogeneous in their response to BMP, with subsets of cells primed to becoming quiescent astrocyte-like stem cells (type B cells), while others were driven to being differentiated astrocytes. However, pervasive cell-cycle re-entry was observed upon re-exposure to GFs, suggesting that neither cell state had undergone proliferative arrest. Better markers that distinguish GFAP-expressing astrocyte stem cells from parenchymal differentiated astrocytes will be needed to study this further. Determining the molecular pathways that control reactivation of dormant or quiescent stem cells will be an important issue to resolve, as these may include new therapeutic targets.
Mapping of chromatin accessibility before and after BMP treatment in both NS cells and GNS cells enabled us to identify SOX-enriched regulatory regions as loci that were failing to be silenced appropriately in GNS cells. This offers a potential mechanistic explanation for the susceptibility to de-differentiation or acquisition of a quiescent astrocyte stem cell phenotype. SOX2 is a clear candidate for the specific factor that could be involved. It is a pioneer factor necessary for NS cell self-renewal that is amplified in a subset of GBMs (Brennan et al., 2013). Whether it is the increased activity or levels of SOX proteins or some of the other developmental transcription factors we identified that limit differentiation will be an interesting question to explore in the future.
Our observations challenge the simple notion that the exit from cell cycle and acquisition of differentiated features is indicative of terminal differentiation and an irreversible exit from cell cycle. The finding that both NS and GNS cells take many weeks or months to accumulate significant changes in DNA methylation was surprising, and it is in stark contrast to similar analyses carried out in the hematopoietic system where many thousands of MVPs occur rapidly (in days). This may reflect contrasting time frames of terminal differentiation of different tissues, which are associated with their physiological demands during embryogenesis (i.e., human CNS continues its maturation, plasticity, and growth for many years postnatally, whereas hematopoietic cell types are needed urgently for embryo and postnatal viability and they turn over rapidly). Clearly, if differentiation commitment and terminal cell-cycle arrest are underlain by epigenetic mechanisms that operate over the course of many weeks or months, this should to be taken into account in the design of any therapeutic approach.
There is little evidence that NS cells generate progeny via a deep hierarchy, i.e., a series of multiple restricted progenitors and precursors (Rowitch and Kriegstein, 2010). This means differentiated glial cells might be especially vulnerable to re-acquiring features of their more primitive ancestors, as several stepwise de-differentiation events would not be required. Indeed, we found following transplantation that BMP-treated GNS cultures remained capable of initiating tumors.
A caveat of our study is that differentiation was assessed in vitro. Whether the high levels of EGF and FGF to which we re-exposed differentiated cells would be encountered in vivo is not clear. However, genetic activation/amplification of EGFR- and FGFR-signaling pathways are hallmarks of the molecular pathology of GBM, and, therefore, sustained high levels of cell-autonomous signaling will be frequent across patient populations. Our study highlights the caution that must be exerted when assuming that non-cycling cells with differentiated features are terminally differentiated. In the future, use of in vivo genetic fate mapping of the behavior of the differentiated progeny of tumor-initiating cells will help shed light on this issue.
Suvà et al. (2014) recently have explored the transcriptional circuits that correlate with the GBM stem cell state versus serum-cultured astrocyte progeny that are non-malignant. The inductive signals specific to serum that limit tumor initiation remain unclear. However, these cells were still proliferative in serum and were cultured long term before tumor initiation potential and molecular characterization were performed, which may risk in vitro selection of acquired genetic changes that adapt cultures to serum.
The focus of our study has been on primary GBM. It will be important to explore whether differentiation therapy might have value for other forms of glioma, particularly those that are driven by mutations in core epigenetic regulators, such as pediatric high-grade glioma (Wu et al., 2012, Schwartzentruber et al., 2012), ependymoma (Mack et al., 2014), and medulloblastoma (Parsons et al., 2011). However, the genetic drivers that have been uncovered in these forms of brain tumors include direct genetic disruptions to the epigenetic memory machinery (e.g., inhibition of PRC2 by H3.3 mutations in pediatric glioma, IDH1/2 mutations, and associated CpG hypermethylator phenotypes for secondary GBM). Thus, the potential for brain tumor stem cells to be permanently driven to a differentiated and non-proliferative state may well be limited.
Our findings highlight inter-tumoral heterogeneity, aberrant changes in DNA methylation patterns, and vulnerability to de-differentiation as major challenges to the effectiveness of differentiation to robustly suppress tumor cell proliferation in primary GBM.
Experimental Procedures
Cell Culture
Briefly, GNS and NS cell lines were propagated under identical culture conditions using adherent culture on laminin substrate in the presence of EGF and FGF-2, as described previously (Pollard, 2013, Sun et al., 2008). Human NS cells were those used in the study by Danovi et al. (2013) and designated 11130. GNS cell lines G144, G166, G179, and GliNS2 have been described previously (Pollard et al., 2009). Remaining lines were generated as described by Fael Al-Mayhani et al. (2009). For DNA methylation analysis, T75 flasks, pre-coated with laminin (10 μg/ml), were seeded with four million cells. Biological duplicates were used with independent cultures. Then 24 hr later, cells were washed with PBS and replaced with media containing BMP4 (20 ng/ml, Sigma-Aldrich), but no EGF or FGF-2. Differentiation media, containing BMP, was replaced every 8 days throughout the time course and supplemented with fresh BMP at 4 days. All samples were stored at −80°C and processed in parallel.
Cell Proliferation Analysis
Cells were counted using the Vi-CELL automated cell counter (Beckman Coulter Genomics). Triplicate wells were used (two independent experiments). EdU incorporation assays were performed using the Click-iT EdU Alexa Fluor 488 Imaging Kit (Invitrogen). Cells were grown in 96-well plates and pulsed with (10 μM) EdU for 16 hr prior to fixing in 4% paraformaldehyde.
Immunocytochemistry
Primary antibodies (GFAP monoclonal mouse antibody, Sigma-Aldrich G3893, 1:1,000; rabbit OLIG2, Chemicon AB9610, 1:300) were incubated overnight at 4°C. O4 staining was performed on live cells (O4 hybridoma). Goat secondary antibodies conjugated to Alexa dyes (Molecular Probes) were added at 1:1,000 for 1 hr at room temperature and DAPI was used as a nuclear counterstain.
DNA Extraction and Bisulfite Modification
DNA extraction was performed using the DNeasy Blood & Tissue Kit (QIAGEN), including an RNA removal step. DNA (500 ng) was used for bisulfite modification using the EZ DNA methylation kit (D5001, Zymo Research).
DNA Methylation Data Bioinformatics Analysis
The bisulfite-modified DNA was applied to the Infinium HumanMethylation450 BeadChips (Illumina) as previously described (Stricker et al., 2013). The data generated by the BeadStudio software were exported and further analyses were performed in R. The R package ChAMP was used for normalization and MVP calling (Morris et al., 2014).
TCGA datasets were used to generate a GBM methylation set of MVPs by comparing to Pilocytic astrocytoma from GEO: GSE44684 and FACS-sorted human brain orbitofrontal cortex (nonneuronal) from GEO: GSE50798. CIMP+ cases were excluded since they harbor a specific methylation profile.
Quantitative Real-Time RT-PCR
Total RNA was extracted with the RNeasy Mini Kit (QIAGEN), including a genomic DNA removal step. cDNA was generated from 1 μg mRNA using Superscript III (Invitrogen). The qRT-PCR was conducted on the ABI 7300 (Applied Biosystems), with the Universal Probe Library system (Roche) or using TaqMan gene cards (Applied Biosystems). The reactions were carried out in technical duplicates on biological replicates.
Gene Expression and Subtyping Analysis
Exon arrays were processed using v1.28 of the xps Bioconductor package normalized using the Robust Multi-chip Average (RMA) method (Irizarry et al., 2003), and probe sets were summarized by median polish in xps. Additional details are provided in the Supplemental Experimental Procedures.
RNA-Seq
RNA quality was assessed using a Bioanalyzer (Agilent Technologies) and all RNA integrity number (RIN) values were above 9.4. The mRNA was isolated with the NEBNext Poly(A) mRNA Magnetic Isolation Module (New England Biolabs), and libraries were prepared with NEBNext mRNA Library Prep Master Mix Set for Illumina (NEB). Samples were multiplexed with seven or eight samples per sequencing lane (samples indexed with NEBNext Multiplex Oligos for Illumina). Samples were run in biological duplicates and sequenced in 100-bp paired-end format. RNA-seq data were processed using RSEM (v1.2.0) (Li and Dewey, 2011). The BioConductor package DESeq was used to detect differential expression (Anders and Huber, 2010).
ATAC-Seq
ATAC library preparation was undertaken according to published protocols (Buenrostro et al., 2013). Eight libraries were derived from cell lines NS-1 and G26, in EGF/FGF and BMP, in biological replicates. Additional details are provided in the Supplemental Experimental Procedures.
Xenotransplantation
GNS cells, proliferating, BMP4 treated, or BMP4 treated and subsequently cultured in EGF and FGF-2 for 4 days (BMP4 + GF), were detached with Accutase, and 50,000 GNS cells were diluted in 1 μl PBS and injected using a stereotaxic frame into 10- to 20-week-old NOD/SCID striatum (Pollard et al., 2009). Animals were maintained and used in a designated facility under licenses issued and approved by the UK Government Home Office.
Acknowledgments
We thank Tomi Bähr-Ivacevic (EMBL Genomics Core Facility, Heidelberg, Germany) for microarray processing. This work was supported by grants from The Brain Tumour Charity (grant 8/105) and Cancer Research UK (A9160). H.C. was supported by the Swedish Research Council and the Wenner-Gren foundation. P.B. was supported by EMBL and Biotechnology and Biological Sciences Research Council (BBSRC). Research in the S.B. lab was supported by Wellcome Trust (WT093855), Royal Society Wolfson Research Merit Award (WM100023), and EU-FP7 projects EPIGENESYS (257082) and BLUEPRINT (282510). S.M.P. was supported by a Wellcome Beit Memorial research fellowship, an Alex Bolt Research fellowship, and the Cancer Research UK Senior Research Fellowship (C25858/A19778).
Published: October 22, 2015
Footnotes
This is an open access article under the CC BY-NC-ND license (http://creativecommons.org/licenses/by-nc-nd/4.0/).
Supplemental Information includes Supplemental Experimental Procedures, two figures, and one table and can be found with this article online at http://dx.doi.org/10.1016/j.stemcr.2015.09.014.
Contributor Information
Stephan Beck, Email: s.beck@ucl.ac.uk.
Steven M. Pollard, Email: steven.pollard@ed.ac.uk.
Accession Numbers
The accession numbers for the data reported in this paper are ArrayExpress: E-MTAB-3864, E-MTAB-3867, and E-MTAB-3868.
Supplemental Information
References
- Adam S.A., Schnell O., Pöschl J., Eigenbrod S., Kretzschmar H.A., Tonn J.C., Schüller U. ALDH1A1 is a marker of astrocytic differentiation during brain development and correlates with better survival in glioblastoma patients. Brain Pathol. 2012;22:788–797. doi: 10.1111/j.1750-3639.2012.00592.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anders S., Huber W. Differential expression analysis for sequence count data. Genome Biol. 2010;11:R106. doi: 10.1186/gb-2010-11-10-r106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonaguidi M.A., McGuire T., Hu M., Kan L., Samanta J., Kessler J.A. LIF and BMP signaling generate separate and discrete types of GFAP-expressing cells. Development. 2005;132:5503–5514. doi: 10.1242/dev.02166. [DOI] [PubMed] [Google Scholar]
- Brennan C.W., Verhaak R.G., McKenna A., Campos B., Noushmehr H., Salama S.R., Zheng S., Chakravarty D., Sanborn J.Z., Berman S.H., TCGA Research Network The somatic genomic landscape of glioblastoma. Cell. 2013;155:462–477. doi: 10.1016/j.cell.2013.09.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buenrostro J.D., Giresi P.G., Zaba L.C., Chang H.Y., Greenleaf W.J. Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat. Methods. 2013;10:1213–1218. doi: 10.1038/nmeth.2688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carén H., Pollard S.M., Beck S. The good, the bad and the ugly: epigenetic mechanisms in glioblastoma. Mol. Aspects Med. 2013;34:849–862. doi: 10.1016/j.mam.2012.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dahlstrand J., Collins V.P., Lendahl U. Expression of the class VI intermediate filament nestin in human central nervous system tumors. Cancer Res. 1992;52:5334–5341. [PubMed] [Google Scholar]
- Danovi D., Folarin A., Gogolok S., Ender C., Elbatsh A.M., Engström P.G., Stricker S.H., Gagrica S., Georgian A., Yu D. A high-content small molecule screen identifies sensitivity of glioblastoma stem cells to inhibition of polo-like kinase 1. PLoS ONE. 2013;8:e77053. doi: 10.1371/journal.pone.0077053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doetsch F., Caillé I., Lim D., Garcia-Verdugo J.M., Alvarez-Buylla A. Subventricular zone astrocytes are neural stem cells in the adult mammalian brain. Cell. 1999;97:703–716. doi: 10.1016/s0092-8674(00)80783-7. [DOI] [PubMed] [Google Scholar]
- Engström P.G., Tommei D., Stricker S.H., Ender C., Pollard S.M., Bertone P. Digital transcriptome profiling of normal and glioblastoma-derived neural stem cells identifies genes associated with patient survival. Genome Med. 2012;4:76. doi: 10.1186/gm377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fael Al-Mayhani T.M., Ball S.L., Zhao J.W., Fawcett J., Ichimura K., Collins P.V., Watts C. An efficient method for derivation and propagation of glioblastoma cell lines that conserves the molecular profile of their original tumours. J. Neurosci. Methods. 2009;176:192–199. doi: 10.1016/j.jneumeth.2008.07.022. [DOI] [PubMed] [Google Scholar]
- Irizarry R.A., Hobbs B., Collin F., Beazer-Barclay Y.D., Antonellis K.J., Scherf U., Speed T.P. Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics. 2003;4:249–264. doi: 10.1093/biostatistics/4.2.249. [DOI] [PubMed] [Google Scholar]
- Ji H., Ehrlich L.I., Seita J., Murakami P., Doi A., Lindau P., Lee H., Aryee M.J., Irizarry R.A., Kim K. Comprehensive methylome map of lineage commitment from haematopoietic progenitors. Nature. 2010;467:338–342. doi: 10.1038/nature09367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J., Son M.J., Woolard K., Donin N.M., Li A., Cheng C.H., Kotliarova S., Kotliarov Y., Walling J., Ahn S. Epigenetic-mediated dysfunction of the bone morphogenetic protein pathway inhibits differentiation of glioblastoma-initiating cells. Cancer Cell. 2008;13:69–80. doi: 10.1016/j.ccr.2007.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li B., Dewey C.N. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics. 2011;12:323. doi: 10.1186/1471-2105-12-323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mack S.C., Witt H., Piro R.M., Gu L., Zuyderduyn S., Stütz A.M., Wang X., Gallo M., Garzia L., Zayne K. Epigenomic alterations define lethal CIMP-positive ependymomas of infancy. Nature. 2014;506:445–450. doi: 10.1038/nature13108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martynoga B., Mateo J.L., Zhou B., Andersen J., Achimastou A., Urbán N., van den Berg D., Georgopoulou D., Hadjur S., Wittbrodt J. Epigenomic enhancer annotation reveals a key role for NFIX in neural stem cell quiescence. Genes Dev. 2013;27:1769–1786. doi: 10.1101/gad.216804.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris T.J., Butcher L.M., Feber A., Teschendorff A.E., Chakravarthy A.R., Wojdacz T.K., Beck S. ChAMP: 450k Chip Analysis Methylation Pipeline. Bioinformatics. 2014;30:428–430. doi: 10.1093/bioinformatics/btt684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parsons D.W., Li M., Zhang X., Jones S., Leary R.J., Lin J.C., Boca S.M., Carter H., Samayoa J., Bettegowda C. The genetic landscape of the childhood cancer medulloblastoma. Science. 2011;331:435–439. doi: 10.1126/science.1198056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piccirillo S.G., Reynolds B.A., Zanetti N., Lamorte G., Binda E., Broggi G., Brem H., Olivi A., Dimeco F., Vescovi A.L. Bone morphogenetic proteins inhibit the tumorigenic potential of human brain tumour-initiating cells. Nature. 2006;444:761–765. doi: 10.1038/nature05349. [DOI] [PubMed] [Google Scholar]
- Pollard S.M. In vitro expansion of fetal neural progenitors as adherent cell lines. Methods Mol. Biol. 2013;1059:13–24. doi: 10.1007/978-1-62703-574-3_2. [DOI] [PubMed] [Google Scholar]
- Pollard S.M., Yoshikawa K., Clarke I.D., Danovi D., Stricker S., Russell R., Bayani J., Head R., Lee M., Bernstein M. Glioma stem cell lines expanded in adherent culture have tumor-specific phenotypes and are suitable for chemical and genetic screens. Cell Stem Cell. 2009;4:568–580. doi: 10.1016/j.stem.2009.03.014. [DOI] [PubMed] [Google Scholar]
- Rowitch D.H., Kriegstein A.R. Developmental genetics of vertebrate glial-cell specification. Nature. 2010;468:214–222. doi: 10.1038/nature09611. [DOI] [PubMed] [Google Scholar]
- Schwartzentruber J., Korshunov A., Liu X.Y., Jones D.T., Pfaff E., Jacob K., Sturm D., Fontebasso A.M., Quang D.A., Tönjes M. Driver mutations in histone H3.3 and chromatin remodelling genes in paediatric glioblastoma. Nature. 2012;482:226–231. doi: 10.1038/nature10833. [DOI] [PubMed] [Google Scholar]
- Sell S. Stem cell origin of cancer and differentiation therapy. Crit. Rev. Oncol. Hematol. 2004;51:1–28. doi: 10.1016/j.critrevonc.2004.04.007. [DOI] [PubMed] [Google Scholar]
- Shackleton M., Quintana E., Fearon E.R., Morrison S.J. Heterogeneity in cancer: cancer stem cells versus clonal evolution. Cell. 2009;138:822–829. doi: 10.1016/j.cell.2009.08.017. [DOI] [PubMed] [Google Scholar]
- Singh S.K., Hawkins C., Clarke I.D., Squire J.A., Bayani J., Hide T., Henkelman R.M., Cusimano M.D., Dirks P.B. Identification of human brain tumour initiating cells. Nature. 2004;432:396–401. doi: 10.1038/nature03128. [DOI] [PubMed] [Google Scholar]
- Stricker S.H., Feber A., Engström P.G., Carén H., Kurian K.M., Takashima Y., Watts C., Way M., Dirks P., Bertone P. Widespread resetting of DNA methylation in glioblastoma-initiating cells suppresses malignant cellular behavior in a lineage-dependent manner. Genes Dev. 2013;27:654–669. doi: 10.1101/gad.212662.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Y., Pollard S., Conti L., Toselli M., Biella G., Parkin G., Willatt L., Falk A., Cattaneo E., Smith A. Long-term tripotent differentiation capacity of human neural stem (NS) cells in adherent culture. Mol. Cell. Neurosci. 2008;38:245–258. doi: 10.1016/j.mcn.2008.02.014. [DOI] [PubMed] [Google Scholar]
- Suvà M.L., Rheinbay E., Gillespie S.M., Patel A.P., Wakimoto H., Rabkin S.D., Riggi N., Chi A.S., Cahill D.P., Nahed B.V. Reconstructing and reprogramming the tumor-propagating potential of glioblastoma stem-like cells. Cell. 2014;157:580–594. doi: 10.1016/j.cell.2014.02.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verginelli F., Perin A., Dali R., Fung K.H., Lo R., Longatti P., Guiot M.C., Del Maestro R.F., Rossi S., di Porzio U. Transcription factors FOXG1 and Groucho/TLE promote glioblastoma growth. Nat. Commun. 2013;4:2956. doi: 10.1038/ncomms3956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu G., Broniscer A., McEachron T.A., Lu C., Paugh B.S., Becksfort J., Qu C., Ding L., Huether R., Parker M., St. Jude Children’s Research Hospital–Washington University Pediatric Cancer Genome Project Somatic histone H3 alterations in pediatric diffuse intrinsic pontine gliomas and non-brainstem glioblastomas. Nat. Genet. 2012;44:251–253. doi: 10.1038/ng.1102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S.C. Defining glial cells during CNS development. Nat. Rev. Neurosci. 2001;2:840–843. doi: 10.1038/35097593. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.