

Abstract
Key Points
Most barbiturates are anaesthetics but unexpectedly a few are convulsants whose mechanism of action is poorly understood.
We synthesized and characterized a novel pair of chiral barbiturates that are capable of photolabelling their binding sites on GABAA receptors. In mice the S‐enantiomer is a convulsant, but the R‐enantiomer is an anticonvulsant.
The convulsant S‐enantiomer binds solely at an inhibitory site. It is both an open state inhibitor and a resting state inhibitor. Its action is pH independent, suggesting the pyrimidine ring plays little part in binding. The inhibitory site is not enantioselective because the R‐enantiomer inhibits with equal affinity.
In contrast, only the anticonvulsant R‐enantiomer binds to the enhancing site on open channels, causing them to stay open longer. The enhancing site is enantioselective.
The in vivo actions of the convulsant S‐enantiomer are accounted for by its interactions with GABAA receptors.
Abstract
Most barbiturates are anaesthetics but a few unexpectedly are convulsants. We recently located the anaesthetic sites on GABAA receptors (GABAARs) by photolabelling with an anaesthetic barbiturate. To apply the same strategy to locate the convulsant sites requires the creation and mechanistic characterization of a suitable agent. We synthesized enantiomers of a novel, photoactivable barbiturate, 1‐methyl‐5‐propyly‐5‐(m‐trifluoromethyldiazirinyl) phenyl barbituric acid (mTFD‐MPPB). In mice, S‐mTFD‐MPPB acted as a convulsant, whereas R‐mTFD‐MPPB acted as an anticonvulsant. Using patch clamp electrophysiology and fast solution exchange on recombinant human α1β3γ2L GABAARs expressed in HEK cells, we found that S‐mTFD‐MPPB inhibited GABA‐induced currents, whereas R‐mTFD‐MPPB enhanced them. S‐mTFD‐MPPB caused inhibition by binding to either of two inhibitory sites on open channels with bimolecular kinetics. It also inhibited closed, resting state receptors at similar concentrations, decreasing the channel opening rate and shifting the GABA concentration–response curve to the right. R‐mTFD‐MPPB, like most anaesthetics, enhanced receptor gating by rapidly binding to allosteric sites on open channels, initiating a rate‐limiting conformation change to stabilized open channel states. These states had slower closing rates, thus shifting the GABA concentration–response curve to the left. Under conditions when most GABAARs were open, an inhibitory action of R‐mTFD‐MPPB was revealed that had a similar IC50 to that of S‐mTFD‐MPPB. Thus, the inhibitory sites are not enantioselective, and the convulsant action of S‐mTFD‐MPPB results from its negligible affinity for the enhancing, anaesthetic sites. Interactions with these two classes of barbiturate binding sites on GABAARs underlie the enantiomers’ different pharmacological activities in mice.
Abbreviations
- C
channel closed, resting state
- CD50
median clonic siezure dose
- CI
confidence interval
- G2O
open channel state bound with two GABA molecules
- GABAAR
GABA receptor Type A
- HEK
human embryonic kidney
- I
peak current amplitude
- Imax
maximal peak current amplitude
- k−1
dissociation rate constant
- k+1
binding rate constant
- kact
activation energy
- LoRR
loss of righting reflex
- MPPB
1‐methyl‐5‐phenyl‐5‐propyl‐barbituric acid
- nAChR
nicotinic acetylcholine receptor
- O
open channel state
- O’
stabilized open channel state
- pK
acid dissociation constant
- PTZ
pentylenetetrazol
- R‐mTFD‐MPAB
R‐5‐allyl‐1‐methyl‐5‐(m‐trifluoromethyl‐diazirynylphenyl) barbituric acid
- R‐mTFD‐MPPB
R‐1‐methyl‐5‐propyly‐5‐(m‐trifluoromethyldiazirinyl) phenyl barbituric acid
- S‐mTFD‐MPAB
S‐5‐allyl‐1‐methyl‐5‐(m‐trifluoromethyl‐diazirynylphenyl) barbituric acid
- S‐mTFD‐MPPB
S‐1‐methyl‐5‐propyly‐5‐(m‐trifluoromethyldiazirinyl) phenyl barbituric acid
- TID
3‐(trifluoromethyl)‐3‐(m‐iodophenyl) diazirine
- α
channel closing rate
- β
channel opening rate
Introduction
Although much progress has been made in understanding the mechanism of action of general anaesthetics (Zeller et al. 2008) and their action on GABAA receptors (GABAARs) (Olsen & Li, 2011), the unwanted excitatory physiology exerted by these agents has received little attention. For example, isoflurane (CF3.CHCl.O.CHF2) is a commonly used general anaesthetic, whereas its structural isomer enflurane (CHFCl.CF2.OCHF2) is no longer in use because it produces spike complexes in the EEG that are typical of seizure activity and it lowers the threshold to seizures (Najjar et al. 2002). Indoklon (flurothyl, (CF3.CH2)2O), formerly used for treatment of depression, is a convulsant as are many volatile agents noted in the early literature on developing volatile anaesthetics (Krantz, 1966). Thus, there is a continuum of actions from anaesthesia to convulsions with the structure–activity relationships of the convulsant action appearing and disappearing seemingly randomly.
Barbiturates provide a good vehicle for studying this convulsant action. Thousands of barbiturate derivatives have been synthesized and their in vivo actions characterized since the introduction of barbital in 1903 (reviewed by Swanson et al. 1955; Richter & Holtman, 1982; Löscher & Rogawski, 2012). Although their anaesthetic action is loosely related to hydrophobicity, it shows more structural selectivity than this implies and in some cases modest enantioselectivity occurs. Furthermore, some barbiturates cause excitation and even convulsions. Thus, the structure–activity relationships of the anaesthetic and the convulsant actions are distinct and many investigators have suggested that different sites are involved (Downes et al. 1970). This is in contrast to the action of benzodiazepines,which cause opposing allosteric modulatory effects on GABAARs by binding to a single site (Sigel & Buhr, 1997).
Subsequent studies on ion channels have rationalized some of these phenomena. The excitatory action of some barbiturates that have chiral centres in the C5 side chain, such as 5‐ethyl‐5‐(3‐methylbut‐2‐enyl) barbituric acid (3M2B), and (+)‐5‐ethyl‐5‐(1,3‐dimethylbutyl) barbituric acid ((+)‐DMBB), are independent of their action on GABAARs in cultured rat hippocampal neurons (Holland et al. 1990). By contrast, some convulsant barbiturates, whose chirality is dependent on N‐methylation of the pyrimidine ring, do exert their excitatory actions through GABAARs. These include mephobarbital and 1‐methyl‐5‐phenyl‐5‐propyl barbituric acid (MPPB; Fig. 1) (Buch et al. 1970; Richter & Holtman, 1982; Harrison & Simmonds, 1983; Ticku et al. 1985; Dunwiddie et al. 1986; Mehta & Ticku, 1999). The enantiomers of MPPB display contrasting pharmacological actions in rodents: R‐MPPB produces anaesthesia whereas S‐MPPB produces convulsions (Ticku et al. 1985). Consistent with their contrasting pharmacological effects, the two MPPB enantiomers also have opposing actions on GABAAR‐mediated currents; R‐MPPB enhances, whereas S‐MPPB inhibits the currents (Maksay et al. 1996; Kamiya et al. 1999). These profound pharmacological differences suggest that the MPPB enantiomers interact with at least two distinct binding sites on GABAARs (Maksay & Ticku, 1985; Rho et al. 1996). The site for anaesthetic action might well be that recently located in the transmembrane domain of GABAARs by photolabelling with [3H]R‐mTFD‐MPAB (Chiara et al. 2013), a site distinct from that for etomidate (Chiara et al. 2012). The location of the convulsant site is unknown, although some suggest it co‐locates with the picrotoxin site (Ticku et al. 1985).
Figure 1.
Chemical structures of S‐enantiomers of mephobarbital, MPPB and mTFD‐MPPB. Star denotes the chiral centre.
To find the convulsant site, we initially chose to synthesize the 5’‐propyl derivatives of mTFD‐MPAB (1‐methyl‐5‐propyly‐5‐(m‐trifluoromethyldiazirinyl) phenyl barbituric acid), the mTFD‐MPPB enantiomers, for a number of reasons (Fig. 1). First, they are photoactivable derivatives of the well‐characterized MPPB enantiomers (Ticku et al. 1985; Allan & Harris, 1986; Kamiya et al. 1999). Second, 5‐allyl to 5‐propyl substitutions often enhance excitatory properties and, third, there are no barbiturates with a 5‐propyl group in anaesthetic practice (Richter & Holtman, 1982). We find that in rodents S‐mTFD‐MPPB is a convulsant, whereas R‐mTFD‐MPPB is an anticonvulsant. In recombinant human α1β3γ2L GABAARs, R‐mTFD‐MPPB produces both enhancing and inhibiting actions on GABA currents, whereas S‐mTFD‐MPPB produces only inhibition. Thus, the mTFD‐MPPB enantiomers retain the pharmacology of the MPPB enantiomers, and S‐mTFD‐MPPB binds selectively to the uncharacterized convulsant site.
Materials and methods
Ethical approval
The mouse facilities were fully accredited by the Association for Assessment and Accreditation of Laboratory Animal Care, and all studies were performed under protocols approved by the University of California, Davis Institutional Animal Care and Use Committee in strict compliance with the Guide for the Care and Use of Laboratory Animals of the National Research Council (http://www.nap.edu/readingroom/books/labrats/). At the end of the observation period, mice were killed via CO2, consistent with the recommendations of the Panel on Euthanasia of the American Veterinary Medical Association.
Tadpole experiments were conducted according to an animal protocol preapproved by the Massachusetts General Hospital (MGH) Subcommittee on Research Animal Care, following previously published protocols (Ge et al. 2014). Tadpoles were killed immediately after each protocol by immersion in a lethal concentration of pentobarbital (1 mm).
Synthesis of mTFD‐MPPB enantiomers
1‐Methyl‐5‐propyl‐pyrimidine‐2,4,6(1H,3H,5H)‐trione was synthesized according to a previously published procedure (Knabe et al. 1982). Colorless crystals, yield 67%. 1H NMR (400 MHz, CDCl3): 8.95 (brs, 1H), 3.49 (t, J = 5.0 Hz, 1H, CH 2), 3.30 (s, 3H, NCH 3), 2.13 (td, J = 8.1, 5.5 Hz, 1H), 1.49–1.29 (m, 2H, CH 2), 0.95 (t, J = 7.2 Hz, CH 3). 13C NMR (CDCl3): δ 169.3, 168.7, 150.6, 48.8, 32.8, 27.8, 19.3, 13.7. High‐resolution mass spectrometry (electrospray ionization): calculated for C8H12N2O3 [M+H]+: 185.09207. Found: 185.0925.
(±)‐1‐Methyl‐5‐propyl‐5‐[3‐(3‐trifluoromethyl‐3H‐diazirin‐3‐yl)‐phenyl]‐pyrimidine‐2,4,6(1H,3H,5H)‐trione [mTFD‐MPPB]. The mixture of 1‐methyl‐5‐propylbarbiturate from above (278 mg, 1.50 mmol), diisopropylamine or triethylamine (1.3 mmol) and (4‐methoxyphenyl)‐[3‐(3‐trifluoromethyl‐3H‐diazirin‐3‐yl)‐phenyl]‐iodonium trifluoroacetate (532 mg, 1.00 mmol) in dry dimethylformamide (0.75 ml) was stirred at 40°C for 72 h using thin layer chromatography to monitor the progress of the reaction. After completion, the reaction mixture was chromatographed on silica gel using ethyl acetate/hexane 1:9 to 1:5 as an eluent. The product was recrystallized from hexane – ethyl acetate to afford pure mTFD‐MPPB as colourless crystals (298 mg, 81%), identical to those obtained by catalytic reduction of the allyl‐derivative (Savechenkov et al. 2012).
R‐(−)‐ and S‐(+)‐1‐Methyl‐5‐propyl‐5‐[3‐(3‐trifluoromethyl‐3H‐diazirin‐3‐yl)‐phenyl]‐pyrimidine‐2,4,6(1H,3H,5H)‐trione. Preparative separation of mTFD‐MPPB enantiomers was performed by chiral chromatography on a Chiralpak IC column (250 × 21 mm, 20 nm particles), and the mobile phase in both cases was 2% ethanol in n‐hexane. Two well‐separated peaks were observed with retention times of 10.0 min for R‐mTFD‐MPPB and 11.8 min for S‐mTFD‐MPPB.
Drug solutions
Aliquots of a 1 m stock of GABA (Sigma‐Aldrich, St. Louis, MO, USA) were prepared in bath solution (see below) and kept at –20°C before dilution to working conditions on the day of experiments. Enantiomers of mTFD‐MPPB were stored as a 100 mm stock in methanol and stored at −80°C. Before each experiment, solutions were made with an appropriate volume of methanol, which was evaporated in a glass vial before the solid barbiturate was dissolved in bath solution by vortexing and sonicating. The saturated concentration of mTFD‐MPPB enantiomers in the bath solution was 46 μm. To eliminate currents mediated via GABAA receptors composed of α1β3 subunits, ZnCl2 (10 μm) was always included in the bath as low concentrations of ZnCl2 (≤30 μM) does not influence the properties α1β2γ2L receptors (Barberis et al. 2002). For in vivo experiments, mTFD‐MPPB enantiomers were freshly dissolved in 100% DMSO (Sigma). The convulsant agent, pentylenetetrazol (PTZ; Sigma) was dissolved in saline immediately before use.
Behavioural studies in animals
Mice
Male NIH Swiss mice (22–30 g) were housed four per cage and kept in a vivarium under controlled environmental conditions (temperature, 22–26°C; 40–50% humidity) with an artificial 12 h light/dark cycle and free access to food and water. Animals were allowed to acclimatize to the vivarium for ≥5 days. Experiments were performed during the light phase of the light/dark cycle after a minimum 30 min period of acclimatization to the experimental room.
The motor impairment test was evaluated by using a modification of the horizontal screen test as previously described (Kokate et al. 1994). Mice were placed on a horizontally orientated grid (consisting of parallel 1.5 mm diameter rods situated 1 cm apart), and the grid was inverted. Animals that fell from the grid within 10 s were scored as impaired.
PTZ seizures in mice
Mice received an intraperitoneal (i.p.) injection of R‐ or S‐mTFD‐MPPB and 5 min later PTZ was administered i.p. at a dose of 80 mg kg−1, sufficient to cause seizures in> 97% of untreated mice. Animals were observed for a period of 30 min following injection. The time to onset of tonic extension was recorded; this endpoint is highly sensitive to GABAAR‐positive modulators (Dhir & Rogawski, 2012). To assess the ability of S‐mTFD‐MPPB to reduce the latency to seizures, PTZ was administered by the s.c. route (15 min after i.p. S‐mTFD‐MPPB) so that seizure onset was more prolonged, which improves the ability to detect a drug effect.
PTZ intravenous seizure threshold test
The thresholds for various seizure signs in response to i.v. PTZ were determined as described previously (Dhir et al. 2011). R‐mTFD‐MPPB was administered i.p. at a dose of 10 mg kg−1 and 5 min later an infusion of PTZ solution (10 mg ml−1) was begun at a constant rate of 0.5 ml min−1 via a 27‐gauge, 0.75‐inch ‘butterfly’ needle inserted into the lateral tail vein. Latencies were measured from the start of the PTZ infusion to the onset of (1) first myoclonic jerk, (2) generalized clonus with loss of righting reflex (LoRR) and (3) tonic extension. The infusion was stopped at the onset of tonic extension. The threshold value (mg kg−1) was determined according to the following formula: (infusion duration [s] × infusion rate [ml min−1] × PTZ concentration [10 mg ml−1] × 1000)/(60 [s] × weight of mouse [g]).
Characterization of convulsant activity
S‐mTFD‐MPPB was administered i.p. and the mice were monitored for seizure activity for 1 h. During this period, the occurrence and time of onset of myoclonic jerks, clonus and tonic extension, and the incidence of lethality was recorded. The CD50 value is the dose estimated to produce clonic seizures in 50% of animals. The duration of seizure activity was determined for animals exhibiting seizure activity during the initial 1 h observation period.
Loss of righting reflex assay in Xenopus tadpoles
General anaesthetic potency was assessed in pre‐limb‐bud stage (1–2 cm in length) Xenopus laevis tadpoles (Xenopus One, Dextor, MI, USA) according to an animal protocol preapproved by the MGH Subcommittee on Research Animal Care, following previously published protocols (Ge et al. 2014). Groups of five tadpoles in 100 ml beakers were exposed to the test compound in 2.5 mmTris‐HCl at pH 7.4, and assayed for LoRR every 5 min until a stable response was achieved (≤30 min). All animals were placed in a recovery beaker overnight. Each animal was assigned a score of either 0 (awake) or 1 (lost righting reflex), and the individual points were plotted against the barbiturate concentration (Waud, 1972). Only animals that fully recovered in fresh water overnight were included in the analysis. There was one death in the presence of each enantiomer but none at the highest concentration, so this was probably unrelated to the agent.
Cell culture and electrophysiology
Previously established tetracycline inducible HEK293 cells expressing human (N)‐FLAG α1β3γ2L–(GGS)3GK‐1D4 GABAA receptors were used in this study (Dostalovaet al. 2014). Cells were seeded on a glass coverslip and expression was induced with tetracycline (2 μg ml−1) for 5–26 h depending upon the required level of current expression. GABAAR‐mediated chloride currents from HEK293 cells were recorded using whole‐cell or outside‐out configuration of patch‐clamp electrophysiology. All experiments were performed at room temperature (20–22°C). The recording chamber was continuously perfused with the bath solution (mm): 145 NaCl, 5 KCl, 10 Hepes, 2 CaCl2, 1 MgCl2 and 10 glucose, pH 7.4 (pH adjusted with N‐methylglucosamine). The pipette solution for whole‐cell recordings contained (in mm) 140 KCl, 10 Hepes, 1 EGTA and 2 MgCl2 at pH 7.3 (pH adjusted with KOH). Mg‐ATP at 2 mm was added to the pipette solution during outside‐out patch recordings to improve patch stability. Open pipette resistances ranged from 1.6 to 3 MΩ. Cells were voltage clamped at –50 mV using the patch clamp amplifier (Axopatch 200A, Molecular Devices Corp., Sunnyvale, CA, USA). For whole‐cell recordings only, series resistance ranged from 1 to 5 MΩ and cell capacitances from 4 to 11 pF. The membrane capacitance and series resistance were compensated for electronically by> 85% with a lag of 10 μs. GABAARs were activated with GABA and/or barbiturate delivered via a quad‐channel superfusion pipette coupled to a piezoelectric element that switched the superfusion solution in< 1 ms (Forman, 1999). Cells were washed with bath solution alone for at least 3 min between each pulse of agonist application to allow the receptors to recover from desensitization. The stability of the recording was ascertained by application of a normalizing GABA (10 μm or 10 mm depending on the experiment) pulse before and after the experimental pulse. The only recordings included in the analysis were those where the peak amplitudes of the pre‐ and post‐normalizing pulses differed by ±5%.
Electrophysiology data acquisition and analysis
Electrophysiology data were acquired using Clampex version 8.1 (Molecular Devices), digitized at 10 kHz and filtered at 5 kHz. Current traces were analysed and curve fitting was performed using Clampfit version 9 (Molecular Devices).
Concentration–response curves were fitted to a Hill equation in the following form:
| (1) |
where I norm is the normalized peak current in the presence of the ligand and the EC50 is the barbiturate concentration that gives a response halfway to the maximum. In the case of inhibition EC50 was replaced by IC50.
Rates of mTFD‐MPPB action on GABAARs
Kinetic phases of current traces were fitted by non‐linear least squares to multi‐exponential equations using the F‐test to determine the number of terms. For bimolecular inhibition, the reciprocal of the τ values obtained from fitting the exponential functions above were plotted against barbiturate concentration, fitted by linear regression to the on‐ and off‐rate (Dillon et al. 1995). Further details are provided in the Results.
Statistical analysis of electrophysiology data
To allow for visual comparison between different cells with dissimilar levels of receptor expression, using Origin 6 software (Originlab, Northampton, MA, USA) current traces were normalized to the value of peak current amplitudes obtained with 10 mm GABA for each cell. GraphPad Prism version 5 software (Graphpad Software, San Diego, CA, USA) was used for all statistical analysis: curve fitting of dose– and concentration–response relationships; determination of 95% confidence intervals (CI); unpaired Student's t tests with Welchs's correction; and one‐way ANOVA with Tukey's multiple comparisons test. All data are reported as mean ± SD, except for CI, which is reported as a range.
Results
In mice, S‐mTFD‐MPPB is a convulsant whereas R‐mTFD‐MPPB is an anticonvulsant
R‐mTFD‐MPPB produced motor impairment in the inverted screen test (Fig. 2 A) at doses above 10 mg kg−1. Impaired animals appeared disorientated but could still right themselves. All animals survived the experiment, but at 80 mg kg−1 remained impaired at the end of the observation period (300 min).
Figure 2. In mice, S‐mTFD‐MPPB is convulsant whereas R‐mTFD‐MPPB is sedative and anticonvulsant .
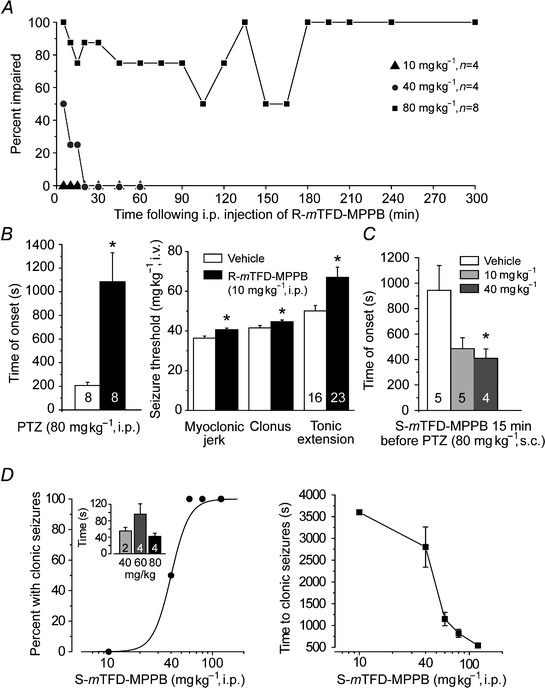
A, time course for motor impairment in the horizontal screen test following injection with various doses of R‐mTFD‐MPPB. Data points represent per cent of animals exhibiting motor impairment at the specified time; n is the number of mice. B (left), delay in the onset of PTZ‐induced tonic seizures by R‐m‐TFD‐MPPB pretreatment indicating an anticonvulsant action. Open bar represents mean ± SEM time to onset in vehicle group (100% DMSO i.p. 5 min before 80 mg kg−1 PTZi.p.); filled bar represents group pretreated with 10 mg kg−1 R‐mTFD‐MPPB. *P = 0.0032 vs. vehicle. Numbers on bars are the total number of mice in group. B (right), mean ± SEM thresholds for various seizure signs in PTZ infusion test. PTZ infusion (10 mg ml−1 at a rate of 0.5 ml min−1) was begun 5 min after i.p. injection of vehicle with or without 10 mg kg−1 R‐mTFD‐MPPB. *P < 0.05. A reduction in seizure threshold was not observed indicating a lack of proconvulsant activity. C, S‐mTFD‐MPPB reduces the time to onset of tonic seizures induced by PTZ. S‐mTFD‐MPPB was administered i.p. 15 min before 80 mg kg−1 s.c. PTZ. Note that the time to onset with s.c. PTZ is substantially longer than with i.p. PTZ (B, left), P = 0.037 by one‐way ANOVA. *P < 0.05 by Tukey HSD test. D (left),dose–response relationship for S‐mTFD‐MPPB's induction of clonic seizures. Points indicate percent of animals exhibiting clonic seizures; four animals were tested at each dose. The sigmoidal curve is a logistic fit by eye. The CD50 is 40 mg kg−1. The inset shows the mean ± SEM seizure duration at the indicated doses; the number of animals is indicated by numerals in bars. D (right), time to onset of clonic seizure for the experiment of D (left); the value for the 10 mg kg−1 group represents the duration of the observation period because no seizures were observed.
R‐mTFD‐MPPB protected against PTZ‐induced seizures at all doses. At the non‐impairing dose (10 mg kg−1), it acted as an anticonvulsant when administered i.p. 5 min prior to PTZ (80 mg kg−1 i.p.) causing an ∼5‐fold delay in the onset of tonic extension (Fig. 2 B, left). R‐mTFD‐MPPB did not exhibit proconvulsant effects in the intravenous PTZ seizure threshold test (Fig. 2 B, right). Rather, the thresholds for myoclonic jerk, clonic seizure and tonic extension were all significantly elevated, confirming the anticonvulsant action.
S‐mTFD‐MPPB, in contrast to R‐mTFD‐MPPB, had no anticonvulsant action. Instead, it was proconvulsant reducing the time to onset of PTZ–induced tonic seizures (Fig. 2 C). Moreover, S‐mTFD‐MPPB alone caused a sequence of seizure signs including immobility, myoclonic body jerks, clonic seizure of the forelimbs and/or hind limbs, and tonic seizures (forelimb tonic contraction and hind limb tonic extension). Animals exhibiting tonic seizures all died. The fraction of animals experiencing clonic seizures increased with dose, with a CD50 of ∼40 mg kg−1 (Fig. 2 D, left), and there was a dose‐dependent reduction in the latency to seizure onset (Fig. 2 D, right). However, as is typical, there was no dependence of seizure duration on dose inasmuch as once triggered seizures tended to have a stereotypical duration (Fig. 2 D, inset). Thus, in mice, S‐mTFD‐MPPB is a convulsant as is S‐MPPB in rats (Ticku et al. 1985).
Anaesthetic potency
To estimate physiologically relevant concentrations at equilibrium, the potencies of the mTFD‐MPPB enantiomers were determined in Xenopus tadpoles. For each enantiomer, 15 animals were used at each concentration. For R‐mTFD‐MPPB, LoRR was examined at four different concentrations between 3 and 60 μm, yielding an EC50 of 27 ± 8 μm (59 animals, data not shown). For S‐mTFD‐MPPB, LoRR was examined at five different concentrations between 10 and 60 μm, yielding an EC50 of 42 ± 2 μm (75 animals, data not shown). LoRR was reversible, as the animals recovered in fresh solution overnight. Unlike R‐mTFD‐MPPB, S‐mTFD‐MPPB caused excitation and sensitivity to touch in addition to LoRR. This behavioural difference between tadpoles and mice might be due either to the tadpole assay failing to distinguish between anaesthesia and seizure activity, or to the difficulty in achieving high plasma concentrations in mammals, as is seen with long chain alcohols (Dildy‐Mayfield et al. 1996).
Enhancing actions of mTFD‐MPPB are enantioselective, but inhibitory actions are not
To reveal actions on open channels, we used a notch protocol (Fig. 3) adding mTFD‐MPPB enantiomers after receptors were activated with GABA. In this protocol, GABA was perfused for 8.8 s during which 46 μm mTFD‐MPPB (the highest concentration attainable in this buffer) was co‐applied after 1 s for a period of 4 s. To best reveal enhancing actions, we used 10 μm GABA, which opens only ∼10% of available channels. Under these conditions, the enantiomers had opposite actions on open channels; S‐mTFD‐MPPB reversibly inhibited currents, whereas R‐mTFD‐MPPB strongly and reversibly potentiated them (Fig. 3 A and B).
Figure 3. S‐mTFD‐MPPB (red) only inhibited whereas R‐mTFD‐MPPB (blue) enhanced and inhibited GABAAR‐mediated currents .
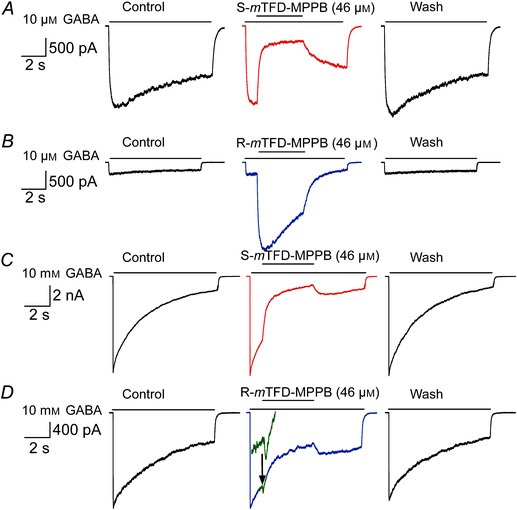
Representative notch current traces in HEK293T cells expressing GABAARs (α1β3γ2L) showing the effects of S‐mTFD‐MPPB and R‐mTFD‐MPPB on currents elicited by 10 μm GABA (A, B) and 10 mm GABA (C, D). Currents in this notch protocol were obtained using a four‐channel superfusion pipette. In each set of three panels: control is an 8.8 spulse of GABA alone; in the central panels 46 μm R‐ or S‐mTFD‐MPPB was co‐applied for 4 s after 1 s of GABA alone, and Wash was an 8.8 s pulse of GABA after cells were held in the bath solution for 5 min. The green inset in the middle panel of D shows on an expanded current scale the brief enhancement before the onset of inhibition.
To study inhibition, we used 10 mm GABA in the notch protocol because it opens ∼90% of available channels, minimizing enhancement (Desai et al. 2009). Under these conditions, both enantiomers rapidly and reversibly inhibited open channels (Fig. 3 C and D). However, currents in the presence of R‐ but not S‐mTFD‐MPPB exhibited a brief enhancement before onset of the slower inhibitory action (Fig. 3 D inset).
The concentration dependence of these actions was established by normalizing the current traces to the GABA‐alone peak amplitude achieved during the first second of the notch perfusion. At 10 μm GABA, inhibition (1 – (I/I max)) by S‐mTFD‐MPPB increased with concentration, reaching 67 ± 3.3% (n = 6) at 46 μm (Fig. 4 C). We estimated an IC50 value of 14 ± 1.7 μm by constraining the slope to be –1 and assuming that 100% inhibition would have been attained if high enough concentrations could have been achieved (Fig. 4 C). The enhancing action of R‐mTFD‐MPPB increased with concentration, reaching 425 ± 200% at 46 μm (Fig. 4 A). The effect did not saturate, but we estimated an EC50 of ∼65 μm by assuming a maximum enhancement of 10‐fold when all the channels would be open (Fig. 4 C).
Figure 4. Concentration dependence of actions of the mTFD‐MPPB enantiomers on GABA‐evoked currents .
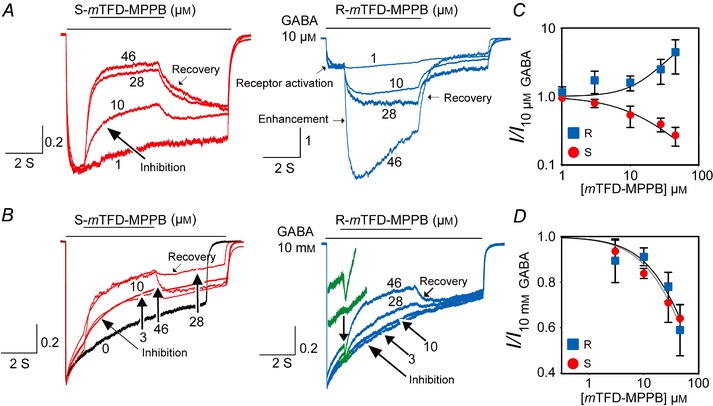
The notch protocol was the same as that in Fig. 3. Current traces were normalized to peak current amplitudes elicited by 10 μm (A) or 10 mm (B) GABA to allow for visual comparison between different cells. The section of the current trace shown in green (right panel of B), where a brief enhancement occurs with R‐mTFD‐MPPB, is enlarged in the inset. The concentration dependence of the actions of R‐ and S‐mTFD‐MPPB on normalized current amplitudes elicited by 10 μm GABA or 10 mm GABA are shown in C and D, respectively. Data in those panels were fit to the Hill equation by assuming that maximum inhibition or enhancement is attainable. The IC50 values are given in the text.
With 10 mm GABA (Fig. 3 C and D), the inhibitory phases of notch currents were fit to a double exponential equation because it has contributions from both inhibition and desensitization. Desensitization was slower than inhibition and was unchanged from that with GABA alone (0.54 ± 0.26 s–1, n = 25). The amplitude of the faster inhibitory phase was similar for both enantiomers at all concentrations (Fig. 4 D). Maximum inhibition at 46 μm was 24 ± 17 vs. 36 ± 6% (P = 0.07) for R‐ and S‐mTFD‐MPPB, respectively. Because of the small degree of inhibition (<40%) and the long extrapolation required, both data sets were fitted simultaneously using the assumptions above to give an IC50 of 73 ± 5.4 μm
GABA concentration–response curves are shifted in opposite directions by R‐ and S‐mTFD‐MPPB
To further characterize the contrasting actions of the mTFD‐MPPB enantiomers, we determined their action on GABA concentration–response curves in a protocol where GABA and mTFD‐MPPB (28 μm) enantiomers were applied simultaneously for 8 s (Fig. 5 A). The peak current amplitudes at each GABA concentration were normalized to their respective 10 mm GABA ± drug currents and fitted by non‐linear least squares (Fig. 5 B). The slopes of the curves did not differ and the results are given in Table 1. The control GABA EC50 of 28 μm is consistent with previous reports for α1β3γ2L GABAARs (Nagaya & Macdonald, 2001). The GABA concentration–response curve was shifted ∼3‐fold to the left by R‐mTFD‐MPPB and ∼2‐fold in the opposite direction by S‐mTFD‐MPPB (Table 1). The GABA concentration–response curves for the amplitude of desensitization gave similar EC50 values (Fig. 5 C), indicating that desensitization proceeds from activated states.
Figure 5. The two enantiomers shift GABA's EC50 in opposite directions .
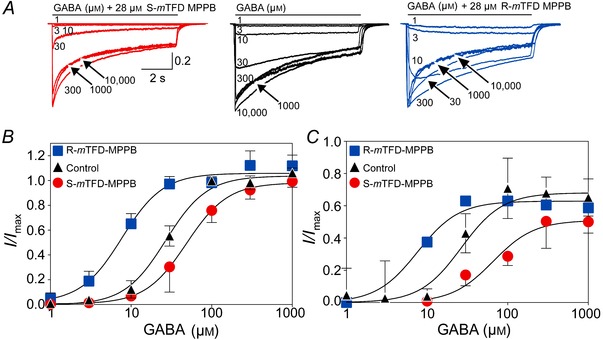
A, representative normalized current traces elicited by an 8 s pulse of varying concentrations of GABA (as indicated) with or without 28 μm of either R‐ or S‐mTFD‐MPPB. B and C, normalized peak current amplitudes (B) and desensitization amplitudes (C) expressed as a function of GABA concentration fitted to the Hill equation (parameters are given in Table1). Data are shown as mean ± SD with lower (R‐mTFD‐MPPB) or upper (S‐mTFD‐MPPB) bars omitted for clarity.
Table 1.
Parameters of the GABA concentration–response curves
| EC50 (μm) (Activation) | Hill coefficient (Activation) | EC50 (μm) (Desensitization) | |
|---|---|---|---|
| GABA | 28 (25–31) | 2.3 (1.8–2.8) | 26.6 (19–36) |
| R‐mTFD‐MPPB | 8.5 (6–10) | 1.8 (1.3–2.3) | 7.6 (5–11) |
| S‐mTFD‐MPPB | 49 (40–60) | 1.6 (1.3–1.9) | 66 (38–112) |
n = 3 – 7 for each concentration tested. 95% CIs are reported in parentheses. The desensitization data were fit with the slope constrained to 1.7.
R‐ but not S‐mTFD‐MPPB activates currents in the absence of GABA
Anaesthetics that enhance GABA's action can often activate GABAARs directly (Akaike et al. 1987; Cottrell et al. 1987; Hales & Lambert, 1991; Hara et al. 1993; Franks & Lieb, 1994; Desai et al. 2009). Consistent with this, R‐mTFD‐MPPB activated currents (Fig. 6 A) whereas S‐mTFD‐MPPB did not even at 46 μm (data not shown). R‐mTFD‐MPPB activated currents with a low efficacy of ∼1% of the 10 mm GABA current. This compares to pentobarbital, which activated up to 20% of channels and also inhibited them at higher concentrations (Rho et al. 1996; Thompson et al. 1996; Wooltorton et al. 1997; Akk & Steinbach, 2000; Akk et al. 2004; Feng et al. 2004). This suggests that R‐mTFD‐MPPB's agonist efficacy is also limited by inhibition. However, when the perfusion of R‐mTFD‐MPPB ceased, we did not see the large rebound or surge currents observed by the above workers with pentobarbital. Pentobarbital's affinity for the inhibitory site is some 100‐fold lower than R‐mTFD‐MPPB's, so it probably dissociates much faster, revealing the surge current.
Figure 6. R‐mTFD‐MPPB directly activates GABAAR‐mediated currents .
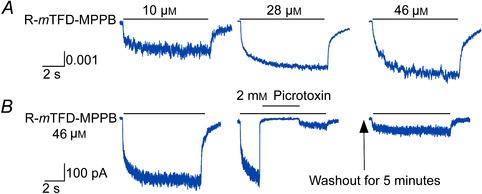
A, representative normalized traces of currents elicited by an 8 s pulse of R‐mTFD‐MPPB at the indicated concentration in HEK293 cells expressing GABAARs (α1β3γ2L). Current traces and amplitudes are normalized to the peak amplitude value of the cell's response to 10 mm GABA. The average rates of direct activation were 1.0 ± 0.6 and 22 ± 17 s−1 (mean ± SD, n = 19) and of recovery were 9.5 ± 4.1 and 0.52 ± 0.26 s–1 (n = 14). B, currents activated by 46 μm R‐mTFD‐MPPB are inhibited by 2 mm picrotoxin. Left panel, control; middle panel, a notch experiment where R‐mTFD‐MPPB directly activated currents for 8.8 s and 2 mm picrotoxin was co‐applied for 4 s after 2 s of R‐mTFD‐MPPB application alone. A small surge current was observed upon removal of picrotoxin, but currents did not fully recover after 5 min washing with bath solution alone (right panel).
Direct activation occurred in two phases of comparable amplitudes with rates that did not vary with R‐mTFD‐MPPB concentration, suggesting that the rate‐limiting processes involve a conformation change. Because of the low efficacy of R‐mTFD‐MPPB and the simultaneous presence of activation and inhibition, we did not investigate the kinetics of this action further.
R‐mTFD‐MPPB‐activated currents are mediated by GABAARs because they were completely blocked by 2 mm picrotoxin (n = 4; Fig. 6 B). A small surge current was observed upon removal of picrotoxin, but currents did not recover to their original peak values even after wash with bath solution alone for 5 min (Fig. 6 B, right panel). Although picrotoxin block recovers rapidly when GABA activates, similar incomplete recovery from picrotoxin block has been observed for currents directly activated by pentobarbital (Rho et al. 1996).
Mechanism of inhibition of GABAARs
In ligand‐gated ion channels, the inhibitor often displays higher affinity for the open than for the closed state, and bimolecular open channel inhibition is observed when the channel opens (Neher & Steinbach, 1978). For barbiturate inhibition of GABA currents, open channel inhibition models similar to that below (Scheme 1) have been proposed, where G is GABA, C, O and Ø are GABAARs that are closed, open and inhibited, respectively, S‐B is S‐mTFD‐MPPB and i represents the inhibitory sites. The annotations above and below the equilibrium arrows are rate constants. Binding of a single inhibitor molecule to either one of two inhibitory sites is sufficient for inhibition. (MacDonald et al. 1989; Rho et al. 1996; Wooltorton et al. 1997; Akk & Steinbach, 2000; Burkat et al. 2001; Steinbach & Akk, 2001; Krampfl et al. 2002; Akk et al. 2004; Gingrich et al. 2009; Forman & Miller, 2011).
Scheme 1
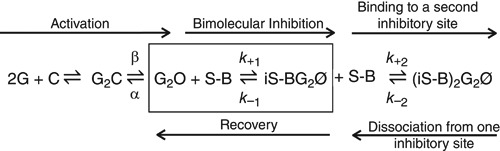
In the notch protocol (Figs 3 and 4), S‐mTFD‐MPPB is added to channels that are already open and the processes in the box of Scheme 1 govern the kinetics of inhibition, where k +1 is S‐mTFD‐MPPB's binding rate constant and k –1 is its dissociation rate constant. These two parameters may be obtained from the concentrationdependence of the measured initial rate, ki (the reciprocal of τfast obtained from fitting the current traces in Fig. 4), from the following equation:
| (2) |
where the square brackets denote concentration.
After correction for desensitization at 10 mm GABA, the initial rate of inhibition depended linearly on concentration under all conditions for which inhibition could be resolved (Fig. 7 A–C; Table 2). For example, with S‐mTFD‐MPPB at 10 μm GABA (Fig. 7 A), k +1 was 1.0 ± 0.1 × 105 m –1 s–1 and k –1 was 2.0 ± 0.3 s–1, giving a K D (k –1/k +1) of 20 ± 4 μm (Table 2). S‐mTFD‐MPPB's observed IC50 and K D were lower at low than at high GABA concentration because k +1 was 2‐fold slower at high GABA whereas the recovery rate did not depend on GABA concentration (Table 2). Our values of k +1 may be compared to that reported for pentobarbital (3.2 × 105 m –1 s–1) (Steinbach & Akk, 2001).
Figure 7. Concentration dependence of the kinetics of mTFD‐MPPB actions on currents evoked by 10 μm or 10 mm GABA .
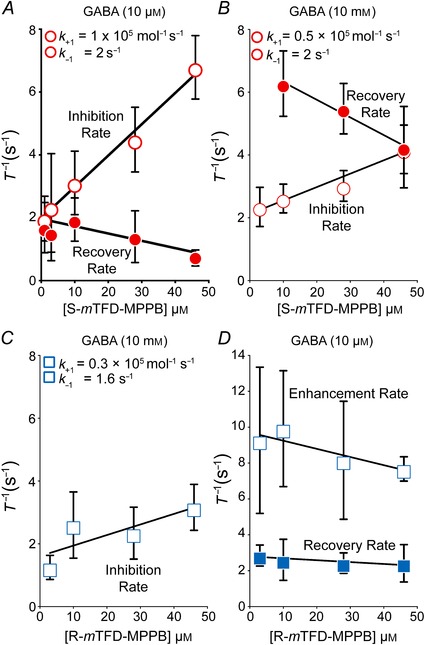
The time constants (τ = (1/rate)) for the enhancing, inhibiting and recovery phases (indicated by arrows on the traces in Fig. 4) were determined from non‐linear least squares fits to exponential equations. The rates so derived were plotted as mean ± SD against S‐ (A and B) or R‐mTFD‐MPPB (C and D) concentration. For inhibition, fitting by linear regression to eqn (2) yielded the observed inhibition (k +1) and recovery (k –1) rates (eqn 2 and Table 2). The slope obtained for R‐mTFD‐MPPB's enhancement rate in D was not significantly different from zero (P = 0.77).
Table 2.
The inhibition site is not enantioselective
| X‐mTFD‐MPPB | k –1 (s–1) | k +1 (×105 m −1 s–1) | K D (μm) | Recovery (s−1) |
|---|---|---|---|---|
| S,10 μmGABA | 2.0 ± 0.32∗ | 1.0 ± 0.12∗∗ | 20 ± 4.0 | 2 ± 0.2 |
| S,10 mmGABA | 2.1 ± 0.24 | 0.43 ± 0.08 | 52 ± 11 | 6.9 ± 0.5 |
| R,10 mmGABA | 1.6 ± 0.38 | 0.34 ± 0.1 | 48 ± 20 | — |
Data are shown as mean ± SD.
*k –1 values do not differ.
**k +1 at 10 μm and 10 mm are different (P < 0.001).
With 10 mm GABA (Fig. 7 B), the inhibitor binding (k +1) and dissociation (k –1) rates of R‐ and S‐mTFD‐MPPB were identical (Table 2). Two conclusions can be drawn from this observation: (1) the enantiomers’ affinities for their inhibitory binding sites (1/ K D) are indistinguishable, and therefore the inhibitory binding sites are not enantioselective; and (2) the enhancing and inhibiting sites do not interact strongly because simultaneous occupation of R‐mTFD‐MPPB's enhancing sites does not change the binding parameters at its inhibitory sites. This suggests that the conformation change that is associated with R‐mTFD‐MPPB's interaction with the open state's enhancing site(s) is not propagated to the inhibitory sites.
Recovery from inhibition
Scheme 1 predicts that the recovery rate should depend on S‐mTFD‐MPPB's concentration because at higher concentrations two molecules of the drug must dissociate before channel inhibition is relieved (Akk & Steinbach, 2000). Consistent with this prediction, at 10 μm GABA when we removed S‐mTFD‐MPPB in the presence of GABA, we observed (Fig. 4) a 2‐fold decrease in recovery rate with increasing S‐mTFD‐MPPB concentration (slope is different from zero, P = 0.0074; Fig. 7 A). Furthermore, the data extrapolated to a recovery rate of 2.0 ± 0.20 s–1 at zero concentration are in good agreement with S‐mTFD‐MPPB's dissociation rate (k –1) obtained independently from the initial rate of inhibition (eqn 2).
However, our observations suggest that Scheme 1 is incomplete because at 10 mm GABA the recovery rate from inhibition by S‐mTFD‐MPPB was unexpectedly 3–4 times faster than that at 10 μm GABA (compare Fig. 7 A and B), even though recovery had a similar concentration dependence. The recovery rate of R‐mTFD‐MPPB at 10 mm GABA was also faster with a mean of 9.9 ± 6.4 s–1 at 28 and 46 μm (data not shown). Overall, we conclude that: (1) during the 4 s of exposure to 10 mm GABA + mTFD‐MPPB, a conformation change must occur to a non‐conducting state from which mTFD‐MPPB can dissociate more rapidly (i.e. the two mTFD‐MPPB binding sites have lower affinity in this state); and (2) dissociation of the inhibitor from this state leads back to an open state (Akk et al. 2004).
S‐mTFD‐MPPB slows channel opening
The S‐mTFD‐MPPB‐induced rightward shift of the GABA concentration–response curve (Fig. 5 A) measures the overall equilibrium 2G + C ⇌ G2O. A rightward shift could result from decreasing any of the forward rates or increasing any of the reverse rates, or a mixture of both, in any of the intervening kinetic steps. Gating kinetics are fast, so we employed outside‐out patches with very rapid solution exchange (∼0.4 –1.2 ms (Forman, 1999) compared to 2–5 ms for whole‐cell experiments (Udgaonkar & Hess, 1987)). We used a reverse notch protocol, in which mTFD‐MPPB is perfused continuously and gating is initiated by a brief pulse of GABA that was long enough to obtain a steady state current but short enough to avoid desensitization. These conditions were fulfilled by a 400 ms pulse of 10 μm GABA or a 5 ms pulse of 10 mm GABA (Fig. 8 A and B).
Figure 8. R‐mTFD‐MPPB slows channel deactivation but S‐mTFD‐MPPB has no effect .
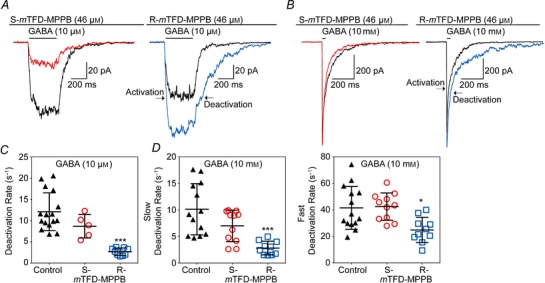
A and B, representative currents elicited in a reverse notch protocol by (A) a 400 ms pulse of 10 μm GABA or (B) a 5 ms pulse of 10 mm GABA (black traces), in the presence or the absence of 46 μm S‐mTFD‐MPPB (red) or R‐mTFD‐MPPB (blue). Outside‐out patches were held in the presence of 46 μm mTFD‐MPPB enantiomers for 30 s prior to the co‐application of GABA. The currents were also allowed to deactivate in the continuing presence of barbiturate. Deactivation rates after GABA was withdrawn were obtained from either single (10 μm) or double exponential (10 mm) fits and are shown as scatter plots (C and D). Horizontal bars indicate mean ± SD. Control values were consistent with those of Scheller & Forman (2002). Significance level compared to control were determined by one‐way ANOVA with Tukey multiple comparison test and are denoted as ***P ≤ 0.0001 and *P < 0.05. The deactivation rate at 10 μm GABA fell 4.4 (± 2.1)‐fold from 12 ± 4.5 to 2.7 ± 0.8 s−1 in the presence of 46 μm R‐mTFD‐MPPB. At 10 mm GABA, deactivation was bi‐exponential, and the faster rate fell 1.7 (± 0.9)‐fold from 42 ± 16 to 25 ± 9.5 s−1 and the slower rate fell 3.6 (± 2.4)‐fold from10 ± 4.8 to 2.8 ± 1.3 s–1 in the presence of R‐mTFD‐MPPB. Activation rates are given in the text.
The activation rate, β, is best determined at 10 mm GABA because under these conditions binding and re‐binding of GABA is extremely fast (30–100 ms–1 based on various literature values (Jones & Westbrook, 1995; Scheller & Forman, 2002)), so the process G2C ⇌ G2O will be rate limiting (β is ∼ 2 ms–1). The measured activation rate is k act = β + α, which reduces to k act ≈ β when β >> α. Under these conditions, current activation was mono‐exponential and k act was 2400 ± 600 s–1 (n = 16), in agreement with literature values of β (for cultured hippocampal neurons, β = 2500 s–1 (Jones & Westbrook, 1995), and in HEK cells expressing α1β2γ2L GABAARs, β = 3400 ± 1500 s–1 (Scheller & Forman, 2002) or 1900 s–1 (Steinbach & Akk, 2001)).
S‐mTFD‐MPPB reduced the channel opening rate, β (see Fig. 9 A, where each curve is normalized to its own maximum to aid comparison). In four paired experiments using 10 mm GABA, the reduction in β was ∼2‐fold from 2000 ± 400 to 1100 ± 200 s–1 (P = 0.0001). This is consistent with the ∼2‐fold increase in EC50 of the GABA concentration–response curve (Table 1; Fig. 5 B), but we cannot rule out action on the GABA binding steps.
Figure 9. S‐mTFD‐MPPB decreases the activation rate of GABAARs whereas R‐mTFD‐MPPB does not .
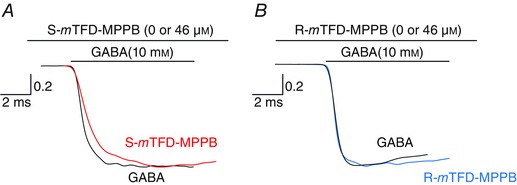
Rising phases from Fig. 8 B are shown on an expanded scale and normalized to their own maximum to aid comparison of the slopes. The first 30 ms of 10 mm GABA traces are shown with the same colour scheme as Fig. 8. All curves could be fitted to a single exponential and the mean rates ± SD for 10 mm GABA are given in the text.
When GABA was removed at the end of these pulses, deactivation occurred in one (10 μm GABA, Fig. 8 C) or two (10 mm GABA, Fig. 8 D) phases. These rates of deactivation were not changed by the presence of S‐mTFD‐MPPB, whereas the decrease in β would be expected to enhance the rate of deactivation because of a reduced return flux, G2C ⇀ G2O (Scheme 1). Clearly other processes are contributing to G2O to attenuate deactivation, suggesting that under conditions of S‐mTFD‐MPPB preincuabtion Scheme 1 is inadequate.
S‐mTFD‐MPPB is a resting state inhibitor
During short GABA pulses (Fig. 8), S‐mTFD‐MPPB (46 μm) decreased (P = 0.02, Student's t test) the integrated current relative to control at both 10 μm and 10 mm GABA to 0.29 ± 0.10 (n = 6) and 0.54 ± 0.21 (n = 8) of control, respectively. This degree of inhibition is not accounted for by open state inhibition, which develops at only 3–4 s–1 at these concentrations (Fig. 7). To test for closed or resting state inhibition, we used the protocol above except that receptors were preincubated with 46 μm S‐mTFD‐MPPB for either 0 or 30 s followed by a test pulse of 10 mm GABA. When S‐mTFD‐MPPB (46 μm) was co‐applied with GABA, the rate of activation was unchanged from control (ratio of rates (control/drug) = 1.5 ± 0.5). Nor were peak currents inhibited ((1 –(I/I max)) was 0 ± 10%, n = 4 (Fig. 10 A)). However, after a 30 s preincubation with S‐mTFD‐MPPB initial currents were significantly inhibited (1 –(I/I max)) = 34 ± 18% (n = 7) P = 0.008; Fig. 10 B). This resting state inhibition increased with S‐mTFD‐MPPB concentration (3–46 μm). Making the same assumptions as before, an IC50 of 48 ± 8 μm was obtained, comparable to that in Fig. 4 for open channel inhibition. Inhibition by R‐mTFD‐MPPB was only increased modestly by preincubation (9 ± 2%, n = 5 (Fig. 10 C)), but the degree of enhancement of peak currents elicited by 10 μm GABA after preincubation with R‐mTFD‐MPPB (Fig. 8A) was less than that when the drug was added in the presence of GABA (Fig. 4A & C; 1.2 ± 0.3 vs. 4 ± 2; P = 0.01), suggesting that enhancement is attenuated by resting state inhibition after preincubation with R‐mTFD‐MPPB.
Figure 10. S‐mTFD‐MPPB inhibits resting‐state receptors .
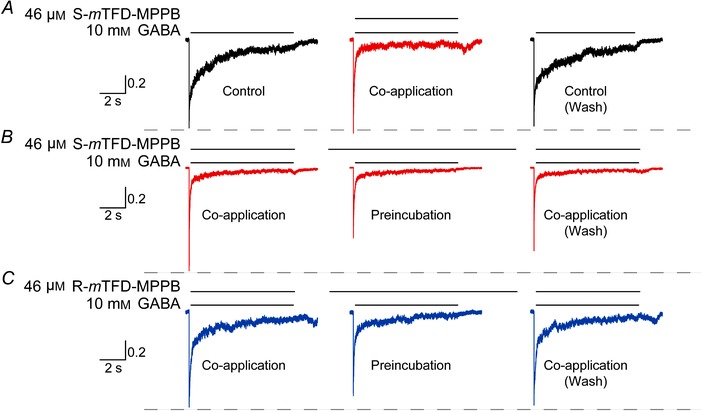
Representative currents elicited by an 8 s pulse of 10 mm GABA in outside‐out patches. Peaks are normalized to the first trace on the left. A, simultaneous addition of GABA and 46 μm S‐mTFD‐MPPB elicits the same peak amplitude as GABA alone. B and C, a 30 s pre‐incubation with 46 μm S‐ (B) or R‐mTFD‐MPPB (C) tests for resting state inhibition. The dashed line is drawn to aid visual comparison between peak current amplitudes of control and barbiturate‐treated traces.
Dependence of inhibition on ionization state of S‐mTFD‐MPPB
Akk & Steinbach (2000) noted that the inhibitory potency of pentobarbital was independent of pH. This is unexpected because the anaesthetic action of barbiturates is commonly considered to be mediated by the lipid soluble uncharged form of these agents (Narahashi et al. 1971). The pK of mTFD‐MPPB is estimated to be 7.65 (Savechenkov et al. 2012), so the concentration of uncharged mTFD‐MPPB should decrease ∼15‐fold between pH 7.4 and 9.0 from 29 to 2 μm, respectively. Using the protocol in Fig. 3, we found that 10 mm GABA currents were inhibited nearly equally by 46 μm S‐mTFD‐MPPB at pH 7 and 9 (pH, (I/I max): 7.4, (0.76 ± 0.04); 9.0, (0.65 ± 0.06; P = 0.012)). We conclude that inhibition is mediated by both uncharged and charged forms of S‐mTFD‐MPPB. Taking advantage of the higher solubility at pH 9, we found that at 100 μm S‐mTFD‐MPPB inhibition (1 – (I/I max)) was 80 ± 5%, consistent with the assumption made in obtaining the IC50 values in Fig. 4 that complete inhibition is possible. Adding these values to the previous analysis yields an IC50 of 47 ± 3 μm and a Hill coefficient 1.4 ± 0.2, in good agreement with values obtained from the initial rate of inhibition analysis (Table 2).
Mechanisms of enhancement and gating of R‐mTFD‐MPPB
There is a clear consensus that anaesthetics stabilize the open state relative to the resting state and consequently increase the opening probability at a given agonist concentration. The simplest scheme can be written (Akk & Steinbach, 2000) as in Scheme 2 where G is GABA, C is the closed, resting state, O is the open state and O’ is the enhanced open state of the GABAAR, R‐B is R‐mTFD‐MPPB and e represents the binding site(s) responsible for enhancement.
Scheme 2
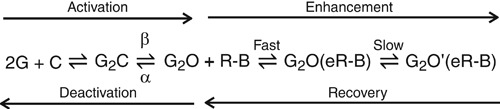
In outside‐out patches briefly exposed to GABA, the normalized integrated current areas increased at both 10 μm and 10 mm GABA by 2.3 ± 0.24‐ (n = 4) and 2.0 ± 0.56‐fold (n = 5), respectively (Fig. 8 A andB). These conditions allowed us to study enhancement because inhibition has no time to develop. R‐mTFD‐MPPB did not change the activation rate, β, in five paired patch experiments (control, 2500 ± 500 s–1; R‐mTFD‐MPPB, 2100 ± 500 s–1; n = 5; Fig. 9 A). This is consistent with the single channel studies of Steinbach & Akk (2001), who found no evidence for an enhanced rate of activation in the presence of pentobarbital.
On the other hand, the deactivation rate was consistently slower in the presence of R‐mTFD‐MPPB. Thus, in patch experiments in the continuing presence of R‐mTFD‐MPPB, after a brief pulse of GABA (Fig. 8), open receptors return to the closed state (the process of deactivation) following one (10 μm GABA) or two (10 mm GABA) exponentials. The slower rate fell 4‐fold and the faster rate 2‐fold in the presence of R‐mTFD‐MPPB compared to control. These observations are consistent with a decrease in α and with the ∼3‐fold decrease in the EC50 of the GABA concentration–response curve (Table 1; Fig. 5 B).
At 10 μm GABA, negligible inhibition is observed with R‐mTFD‐MPPB, enabling its enhancing action to be resolved (Fig. 4 A, left panel; top right arrow in Scheme 2). Enhancement followed a single exponential time course and the rate was independent of R‐mTFD‐MPPB's concentration (8.8 ± 3.2 s–1; P = 0.24; Fig. 7 D). Thus, the rate‐limiting step for enhancement is the conformational change (G2O(eR‐B)⇌G2O’(eR‐B)) not the faster drug‐binding step (Scheme 2). Similarly, when R‐mTFD‐MPPB is removed in the continuing presence of 10 μm GABA (Fig. 4 A, right panel), the rate of recovery from enhancement (G2O’(eR‐B) ⇀ G2O + R‐B; Scheme 2) was mono‐exponential. The average recovery rate was 2.4 ± 0.8 s–1(n = 25; Fig. 7 D) and did not vary with concentration (P = 0.3). It is unclear if there is a rate‐limiting step during recovery.
Desensitization
Desensitization was studied with 8 s pulses of 10 mm GABA in outside‐out patches with very rapid solution exchange. To ensure stable conditions for measuring the desensitization rate, currents were first elicited with 10 mm GABA for 8 s until peak amplitudes stabilized after 3–5 pulses. Furthermore, only patches whose current amplitudes recovered to their original 10 mm control values after washout were used in the analysis.
After 2 min of preincubation, integrated current areas were inhibited ∼50% by both enantiomers because of inhibition and desensitization. The desensitizing phase was usually best fit with a three‐term exponential equation (F test), but in some traces the fastest component was not resolved. The control rates of the fast, medium and slow components were, respectively, 63 ± 40, 4.1 ± 2.5 and 0.50 ± 0.22 s–1, and the corresponding amplitudes were 45 ± 17, 32 ± 10 and 23 ± 7.2% of total desensitization peak amplitude.
The enantiomers did not alter the rate of fast and slow desensitization. This is consistent with Akk & Steinbach (2000) who reported that pentobarbital did not change the desensitization rates and contrary to the observations of Feng et al. (2004) who observed an increase in desensitization rates in the presence of pentobarbital. The medium desensitization rate has the same magnitude as that for inhibition, so deconvolution is more uncertain. Both enantiomers tended to increase the medium desensitization rate 2–3‐fold, but only R‐mTFD‐MPPB caused a significant change.
Discussion
Summary
The barbiturate S‐mTFD‐MPPB is a convulsant and proconvulsant in mice, and in human α1β3γ2L GABAARs it inhibits GABA‐stimulated currents. The latter action is sufficient to account for its in vivo effects. It binds to both the resting and the open states, but has lower affinity for the desensitized states. Occupation of a single site on the open state is sufficient to inhibit the current, but the existence of a second site is indicated by the recovery kinetics. Two observations suggest the pyrimidine ring does not interact with the binding pocket. First, both the uncharged and the anionic forms of S‐mTFD‐MPPB bind to the inhibitory sites. Second, both enantiomers inhibit with equal potency, which is consistent with the pyrimidine ring being responsible for chirality. In contrast to the inhibitory sites, the separate enhancing/anaesthetic sites are enantioselective; only the R‐enantiomer binds to these sites and only when they are in the open state, stabilizing that state and slowing channel closing. Overall, S‐mTFD‐MPPB is a convulsant because it has lower affinity for the enhancing/anaesthetic site and not because it has high affinity for the inhibitory/convulsant sites.
The in vivo actions
For the most part, the in vivo pharmacology of the mTFD‐MPPB enantiomers mirrors that of the MPPB enantiomers. S‐mTFD‐MPPB in mice and S‐MPPB in rats both cause convulsions by themselves and both are proconvulsant in the presence of a subthreshold challenge by the convulsant PTZ (Skerritt & Macdonald, 1984; Ticku et al. 1985). In contrast, both R‐mTFD‐MPPB and R‐MPPB protect against PTZ‐induced convulsions, and both cause motor impairment. R‐MPPB was more strongly behaviourally depressant in rats than R‐mTFD‐MPPB was in mice because it alone produced dose‐dependent LoRR (Ticku et al. 1985).
The in vitro actions
The contrasting actions of the mTFD‐MPPB enantiomers on heterologously expressed human α1β3γ2 GABAARs in vitro parallels their opposing actions in vivo. When a brief pulse of GABA was applied in the presence of either drug, a protocol that simulates synaptic action, the net current was decreased by S‐mTFD‐MPPB and increased by R‐mTFD‐MPPB at both high and low GABA concentrations (Figs 8 and 9). S‐mTFD‐MPPB appears to be devoid of any enhancing, anaesthetic‐like action. It is unnecessary to invoke other targets for the convulsant action, although we cannot rule out their existence. In this regard, we note that MPPB's actions on other targets often occur at supra‐physiological concentrations and are either not enantioselective (nicotinic acetylcholine receptor (nAChR); Holtman & Richter, 1983) or only weakly so (AMPA and NMDA receptors; Daniell, 1994; Kamiya et al. 1999).
R‐mTFD‐MPPB, like other general anaesthetics, shifts the GABA concentration–response curve to the left, whereas S‐mTFD‐MPPB shifts it to the right. This suggests that the volatile convulsants may share the same underlying mechanisms with S‐mTFD‐MPPB because Indoklon produces a rightward shift in the GABA concentration–response curves in dissociated rat nucleus tractus neurons, whereas halothane causes a leftward shift (Wakamori et al. 1991).
The mechanism of inhibition by S‐mTFD‐MPPB
The model proposed by Akk and colleagues (Akk & Steinbach, 2000; Akk et al. 2004) to account for the inhibitory action of pentobarbital is consistent with much of our data. We were able to extend their model because S‐mTFD‐MPPB was a pure inhibitor whereas pentobarbital both enhanced and inhibited, thus complicating their analysis. S‐mTFD‐MPPB interacts with the inhibitory site on GABAARs as an allosteric effector. It has approximately equal affinity for both the resting closed state (C) and the open state (O), and a lower affinity for the desensitized state (D). The overall kinetic map in Scheme 3 is sufficient to qualitatively account for most of these actions, but the energy landscape surrounding gating is undoubtedly more complex than the scheme implies.
Scheme 3
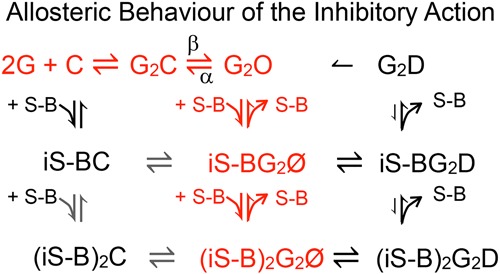
Consider first the red parts of the scheme that corresponds to the proposal of Akk & Steinbach (2000). The data show that when drug is added to open channels (Figs 3, 4 and 7), inhibition occurs by the well‐established open channel inhibition model (Neher & Steinbach, 1978). The kinetics are bimolecular with an on‐rate that increases linearly with concentration, indicating that binding to a single site at a rate of 0.4 ×105 m –1 s–1 is sufficient to inhibit the current and that the off‐rate is 2 s–1 (Fig. 7, and Scheme 3, middle column, rows 1 and 2). This rate is nearly an order of magnitude slower than that of 3.2 × 105 m −1 s−1 obtained for pentobarbital by Steinbach & Akk (2001). Drug binding is the rate‐limiting step and inhibition results either from the bound drug occluding the conduction pathway or from binding being followed by a very rapid conformation change to an inhibited state (not shown in Scheme 3). However, the kinetics of recovery determined independently after a few seconds of inhibition at low GABA are some 2‐fold slower (∼1 s–1; Fig. 7), suggesting that during this time the drug bound to a second inhibitory site and that both must unbind for the current to be restored (Scheme 3, middle column, rows 3 to 1). Although Scheme 3 depicts this binding as sequential, it is also possible that both sites are equivalent, and binding to either is sufficient for inhibition.
Scheme 4
A number of our observations led us to extend Akk and Steinbach's model as shown in black in Scheme 3. First, S‐mTFD‐MPPB has lower affinity for the desensitized state because the rate of recovery from inhibition after exposure to high GABA concentration was 3–4 times faster than that estimated from the initial rate of inhibition (eqn 2), whereas with low GABA concentration there was no such inconsistency (compare Fig. 7 A and B). The major difference between low and high GABA concentrations is the degree of desensitization, so that the states in the right column of Scheme 3 are only highly populated at high GABA concentration. If we assume that S‐mTFD‐MPPB has lower affinity for the desensitized states, it will dissociate leaving G2D, which may reopen. This hypothesis is consistent both with the conclusion of Akk & Steinbach (2000) that desensitization can proceed from the barbiturate‐blocked state and with prior suggestions that desensitized receptors may return directly to the open state (Jones & Westbrook, 1995; Haas & Macdonald, 1999).
The second extension to Akk & Steinbach's model is that S‐mTFD‐MPPB binds to receptors in the absence of GABA (resting state inhibition; left column in Scheme 3). Thus, following a brief pulse of GABA applied during continuous perfusion of S‐mTFD‐MPPB, inhibition is immediately present and on too fast a time scale for open channel inhibition to have developed (Fig. 8 A and B). When drug and 10 mm GABA are co‐applied, initial currents are unchanged, but when receptors are preincubated with S‐mTFD‐MPPB the initial current is inhibited (Fig. 10 A and B). Thus, when GABA binds to a receptor whose inhibitory sites are already occupied, Scheme 3 predicts that the receptor opens to an inhibited state whereas those resting state receptors that are not bound to an inhibitor open normally. Resting state inhibition is also consistent with the observed on‐rate of open channel block at 10 μm GABA being faster than at 10 mm GABA (compare Fig. 7 A and B) because at 10 mm GABA ∼90% of channels are open. When S‐mTFD‐MPPB is added in a notch experiment to open channels, the kinetics are then dominated by open channel inhibition, whereas at 10 μm GABA, a high proportion of the channels are in the resting, closed state, so that resting and open state inhibition can occur simultaneously.
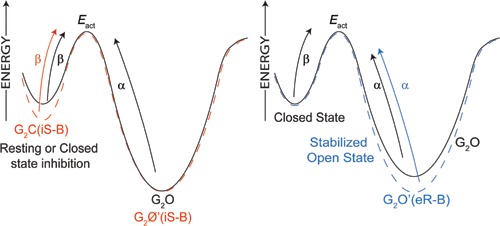
Third, when GABA is added after preincubation with S‐mTFD‐MPPB, those channels that do open do so more slowly (Fig. 9). This observation accounts for the 2‐fold shift to the right of the GABA concentration–response curve (Fig. 5) because there is a 2‐fold decrease in the opening rate, β, in the presence of S‐mTFD‐MPPB. However, Scheme 3 does not include a mechanism for this action, nor does it explain why the opening rate does not change when GABA and S‐mTFD‐MPPB are added simultaneously.
Because gating is poorly understood (see below), we first consider the above questions in a pathway‐independent manner through a free energy diagram (Scheme 4). The decrease in β after preincubation suggests that S‐mTFD‐MPPB increases the activation energy for opening. It must do so by slowly binding to and stabilizing some pre‐open state without altering the energy of the transition state because α is unaffected. In contrast, the enhancing action of R‐mTFD‐MPPB, like that of other anaesthetic barbiturates (Macdonald et al. 1989 b; Steinbach & Akk, 2001), is related to stabilization of the open state, which decreases α without altering the energy of the transition state because β is unaffected. It is likely that the resting state action of S‐mTFD‐MPPB involves the inhibitory sites, but an intriguing alternative is that the enhancing sites switch enantioselectivity in a state‐dependent manner, so that S‐mTFD‐MPPB binds to them selectively in the resting state, whereas R‐mTFD‐MPPB does so in the open state.
A more mechanistic hypothesis can be provided by analogy with the thoroughly studied nAChR resting state inhibitor 3‐(trifluoromethyl)‐3‐(m‐iodophenyl) diazirine (TID), which has been studied by rapid perfusion electrophysiology and by time‐resolved photolabelling. Like S‐mTFD‐MPPB with GABAARs, TID causes no immediate inhibition of nAChRs when added simultaneously with agonist. However, when TID was preincubated, addition of agonist was followed by an extremely rapid concentration‐dependent inhibition of open channels with a very fast on‐rate (k on) of 2 × 108 m –1 s–1, which is close to the diffusion limit (Forman, 1999).
TID's rapid inhibitory phase could be observed because the apparent dissociation constant was 150 nm. For example at 0.5 μm TID, inhibition would be well developed and the observed inhibition rate would be ∼100 s–1 (recall eqn 2). If S‐mTFD‐MPPB (K d = 50 μm) exerted this mechanism on GABAARs with the same k on, the observed on‐rate for inhibition at 50 μm might approach 10,000 s–1. This is well beyond our equipment's time resolution, and could result in an apparent decrease in the activation rate, β.
The molecular mechanism of TID's action has been elucidated by time‐resolved photolabelling studies that revealed two classes of site in the transmembrane domain. The first, activation‐independent class, was in the channel lumen where occupation occurred slowly during TID preincubation. The second, activation‐dependent class, was mediated by an intrasubunit site within the four‐helix bundle of the δ‐subunit's transmembrane domain that was occupied within milliseconds of ACh addition (Arevalo et al. 2005). Although applying this mechanism to S‐mTFD‐MPPB is speculative, the hypothesis is open to testing both by time‐resolved photolabeling with S‐mTFD‐MPPB and by developing inhibitors with higher potency.
The gating kinetics of ligand‐gated ion channels are complex and incompletely understood (Mukhtasimova et al. 2009; Lape et al. 2012; Szczot et al. 2014). In GABAARs, GABA activates three open states, but their interconnectivity is not well established. A comprehensive single channel study rejected many proposed models. A model with three GABA‐occupied pre‐open states and the long recognized three open states was adequate (Lema & Auerbach, 2006). However, this model was only consistent with the data if it occurred in three modes, each with its unique set of rate constants. This implies a very complex free energy landscape, which is not surprising considering three different subunits comprise the pentameric channel, and there is no requirement for symmetric motions (Mukhtasimova et al. 2009; Unwin & Fujiyoshi, 2012). Thus, the pathways proposed in the schemes above should be regarded as conceptual rather than providing a detailed kinetic model.
Chirality of the sites of action
The enhancing site was enantioselective because S‐mTFD‐MPPB was without action. On the other hand, the inhibitory site exhibited no enantioselectivity because both R‐ and S‐mTFD‐MPPB caused inhibition with similar IC50 values. S‐mTFD‐MPPB binds to the inhibitory site in both the uncharged and anionic form because inhibition is independent of pH. This implies that when S‐mTFD‐MPPB is transferred from the aqueous phase to the inhibitory binding site the charged moiety on the pyrimidine ring does not contribute to the binding energy and therefore must remain protruding into the aqueous phase while the hydrophobic tail groups are in the binding pocket. This is consistent with the lack of enantioselectivity at the inhibitory site because chirality resides in the asymmetry of the pyrimidine ring, which does not interact with the protein. Possible protein–aqueous interface sites include the channel lumen, where the hydrophobic tail can insert between hydrophobic helices, and the extracellular domain where anaesthetic sites on homologous channels have been found for ketamine (Mowrey et al. 2013) and bromoform (Spurny et al. 2013).
Therapeutic implications
The excitatory actions of general anaesthetics are generally considered to be an undesirable side effect. It would be an advantage in the development of new anaesthetics to be able to design agents with low affinity for the sites that inhibit GABA's inhibitory action. S‐mTFD‐MPPB appears to interact with high selectivity with such inhibitory sites, imbuing it with the potential for establishing structure–activity relationships for inhibiting sites on GABAARs in much the same way that R‐mTFD‐MPAB and azi‐etomidate have for enhancing sites (Li et al. 2006; Savechenkov et al. 2012).
Furthermore, once established, knowledge of the structure–activity relationships of inhibitory sites would provide the ability to design useful agents by rationally adjusting the balance of GABAAR excitation (EC50) and inhibition (IC50). Such agents might be sedative without being full anaesthetics (partial anaesthetics), or anticonvulsant without being sedative. An efficacious general anaesthetic would have an EC50/IC50≪1, a good sedative an EC50/IC50< 1, a non‐sedating anticonvulsant an EC50/IC50 ≤1 and an effective convulsant an EC50/IC50> 1. Such concepts have been invoked to account for phenobarbital being a less sedating anticonvulsant than pentobarbital; their EC50/IC50 ratios are 0.07 vs. 0.03, respectively (Rho et al. 1996). R‐mTFD‐MPPB has an EC50/IC50 ratio of ∼1, and we suspect that this is why at the lowest dose examined, it protected against PTZ‐induced seizures without impairing the mice (Fig. 2A and B). Thus, it is plausible that compounds with similar properties to R‐mTFD‐MPPB may have potential as anticonvulsants with minimum sedative side effects.
Additional Information
Competing interests
The authors have no conflicts of interest.
Author contributions
All experiments were performed at the MGH unless otherwise noted. R.D. collected and analysed the electrophysiological data under the supervision of S.A.F., D.E.R. and K.W.M. P.Y.S. synthesized the mTFD‐MPPB enantiomers under the supervision of K.S.B. at the University of Illinois, Chicago. D.Z. conducted the rodent experiments under the supervision of M.A.R. at the University of California, Davis. RLG conducted the amphibian experiments under the supervision of D.E.R. and K.W.M. R.D. and K.W.M. wrote the manuscript. K.W.M. initiated the project and was responsible for overall design.
Funding
Funded by grants from the National Institute of General Medical Sciences (GM 58448 to K.W.M.) and the National Institute of Neurological Disorders and Stroke (NS079202 to M.A.R.), and by the Department of Anaesthesia, Critical Care and Pain Medicine, MGH.
References
- Akaike N, Maruyama T & Tokutomi N (1987). Kinetic properties of the pentobarbitone‐gated chloride current in frog sensory neurones. J Physiol 394, 85–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akk G, Bracamontes J & Steinbach JH (2004). Activation of GABAA receptors containing the α4 subunit by GABA and pentobarbital. J Physiol 556, 387–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akk G & Steinbach JH (2000). Activation and block of recombinant GABAA receptors by pentobarbitone: a single‐channel study. Br J Pharmacol 130, 249–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allan AM & Harris RA (1986). Anesthetic and convulsant barbiturates alter gamma‐aminobutyric acid‐stimulated chloride flux across brain membranes. J Pharmacol Exp Ther 238, 763–768. [PubMed] [Google Scholar]
- Arevalo E, Chiara DC, Forman SA, Cohen JB & Miller KW (2005). Gating‐enhanced accessibility of hydrophobic sites within the transmembrane region of the nicotinic acetylcholine receptor's δ‐subunit. A time‐resolved photolabeling study. J Biol Chem 280, 13631–13640. [DOI] [PubMed] [Google Scholar]
- Barberis A, Petrini EM, Cherubini E & Mozrzymas JW (2002). Allosteric interaction of zinc with recombinant α1β2γ2 and α1β2 GABAA receptors. Neuropharmacology 43, 607–618. [DOI] [PubMed] [Google Scholar]
- Buch H, Knabe J, Buzello W & Rummel W (1970). Stereospecificity of anesthetic activity, distribution, inactivation and protein binding of the optical antipodes of two N‐methylated barbiturates. J Pharmacol Exp Ther 175, 709–716. [PubMed] [Google Scholar]
- Burkat PM, Yang J & Gingrich KJ (2001). Dominant gating governing transient GABAA receptor activity: a first latency and Po/o analysis. J Neurosci 21, 7026–7036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiara DC, Dostalova Z, Jayakar SS, Zhou X, Miller KW & Cohen JB (2012). Mapping general anesthetic binding site(s) in human α1β3 gamma‐aminobutyric acid type A receptors with [3H]TDBzl‐etomidate, a photoreactive etomidate analogue. Biochemistry 51, 836–847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiara DC, Jayakar SS, Zhou X, Zhang X, Savechenkov PY, Bruzik KS, Miller KW & Cohen JB (2013). Specificity of intersubunit general anesthetic‐binding sites in the transmembrane domain of the human α1β3γ2 gamma‐aminobutyric acid type A (GABAA) receptor. J Biol Chem 288, 19343–19357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cottrell GA, Lambert JJ & Peters JA (1987). Modulation of GABAA receptor activity by alphaxalone. Br J Pharmacol 90, 491–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daniell LC (1994). Effect of anesthetic and convulsant barbiturates on N‐methyl‐d‐aspartate receptor‐mediated calcium flux in brain membrane vesicles. Pharmacology 49, 296–307. [DOI] [PubMed] [Google Scholar]
- Desai R, Ruesch D & Forman SA (2009). Gamma‐amino butyric acid type A receptor mutations at β2N265 alter etomidate efficacy while preserving basal and agonist‐dependent activity. Anesthesiology 111, 774–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhir A & Rogawski MA (2012). Role of neurosteroids in the anticonvulsant activity of midazolam. Br J Pharmacol 165, 2684–2691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhir A, Zolkowska D, Murphy RB & Rogawski MA (2011). Seizure protection by intrapulmonary delivery of propofol hemisuccinate. J Pharmacol Exp Ther 336, 215–222. [DOI] [PubMed] [Google Scholar]
- Dildy‐Mayfield JE, Mihic SJ, Liu Y, Deitrich RA & Harris RA (1996). Actions of long chain alcohols on GABAA and glutamate receptors: relation to in vivo effects. Br J Pharmacol 118, 378–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dillon GH, Im WB, Carter DB & McKinley DD (1995). Enhancement by GABA of the association rate of picrotoxin and tert‐butylbicyclophosphorothionate to the rat cloned α1β2γ2 GABAA receptor subtype. Br J Pharmacol 115, 539–545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dostalova Z, Zhou X, Liu A, Zhang X, Zhang Y, Desai R, Forman SA & Miller KW (2014). Human α1β3γ2L gamma‐aminobutyric acid type A receptors: high‐level production and purification in a functional state. Protein Sci 23, 157–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Downes H, Perry RS, Ostlund RE & Karler R (1970). A study of the excitatory effects of barbiturates. J Pharmacol Exp Ther 175, 692–699. [PubMed] [Google Scholar]
- Dunwiddie TV, Worth TS & Olsen RW (1986). Facilitation of recurrent inhibition in rat hippocampus by barbiturate and related nonbarbiturate depressant drugs. J Pharmacol Exp Ther 238, 564–575. [PubMed] [Google Scholar]
- Feng HJ, Bianchi MT & Macdonald RL (2004). Pentobarbital differentially modulates α1β3δ and α1β3γ2L GABAA receptor currents. Mol Pharmacol 66, 988–1003. [DOI] [PubMed] [Google Scholar]
- Forman SA (1999). A hydrophobic photolabel inhibits nicotinic acetylcholine receptors via open‐channel block following a slow step. Biochemistry 38, 14559–14564. [DOI] [PubMed] [Google Scholar]
- Forman SA & Miller KW (2011). Anesthetic sites and allosteric mechanisms of action on Cys‐loop ligand‐gated ion channels. Can J Anaesth 58, 191–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franks NP & Lieb WR (1994). Molecular and cellular mechanisms of general anaesthesia. Nature 367, 607–614. [DOI] [PubMed] [Google Scholar]
- Ge R, Pejo E, Gallin H, Jeffrey S, Cotten JF & Raines DE (2014). The pharmacology of cyclopropyl‐methoxycarbonyl metomidate: a comparison with propofol. Anesth Analg 118, 563–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gingrich KJ, Burkat PM & Roberts WA (2009). Pentobarbital produces activation and block of α1β2γ2S GABAA receptors in rapidly perfused whole cells and membrane patches: divergent results can be explained by pharmacokinetics. J Gen Physiol 133, 171–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haas KF & Macdonald RL (1999). GABAA receptor subunit gamma2 and delta subtypes confer unique kinetic properties on recombinant GABAA receptor currents in mouse fibroblasts. J Physiol 514, 27–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hales TG & Lambert JJ (1991). The actions of propofol on inhibitory amino acid receptors of bovine adrenomedullary chromaffin cells and rodent central neurones. Br J Pharmacol 104, 619–628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hara M, Kai Y & Ikemoto Y (1993). Propofol activates GABAA receptor‐chloride ionophore complex in dissociated hippocampal pyramidal neurons of the rat. Anesthesiology 79, 781–788. [DOI] [PubMed] [Google Scholar]
- Harrison NL & Simmonds MA (1983). Two distinct interactions of barbiturates and chlormethiazole with the GABAA receptor complex in rat cuneate nucleus in vitro . Br J Pharmacol 80, 387–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holland KD, Canney DJ, Rothman SM, Ferrendelli JA & Covey DF (1990). Physiological modulation of the GABA receptor by convulsant and anticonvulsant barbiturates in cultured rat hippocampal neurons. Brain Res 516, 147–150. [DOI] [PubMed] [Google Scholar]
- Holtman JR, Jr & Richter JA (1983). Increased release of [3H]acetylcholine in vitro from the mouse hippocampus by a convulsant barbiturate. Neuropharmacology 22, 1101–1108. [DOI] [PubMed] [Google Scholar]
- Jones MV & Westbrook GL (1995). Desensitized states prolong GABAA channel responses to brief agonist pulses. Neuron. 15, 181–191. [DOI] [PubMed] [Google Scholar]
- Kamiya Y, Andoh T, Furuya R, Hattori S, Watanabe I, Sasaki T, Ito H & Okumura F (1999). Comparison of the effects of convulsant and depressant barbiturate stereoisomers on AMPA‐type glutamate receptors. Anesthesiology 90, 1704–1713. [DOI] [PubMed] [Google Scholar]
- Knabe J, Buch HP & Reinhardt J (1982). [Derivatives of barbituric acid, 32. Central nervous activity of racemic and optically active barbituric acids with basic substituents]. Arch Pharma 315, 832–839. [DOI] [PubMed] [Google Scholar]
- Kokate TG, Svensson BE & Rogawski MA (1994). Anticonvulsant activity of neurosteroids: correlation with gamma‐aminobutyric acid‐evoked chloride current potentiation. J Pharmacol Exp Ther 270, 1223–1229. [PubMed] [Google Scholar]
- Krampfl K, Wolfes H, Dengler R & Bufler J (2002). Kinetic analysis of the agonistic and blocking properties of pentobarbital on recombinant rat α1β2γ2S GABAA receptor channels. Eur J Pharmacol 435, 1–8. [DOI] [PubMed] [Google Scholar]
- Krantz J, Rudo, FG (1966). The fluorinated anesthetics In Handbuch der experimentellen Pharmakologie New series, ed. Smith F, pp. 501–563. Springer, Berlin. [Google Scholar]
- Lape R, Plested AJ, Moroni M, Colquhoun D & Sivilotti LG (2012). The α1K276E startle disease mutation reveals multiple intermediate states in the gating of glycine receptors. J Neurosci 32, 1336–1352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lema GM & Auerbach A (2006). Modes and models of GABAA receptor gating. J Physiol 572, 183–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li GD, Chiara DC, Sawyer GW, Husain SS, Olsen RW & Cohen JB (2006). Identification of a GABAA receptor anesthetic binding site at subunit interfaces by photolabeling with an etomidate analog. J Neurosci 26, 11599–11605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Löscher W & Rogawski MA (2012). How theories evolved concerning the mechanism of action of barbiturates. Epilepsia 53Suppl 8, 12–25. [DOI] [PubMed] [Google Scholar]
- MacDonald RL, Rogers CJ & Twyman RE (1989). Barbiturate regulation of kinetic properties of the GABAA receptor channel of mouse spinal neurones in culture. J Physiol 417, 483–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maksay G, Molnar P & Simonyi M (1996). Thermodynamics and kinetics of t‐butylbicyclophosphorothionate binding differentiate convulsant and depressant barbiturate stereoisomers acting via GABAA ionophores. Naunyn Schmiedebergs Arch Pharmacol 353, 306–313. [DOI] [PubMed] [Google Scholar]
- Maksay G & Ticku MK (1985). Dissociation of [35S]t‐butylbicyclophosphorothionate binding differentiates convulsant and depressant drugs that modulate GABAergic transmission. J Neurochem 44, 480–486. [DOI] [PubMed] [Google Scholar]
- Mehta AK & Ticku MK (1999). Prevalence of the GABAA receptor assemblies containing alpha1‐subunit in the rat cerebellum and cerebral cortex as determined by immunoprecipitation: lack of modulation by chronic ethanol administration. Brain ResMol Brain Res 67, 194–199. [DOI] [PubMed] [Google Scholar]
- Mowrey D, Chen Q, Liang Y, Liang J, Xu Y & Tang P (2013). Signal transduction pathways in the pentameric ligand‐gated ion channels. PLoS One 8, e64326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukhtasimova N, Lee WY, Wang HL & Sine SM (2009). Detection and trapping of intermediate states priming nicotinic receptor channel opening. Nature 459, 451–454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagaya N & Macdonald RL (2001). Two γ2L subunit domains confer low Zn2+ sensitivity to ternary GABAA receptors. J Physiol 532, 17–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Najjar S, Devisnsky O & Rosenberg AD (2002). Procedures in epilepsy patients In Managing Epilepsy and Co‐Existing Disorders, ed. Ettinger AB. & Devinsky O, pp. 499–513. Butterworth‐Heinemann, Boston. [Google Scholar]
- Narahashi T, Frazier DT, Deguchi T, Cleaves CA & Ernau MC (1971). The active form of pentobarbital in squid giant axons. J Pharmacol Exp Ther 177, 25–33. [PubMed] [Google Scholar]
- Neher E & Steinbach JH (1978). Local anaesthetics transiently block currents through single acetylcholine‐receptor channels. J Physiol 277, 153–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olsen RW & Li GD (2011). GABAA receptors as molecular targets of general anesthetics: identification of binding sites provides clues to allosteric modulation. Can J Anaesth 58, 206–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rho JM, Donevan SD & Rogawski MA (1996). Direct activation of GABAA receptors by barbiturates in cultured rat hippocampal neurons. J Physiol 497, 509–522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richter JA & Holtman JR, Jr (1982). Barbiturates: their in vivo effects and potential biochemical mechanisms. Progr Neurobiol 18, 275–319. [DOI] [PubMed] [Google Scholar]
- Savechenkov PY, Zhang X, Chiara DC, Stewart DS, Ge R, Zhou X, Raines DE, Cohen JB, Forman SA, Miller KW & Bruzik KS (2012). Allyl m‐trifluoromethyldiazirine mephobarbital: an unusually potent enantioselective and photoreactive barbiturate general anesthetic. J Med Chem 55, 6554–6565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheller M & Forman SA (2002). Coupled and uncoupled gating and desensitization effects by pore domain mutations in GABAA receptors. J Neurosci 22, 8411–8421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sigel E & Buhr A (1997). The benzodiazepine binding site of GABAA receptors. Trends Pharmacol Sci 18, 425–429. [DOI] [PubMed] [Google Scholar]
- Skerritt JH & Macdonald RL (1984). Multiple actions of convulsant barbiturates on mouse neurons in cell culture. J Pharmacol Exp Ther 230, 82–88. [PubMed] [Google Scholar]
- Spurny R, Billen B, Howard RJ, Brams M, Debaveye S, Price KL, Weston DA, Strelkov SV, Tytgat J, Bertrand S, Bertrand D, Lummis SC & Ulens C (2013). Multisite binding of a general anesthetic to the prokaryotic pentameric Erwinia chrysanthemi ligand‐gated ion channel (ELIC). J Biol Chem 288, 8355–8364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinbach JH & Akk G (2001). Modulation of GABAA receptor channel gating by pentobarbital. J Physiol 537, 715–733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swanson EE, Gibson WR & Doran WJ (1955). The pharmacological relationship of a series of pentenyl substituted barbituric acid derivatives. J Am Pharm Assoc Am Pharm Assoc 44, 152–155. [DOI] [PubMed] [Google Scholar]
- Szczot M, Kisiel M, Czyzewska MM & Mozrzymas JW (2014). α1F64 Residue at GABAA receptor binding site is involved in gating by influencing the receptor flipping transitions. J Neurosci 34, 3193–3209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson SA, Whiting PJ & Wafford KA (1996). Barbiturate interactions at the human GABAA receptor: dependence on receptor subunit combination. Br J Pharmacol 117, 521–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ticku MK, Rastogi SK & Thyagarajan R (1985). Separate site(s) of action of optical isomers of 1‐methyl‐5‐phenyl‐5‐propylbarbituric acid with opposite pharmacological activities at the GABA receptor complex. Eur J Pharmacol 112, 1–9. [DOI] [PubMed] [Google Scholar]
- Udgaonkar JB & Hess GP (1987). Chemical kinetic measurements of a mammalian acetylcholine receptor by a fast‐reaction technique. Proc Natl Acad Sci USA 84, 8758–8762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Unwin N & Fujiyoshi Y (2012). Gating movement of acetylcholine receptor caught by plunge‐freezing. J Mol Biol 422, 617–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wakamori M, Ikemoto Y & Akaike N (1991). Effects of two volatile anesthetics and a volatile convulsant on the excitatory and inhibitory amino acid responses in dissociated CNS neurons of the rat. J Neurophysiol 66, 2014–2021. [DOI] [PubMed] [Google Scholar]
- Waud DR (1972). On biological assays involving quantal responses. J Pharmacol Exp Ther 183, 577–607. [PubMed] [Google Scholar]
- Wooltorton JR, Moss SJ & Smart TG (1997). Pharmacological and physiological characterization of murine homomeric β3 GABAA receptors. Eur JNeurosci 9, 2225–2235. [DOI] [PubMed] [Google Scholar]
- Zeller A, Jurd R, Lambert S, Arras M, Drexler B, Grashoff C, Antkowiak B & Rudolph U (2008). Inhibitory ligand‐gated ion channels as substrates for general anesthetic actions. Handb Exp Pharmacol 182, 31–51. [DOI] [PubMed] [Google Scholar]