

Abstract
Key points
This study explores the molecular mechanisms that regulate the recycling of chromophore required for pigment regeneration in mammalian cones.
We report that two chromophore binding proteins, retinol dehydrogenase 8 (RDH8) and photoreceptor‐specific ATP‐binding cassette transporter (ABCA4) accelerate the dark adaptation of cones, first, directly, by facilitating the processing of chromophore in cones, and second, indirectly, by accelerating the turnover of chromophore in rods, which is then recycled and delivered to both rods and cones.
Preventing competition with the rods by knocking out rhodopsin accelerated cone dark adaptation, demonstrating the interplay between rod and cone pigment regeneration driven by the retinal pigment epithelium (RPE).
This novel interdependence of rod and cone pigment regeneration should be considered when developing therapies targeting the recycling of chromophore for rods, and evaluating residual cone function should be a critical test for such regimens targeting the RPE.
Abstract
Rapid recycling of visual chromophore and regeneration of the visual pigment are critical for the continuous function of mammalian cone photoreceptors in daylight vision. However, the molecular mechanisms modulating the supply of visual chromophore to cones have remained unclear. Here we explored the roles of two chromophore‐binding proteins, retinol dehydrogenase 8 (RDH8) and photoreceptor‐specific ATP‐binding cassette transporter 4 (ABCA4), in dark adaptation of mammalian cones. We report that young adult RDH8/ABCA4‐deficient mice have normal M‐cone morphology but reduced visual acuity and photoresponse amplitudes. Notably, the deletion of RDH8 and ABCA4 suppressed the dark adaptation of M‐cones driven by both the intraretinal visual cycle and the retinal pigmented epithelium (RPE) visual cycle. This delay can be caused by two separate mechanisms: direct involvement of RDH8 and ABCA4 in cone chromophore processing, and an indirect effect from the delayed recycling of chromophore by the RPE due to its slow release from RDH8/ABCA4‐deficient rods. Intriguingly, our data suggest that RDH8 could also contribute to the oxidation of cis‐retinoids in cones, a key reaction of the retina visual cycle. Finally, we dissected the roles of rod photoreceptors and RPE for dark adaptation of M‐cones. We found that rods suppress, whereas RPE promotes, cone dark adaptation. Thus, therapeutic approaches targeting the RPE visual cycle could have adverse effects on the function of cones, making the evaluation of residual cone function a critical test for regimens targeting the RPE.
Abbreviations
- ABCA4
ATP‐binding cassette transporter 4
- COS
cone outer segment
- ERG
electroretinogram
- Gnat1
rod transducin α‐subunit
- PNA
peanut agglutinin
- RDH8
retinol dehydrogenase 8
- RPE
retinal pigmented epithelium
- RT
room temperature
- TKO
triple knockout
- WT
wild‐type
Introduction
Vertebrate rods and cones initiate vision by photoisomerization of the visual chromophore retinal from the 11‐cis to the all‐trans conformation. The resulting activation of the visual pigment triggers a phototransduction cascade that ultimately produces a response to light. Continuous function of photoreceptors requires release of the spent chromophore all‐trans‐retinal and its recycling back to 11‐cis‐retinal via either the retinal pigmented epithelium (RPE visual cycle, for both rods and cones) or Müller cells (retina visual cycle, for cones only) (reviewed by Wang & Kefalov, 2011; Tang et al. 2013; Kiser et al. 2014). An important step in the visual cycle is the conversion of all‐trans‐retinal to retinol in photoreceptor outer segments. In rods, this process is catalysed by retinol dehydrogenase 8 (RDH8) and presumably facilitated by a photoreceptor‐specific ATP‐binding cassette transporter (ABCA4).
ABCA4 is an ATP‐driven flippase that transports all‐trans‐retinal from the extracellular to the cytosolic side of internal rod disc membranes (Weng et al. 1999), where it is reduced to retinol and then removed from the outer segments (Sun & Nathans, 1997). ABCA4 can also transport 11‐cis‐retinal (Sun et al. 1999) suggesting its possible involvement in the uptake of recycled chromophore (Quazi & Molday, 2014). Nevertheless, the role of ABCA4 remains controversial, as its deletion causes accumulation of bis‐retinoid adducts (Kim et al. 2007) without affecting the clearance of all‐trans‐retinal from rods (Blakeley et al. 2011) or dark adaptation (Pawar et al. 2008).
Even less is known about ABCA4 in cone photoreceptors. Patients with Abca4 mutations experience reduced and delayed electroretinographic (ERG) cone responses and poor visual acuity (Birch et al. 2001) indicating a possible direct effect of ABCA4 on cone function. Indeed, the overall time course of adaptation in one patient with ABCA4‐related cone–rod dystrophy revealed a delayed cone visual threshold recovery (Fig. 4 in Birch et al. 2001).
Figure 4. M‐cone dark adaptation in the absence of RDH8 and/or ABCA4 in 2‐month‐old mice .
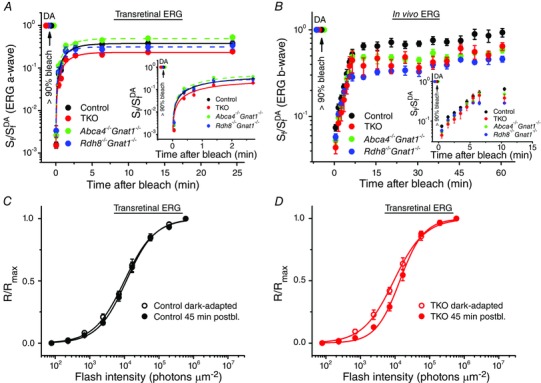
A, recovery of cone a‐wave flash sensitivity (S f) in isolated retinas of control (n = 7), Abca4−/−Gnat1−/− (n = 8), Rdh8−/−Gnat1−/− (n = 6) and TKO (n = 14) mice after bleaching >90% of cone pigment at time 0 with 505 nm LED light. Data were fitted with single‐exponential functions for better visualization of the relative cone sensitivity recovery. S f DA denotes the sensitivity of dark‐adapted cones. The inset shows the initial S f recovery on expanded time scale. B, recovery of photopic ERG b‐wave S f in vivo in control (n = 4), Abca4−/−Gnat1−/− (n = 6), Rdh8−/−Gnat1−/− (n = 6) and TKO (n = 3) mice after bleaching >90% of cone pigment at time 0 with 520 nm LED light. S f DA designates the sensitivity of dark‐adapted cones. The inset shows the initial S f recovery on expanded time scale. C, normalized averaged cone intensity–response relationships of isolated retinas from control mice. Live animals were either dark‐adapted or illuminated with 30 s 520 nm LED light bleaching >90% of M‐cone pigment and kept in the dark for 45 min, prior to ex vivo ERG recordings. Data points were fitted with Naka–Rushton hyperbolic functions. The average cone sensitivities (S f) were 0.21 (dark‐adapted, n = 5) and 0.16 (45 min postbleach (postbl.), n = 10). D, normalized averaged cone intensity–response functions of isolated retinas from TKO mice. The experimental protocol was the same as in C. The average cone sensitivities (S f) were 0.23 (dark‐adapted, n = 5) and 0.13 (45 min postbleach, n = 9), respectively. Error bars represent SEMs (smaller than the symbol size for some data points).
In primates, RDH8 is expressed in the outer segments of rods and cones (Rattner et al. 2000). Moreover, in mouse rods this enzyme reduces ∼70% of all‐trans‐retinal to all‐trans‐retinol in the visual cycle (Maeda et al. 2007). Yet, RDH8 deletion delays rod dark adaptation only mildly (Maeda et al. 2005) and seems not to affect photopic ERG light responses (Maeda et al. 2007). Effects of RDH8 on cone function in bright light and cone dark adaptation have not been reported. Also unknown is whether RDH8 participates in the oxidation of 11‐cis‐retinol to 11‐cis‐retinal, a key cone‐specific reaction of the retina visual cycle (Jones et al. 1989; Mata et al. 2002; Ala‐Laurila et al. 2009; Wang & Kefalov, 2009).
ABCA4/RDH8 double knockout mouse has recently served as a model of human retinal degeneration in which to test candidate therapeutics (Maeda et al. 2009 a). The pathology of the retinoid visual cycle and rod function in these animals were studied extensively (Maeda et al. 2008). Although these mice eventually develop cone–rod dystrophy, it remains unknown how deletion of ABCA4 and RDH8 affects the light response or dark adaptation of cones. Here we addressed these questions by using mice lacking ABCA4, RDH8, or both.
Methods
Animals
Wild‐type (WT) mice with a C57BL/6 background used for RNA‐sequencing and quantitative Reverse Transcription (qRT)‐PCR analyses were obtained from The Jackson Laboratory (Bar Harbor, ME, USA). Rod transducin α‐subunit knockout (Gnat1−/−) mice lacking rod signalling (Calvert et al. 2000) were used as controls in all physiological experiments. Rdh8−/−Gnat1−/−, Abca4−/−Gnat1−/− and Rdh8−/−Abca4−/−Gnat1−/− triple knockout (TKO) mouse lines were generated as described previously (Maeda et al. 2007, 2009 b). All mice used in this study were homozygous for the Leu450 allele of Rpe65 as determined by a genotyping protocol published elsewhere (Grimm et al. 2004) and free of the Crb1/rd8 mutation (Mattapallil et al. 2012). Rho−/− mice were described earlier (Lem et al. 1999), as were Nrl−/− mice (Mears et al. 2001). Young adult animals of either sex (1.5–3 months old) were used in all experiments. Animals were provided with standard chow (LabDiet 5053; LabDiet, Purina Mills, St. Louis, MO, USA) and maintained under a 12 h light (10–20 lx)/12 h dark cycle. Mice were dark‐adapted overnight prior to physiological recordings. A cohort of TKO mice was dark‐reared from postnatal day 10 until they reached the age of 2 months. For all experiments in this study, animals were killed by asphyxiation with a rising concentration of CO2. All experiments were performed in accordance with the principles of UK regulations as described in Grundy (2015), and were approved by the Washington University Animal Studies and Case Western Reserve University Animal Care Committees.
Antibodies
Mouse hybridoma culture supernatant containing the monoclonal Rim 3F4 anti‐ABCA4 antibody, a generous gift from R. Molday (University of British Columbia, Vancouver, Canada), was used at a 1:10 dilution (Illing et al. 1997). Monoclonal antibody TMR1 (Zhang et al. 2015) (1:400) was also used to probe for ABCA4 protein. The polyclonal anti‐RDH8 antibody was raised by immunizing BALB/c mice with C‐terminal fusion protein from Escherichia coli and the C‐terminal 16 amino acid‐long peptide (CGCLPTRVWPRQTEQN) conjugated with keyhole limpet haemocyanin (Pierce, Grand Island, NY, USA) (Maeda et al. 2005). Peroxidase‐labelled goat anti‐rabbit IgG secondary antibody was from (Vector Laboratories, Burlingame, CA, USA). Anti‐M‐cone opsin antibody (0.5 μg ml−1) was from (Millipore, Billerica, MA, USA). The anti‐S‐cone opsin antibody (0.4 μg ml−1) was obtained from (Santa Cruz Biotechnology, Dallas, TX, USA). Alexa‐488 secondary antibodies (0.1 μg ml−1) were from (Invitrogen, Grand Island, NY, USA). Fluorescein isothiocyanate (FITC)‐conjugated peanut agglutinin (PNA) lectin (0.2 mg ml−1) was from (Sigma‐Aldrich, St. Louis, MO, USA). Mouse monoclonal anti‐β‐actin antibody (1:1000) was from Santa Cruz Biotechnology.
Immunohistochemistry
After removal of the cornea and lens, the remaining mouse eyecup was fixed in freshly prepared 4% paraformaldehyde in 0.1 m phosphate buffered saline (PBS) at pH 7.4 for 1.3–2 h at room temperature (RT). The eyecup was then washed once in PBS for 10 min and incubated in 30% sucrose buffered with PBS overnight at 4°C. Next, the eyecup was embedded in Optimal Cutting Temperature compound (Ted Pella, Redding, CA, USA), flash‐frozen in 2‐methylbutane (Sigma‐Aldrich, St. Louis, MO, USA) on dry ice, and cut with a cryo‐microtome to produce 8 μm sections from the central retinal region immediately ventral to the optic nerve head. Sections were dried for 30 min at RT, gently washed in deionized water for 10 min, dried again for 10 min at RT, and blocked for 1 h at RT with a solution containing 1% bovine serum albumin (Sigma), 1% donkey serum (Sigma), or 1.5% goat serum, and 0.1–0.25% Triton X‐100 (Sigma) in PBS (PBST). Sections were then incubated overnight at 4°C with appropriate primary antibodies or anti‐ABCA4 serum diluted in a solution containing 0.1% Triton X‐100 in PBS (PBST) or 1% Tween‐20 (Bio‐Rad, Hercules, CA, USA) and 1% Triton X‐100 in PBS. Next, sections were washed once in PBS or PBST and then incubated with secondary antibodies diluted in PBS (PBST) containing propidium iodide, 4’,6‐diamidino‐2‐phenylindole (DAPI), or Hoechst 33342, for 2 h at RT, washed with PBS or PBST (×2, 10 min, RT), and mounted on coverslips with Fluoromount‐G or Immu‐Mount (Thermo Scientific, Grand Island, NY, USA) for analysis by confocal (Olympus FV1000 confocal microscope) or two‐photon microscopy (Leica SP2 confocal/two‐photon microscope). Images were acquired from the central region of the retina near the optic nerve head.
Western blotting
Retinas from both mouse eyes were homogenized in 80 μl of ice‐cold radioimmunoprecipitation assay (RIPA) buffer (150 mm NaCl, 1.0% NP‐40, 0.5% sodium deoxycholate, 0.1% SDS, 50 mm Tris, pH 8.0) with 2% protease inhibitors (Sigma‐Aldrich). Ten microlitres of each protein sample was loaded onto SDS‐PAGE gel and transferred to polyvinylidene difluoride (PVDF) membranes. Membranes were incubated with primary anti‐ABCA4 TMR1 (1:400) and anti‐β‐actin (1:1000) antibodies overnight followed by secondary antibody (1:5000) treatment for 1 h. The antibody binding was detected by using NBT and BCIP solutions (Promega Corporation, Madison, WI, USA).
Cone cell counts
Cone photoreceptors of 1.5‐ to 2.5‐month‐old control, Abca4−/−Gnat1−/− and TKO mice were stained with PNA and manually counted in four zones of both dorsal and ventral retina areas (Z1–Z4): Z1, 400–500 μm; Z2, 900–1000 μm; Z3, 1400–1500 μm, and Z4, 1900–2000 μm from the optic nerve head. Zones in the dorsal and ventral retina were counted separately. Numbers of cones in each zone were averaged and the data statistically analysed by one‐way ANOVA.
Transmission electron microscopy
Transmission electron microscopy (TEM) was performed as follows. Mice were deeply anesthetized with ketamine/xylazine cocktail as described below and fixed by intracardiac perfusion with 2% glutaraldehyde in PBS (pH 7.0) with the addition of 2 mm CaCl2. Eyes were removed and placed into a dish containing the fixative, enucleated, and cornea and lens were gently removed. After 2 h of fixation, eyecups were washed three times with 0.1 m sodium cacodylate buffer (pH 7.0) for 10 min each and placed into a vial with 1% osmium tetroxide in 0.1 m sodium cacodylate for 45 min. Samples were subsequently rinsed once in 0.1 m sodium cacodylate, followed by three 5 min exchanges in 50 mm sodium acetate (pH 5.2). Eyecups were then stained with 2% uranyl acetate in 50 mm sodium acetate for 45 min in the dark. After staining, samples were rinsed with two exchanges of sodium acetate for 15 min each, placed briefly in dH2O, and gradually dehydrated with ethanol in increments of 20, 40, 60, 80 and 100% for 10 min in each step. Samples were kept at 4°C for 12 h, brought to room temperature and transferred to 100% propylene oxide for 10 min, and infiltrated with araldite resin in the following araldite/propylene oxide progressions: 30%:70% for 1 h, 50%:50% for 2 h and 70%:30% for 1 h. Eyecups were placed in 100% araldite, trimmed into halves and rotated overnight in fresh 100% araldite. Eyecup halves were transferred to moulds, kept in a desiccator for 8 h and then polymerized in an oven at 80°C for 48 h. Longitudinal sections were cut approximately 90 nm thick with a diamond knife, stained with aqueous 4% uranyl acetate and Reynolds lead citrate. Both superior and inferior portions of the retina were chosen for TEM sampling. Imaging was performed on a JEOL 1400 CX transmission electron microscope using an AMT XR111 bottom mount digital camera (Advanced Microscopy Techniques, Woburn, MA, USA). Cone outer segment (COS) length was measured in ImageJ 1.40 g (http://imagej.nih.gov/ij/). Because the orientation of COS profiles exhibited a large variability across the retina in individual samples, only COSs whose length exceeded 6 μm were selected for analysis.
RNA‐sequencing analyses
RNA sequencing library construction, sample runs and data analyses were performed as described previously (Mustafi et al. 2011). Each reported measurement represented an average of at least three biological replicates.
qRT‐PCR
Total RNA was isolated from two retinas with the RNeasy Kit (Qiagen, Valencia, CA, USA), and cDNA was synthesized with SuperScript II Reverse Transcriptase (Invitrogen) from 500 ng total RNA following the manufacturer's instructions. Real‐time PCR amplification was accomplished with iQ SYBR Green Supermix (Bio‐Rad). qRT‐PCR analyses were performed with the following primers: mouse M‐cone opsin, forward 5′‐CGTGCTCTGCTACCTCCAAG‐3′, reverse 5′‐CGAAGACCATCACCACCACC‐3′; mGapdh, forward 5′‐GTGTTCCTACCCCCAATGTG‐3′, reverse 5′‐GGAGACAACCTGGTCCTCAG‐3′. The expression of M‐cone opsin in retinas of mutant mice against C57BL/6 mice was normalized to that of the housekeeping gene Gapdh.
Transretinal (ex vivo ERG) recordings from isolated retinas
Mice were dark‐adapted overnight, killed by CO2 asphyxiation, and a whole retina was removed from each mouse eyecup under infrared illumination and stored in either oxygenated aqueous L15 (13.6 mg ml−1, pH 7.4, Sigma) solution containing 0.1% bovine serum albumin (BSA) or Locke solution at RT. The retina was mounted on filter paper with the photoreceptor side up and placed in a perfusion chamber between two electrodes connected to a differential amplifier. The preparation was perfused with Locke solution containing 112.5 mm NaCl, 3.6 mm KCl, 2.4 mm MgCl2, 1.2 mm CaCl2, 10 mm Hepes, pH 7.4, 20 mm NaHCO3, 3 mm sodium succinate, 0.5 mm sodium glutamate, 0.02 mm EDTA, and 10 mm glucose. This solution was supplemented with 2 mm l‐glutamate and 40 μm d,l‐2‐amino‐4‐phosphonobutyric acid to block postsynaptic components of the photoresponse (Sillman et al. 1969), and with 70 μm BaCl2 to suppress the slow glial PIII component (Nymark et al. 2005). The perfusion solution was continuously bubbled with a 95% O2 / 5% CO2 mixture and heated to 36–37°C.
Light stimulation was applied in 20 ms test flashes of calibrated 505 nm light from a light‐emitting diode (LED). The stimulating light intensity was controlled by a computer in 0.5 log unit steps. Intensity–response relationships (for raw data) were fitted with Naka–Rushton hyperbolic functions, as follows:
where R is the transient‐peak amplitude of the response, R max is the maximal response amplitude, I is the flash intensity, n is the Hill coefficient (exponent), and I 1/2 is the half‐saturating light intensity. In experiments designed to monitor the postbleach recovery of cone a‐wave flash sensitivity (S f, see definition below), >90% of M‐cone visual pigment was bleached with a 30 s exposure to 505 nm light. The bleached fraction was estimated from the following equation:
where F is the fraction of pigment bleached, t is the duration of the light exposure (s), I is the bleaching light intensity of 505 nm LED light (1.6 × 108 photons μm−2 s−1), and P is the photosensitivity of mouse cones at the wavelength of peak absorbance (7.5 × 10−9 μm2), adopted from Nikonov et al. (2006). The correction for a factor of ∼10% of light intensity reduction in the mouse eye before reaching the cones (Vinberg et al. 2014) was included in the estimation of the bleaching level. Photoresponses were amplified by a differential amplifier (DP‐311, Warner Instruments, Hamden, CT, USA), low‐pass filtered at 300 Hz (8‐pole Bessel), digitized at 1 kHz and stored on a computer for further analysis. Cone a‐wave flash sensitivity (S f) was calculated from the linear part of the intensity–response curve, as follows:
where R is the cone a‐wave dim flash response amplitude, R max is the maximal response amplitude for that retina, and I is the flash strength. The time constant of dim flash response recovery (τrec) was obtained from a single‐exponential fit to the falling phase of the response. Data were analysed with Clampfit 10.4 (Molecular Devices, Sunnyvale CA, USA) and Origin 8.5 software (OriginLab Corporation, Northampton, MA, USA).
Application of exogenous 11‐cis‐retinal to dark‐adapted TKO retinas was performed as follows: 300 μg of dried retinoid was dissolved in 8 μl of absolute ethanol and diluted to 8 ml with L15 solution containing 1% BSA. The final concentrations of 11‐cis‐retinal and ethanol were ∼130 μm and 0.1%, respectively. Before transfer to the perfusion chamber for recordings, a whole retina on filter paper was incubated in a Petri dish with 4–5 ml of this oxygenated solution for 1 h in the dark at RT.
Regeneration experiments with exogenous 9‐cis‐retinol were performed as follows: a whole retina on filter paper was fully bleached for 1.5 min with 520 nm light in 0.1% BSA (L15) solution and kept in a Petri dish with 2 ml of the same oxygenated solution for 15 min in the dark at RT. Then it was transferred to 1% BSA (L15) solution supplemented with ∼130 μm 9‐cis‐retinol (Santa Cruz Biotechnology) and 50 μm RDH8 co‐factor NADP+ (Sigma) for 3.5 h in the dark at RT, prior to transretinal recordings. Control bleached retina was processed similarly but the 3.5 h incubation was carried out in the absence of retinoid and co‐factor.
Electroretinography (ERG)
Dark‐adapted mice were anaesthetized with an i.p. injection of a mixture of ketamine (100 mg kg−1) and xylazine (20 mg kg−1). Pupils were dilated with a drop of 1% atropine sulfate. Mouse body temperature was maintained at 37°C with a heating pad. ERG responses were measured from both eyes by contact corneal electrodes held in place by a drop of Gonak solution. Full‐field ERGs were recorded with the UTAS BigShot apparatus (LKC Technologies, Gaithersburg, MD, USA) using Ganzfeld‐derived test flashes of calibrated green 530 nm LED light (within a range from 0.24 to 7.45 cd s m−2). Cone b‐wave flash sensitivity (S f, calculated similarly to the a‐wave S f in ex vivo ERG recordings) was first determined in the dark (from the average of up to 20 flash responses) and normalized to the maximal b‐wave amplitude obtained with the brightest white light stimulus of the xenon flash tube (700 cd s m−2). Bright green background Ganzfeld illumination (300 cd m−2; estimated to bleach ∼0.8% M‐cone pigment s−1) was then applied continuously for 15 min and the cone b‐wave S f change was monitored during this period of light exposure. At the end of this illumination period, mice were re‐anaesthetized once with a smaller dose of ketamine (∼1/2 of the initial dose) and a 1:1 mixture of PBS and Gonak solutions was gently applied to the eyes with a plastic syringe to protect them from drying and to maintain electrode contacts. Finally, the remaining M‐cone pigment was near‐completely bleached by a 30 s exposure to additional bright light delivered by a 520 nm LED focused at the surface of mouse eye cornea that produced ∼1.3 × 108 photons μm−2 s−1. The bleaching fractions were estimated by the formula F = 1 – exp(–IPt) defined above. After the bleach, the recovery of cone b‐wave S f was followed in darkness for up to 1 h (with one more re‐anaesthesia in the middle of that period). In a subset of experiments, the 15 min Ganzfeld illumination step was omitted and S f recovery was followed after a >90% cone pigment bleach (520 nm LED, 30 s) that was applied to dark‐adapted animals.
Retinylamine, prepared as per Golczak et al. (2005), was dissolved before each experiment in DMSO to 5 μg μl−1, and 50 μl of this solution was administered by intraperitoneal injection in the dark, followed by a period of 15–18 h of dark adaptation prior to physiological recordings.
Photopic visual function tests
Visual acuity and contrast sensitivity of 2‐month‐old mice were measured using a two‐alternative forced‐choice protocol (Umino et al. 2008) with the OptoMotry system (Cerebral Mechanics Inc., Lethbride, AB, Canada). A mouse was placed on a pedestal surrounded by a square array of four computer monitors. A television camera (Sony) was mounted above the animal to allow its observation but not that of the stimulus displayed by the monitors. Rotating sine‐wave vertical gratings were applied to the monitors where they formed a virtual cylinder around the mouse (Prusky et al. 2004). The direction of the gratings movement for each trial was randomly selected by a computer‐controlled protocol. Mice responded to stimuli by reflexively rotating their heads in the corresponding direction. The observer registered either a clockwise or counterclockwise direction of mouse optomotor responses.
Based on the observer's choice and using a staircase protocol, the computer changed the spatial frequency (F s) of the stimuli, starting from 0.128 cycles deg−1 until reaching its threshold (visual acuity), defined as the correctness of the experimenter's responses in 70% of trials. In this setting, the speed of gratings (S p) having 100% contrast was set at its optimal value of 12.0 deg s−1. Temporal frequency (F t) was automatically adjusted by the computer program, based on the following equation: F t = S p × F s (Umino et al. 2008). Contrast sensitivity was defined as the inverse of the obtained contrast threshold values. In this mode, contrast of the stimuli was gradually decreased by the computer protocol until its threshold (70% correct answers) was determined. F s was fixed at 0.128 cycles deg−1, F t was set to 1.5 Hz, and S p was 12.0 deg s−1. Behavioural tests were performed under standard photopic conditions (unattenuated monitor luminance of 70 cd m−2 at the mouse eye level) or in dim light photopic conditions (0.018 cd m−2 at the mouse eye level). In the latter case, the background monitor luminance was adjusted by four layers of neutral density film filters (0.9 ND, Rosco Laboratories, Stamford, CT, USA). A camera was switched to night mode detecting light from a round array of six infrared LEDs mounted above the animal to visualize it.
Statistics
For all experiments, data were expressed as means ± SEMs. Unless stated otherwise, data were analysed using the independent two‐tailed Student's t test, with an accepted significance level of P < 0.05.
Results
Expression profile and morphology of RDH8‐ and/or ABCA4‐deficient mouse cones
To determine the expression levels of Rdh8 and Abca4 genes in mouse eye, we mined our recently obtained RNA‐Seq transcriptome data (Mustafi et al. 2011). In whole eyes and retinas of wild‐type (WT) mice the Abca4 transcript was twice as abundant as that of Rdh8 and expressed at levels about 10% of Gcap1, an important component of the photoreceptor phototransduction pathway (Table 1). Comparison of expression levels from mice of various genetic backgrounds, including those with varying numbers of rods and cones (Rho−/− and Nrl−/−), suggests that these proteins are expressed in both rod and cone photoreceptors. We then characterized the localization of RDH8 and ABCA4 proteins in cones of both control mice and their respective knockout counterparts generated with a Gnat1−/− background to eliminate interference by rod signalling in behavioural and physiological experiments (see below). RDH8 was abundantly expressed in photoreceptor outer segments of retinas from Gnat1−/− mice (referred to as control(s) throughout the text) (Fig. 1 A, top panel). As expected, immunostaining of Rdh8−/−Abca4−/−Gnat1−/− (TKO) retinas with anti‐RDH8 antibody yielded negative results (Fig. 1 A, middle panel). Cone expression of RDH8 was evidenced by its co‐localization with M‐cone opsin in WT mouse retinas (Fig. 1 A, bottom panel). In contrast, the ABCA4 labelling with the commonly used anti‐ABCA4 antibody 3F4 was far less pronounced in M‐cones compared to RDH8 labelling there (Fig. 1 B, top panels and their insets, white arrows). As expected, both Abca4−/−Gnat1−/− and TKO retinas showed no ABCA4 expression (Fig. 1 B, middle and bottom panels). No obvious overall retinal degeneration was observed in any of the mutant mice at the age of 1.5–2.5 months.
Table 1.
Gene expression analysis of 1‐month‐old mouse eyes by RNA sequencing
| WT (whole eye) | WT (retina) | Rho−/− (whole eye) | Nrl−/− (whole eye) | |
|---|---|---|---|---|
| Abca4 | 59 | 140 | 16 | 20 |
| Rdh8 | 32 | 73 | 7.6 | 12 |
| Gcap1 | 508 | 955 | 206 | 508 |
Values are represented as fragments per kilobase per million reads (FPKM).
Figure 1. Expression of RDH8 and ABCA4 in rods and cones of 2‐month‐old mice .
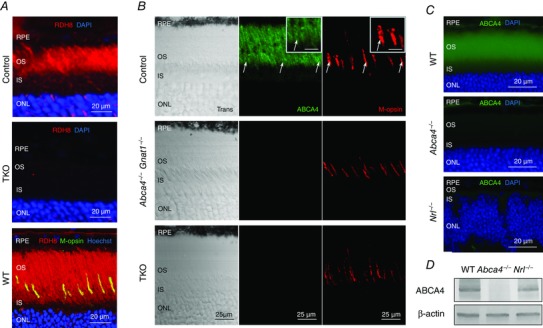
A, RDH8 expression was examined by immunohistochemistry with anti‐RDH8 antibody. RDH8 (red) was present in Gnat1−/− (control) outer segments of photoreceptors, whereas no signal was detected in the retinas of Abca4−/−Rdh8−/−Gnat1−/− (TKO) mice. WT retinas revealed that mouse cones express RDH8 manifested as a yellow colour after co‐staining with anti‐RDH8 antibody (red) and anti‐M‐cone opsin antibody (M‐opsin, green). Cell nuclei (blue) were stained with DAPI or Hoechst 33342. Scale bar, 20 μm. B, immunostaining of control, Abca4−/−Gnat1−/− and TKO retinas (central location near the optic nerve) with Rim 3F4 anti‐ABCA4 antibody (green) or anti‐M‐opsin antibody (red). Scale bar, 25 μm. Trans, confocal images in transmitted light. Cell nuclei were stained with DAPI. White arrows indicate cone outer segments. Inset, higher magnification of cone outer segments. Scale bar, 10 μm. RPE, retinal pigmented epithelium; OS, outer segments; IS, inner segments; ONL, outer nuclear layer. C, immunostaining of WT, Abca4−/− and Nrl−/− retinas with TMR1 anti‐ABCA4 antibody (green). Scale bar, 20 μm. Cell nuclei were stained with DAPI. D, immunoblotting of WT, Abca4−/− and Nrl−/− retina samples using TMR1 anti‐ABCA4 antibody demonstrates that ABCA4 protein is expressed in Nrl−/− retinas. β‐actin served as a loading control.
To further investigate the expression of ABCA4 in cones, we performed immunostainings with another antibody, TMR1. This antibody labelled strongly and specifically ABCA4 in the outer segment layer of wild‐type retina sections (Fig. 1 C, top panel). In contrast, immunoblotting with TMR1 antibody of sections from Nrl−/− retina, populated exclusively with cone‐like photoreceptors, showed no ABCA4 signal (Fig. 1 C, bottom panel). However, Western blot analysis of Nrl−/− retinas with TMR1 revealed robust expression of the ABCA4 protein in these photoreceptors (Fig. 1 D). We conclude that the detection of ABCA4 in mouse cones by immunocytochemistry is problematic, possibly due to a masked ABCA4 epitope in cone discs. However, the presence of the Abca4 transcript in Rho−/− and Nrl−/− retinas (Table 1), together with the robust detection of ABCA4 in the cone‐only retinas of Nrl−/− mice demonstrate that this protein is indeed expressed in mouse cones.
To investigate if the deletion of RDH8 and/or ABCA4 induces degeneration specifically of cone photoreceptors, we immunostained M‐ and S‐cone opsins in retinal cross sections. Both M‐cones (Fig. 2 A) and S‐cones (Fig. 2 B) had normal morphology in retinas of 2‐month‐old ABCA4‐deficient mice (middle panels) as well as in those of TKO mice (bottom panels). Furthermore, for both mutant lines M‐cone (Fig. 2 A) and S‐cone (Fig. 2 B) pigments were localized properly in the outer segments. Cone density was also comparable in control, Abca4−/−Gnat1−/− and TKO retinas of 2.5‐month‐old mice (Fig. 2 C and D). The morphology of cones in 2.5‐month‐old mice was also analysed by electron microscopy (Fig. 3 A and B). The average length of cone outer segments in longitudinal sections was normal in TKO animals (7.7 ± 0.5 μm, n = 16, vs. 7.7 ± 0.4 μm, n = 9, in control mice, P > 0.05) at this age.
Figure 2. Normal cone density, morphology and opsin localization in 2‐month‐old mice lacking either ABCA4 or both ABCA4 and RDH8 .
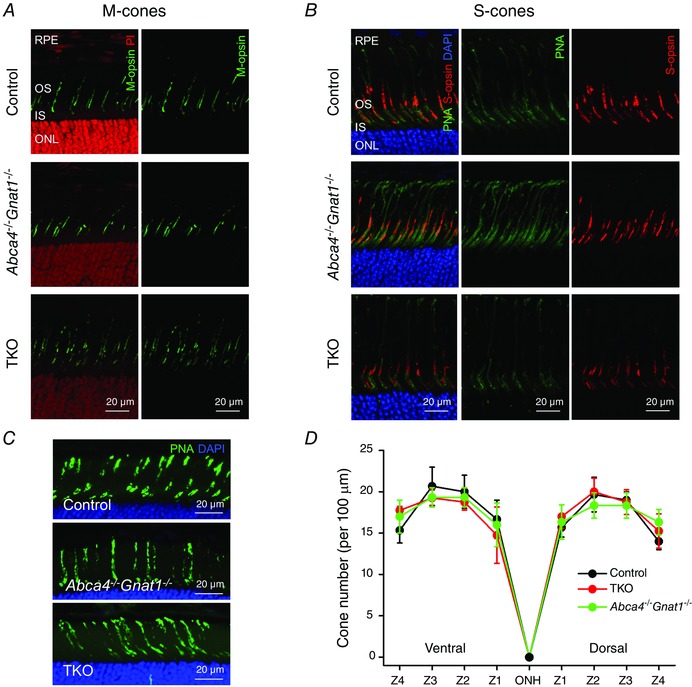
Both M‐opsin (A), and S‐opsin (B) were properly localized in cone outer segments of Abca4−/−Gnat1−/− and TKO mice. Cross section images of central retina (near the optic nerve head) are shown. RPE, retinal pigmented epithelium; OS, outer segments; IS, inner segments; ONL, outer nuclear layer. Cell nuclei were stained with either propidium iodide (PI) or DAPI. Cone glycoprotein sheaths in B were additionally stained with PNA (green). Scale bar, 20 μm. C, cones in dorsal‐to‐ventral retina cross‐sections were stained with PNA (green) and nuclei were stained with DAPI (blue). Representative images from ventral retina (zone 2) are shown. Scale bar, 20 μm. D, PNA‐positive cells in 100 μm retina width were counted in four zones (Z1–Z4) of the dorsal and ventral retina areas. Z1, 400–500 μm; Z2, 900–1000 μm; Z3, 1400–1500 μm; and Z4, 1900–2000 μm from the optic nerve head (ONH). Error bars are SDs (control, n = 3; TKO, n = 4; Abca4−/−Gnat1−/−, n = 3).
Figure 3. Transmission electron microphotographs of cones in retina cross sections from 2.5‐month‐old mice .
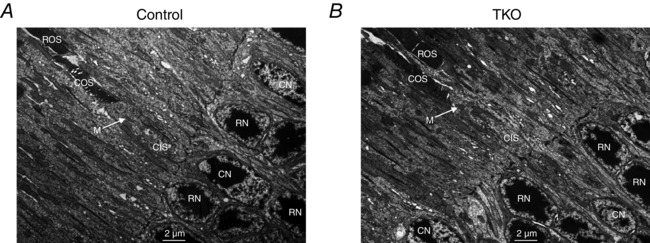
A, retina from control mouse. B, retina from TKO mouse. The images were taken at ×2500 magnification. Scale bar, 2 μm. COS, cone outer segment; ROS, rod outer segment; CIS, cone inner segment; CN, cone nuclei; RN, rod nuclei; M, mitochondria in the cone inner segment (white arrows).
Taken together, the normal cone density and morphology as well as the proper cone opsin localization imply the absence of detectable cone degeneration in young adult TKO animals. Previous studies with these mice had demonstrated that they eventually develop severe cone–rod dystrophy (Maeda et al. 2008). However, the lack of detectable morphological changes or loss of cones in 1.5‐ to 2.5‐month‐old TKO mice allowed us to investigate the roles of RDH8 and ABCA4 in dark adaptation of cones as well as their function in bright light.
Suppressed M‐cone dark adaptation in mice lacking RDH8 and/or ABCA4
To address the possible role of RDH8 and ABCA4 in cone dark adaptation, we performed a series of physiological experiments in both isolated retinas and live animals (Fig. 4). First, the recovery of M‐cone ERG a‐wave flash sensitivity (S f) after almost complete (>90%) bleaching of cone visual pigment with 505 nm LED light (3 s exposure) was monitored in isolated mouse retinas with transretinal ERG recordings. In this test, the presence of postsynaptic inhibitors blocked contributions of higher order response components, and the lack of α‐subunit of rod G‐protein transducin in these mice completely eliminated interference from rod signalling (Calvert et al. 2000). The classical RPE visual cycle is not functional due to the absence of the RPE under these conditions, and the recovery of cone sensitivity relies exclusively on chromophore recycling within the retina (Kolesnikov et al. 2011). First, we determined the dark‐adapted cone sensitivity (S f DA), and used this value to normalize all subsequent measurements and obtain relative recovery time courses. Following a nearly complete pigment bleach, cones initially were desensitized by >2 log units and then gradually recovered most of their sensitivity. We found that the lack of RDH8 alone did not result in substantial changes in postbleach cone sensitivity recovery (Fig. 4 A and its inset, P > 0.05 for all data points, compared to control). Similarly, the cone recovery in ABCA4‐deficient retinas was comparable to that in control retinas. However, when both proteins were removed from photoreceptors in TKO retinas, the final postbleach cone a‐wave flash sensitivity level was ∼2 times lower (P < 0.05) than in control retinas, and the initial recovery phase was also significantly suppressed (Fig. 4 A inset, P < 0.01 for all postbleach data points). Together, these results indicate that recycling of 11‐cis‐retinal through the retina visual cycle was essentially normal in the absence of either RDH8 or ABCA4, but was compromised when both were deleted simultaneously in the TKO retinas.
We then performed a similar bleaching experiment (with a 30 s exposure to 520 nm LED light) in live animals where the photoreceptors were in their native environment and both retina and RPE visual cycles could contribute to the dark adaptation of cones, as suggested previously (Kolesnikov et al. 2011). In this case, the effect of RDH8 and/or ABCA4 deletion on the restoration of photosensitivity during dark adaptation of cones was tested using in vivo full‐field ERG recordings. Because the small amplitudes of photopic ERG a‐waves in mice are masked by much larger cone bipolar cell‐driven b‐waves of inverse polarity, we used cone ERG b‐waves to monitor postbleach cone sensitivity changes in vivo. Consistent with the results obtained with isolated retinas, we found that the initial S f recovery phase, presumably driven by the retina visual cycle (Kolesnikov et al. 2011) lagged behind in the absence of RDH8 and ABCA4 in TKO mice (Fig. 4 B and its inset). Notably, in the presence of the RPE visual cycle, the second slow component of cone dark adaptation was also suppressed in RDH8‐deficient, ABCA4‐deficient and TKO cones, resulting in ∼2 times lower (P < 0.05) final recovery level even 60 min after the bleach. Thus, unlike the case of isolated retina, where only the simultaneous deletion of RDH8 and ABCA4 affected cone dark adaptation, in the intact eye the removal of either RDH8 or ABCA4 was sufficient to suppress the RPE‐driven component of cone dark adaptation.
It was possible that administration of anaesthesia prior to in vivo ERG recordings could affect the RPE and retina visual cycles (Keller et al. 2001) thereby introducing an unappreciated bias in our results. Indeed, the average cone sensitivity in our control anaesthetized mice did not recover fully even 1 h following the bleach (Fig. 4 B). To characterize cone dark adaptation in live control and TKO animals not subjected to anaesthesia, we illuminated unanaesthetized mice instead using the same bleaching protocol. Following a 45 min period of dark adaptation, these mice were killed, their retinas were quickly isolated, and M‐cone a‐wave fractional sensitivities (S f) were determined by ex vivo ERG recordings (Fig. 4 C and D). Importantly, despite its limited time resolution, this experiment also allowed us to analyse the extent of cone sensitivity recovery directly (as opposed to using cone b‐waves as in the test described in Fig. 4 B). As expected, the sensitivity of bleached control cones was fully restored (P > 0.05) after 45 min in the dark (Fig. 4 C). In contrast, TKO cones remained desensitized by ∼1.8‐fold (P < 0.05) over the same period (Fig. 4 D) confirming that cone pigment regeneration was compromised in the absence of RDH8 and ABCA4.
Prolonged bright light exposure and subsequent dark adaptation of M‐cones in mice lacking RDH8 and/or ABCA4
To obtain further insight into the significance of RDH8 and ABCA4 for cone function in response to bright light in vivo, we performed the following ERG experiment. After recording dark‐adapted M‐cone b‐wave flash sensitivity, a bright 530 nm Ganzfeld light (300 cd m−2) estimated to bleach ∼0.8% M‐cone pigment s−1 was turned on to induce a 2 log unit cone desensitization immediately (Fig. 5 A). During this initial 15 min irradiation period, we observed a small transient increase in cone b‐wave S f (presumably, as a consequence of retina network adaptation) in both control and TKO animals. The short period of S f stability observed after 5–6 min of continuous illumination was then followed by its gradual decline. After 15 min of light exposure, the cones of TKO mice were substantially more desensitized (by ∼1.7‐fold, P < 0.01) than those in control animals. A similar reduction of cone sensitivity (∼1.6‐fold, P < 0.001) was observed in Rdh8−/−Gnat1−/− mice at this time. Elimination of ABCA4 alone resulted in a smaller initial cone b‐wave desensitization (the reason for which is currently unclear), followed by its monotonic decline for the duration of the 15 min illumination (by approximately the same amount as in the two other mutant strains), although its final level was indistinguishable from that in control mice (P > 0.05).
Figure 5. Effects of extended light exposure on dark adaptation of M‐cones in 2‐month‐old RDH8‐ and/or ABCA4‐deficient mice .
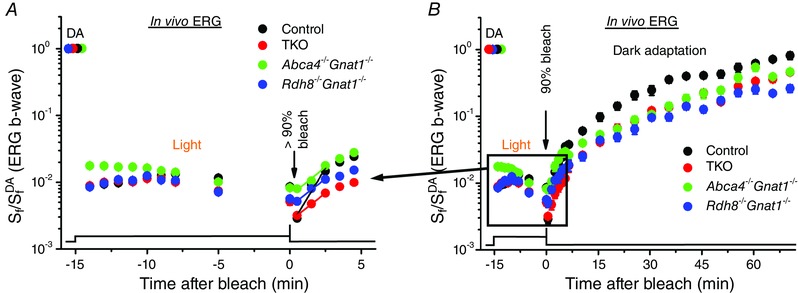
A, change of photopic ERG b‐wave S f in vivo following illumination with green 530 nm Ganzfeld background light (300 cd m−2, 15 min), and its subsequent recovery in the dark after bleaching the bulk of the remaining cone pigment (B) in control (n = 5), Abca4−/−Gnat1−/− (n = 6), Rdh8−/−Gnat1−/− (n = 6) and TKO (n = 5) mice. S f was normalized to the corresponding dark‐adapted value (S f DA) in each case. Initial rates of S f recovery were determined from linear fits (shown in A) that yielded the following slopes: 0.36 min−1 (control), 0.17 min−1 (Abca4−/−Gnat1−/−), 0.17 min−1 (Rdh8−/−Gnat1−/−) and 0.17 min−1 (TKO). Bleaching was induced by a 30 s illumination with 520 nm LED light at time 0. The time course of light exposure is shown at the bottom. Error bars represent SEMs (smaller than symbol size for most data points).
We then applied an additional bright 30 s 520 nm LED light to bleach the bulk of the remaining M‐cone pigment and monitored the recovery of photopic b‐wave sensitivity in the dark (Fig. 5 B). This allowed us to dissect the roles of RDH8 and ABCA4 proteins in cone dark adaptation under conditions where any potential sources of cis‐retinoids available to dark‐adapted cones (e.g. those in Müller cells and/or RPE) should already be exhausted. Indeed, similar to the case in human cones (Mahroo & Lamb, 2012), the overall rate of dark adaptation of control mouse cones decelerated substantially under these conditions, as compared to those when equivalent 30 s 520 nm LED bleaching was applied to dark‐adapted cones (compare Fig. 5 B with Fig. 4 B). Notably, both the retina‐ and RPE‐driven phases of cone dark adaptation were greatly delayed (by ∼2‐fold) in RDH8‐deficient, ABCA4‐deficient and TKO mice (see Fig. 5 legend for comparison of initial S f recovery rates). Thus, our results demonstrate that both of these proteins are required for the timely completion of cone dark adaptation after intense illumination.
Lack of cis‐retinol oxidase activity in RDH8/ABCA4‐deficient mouse M‐cones
The intermediate precursor form of visual chromophore produced in the retina visual cycle is 11‐cis‐retinol which then must be oxidized to 11‐cis‐retinal to complete cone pigment regeneration. This reaction is cone specific as only cones and not rods can utilize exogenous 11‐cis‐retinol for pigment regeneration and dark adaptation (Jones et al. 1989; Wang & Kefalov, 2009). However, the nature of the enzyme(s) responsible for 11‐cis‐retinol oxidation in cones is unknown. To test the possible contribution of RDH8 in this process, we exogenously applied 9‐cis‐retinol (a commercially available more stable analogue of 11‐cis‐retinol) supplemented with RDH8 co‐factor NADP+ to isolated fully bleached retinas of control (Gnat1−/−) and TKO animals and measured their M‐cone sensitivity recovery with transretinal ERG recordings at steady state, following a 3.5 h period of dark adaptation (Fig. 6). While control cones had robust retinol oxidative activity which recovered their sensitivity by an extra ∼2‐fold in the presence of 9‐cis‐retinol (Fig. 6 A), cones of TKO mice were unaffected by the application of 9‐cis‐retinol (Fig. 6 B). Thus, the deletion of RDH8 blocked the ability of M‐cones to oxidize 9‐cis‐retinol and use this retinoid for pigment regeneration and dark adaptation.
Figure 6. Inability to oxidize exogenous 9‐cis‐retinol by isolated bleached retinas of RDH8‐ and ABCA4‐deficient mice .
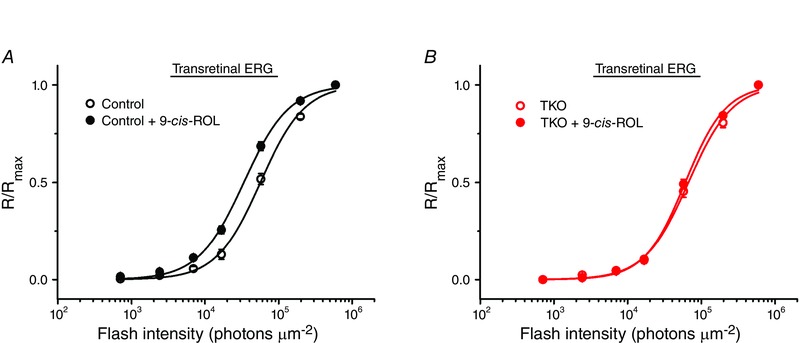
A, normalized averaged cone intensity–response relationships of isolated retinas from control mice. The average cone sensitivities (I 1/2) derived from fitting the data with a Naka–Rushton hyperbolic functions were 5.6 × 104 photons μm−2 (3.5 h postbleach, n = 10) and 3.3 × 104 photons μm−2 (3.5 h postbleach, with 9‐cis‐retinol (9‐cis‐ROL) and NADP+, n = 11). B, normalized averaged cone intensity–response relationships of isolated retinas from TKO mice. The average cone sensitivities (I 1/2) were 6.6 × 104 photons μm−2 (3.5 h postbleach, n = 6) and 6.0 × 104 photons μm−2 (3.5 h postbleach, with 9‐cis‐retinol and NADP+, n = 6). Error bars represent SEMs (smaller than the symbol size for some data points).
Impact of rods and the RPE on dark adaptation of M‐cones
The above experiments indicate that deletion of RDH8 and ABCA4 delays the RPE visual cycle‐driven cone dark adaptation in vivo (Fig. 4 B). However, they do not reveal whether this delay in cone dark adaptation is a direct result of the slower turnover of chromophore in cones, or whether cone dark adaptation is also indirectly affected by the economy of the chromophore in rods. To address this question, we used a rhodopsin‐deficient mouse line lacking rod outer segments but with preserved functional cones up to 5–6 weeks of age (Jaissle et al. 2001). By performing in vivo ERG recordings, we found that 4‐ to 5‐week‐old Rho−/− animals indeed recovered their M‐cone b‐wave sensitivity substantially faster and to a greater extent than control (Gnat1−/−) mice after a nearly complete cone pigment bleach (Fig. 7 A). In fact, we noted a 100% recovery of cone sensitivity in the absence of functional rods in vivo, whereas control cones still remained desensitized by ∼1.7‐fold even 1 h after the bleach. Thus, the turnover of chromophore in the rod photoreceptors can affect the pigment regeneration and dark adaptation in cones.
Figure 7. Modulation of cone dark adaptation by rods and the RPE visual cycle .
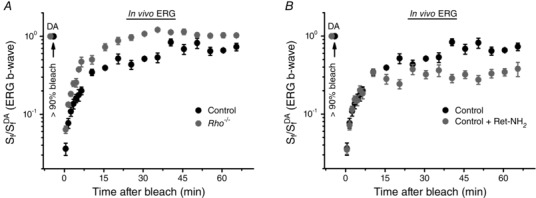
A, recovery of photopic ERG b‐wave S f in vivo in control (n = 5) and Rho−/− (n = 5) mice after bleaching >90% of their cone pigment at time 0 with 520 nm LED light. S f DA denotes the sensitivity of dark‐adapted cones. Error bars represent SEMs (smaller than symbol size for some data points). B, recovery of photopic ERG b‐wave S f in vivo in untreated (n = 5) and retinylamine (Ret‐NH2)‐treated (n = 4) control mice after bleaching >90% of their cone pigment at time 0 with 520 nm LED light. S f DA is the sensitivity of dark‐adapted cones. Error bars represent SEMs.
Mouse M‐cone dark adaptation in the RPE‐attached retina is biphasic and it was suggested that the second, slow phase reflects pigment regeneration driven by the RPE visual cycle (Kolesnikov et al. 2011). However, direct experimental evidence for the role of the RPE in the recovery of cone sensitivity was still missing. To resolve this issue and determine how suppression of the RPE visual cycle affects cone dark adaptation, we used the potent RPE visual cycle inhibitor retinylamine (Golczak et al. 2005). Control (Gnat1−/−) mice were injected intraperitoneally with retinylamine and then dark adapted for 15–18 h. Their eyes were then exposed to bright 520 nm light that bleached >90% of their M‐cone pigment and the recovery of photopic b‐wave S f was followed by ERG in the dark. We found that whereas the initial rapid phase of postbleach cone sensitivity recovery in vivo was unchanged upon retinylamine treatment, its later phase was essentially abolished by the drug which lowered the final postbleach S f level by ∼2‐fold as compared with that in untreated mice (Fig. 7 B). These results directly demonstrate the distinct roles of the retina and RPE visual cycles in the fast and slow phases of cone dark adaptation, respectively. They also suggest that inhibition of the RPE visual cycle, either directly or by slowing down the turnover of chromophore in rods by deleting RDH8 and ABCA4, suppresses cone dark adaptation.
Impaired photopic visual function in RDH8/ABCA4‐deficient mice
To determine the combined effect of the lack of RDH8 and ABCA4 proteins on cone visual function, we evaluated mouse optomotor responses (Fig. 8). This behavioural test is based on the ability of mice to reflexively track rotating gratings generated by computer (Prusky et al. 2004). We found that in standard photopic conditions (70 cd m−2) the visual acuity in adult TKO mice (0.74 ± 0.03 cycles deg−1, n = 5) was reduced by 11% (P < 0.05) compared with that in the control (Gnat1−/−) group (0.84 ± 0.02 cycles deg−1, n = 5) (Fig. 8 A, right). Interestingly, in dimmer‐light photopic conditions (0.018 cd m−2) the difference in the visual acuity between the two groups was not observed (Fig. 8 A, left). Thus, the deletion of RDH8 and ABCA4 compromised cone‐driven visual acuity in bright photopic light conditions. In contrast, photopic contrast sensitivities of TKO (41.3 ± 6.4, n = 5) and control mice (40.4 ± 4.2, n = 5) were similar for both light conditions (Fig. 8 B) indicating that the lack of RDH8 and ABCA4 had no effect on the ability of these animals to discriminate low‐contrast stimuli.
Figure 8. Evaluation of mouse vision from optomotor responses in behavioural task .
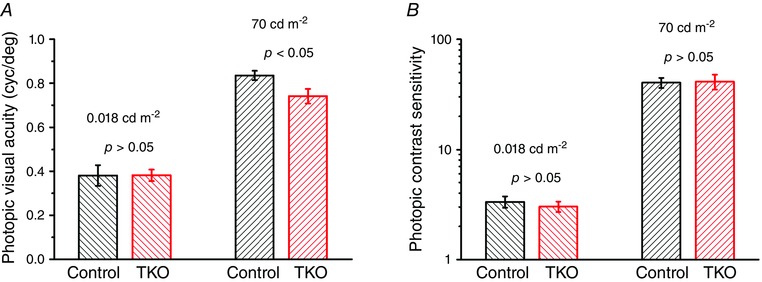
Photopic visual acuity (A) and contrast sensitivity (B) of control and TKO animals were determined at light levels of 0.018 cd m−2 (left) and 70 cd m−2 (right) at the mouse eyes. Mice of age 2–3 months were tested (n ≥ 5 for each group). Error bars represent SEMs.
To address the possible reasons for diminished photopic visual acuity in TKO mice in bright light conditions and directly examine whether individual or combined deletion of RDH8 and ABCA4 affects cone photoresponses, we recorded a series of M‐cone transretinal ERG responses elicited by test flashes of increasing light intensity (Fig. 9 A and B). We found that the maximal M‐cone response amplitude was, on average, ∼20% lower (P < 0.001) in retinas of 2‐month‐old TKO mice compared with that in age‐matched controls (Fig. 9 A–C). Neither Rdh8 −/− Gnat1 −/− nor Abca4−/−Gnat1−/− animals revealed significant changes in their cone maximal response amplitudes or photosensitivities (Fig. 9 C). The M‐cone sensitivity in TKO mice (defined as the half‐saturating light intensity, I 1/2) was also unaltered, as observed after response normalization (Fig. 9 D).
Figure 9. Sensitivity (I1/2) and kinetics of M‐cone photoresponses in mouse lines lacking RDH8 and/or ABCA4 .
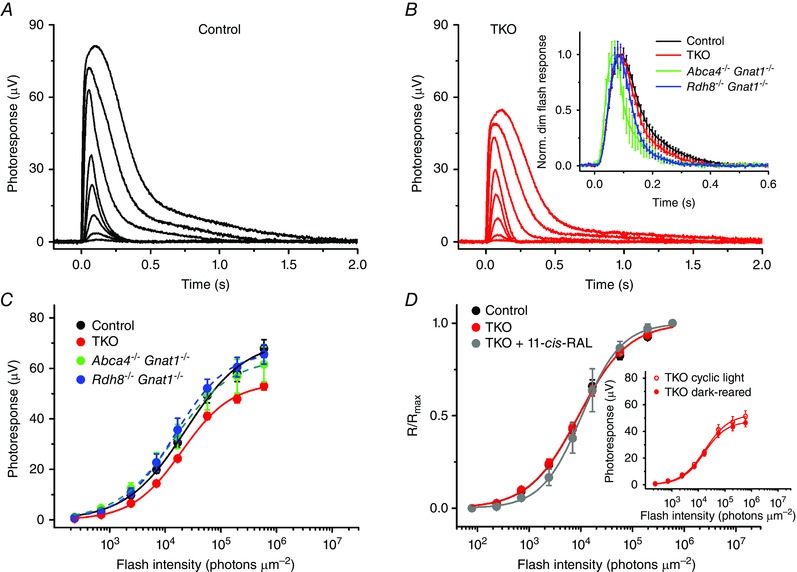
Representative families of M‐cone transretinal ERG responses from 2‐month‐old control (A) and TKO (B) animals. Flash strengths were increased from 2.4 × 102 to 6.0 × 105 photons μm−2 by steps of ∼0.5 log units (505 nm light). The inset in B shows population‐averaged normalized cone responses to test stimuli of 2.4 × 103 photons μm−2 in the four mouse lines. Error bars represent SEMs. C, averaged intensity–response functions of isolated control (n = 15), TKO (n = 15), Abca4−/−Gnat1−/− (n = 10) and Rdh8−/−Gnat1−/− (n = 12) retinas. Points were fitted with Naka–Rushton hyperbolic functions (see Methods). Error bars represent SEMs. D, normalized averaged cone intensity–response relationships of isolated retinas from control (I 1/2 = 9.1 × 103 photons μm−2, n = 8) and TKO (I 1/2 = 9.0 × 103 photons μm−2, n = 13) mice. Also shown (in grey) is the intensity–response relationship for TKO retinas exogenously treated with 130 μm 11‐cis‐retinal (11‐cis‐RAL) (1 h, at RT) prior to the recordings (I 1/2 = 10.0 × 103 photons μm−2, n = 5). Inset, averaged intensity–response functions of isolated retinas from cyclic light‐reared (n = 10) or dark‐reared (n = 11) 2‐month‐old TKO mice. Points were fitted with Naka–Rushton hyperbolic functions. Error bars indicate SEMs.
The kinetics of dim flash responses in control and TKO cones, as well as those in Abca4−/−Gnat1−/− and Rdh8−/−Gnat1−/− cones were also similar (Fig. 9 B, inset). The activation phase of phototransduction estimated from the rising part of the responses was largely unchanged in cones of all three mutant strains. Although the inactivation phase of the cascade tended to be somewhat faster in all mutant cones as compared to control cells, this did not cause marked cone desensitization (Fig. 9 C and D). The average dim flash recovery time constants (τrec) determined from single‐exponential fits to the falling phase of cone photoresponses after their peak were 87 ± 8 ms (control, n = 15), 54 ± 6 ms (Abca4−/−Gnat1−/−, n = 10, P < 0.05), 57 ± 4 ms (Rdh8−/−Gnat1−/−, n = 12, P < 0.05), and 74 ± 7 ms (TKO, n = 15, P > 0.05). The dim flash responses completely recovered to a baseline within 400 ms following the test flash of 2.4 × 103 photons μm−2. The time‐to‐peak values of cone dim flash responses were in the range of 70–90 ms in all four mouse lines, comparable to previously published results (Nikonov et al. 2006). Treatment of dark‐adapted TKO retinas with exogenous 11‐cis‐retinal had no effect on their cone sensitivity indicating that the mutant cones did not have excessive free opsin due to chromophore deficiency after overnight dark adaptation (Fig. 9 D). Consistent with this notion, the dark‐rearing of TKO animals from postnatal day 10 did not rescue M‐cone function (Fig. 9 D, inset). Together, these results imply that chronic chromophore deficiency was not the cause of the diminished cone responses in TKO retinas.
In a further attempt to determine the reason for reduced overall cone response amplitudes in TKO animals we evaluated the total M‐cone opsin expression by qRT‐PCR. Even though there were no obvious histological changes in 2‐ to 2.5‐month‐old mice used for this study (Figs 1, 2, 3), the retinas of RDH8/ABCA4‐deficient mice expressed ∼16% less (P < 0.05) M‐cone opsin than control retinas, which correlated well with the magnitude of cone response compression (Fig. 9 C). M‐cone opsin expression was normal in mouse retinas lacking either ABCA4 or RDH8 alone.
Overall, the absence of RDH8 and ABCA4 (but not either one of these proteins alone) in young adult mice compromised M‐cone transretinal responses in dark‐adapted animals. The smaller cone responses in TKO animals correlated with the reduced M‐cone pigment expression and could be the cause of the lower photopic visual acuity in these mutant mice.
Discussion
Role of RDH8 and ABCA4 in mammalian cone dark adaptation
Here we show that deletion of RDH8 and ABCA4 from mouse M‐cone photoreceptors results in a reduced cone response (Fig. 9), substantial loss of photosensitivity during prolonged exposure to bright light (Fig. 5 A), and suppressed cone dark adaptation following a nearly complete pigment bleach (Figs 4 and 5). These results clearly demonstrate that RDH8 and ABCA4 cooperatively mediate the efficient processing of chromophore in mammalian M‐cones. Mutations in human ABCA4 have been associated with retinal disorders primarily affecting cone function. These include recessive cone–rod dystrophy (Birch et al. 2001) and a subset of individuals with Stargardt disease (Lois et al. 1999). No treatments currently exist for these visual disorders. Better understanding of the mechanisms by which visual chromophore is recycled and supports cone function will provide insights into the aetiology of these disorders. Our experiments lay the foundation for investigating how specific defects in the intraretinal visual cycle produce cone‐related retinal disorders, as well as for targeting new compounds for their treatment.
In contrast to rods, most outer segment discs of mammalian cones are found to be continuous with the plasma membrane and feature open configurations (Carter‐Dawson & LaVail, 1979). However, some cone discs are closed (Anderson et al. 1978), creating the need to flip the released all‐trans‐retinal in these discs, as in rods. Furthermore, even in open discs, at least part of all‐trans‐retinal is released from bleached pigment in the same orientation as in rods, facing the extracellular side. Thus, flipping hydrophobic all‐trans‐retinal could also be required to accelerate chromophore reduction by RDH8 in the cone cytoplasm along with its subsequent release from cones. The expression pattern of ABCA4 has been a point of debate. Several early studies found no expression of ABCA4 in human (Allikmets et al. 1997) or macaque cone photoreceptors (Sun & Nathans, 1997). However, by using immunofluorescence microscopy and immunoblotting analyses, Molday and colleagues more recently found that ABCA4 is indeed expressed in human foveal and peripheral cones (Molday et al. 2000). Although ABCA4 protein was not specifically documented in mouse cones, its gene was found abundantly expressed in cone‐like photoreceptors of Nrl transcription factor knockout (Nrl−/−) mice (Mears et al. 2001). Our results support the notion that mouse cones express ABCA4 (Fig. 1, Table 1) and demonstrate that deletion of ABCA4 affects cone dark adaptation in vivo.
RDH8 contribution to oxidation of cis‐retinoids in mouse M‐cones
The unique capability of cones (but not rods) to oxidize 11‐cis‐retinol to 11‐cis‐retinal to promote cone pigment regeneration is well documented from studies of both amphibian (Jones et al. 1989; Ala‐Laurila et al. 2009) and mammalian (Wang & Kefalov, 2009) photoreceptors. This reaction also occurs in cone‐like photoreceptors of Nrl −/− mice (Parker et al. 2011), hybrid rd7 rods (Wang et al. 2014), and even in adult transdifferentiated mouse rods (Montana et al. 2013). The substrate 11‐cis‐retinol is presumably produced from all‐trans‐retinol in Müller glial cells that surround cones (Mata et al. 2002, 2005; Kaylor et al. 2013). The mouse eye contains several retinol dehydrogenases potentially capable of oxidizing the delivered 11‐cis‐retinol (Parker & Crouch, 2010; Kiser et al. 2012; Sato et al. 2015), though the enzyme(s) responsible for this reaction in cones remain unknown. TKO mice provided us with a unique opportunity to test the possible contribution of RDH8 in the oxidation of cis‐retinoids. Intriguingly, we observed that RDH8/ABCA4‐deficient bleached cones were unable to utilize 9‐cis‐retinol to regenerate visual pigment and recover their photosensitivity, even in the presence of the RDH co‐factor NADP+ (Fig. 6 B). Thus, RDH8 can potentially contribute to the oxidation of cis‐retinoids in mouse M‐cones and promote their dark adaptation following exposure to bright bleaching light.
Interplay between the rod and cone visual cycles
Our results clearly show that cone dark adaptation is suppressed in TKO mice. At first glance, this could be attributed entirely to the deletion of RDH8 and ABCA4 in cones and the subsequent slowing of the release of spent chromophore from these cells. However, the same RDH8 and ABCA4 proteins are also expressed in rods and their simultaneous elimination greatly delays the release of all‐trans‐retinal from these photoreceptors (Maeda et al. 2008). Accordingly, the delivery of chromophore from rods to the RPE is compromised in our TKO mice, in turn delaying its recycling and supply back to photoreceptors in the retina. One previously unappreciated consequence could be that cone dark adaptation is delayed as a secondary effect of the slower visual cycle in rods. Consistent with the interplay between the visual cycles of rods and cones, we found that cone dark adaptation can be accelerated substantially in the absence of rod opsin in Rho−/− mice (Fig. 7 A). This novel finding could have clinical implications leading to the development of therapeutic approaches whereby lowering the consumption of chromophore by rods could enhance its supply to cones. We also found that acute selective inhibition of the RPE visual cycle with retinylamine failed to affect the initial component of M‐cone dark adaptation but instead blocked the late recovery of cone photosensitivity (Fig. 7 B). This result reveals a potential flaw in some therapeutic approaches targeting the RPE visual cycle that are currently being developed. Although slowing the RPE visual cycle with pharmacological inhibitors might modulate the accumulation of toxic retinoid byproducts (Radu et al. 2003; Maeda et al. 2008), the resulting lower availability of chromophore could adversely affect the function of cones, eventually causing their degeneration (Xue et al. 2015). Thus, evaluating residual cone function should be a critical test for regimens targeting the RPE.
Possible mechanisms of cone degeneration in RDH8/ABCA4‐deficient mice
ABCA4 plays an important role in preventing the accumulation of hazardous bis‐retinoid adducts of all‐trans‐retinal released from visual pigment following photoexcitation (Kim et al. 2007) as well as clearing the excess 11‐cis‐retinal delivered to photoreceptors during pigment regeneration (Boyer et al. 2012). The all‐trans‐retinal released from bleached pigment or produced from extra 11‐cis‐retinal (Quazi & Molday, 2014) is then reduced to all‐trans‐retinol by RDH8, the major retinoid dehydrogenase in the outer segments (Maeda et al. 2005, 2007; Chen et al. 2012). The less toxic and more soluble alcohol form of the chromophore is then released from the outer segments of photoreceptors (Chen et al. 2005, 2009; Blakeley et al. 2011) and recycled by the RPE and retina visual cycles (Saari, 2012). Mutations in the human ABCA4 gene lead to Stargardt disease, a recessive form of macular degeneration with an onset in childhood (Allikmets et al. 1997). However, mutations in RDH8 are yet to be identified in patients and its deletion from mouse photoreceptors caused only a mild delay in rod dark adaptation (Maeda et al. 2005), probably indicating a redundancy in photoreceptor dehydrogenases (Maeda et al. 2005, 2007; Chen et al. 2012).
The combined deletion of RDH8 and ABCA4 causes progressive rod and cone degeneration in mice. Accumulation of toxic retinoid byproducts in lipofuscin and drusen deposits (Sparrow et al. 2000, 2003; Mata et al. 2001) and/or all‐trans‐retinal itself (Maeda et al. 2008, 2009 b) is believed to drive this process. Our morphological analysis demonstrates that cones in 1.5‐ to 2.5‐month‐old TKO mice still retain normal density and appearance (Figs 2 and 3) but already produce decreased responses to light (Fig. 9) accompanied by reduced expression of M‐opsin. The milder morphological phenotype of our mice compared to previous results (Maeda et al. 2008) is probably due to the absence of the Crb1/rd8 mutation which can produce a degenerative phenotype (Mattapallil et al. 2012).
Growing evidence suggests that a reduced supply of chromophore can affect cone opsin expression and eventually lead to cone loss. For instance, the ablation of the RPE visual cycle in RPE65 knockout mice results in rapid cone degeneration (Znoiko et al. 2005; Fan et al. 2008) which is largely prevented by supplementing animals with exogenous retinoids (Rohrer et al. 2005; T. Maeda et al. 2009; Tang et al. 2010). Similarly, expression of mutant mouse S‐opsin can be enhanced by exogenous 11‐cis‐retinal suggesting that the chromophore is important for cone opsin biosynthesis (Insinna et al. 2012). Retinoid deficiency also impairs cone function in mice lacking either the interphotoreceptor retinoid‐binding protein IRBP (Jin et al. 2009; Parker et al. 2009; Kolesnikov et al. 2011), or the cellular retinaldehyde‐binding protein CRALBP (Xue et al. 2015). However, unlike in CRALBP‐deficient mice, dark‐rearing did not correct the cone response magnitude in TKO animals (Fig. 9 D), indicating that chronic chromophore deficiency is unlikely to cause the eventual degeneration of M‐cones. Thus, we conclude that the most likely explanation for the age‐related degeneration of TKO cones is the accumulation of toxic retinoid byproducts resulting from slower turnover of chromophore in RDH8/ABCA4‐deficient photoreceptors. As the early survival of cones in TKO mice and their eventual degeneration mimic the clinical phenotypes of Stargardt disease and age‐related macular degeneration, TKO mutant animals represent an excellent model for investigating the mechanisms of cone loss in these visual disorders.
Additional information
Competing interests
The authors have no competing interests to declare.
Author contributions
Conception and design of the experiments: A.V.K., K.P. and V.J.K. Collection, assembly, analysis and interpretation of data: A.V.K., A.M., P.H.T., Y.I. and V.J.K. Drafting the article or revising it critically for important intellectual content: A.V.K. and V.J.K. All authors have approved the final version of the manuscript and agree to be accountable for all aspects of the work. All persons designated as authors qualify for authorship, and all those who qualify for authorship are listed. All experiments were carried out at Washington University School of Medicine or Case Western Reserve University.
Funding
This work was supported by NIH grants EY019312 and EY21126 (V.J.K.), EY009339 and EY021126 (K.P.), EY022658 (A.M.) and EY002687 to the Department of Ophthalmology and Visual Sciences at Washington University, and by Research to Prevent Blindness. K.P. is John H. Hord Professor of Pharmacology.
Acknowledgements
We thank Drs Janis Lem (Tufts University School of Medicine) for providing Rho−/− and Gnat1−/− animals, Anand Swaroop (National Eye Institute) for providing Nrl−/− mice, Debarshi Mustafi for the RNA‐Seq data, Robert Molday (University of British Columbia) for providing ABCA4 antibody, Rosalie Crouch (Medical University of South Carolina) and the NEI for 11‐cis‐retinal, Marcin Golczak (Case Western Reserve University) for retinylamine, Jinshan Wang for preliminary experiments with ABCA4‐deficient mice, Mr Richard Lee for technical assistance in immunofluorescence microscopy, and Ms Robyn Roth and Ms Teresa Doggett for their help with electron microscopy analysis. We are also grateful to Dr Rosalie Crouch for her comments on the manuscript.
References
- Ala‐Laurila P, Cornwall MC, Crouch RK & Kono M (2009). The action of 11‐cis‐retinol on cone opsins and intact cone photoreceptors. J Biol Chem 284, 16492–16500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allikmets R, Singh N, Sun H, Shroyer NF, Hutchinson A, Chidambaram A, Gerrard B, Baird L, Stauffer D, Peiffer A, Rattner A, Smallwood P, Li Y, Anderson KL, Lewis RA, Nathans J, Leppert M, Dean M & Lupski JR (1997). A photoreceptor cell‐specific ATP‐binding transporter gene (ABCR) is mutated in recessive Stargardt macular dystrophy. Nat Genet 15, 236–246. [DOI] [PubMed] [Google Scholar]
- Anderson DH, Fisher SK & Steinberg RH (1978). Mammalian cones: disc shedding, phagocytosis, and renewal. Invest Ophthalmol Vis Sci 17, 117–133. [PubMed] [Google Scholar]
- Birch DG, Peters AY, Locke KL, Spencer R, Megarity CF & Travis GH (2001). Visual function in patients with cone‐rod dystrophy (CRD) associated with mutations in the ABCA4(ABCR) gene. Exp Eye Res 73, 877–886. [DOI] [PubMed] [Google Scholar]
- Blakeley LR, Chen C, Chen CK, Chen J, Crouch RK, Travis GH & Koutalos Y (2011). Rod outer segment retinol formation is independent of Abca4, arrestin, rhodopsin kinase, and rhodopsin palmitylation. Invest Ophthalmol Vis Sci 52, 3483–3491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyer NP, Higbee D, Currin MB, Blakeley LR, Chen C, Ablonczy Z, Crouch RK & Koutalos Y (2012). Lipofuscin and N‐retinylidene‐N‐retinylethanolamine (A2E) accumulate in retinal pigment epithelium in absence of light exposure: their origin is 11‐cis‐retinal. J Biol Chem 287, 22276–22286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calvert PD, Krasnoperova NV, Lyubarsky AL, Isayama T, Nicolo M, Kosaras B, Wong G, Gannon KS, Margolskee RF, Sidman RL, Pugh EN Jr, Makino CL & Lem J (2000). Phototransduction in transgenic mice after targeted deletion of the rod transducin α‐subunit. Proc Natl Acad Sci U S A 97, 13913–13918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter‐Dawson LD & LaVail MM (1979). Rods and cones in the mouse retina. I. Structural analysis using light and electron microscopy. J Comp Neurol 188, 245–262. [DOI] [PubMed] [Google Scholar]
- Chen C, Blakeley LR & Koutalos Y (2009). Formation of all‐trans retinol after visual pigment bleaching in mouse photoreceptors. Invest Ophthalmol Vis Sci 50, 3589–3595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C, Thompson DA & Koutalos Y (2012). Reduction of all‐trans‐retinal in vertebrate rod photoreceptors requires the combined action of RDH8 and RDH12. J Biol Chem 287, 24662–24670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C, Tsina E, Cornwall MC, Crouch RK, Vijayaraghavan S & Koutalos Y (2005). Reduction of all‐trans retinal to all‐trans retinol in the outer segments of frog and mouse rod photoreceptors. Biophys J 88, 2278–2287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan J, Rohrer B, Frederick JM, Baehr W & Crouch RK (2008). Rpe65−/− and Lrat−/− mice: comparable models of Leber congenital amaurosis. Invest Ophthalmol Vis Sci 49, 2384–2389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golczak M, Kuksa V, Maeda T, Moise AR & Palczewski K (2005). Positively charged retinoids are potent and selective inhibitors of the trans‐cis isomerization in the retinoid (visual) cycle. Proc Natl Acad Sci U S A 102, 8162–8167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimm C, Wenzel A, Stanescu D, Samardzija M, Hotop S, Groszer M, Naash M, Gassmann M & Reme C (2004). Constitutive overexpression of human erythropoietin protects the mouse retina against induced but not inherited retinal degeneration. J Neurosci 24, 5651–5658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grundy D (2015). Principles and standards for reporting animal experiments in The Journal of Physiology and Experimental Physiology . J Physiol 593, 2547–2549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Illing M, Molday LL & Molday RS (1997). The 220‐kDa rim protein of retinal rod outer segments is a member of the ABC transporter superfamily. J Biol Chem 272, 10303–10310. [DOI] [PubMed] [Google Scholar]
- Insinna C, Daniele LL, Davis JA, Larsen DD, Kuemmel C, Wang J, Nikonov SS, Knox BE & Pugh EN Jr (2012). An S‐opsin knock‐in mouse (F81Y) reveals a role for the native ligand 11‐cis‐retinal in cone opsin biosynthesis. J Neurosci 32, 8094–8104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaissle GB, May CA, Reinhard J, Kohler K, Fauser S, Lutjen‐Drecoll E, Zrenner E & Seeliger MW (2001). Evaluation of the rhodopsin knockout mouse as a model of pure cone function. Invest Ophthalmol Vis Sci 42, 506–513. [PubMed] [Google Scholar]
- Jin M, Li S, Nusinowitz S, Lloyd M, Hu J, Radu RA, Bok D & Travis GH (2009). The role of interphotoreceptor retinoid‐binding protein on the translocation of visual retinoids and function of cone photoreceptors. J Neurosci 29, 1486–1495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones GJ, Crouch RK, Wiggert B, Cornwall MC & Chader GJ (1989). Retinoid requirements for recovery of sensitivity after visual‐pigment bleaching in isolated photoreceptors. Proc Natl Acad Sci U S A 86, 9606–9610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaylor JJ, Yuan Q, Cook J, Sarfare S, Makshanoff J, Miu A, Kim A, Kim P, Habib S, Roybal CN, Xu T, Nusinowitz S & Travis GH (2013). Identification of DES1 as a vitamin A isomerase in Muller glial cells of the retina. Nat Chem Biol 9, 30–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keller C, Grimm C, Wenzel A, Hafezi F & Reme C (2001). Protective effect of halothane anesthesia on retinal light damage: inhibition of metabolic rhodopsin regeneration. Invest Ophthalmol Vis Sci 42, 476–480. [PubMed] [Google Scholar]
- Kim SR, Jang YP, Jockusch S, Fishkin NE, Turro NJ & Sparrow JR (2007). The all‐trans‐retinal dimer series of lipofuscin pigments in retinal pigment epithelial cells in a recessive Stargardt disease model. Proc Natl Acad Sci U S A 104, 19273–19278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiser PD, Golczak M, Maeda A & Palczewski K (2012). Key enzymes of the retinoid (visual) cycle in vertebrate retina. Biochim Biophys Acta 1821, 137–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiser PD, Golczak M & Palczewski K (2014). Chemistry of the retinoid (visual) cycle. Chem Rev 114, 194–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolesnikov AV, Tang PH, Parker RO, Crouch RK & Kefalov VJ (2011). The mammalian cone visual cycle promotes rapid M/L‐cone pigment regeneration independently of the interphotoreceptor retinoid‐binding protein. J Neurosci 31, 7900–7909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lem J, Krasnoperova NV, Calvert PD, Kosaras B, Cameron DA, Nicolo M, Makino CL & Sidman RL (1999). Morphological, physiological, and biochemical changes in rhodopsin knockout mice. Proc Natl Acad Sci U S A 96, 736–741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lois N, Holder GE, Fitzke FW, Plant C & Bird AC (1999). Intrafamilial variation of phenotype in Stargardt macular dystrophy – Fundus flavimaculatus. Invest Ophthalmol Vis Sci 40, 2668–2675. [PubMed] [Google Scholar]
- Maeda A, Golczak M, Maeda T & Palczewski K (2009. a). Limited roles of Rdh8, Rdh12, and Abca4 in all‐trans‐retinal clearance in mouse retina. Invest Ophthalmol Vis Sci 50, 5435–5443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maeda A, Maeda T, Golczak M, Chou S, Desai A, Hoppel CL, Matsuyama S & Palczewski K (2009. b). Involvement of all‐trans‐retinal in acute light‐induced retinopathy of mice. J Biol Chem 284, 15173–15183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maeda A, Maeda T, Golczak M & Palczewski K (2008). Retinopathy in mice induced by disrupted all‐trans‐retinal clearance. J Biol Chem 283, 26684–26693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maeda A, Maeda T, Imanishi Y, Kuksa V, Alekseev A, Bronson JD, Zhang H, Zhu L, Sun W, Saperstein DA, Rieke F, Baehr W & Palczewski K (2005). Role of photoreceptor‐specific retinol dehydrogenase in the retinoid cycle in vivo . J Biol Chem 280, 18822–18832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maeda A, Maeda T, Sun W, Zhang H, Baehr W & Palczewski K (2007). Redundant and unique roles of retinol dehydrogenases in the mouse retina. Proc Natl Acad Sci U S A 104, 19565–19570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maeda T, Cideciyan AV, Maeda A, Golczak M, Aleman TS, Jacobson SG & Palczewski K (2009). Loss of cone photoreceptors caused by chromophore depletion is partially prevented by the artificial chromophore pro‐drug, 9‐cis‐retinyl acetate. Hum Mol Genet 18, 2277–2287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahroo OA & Lamb TD (2012). Slowed recovery of human photopic ERG a‐wave amplitude following intense bleaches: a slowing of cone pigment regeneration? Doc Ophthalmol 125, 137–147. [DOI] [PubMed] [Google Scholar]
- Mata NL, Radu RA, Clemmons RC & Travis GH (2002). Isomerization and oxidation of vitamin A in cone‐dominant retinas: a novel pathway for visual‐pigment regeneration in daylight. Neuron 36, 69–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mata NL, Ruiz A, Radu RA, Bui TV & Travis GH (2005). Chicken retinas contain a retinoid isomerase activity that catalyzes the direct conversion of all‐trans‐retinol to 11‐cis‐retinol. Biochemistry 44, 11715–11721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mata NL, Tzekov RT, Liu X, Weng J, Birch DG & Travis GH (2001). Delayed dark‐adaptation and lipofuscin accumulation in abcr+/− mice: implications for involvement of ABCR in age‐related macular degeneration. Invest Ophthalmol Vis Sci 42, 1685–1690. [PubMed] [Google Scholar]
- Mattapallil MJ, Wawrousek EF, Chan CC, Zhao H, Roychoudhury J, Ferguson TA & Caspi RR (2012). The Rd8 mutation of the Crb1 gene is present in vendor lines of C57BL/6N mice and embryonic stem cells, and confounds ocular induced mutant phenotypes. Invest Ophthalmol Vis Sci 53, 2921–2927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mears AJ, Kondo M, Swain PK, Takada Y, Bush RA, Saunders TL, Sieving PA & Swaroop A (2001). Nrl is required for rod photoreceptor development. Nat Genet 29, 447–452. [DOI] [PubMed] [Google Scholar]
- Molday LL, Rabin AR & Molday RS (2000). ABCR expression in foveal cone photoreceptors and its role in Stargardt macular dystrophy. Nat Genet 25, 257–258. [DOI] [PubMed] [Google Scholar]
- Montana CL, Kolesnikov AV, Shen SQ, Myers CA, Kefalov VJ & Corbo JC (2013). Reprogramming of adult rod photoreceptors prevents retinal degeneration. Proc Natl Acad Sci U S A 110, 1732–1737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mustafi D, Kevany BM, Genoud C, Okano K, Cideciyan AV, Sumaroka A, Roman AJ, Jacobson SG, Engel A, Adams MD & Palczewski K (2011). Defective photoreceptor phagocytosis in a mouse model of enhanced S‐cone syndrome causes progressive retinal degeneration. FASEB J 25, 3157–3176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikonov SS, Kholodenko R, Lem J & Pugh EN Jr (2006). Physiological features of the S‐ and M‐cone photoreceptors of wild‐type mice from single‐cell recordings. J Gen Physiol 127, 359–374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nymark S, Heikkinen H, Haldin C, Donner K & Koskelainen A (2005). Light responses and light adaptation in rat retinal rods at different temperatures. J Physiol 567, 923–938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker R, Wang JS, Kefalov VJ & Crouch RK (2011). Interphotoreceptor retinoid‐binding protein as the physiologically relevant carrier of 11‐cis‐retinol in the cone visual cycle. J Neurosci 31, 4714–4719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker RO & Crouch RK (2010). Retinol dehydrogenases (RDHs) in the visual cycle. Exp Eye Res 91, 788–792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker RO, Fan J, Nickerson JM, Liou GI & Crouch RK (2009). Normal cone function requires the interphotoreceptor retinoid binding protein. J Neurosci 29, 4616–4621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pawar AS, Qtaishat NM, Little DM & Pepperberg DR (2008). Recovery of rod photoresponses in ABCR‐deficient mice. Invest Ophthalmol Vis Sci 49, 2743–2755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prusky GT, Alam NM, Beekman S & Douglas RM (2004). Rapid quantification of adult and developing mouse spatial vision using a virtual optomotor system. Invest Ophthalmol Vis Sci 45, 4611–4616. [DOI] [PubMed] [Google Scholar]
- Quazi F & Molday RS (2014). ATP‐binding cassette transporter ABCA4 and chemical isomerization protect photoreceptor cells from the toxic accumulation of excess 11‐cis‐retinal. Proc Natl Acad Sci U S A 111, 5024–5029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radu RA, Mata NL, Nusinowitz S, Liu X, Sieving PA & Travis GH (2003). Treatment with isotretinoin inhibits lipofuscin accumulation in a mouse model of recessive Stargardt's macular degeneration. Proc Natl Acad Sci U S A 100, 4742–4747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rattner A, Smallwood PM & Nathans J (2000). Identification and characterization of all‐trans‐retinol dehydrogenase from photoreceptor outer segments, the visual cycle enzyme that reduces all‐trans‐retinal to all‐trans‐retinol. J Biol Chem 275, 11034–11043. [DOI] [PubMed] [Google Scholar]
- Rohrer B, Lohr HR, Humphries P, Redmond TM, Seeliger MW & Crouch RK (2005). Cone opsin mislocalization in Rpe65−/− mice: a defect that can be corrected by 11‐cis retinal. Invest Ophthalmol Vis Sci 46, 3876–3882. [DOI] [PubMed] [Google Scholar]
- Saari JC (2012). Vitamin A metabolism in rod and cone visual cycles. Ann Rev Nutr 32, 125–145. [DOI] [PubMed] [Google Scholar]
- Sato S, Miyazono S, Tachibanaki S & Kawamura S (2015). RDH13L, an enzyme responsible for the aldehyde‐alcohol redox coupling reaction (AL‐OL coupling reaction) to supply 11‐cis retinal in the carp cone retinoid cycle. J Biol Chem 290, 2983–2992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sillman AJ, Ito H & Tomita T (1969). Studies on the mass receptor potential of the isolated frog retina. I. General properties of the response. Vision Res 9, 1435–1442. [DOI] [PubMed] [Google Scholar]
- Sparrow JR, Cai B, Fishkin N, Jang YP, Krane S, Vollmer HR, Zhou J & Nakanishi K (2003). A2E, a fluorophore of RPE lipofuscin: can it cause RPE degeneration? Adv Exp Med Biol 533, 205–211. [DOI] [PubMed] [Google Scholar]
- Sparrow JR, Nakanishi K & Parish CA (2000). The lipofuscin fluorophore A2E mediates blue light‐induced damage to retinal pigmented epithelial cells. Invest Ophthalmol Vis Sci 41, 1981–1989. [PubMed] [Google Scholar]
- Sun H, Molday RS & Nathans J (1999). Retinal stimulates ATP hydrolysis by purified and reconstituted ABCR, the photoreceptor‐specific ATP‐binding cassette transporter responsible for Stargardt disease. J Biol Chem 274, 8269–8281. [DOI] [PubMed] [Google Scholar]
- Sun H & Nathans J (1997). Stargardt's ABCR is localized to the disc membrane of retinal rod outer segments. Nat Genet 17, 15–16. [DOI] [PubMed] [Google Scholar]
- Tang PH, Fan J, Goletz PW, Wheless L & Crouch RK (2010). Effective and sustained delivery of hydrophobic retinoids to photoreceptors. Invest Ophthalmol Vis Sci 51, 5958–5964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang PH, Kono M, Koutalos Y, Ablonczy Z & Crouch RK (2013). New insights into retinoid metabolism and cycling within the retina. Prog Retin Eye Res 32, 48–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Umino Y, Solessio E & Barlow RB (2008). Speed, spatial, and temporal tuning of rod and cone vision in mouse. J Neurosci 28, 189–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vinberg F, Kolesnikov AV & Kefalov VJ (2014). Ex vivo ERG analysis of photoreceptors using an in vivo ERG system. Vision Res 101, 108–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang JS & Kefalov VJ (2009). An alternative pathway mediates the mouse and human cone visual cycle. Curr Biol 19, 1665–1669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang JS & Kefalov VJ (2011). The cone‐specific visual cycle. Prog Retin Eye Res 30, 115–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang JS, Nymark S, Frederiksen R, Estevez ME, Shen SQ, Corbo JC, Cornwall MC & Kefalov VJ (2014). Chromophore supply rate‐limits mammalian photoreceptor dark adaptation. J Neurosci 34, 11212–11221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weng J, Mata NL, Azarian SM, Tzekov RT, Birch DG & Travis GH (1999). Insights into the function of Rim protein in photoreceptors and etiology of Stargardt's disease from the phenotype in abcr knockout mice. Cell 98, 13–23. [DOI] [PubMed] [Google Scholar]
- Xue Y, Shen SQ, Jui J, Rupp AC, Byrne LC, Hattar S, Flannery JG, Corbo JC & Kefalov VJ (2015). CRALBP supports the mammalian retinal visual cycle and cone vision. J Clin Invest 125, 727–738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang N, Tsybovsky Y, Kolesnikov AV, Rozanowska M, Swider M, Schwartz SB, Stone EM, Palczewska G, Maeda A, Kefalov VJ, Jacobson SG, Cideciyan AV & Palczewski K (2015). Protein misfolding and the pathogenesis of ABCA4‐associated retinal degenerations. Hum Mol Genet 24, 3220–3237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Znoiko SL, Rohrer B, Lu K, Lohr HR, Crouch RK & Ma JX (2005). Downregulation of cone‐specific gene expression and degeneration of cone photoreceptors in the Rpe65−/− mouse at early ages. Invest Ophthalmol Vis Sci 46, 1473–1479. [DOI] [PubMed] [Google Scholar]