

Abstract
As a gram-positive, spore-forming anaerobic bacillus, Clostridium difficile (C. difficile) is responsible for severe and fatal pseudomembranous colitis, and poses the most urgent antibiotic resistance threat worldwide. Epidemic C. difficile is the leading cause of antibiotic-associated diarrhoea globally, especially diarrhoea due to the emergence of hypervirulent strains associated with high mortality and morbidity. TcdB, one of the key virulence factors secreted by this bacterium, enters host cells through a poorly understood mechanism to elicit its pathogenic effect. Here we report the first identification of the TcdB cellular receptor, chondroitin sulfate proteoglycan 4 (CSPG4). CSPG4 was initially isolated from a whole-genome human shRNAmir library screening, and its role was confirmed by both TALEN- and CRISPR/Cas9-mediated gene knockout in human cells. CSPG4 is critical for TcdB binding to the cell surface, inducing cytoskeleton disruption and cell death. A direct interaction between the N-terminus of CSPG4 and the C-terminus of TcdB was confirmed, and the soluble peptide of the toxin-binding domain of CSPG4 could protect cells from the action of TcdB. Notably, the complete loss of CSPG4/NG2 decreased TcdB-triggered interleukin-8 induction in mice without significantly affecting animal mortality. Based on both the in vitro and in vivo studies, we propose a dual-receptor model for TcdB endocytosis. The discovery of the first TcdB receptor reveals a previously unsuspected role for CSPG4 and provides a new therapeutic target for the treatment of C. difficile infection.
Keywords: CSPG4, Clostridium difficile, TcdB, toxin, receptor, CROPs
Introduction
Clostridium difficile (C. difficile) produces two large proteins to elicit its toxic effect: enterotoxin TcdA (308 kDa) and cytotoxin TcdB (270 kDa)1. Although clinical isolates of C. difficile that lack these two toxins are non-pathogenic1, the individual significance of TcdA and TcdB in C. difficile infection (CDI) has been controversial. Nevertheless, TcdB proved essential for high virulence2,3. Both TcdA and TcdB are single-chain proteins possessing a similar primary structure, which includes a C-terminal receptor-binding domain featuring repetitive peptide elements called combined repetitive oligopeptides (CROPs), an intermediate cysteine protease domain, a transmembrane domain (TD), and an N-terminal glucosyltransferase domain (GTD) that exhibits mono-glucosyltransferase activity4. The catalytic GTD of TcdA or TcdB utilizes the nucleotide sugar UDP-glucose as a cosubstrate to transfer the glucose moiety onto Rho GTPases, leading to cytoskeleton disruption and cell rounding5.
The endocytic uptake of TcdA/B is clathrin-dependent6, and both toxins enter the cells through receptor-mediated endocytosis that requires acidified endosomes for translocation7 and exert their cytotoxic effect intracellularly8. Whereas the toxin A receptor has been partially characterised9,10, nothing is known about the toxin B receptor(s), except that it is different from the TcdA receptor11.
Here we report the first identification of the TcdB functional receptor, chondroitin sulfate proteoglycan 4 (CSPG4). Through direct binding, CSPG4 mediates the endocytosis of TcdB, and consequently its cytopathic effects post internalization.
Results
Identification of CSPG4, a cell surface protein involved in TcdB toxicity
To identify host cellular proteins that specifically affect TcdB toxicity, we designed a functional screening procedure in HeLa cells (Supplementary information, Figure S1A). An shRNAmir library targeting about 20 000 human genes was constructed through lentiviral infection, and this library of HeLa cells was exposed to the TcdB for 8 h; majority of the cells became loosely attached and were removed by repeated pipetting. After changing to fresh DMEM medium, few survival cells remained spindle-shaped, and these cells were grown and expanded from the library (Supplementary information, Figure S1B). These cells were again challenged with TcdB, and this cycle was repeated six times until the survival cells no longer turned round after toxin exposure. The genomic DNAs isolated from these toxin-resistant cells, as well as the original HeLa library cells, were used as templates for subsequent PCR amplification, and the regions harboring the shRNA-coding DNA were amplified using a pair of specific primers (Supplementary information, Figure S1C). The PCR products were then subjected to deep-sequencing analysis.
A few thousand distinct shRNA sequences from the library screening were revealed using high-throughput sequencing analysis, and the targeted genes corresponding to each individual shRNA were obtained from Blast analysis. Two different shRNAs targeting the same gene that encodes a cell surface receptor protein, CSPG4, were enriched and ranked among the top hits from the screening (Figure 1A). Interestingly, the cytoplasmic domain of CSPG4 is involved in the activation of the Rho family GTPases Rac and Cdc4212.
Figure 1.

CSPG4 is essential for TcdB toxicity in HeLa cells. (A) Ranking of shRNA abundance of the TcdB-resistant cells after library screening. The x-axis labels indicate the serial number of the distinct shRNAs, and the y-axis labels indicate the log2 ratio of the reads of any given shRNA in the TcdB survival pools vs the controls according to the deep-sequencing analysis. All of the data for the shRNAs that remained in the survival pools were plotted in descending order. shRNAs targeting the CSPG4 gene are highlighted. The arrows point to the ranking position of two distinct CSPG4-targeting shRNAs. (B) Partial coding sequences of the CSPG4 gene in the genome containing the TALEN binding regions (overlined for TALENL and underlined for TALENR) and the sequencing analysis of the mutated alleles from 8 randomly selected TALEN clones. The dashes indicate deletions. (C) Immunoblot analysis of the indicated HeLa cell lysates. A rabbit monoclonal antibody against human CSPG4 was used. The β-tubulin bands were used as internal controls in this and other figures. (D) Microscopic images of the indicated HeLa cells exposed to 0.1 ng ml−1 of TcdB for 8 h. The images were taken using an ImageXpress Micro XLS System (Molecular Device). Scale bar, 100 μm. (E) Immunoblot analysis of total Rac1 and non-glucosylated (non-glu.) Rac1 in the indicated HeLa cells. The cells were treated for increasing time intervals with three different concentrations of TcdB (0.1, 0.5 and 1 ng ml−1). Total Rac1 was detected using an antibody targeting both glucosylated and non-glucosylated Rac1. (F) Cells were treated with different concentrations of TcdB as indicated and were imaged every 2 h over an 8-h time course. Cell rounding was defined by an area less than 500 μm2 and a shape factor above 0.95. The percentage of round cells was normalized to the percentage of round cells at the initial time point. The images were acquired and analyzed using the ImageXpress Micro XLS System. The data are the mean ± SD, n = 12. (G) The indicated HeLa cells were treated with serially increasing amounts of TcdB (0-10 000 ng ml−1) for 8 h prior to the LDH cytotoxicity assay. The data are the mean ± SD, n = 3.
Deficiency of CSPG4 gene conferred cell resistance to TcdB, but not to TcdA
To confirm the role of CSPG4 in the cytotoxicity of TcdB, we generated CSPG4-knockout HeLa cells using the TALEN technique13. A pair of TALEN constructs was designed to target the first exon of the CSPG4 gene (Figure 1B). After transfection and antibiotic selection, individual colonies were challenged with 70 pg ml−1 TcdB. Most cells turned round after 8 hours of toxin exposure; however, some colonies appeared resistant and maintained a healthy morphology (Supplementary information, Figure S2A). Immunoblotting analysis demonstrated that the expression of CSPG4 protein completely disappeared from five randomly selected TcdB-resistant clones (Supplementary information, Figure S2B). We selected one of the TALEN clones for further study. Sequencing analysis revealed that the CSPG4-targeting TALENs in this clone generated two kinds of deletions in the coding region (7 and 8 nt deletions), both of which resulted in a frame shift (Figure 1B) and the complete inhibition of protein production (Figure 1C). We cloned the full-length human CSPG4 gene into a lentiviral expression vector. Through viral infection, a CSPG4 stably expressing clone was isolated from HeLa/CSPG4−/− cells. The loss of CSPG4 protein production was completely restored to a level that was even higher than that in wild-type HeLa cells (Figure 1C). We then examined the role of CSPG4 in TcdB-mediated cytopathic effect. Eight hours of TcdB exposure at a concentration of 0.1 ng ml−1 induced typical cell rounding in both wild-type and HeLa/CSPG4−/−/CSPG4 cells, but not in CSPG4-null HeLa cells (Figure 1D). We further examined the status of Rac1 glucosylation after toxin exposure. After treatment with TcdB toxin, Rac1 glucosylation clearly increased in both wild-type and HeLa/CSPG4−/−/CSPG4 cells in a timely manner, as indicated by the decreased amount of the non-glucosylated (Non-glu.) Rac1. The higher the concentration of TcdB used, the faster the rate of Rac1 glucosylation was. Consistently, Rac1 glucosylation was significantly inhibited in HeLa/CSPG4−/− cells when treated with all three doses of toxin (Figure 1E).
To quantitatively measure the effect of CSPG4 on the cytotoxic activity of TcdB, we recorded the cell morphology changes every 2 h for an 8-h period. TcdB-induced cell rounding was significantly inhibited in HeLa/CSPG4−/− cells at all three concentrations of toxins tested, although the difference was reduced as the dose of TcdB increased. Exogenous expression of CSPG4 completely restored the loss of the TcdB-caused cell-rounding phenotype in HeLa/CSPG4−/− cells (Figure 1F). Because CSPG4 could be covalently attached to chondroitin sulphate glycosaminoglycan (GAG)14, we wanted to know whether this modification is critical for TcdB toxicity. We constructed a CSPG4-expressing plasmid that contains a mutation at the GAG acceptor site (S995A, CSPG4(GAG*); Supplementary information, Figure S3A). After introducing this plasmid into the HeLa/CSPG4−/− cells, the lost sensitivity to TcdB was fully restored in the cell-rounding assay (Supplementary information, Figure S3B). This result confirmed that CSPG4 is critical for TcdB's function in inducing the cell-rounding phenotype, and the GAG side chain frequently associated with CSPG4 is irrelevant in mediating TcdB toxicity.
As high doses of TcdB could induce necrotic cell death15,16, we investigated whether CSPG4 also affects TcdB-triggered cell death. HeLa/CSPG4−/− cells showed significantly decreased levels of TcdB-induced cell death compared with wild-type cells until the TcdB dose reached 10 000 ng ml−1, and CSPG4 overexpression significantly increased the sensitivity of the cells to TcdB-induced cell death (Figure 1G).
We further investigated CSPG4-mediated effects in a colon cell line, HT29, which is widely used to study C. difficile toxin activities2,3,10. We generated a CSPG4-knockout HT29 cell line using the CRISPR/Cas9 technique17. Sequencing analysis of one isolated clone revealed three kinds of indels, all of which resulted in a frame shift in the gene and a complete loss of CSPG4 protein expression. Again, ectopic expression of CSPG4 protein wild-type HT29 cells increased the total expression level of this protein. We conducted the Rac1 glucosylation assay after TcdB exposure (1, 5, 10, and 50 ng ml−1) in these three types of HT29 cells. Consistent with the data obtained from the HeLa cells, the loss of CSPG4 dramatically inhibited TcdB-triggered Rac1 glucosylation, and CSPG4 overexpression increased the sensitivity of HT29 cells to TcdB toxicity (Supplementary information, Figure S4).
The effect of CSPG4 was only specific to TcdB, as HeLa/CSPG4−/− cells behaved the same as wild-type cells in terms of toxin-induced cell rounding, Rac1 glucosylation, and cell death after exposure to TcdA, a closely related C. difficile toxin (Supplementary information, Figure S5).
CSPG4 mediates both binding and internalization of TcdB to cells
Because CSPG4 is a cell surface protein, we hypothesized that it is a functional TcdB receptor or co-receptor. To test this hypothesis, we investigated the role of CSPG4 in the binding and uptake of TcdB. When cells were incubated with TcdB on ice, the amount of the toxin bound to the cell surface correlated with the level of CSPG4 expression as measured by immunoblotting (Figure 2A). When the temperature was shifted to 37 °C, the same pattern was observed and the toxin uptake paralleled the CSPG4 expression level (Figure 2B). To further confirm that CSPG4 acts as a cell surface receptor and mediates the binding of TcdB to the membrane, HeLa cells were incubated with Alexa Fluor 488-labelled TcdB on ice, and cell surface-bound fluorescence was subsequently detected by flow cytometry. Consistent with the immunoblotting results, the intensity of cell surface-bound fluorescence correlated with the level of CSPG4 expression (Figure 2C). Immunofluorescence microscopy further confirmed the strong correlation of the amount of CSPG4 and the level of TcdB binding to the cell surface, as well as the co-localization of membrane-bound TcdB and CSPG4 (Figure 2D).
Figure 2.

Effect of CSPG4 on the binding and internalization of TcdB. (A-B) Immunoblot analysis of the amount of TcdB bound to the cell surface at 4 °C (A) and associated with the cell membrane at 37 °C (B) in the different HeLa cells as indicated. The cells were exposed to 10 μg ml−1 TcdB for 1 h (A) or 30 min (B) before being lysed for analysis. A mouse monoclonal antibody against TcdB and a rabbit monoclonal antibody against human CSPG4 were used. The β-tubulin (A) and Claudin-1 (B) bands were measured as internal controls. (C) The indicated HeLa cells were incubated with or without 5 μg of TcdB-Alexa 488 protein for 30 min on ice, and washed twice before subjected to FACS analysis. Single cell events were plotted against the intensity of bound fluorescence (in log phase). (D) Immunofluorescence staining of TcdB (green) and CSPG4 (red) in the indicated HeLa cells.
CSPG4 N-terminus directly binds to TcdB C-terminal non-CROPs domain
CSPG4 is a surface protein with a single transmembrane domain. The N-terminal domain contains the secretion signal (amino acids 1-29) and two laminin G-type motifs, whereas the middle domain features 15 CSPG repeats. To determine whether CSPG4 can interact with TcdB and which domain(s) is responsible for this interaction, we created constructs expressing either full-length or truncated forms of CSPG4, as indicated (Figure 3A). A co-immunoprecipitation (Co-IP) assay showed that TcdB co-precipitated with the native CSPG4 protein (Figure 3B). Similarly, full-length CSPG4 protein co-precipitated with TcdB, whereas the CSPG4 protein lacking amino acids 30-640 (CSPG4ΔN30-640) lost the ability to bind to the toxin (Figure 3C). In agreement with this binding result, CSPG4ΔN30-640, unlike the full-length protein, failed to rescue the lost phenotype of the HeLa/CSPG4−/− cells in response to TcdB exposure in the cell rounding assay (Figure 3D), suggesting that the N-terminal domain (amino acids 30-640) is critical for both toxin binding and the function of CSPG4 in mediating TcdB toxicity. We then made a construct that expresses only the first 640 amino acids of CSPG4 (CSPG4-N1-640, Figure 3A), and the Fc-tagged CSPG4-N30-640 was purified using protein G agarose. Both the negative control Fc-tagged ANTXR1N (N-terminal anthrax toxin receptor 1 (ANTXR1)/TEM8) protein18 and the His-tagged TcdB protein19 were also purified and shown on a Coomassie blue-stained SDS-PAGE gel (Supplementary information, Figure S6A). The purified CSPG4-N30-640 and TcdB were mixed for the pull-down assay. The TcdB protein co-precipitated only with CSPG4-N30-640, and CSPG4-N30-640 only co-precipitated with TcdB. In contrast, ANTXR1N-Fc was not found to co-precipitate with TcdB (Figure 3E). This result demonstrates that TcdB directly binds to the N-terminal domain of CSPG4. Importantly, because no interaction was detected between CSPG4ΔN30-640 and TcdB (Figure 3C), the N-terminal region (CSPG4-N30-640) is the only site for TcdB binding.
Figure 3.

The N-terminal domain of CSPG4 directly binds to the C-terminal non-CROPs domain of TcdB. (A) A multi-domain schematic diagram of CSPG4 and two recombinant CSPG4 with partial deletions, CSPG4ΔN30-640 and CSPG4-N1-640. The N-terminus of the first 640 amino acids contains a secretion signal (amino acids 1-29) and two laminin G-type domains. Fifteen CSPG repeats are located in the middle, along with a GAG acceptor site that can bind to the GAG side chain. A single transmembrane domain and a short intracellular domain are located at the C-terminus14. (B) Co-IP of endogenous human CSPG4 with the TcdB toxin in HeLa cells. WCL: whole cell lysate. (C) Co-IP of TcdB-His with the indicated CSPG4 proteins in HEK293T cells transfected with the indicated Flag-CSPG4 constructs. Immunoblot analysis was conducted using anti-Flag and anti-TcdB antibodies after co-incubation of the cell lysate with 5 μg of TcdB at 4 °C overnight. (D) Effect of CSPG4ΔN30-640 expression on the restoration of the lost CSPG4 function in HeLa/CSPG4−/− cells in response to TcdB treatment. Cell rounding analysis was the same as described in Figure 1F. The data are the mean ± SD, n = 12. (E) Co-IP of 10 μg of purified CSPG4-N30-640-Fc with 10 μg of TcdB-His. ANTXR1N-Fc (10 μg) pure protein was used as a control. (F) A multi-domain schematic diagram of TcdB and two recombinant TcdB with partial deletions, TcdB-C1852-2366 and TcdB-C1500-2366. (G) The pull-down assay of CSPG4-N30-640 with purified TcdB protein in truncated form as indicated. (H-I) Effect of the addition of extracellular CSPG4-N30-640-Fc (H) and CSPG4-N401-560-Fc (I) on cell's susceptibility to TcdB. TcdB (1 ng ml−1) was pre-incubated with different molar ratio of indicated proteins for 1 h at 4 °C before applied to HeLa cells at 37 °C. Cell rounding was monitored as described in Figure 1F. ANTXR1N-Fc was used as the control. The data are the mean ± SD, n = 12. (J-K) Immunoblot analysis of non-glucosylated Rac1 was performed in HeLa cells after exposure to TcdB toxin for 2 h, which was pre-incubated with CSPG4-N30-640-Fc (J) or CSPG4-N401-560-Fc (K) for 1 h at 4 °C. Total Rac1 was detected using an antibody targeting both glucosylated and non-glucosylated Rac1. ANTXR1N-Fc was used as the control. +, 1:1 200; ++, 1:6 000; +++, 1:30 000 (molar ratio of TcdB and peptides).
TcdB consists of four domains with distinct functions, and the putative receptor-binding domain is located at the C-terminus and contains the CROPs4,20. To identify the specific region within TcdB that interacts with CSPG4, we expressed two truncated forms of TcdB fragments, TcdB-C1500-2366 and TcdB-C1852-2366 (Figure 3F). Both GST-tagged truncated TcdB proteins were purified and shown on a Coomassie blue-stained SDS-PAGE gel (Supplementary information, Figure S6B). A pull-down assay showed that only TcdB-C1500-2366 co-precipitated with the purified CSPG4-N30-640 protein (Figure 3G), indicating that CSPG4 binds to some amino acids from 1 500 to 1 851 within TcdB, a region just upstream of CROPs. Consistently, the presence of an alternative receptor-binding domain in addition to the CROPs has also been proposed by a previous study using truncated forms of TcdB20.
Based on these results, we predicted that the extracellular addition of CSPG4-N30-640 proteins might compete with the full-length CSPG4 protein for TcdB binding, potentially resulting in an inhibitory effect. To test this hypothesis, we incubated HeLa cells with a fixed amount of TcdB and a series of doses of CSPG4-N30-640 proteins. Indeed, CSPG4-N30-640, but not ANTXR1N, protected cells from TcdB-induced cell rounding (Figure 3H) and significantly inhibited TcdB-triggered Rac1 glucosylation (Figure 3J). We further narrowed down the toxin-binding domain to a region spanning amino acids 401-560 within the N-terminus of CSPG4. The binding affinity of CSPG4-N401-560 to TcdB is higher than that of CSPG4-N30-640, based on the Co-IP assay (Supplementary information, Figure S7). This smaller peptide, CSPG4-N401-560, could protect cells from TcdB toxicity (Figures 3I and 3K) with even better efficiency. To characterize the kinetics of the interaction between CSPG4-N401-560 and TcdB, we performed surface plasmon resonance (SPR) spectroscopy, and an equilibrium dissociation constant of 28.9 ± 0.7 nM was obtained (Supplementary information, Figure S8), whereas no direct binding was observed between ANTXR1N and TcdB under the same condition.
Complete loss of CSPG4/NG2 impaired TcdB-triggered interleukin-8 (IL-8) induction in mice
CSPG4 is a human homolog of rat neuronal/glial 2 (NG2)21. To further investigate the physiological role of CSPG4, we examined the effect of NG2 knockout22 in response to systemic TcdB challenge, as the toxin is disseminated into systemic circulation in severe infection23 and the disease severity can be precisely controlled by the amount of challenged toxin24. Both wild-type and NG2-null C57BL/6 mice were challenged with TcdB toxin intraperitoneally and monitored for either IL-8 induction or animal survival. The plasma levels of IL-8 increased dramatically in wild-type mice 5 h post toxin injection — a result consistent with prior reports25,26,27, whereas the induced IL-8 levels in NG2-null mice were notably lower (Figure 4A). However, the difference in animal viability was marginal between wild-type and NG2-null mice (Figure 4B). These results are in congruent with our prior data suggesting the existence of a CSPG4-independent pathway as CSPG4-deficient cells appeared susceptible to high concentration of TcdB (Figure 1 and Supplementary information, Figure S4).
Figure 4.

Effects of NG2 knockout in mice in response to TcdB intoxication. (A) Plasma levels of IL-8 in wild-type (n = 7, 5 female and 2 male) and NG2-knockout (n = 8, 6 female and 2 male) mice with or without TcdB challenge intraperitoneally (10 ng per mouse, 5 h). The data are the mean ± SEM; *P < 0.05; N.S., not significant; Welch's t-Test. (B) Kaplan-Meier survival curves of wild-type (n = 6, 3 female and 3 male) and NG2-knockout (n = 6, 3 female and 3 male) mice challenged intraperitoneally with TcdB (20 ng per mouse). P = 0.067; Log-rank test. (C) A dual-receptor model for TcdB endocytosis. Both CSPG4 and an unknown alternative receptor are capable of mediating TcdB binding to cell surface and internalization.
Discussion
We identified CSPG4 as the cellular receptor of TcdB based on the following evidence: (1) shRNA-mediated knockdown of CSPG4 in two independent cases conferred increased resistance of HeLa cells to TcdB intoxication; (2) CSPG4 gene knockout by either TALENs or CRISPR/Cas9 conferred significantly increased cell resistance to pathogenicity effects caused by TcdB challenge; (3) CSPG4 affects the specific binding and internalization of TcdB to host cells; (4) the direct interaction was detected between CSPG4 N-terminus and TcdB C-terminal non-CROPs domain; and (5) the soluble peptide of the toxin-binding domain of CSPG4 protects cells from the action of TcdB.
CSPG4, a human homolog of the rat NG2 CSPG molecule21, also known as high molecular weight-melanoma associated antigen or melanoma CSPG, is a surface antigen frequently expressed in human melanoma cells, and is involved in cell adhesion, invasion and spreading, angiogenesis, complement inhibition and signaling14,28. CSPG4 typically consists of a 250-kD core glycoprotein and many characteristic GAG side chains, and contains an open reading frame of 2 322 amino acids, encompassing a large extracellular domain, a hydrophobic transmembrane region, and a relatively short cytoplasmic tail29 (Figure 3A). CSPG4/NG2 interacts with a number of proteins, such as type V and VI collagens30,31, PDGF α-receptor32, multi-PDZ domain protein MUPP133, kringle domain-containing plasminogen34, glutamate receptor interaction protein GRIP135, galectin-336, syntenin-137 and NEDD938. The cytoplasmic domain of CSPG4/NG2 is involved in activation of the Rho family GTPases Rac and Cdc4212,39,40. It remains to be determined whether any of its interacting proteins participates in CSPG4's function in mediating TcdB endocytosis pathway.
CSPG4/NG2 was found abundantly distributed in mouse and human intestines41. CSPG4 is also highly expressed on pericytes in angiogenic vasculature within the tumor microenvironment as well as angiogenic blood vessels in normal tissues42. Interestingly, TcdB toxin has been reported to cause cardiovascular damage in zebrafish embryos43. Given that CSPG4 is evolutionarily conserved from worm to human44, it is important to find out whether CSPG4 is responsible for TcdB-caused cardiovascular damage in zebrafish. Coincidentally, two anthrax toxin receptors, ANTXR1/TEM8 and ANTXR2/CMG245,46, are also involved in the regulation of tumor angiogenesis47. It remains to be elucidated why the pathogenic bacteria preferentially target angiogenesis markers to gain their entry into host cells.
Our results demonstrate that CSPG4 is the receptor that mediates TcdB endocytosis and the subsequent cytopathic and cytotoxic effects, including glucosylation of small GTPase Rho family proteins that results in cellular cytoskeleton disruption and cell rounding, and necrotic cell death. However, it is likely that an alternative CSPG4-independent pathway exists, which could also mediate TcdB endocytosis. The evidence in support of this alternative pathway includes: (1) the high concentration of TcdB was able to cause cell rounding in CSPG4-deficient cells (HeLa/CSPG4−/− and HT29/CSPG4−/−); and (2) high concentration of TcdB was able to trigger cell death in HeLa/CSPG4−/− cells. In consistent with our results, the presence of an alternative receptor-binding domain besides the CROPs has recently been proposed based on truncation study of TcdB toxin20,48. We therefore propose a dual-receptor model for the cellular binding of TcdB, in which CSPG4 binds to the C-terminal non-CROPs domain of TcdB through its N-terminal domain (amino acids 401-560), and an unknown alternative receptor interacts with TcdB possibly through CROPs (Figure 4C). The existence of an alternative receptor might explain the marginal difference we observed in animal viability between wild-type and NG2-knockout mice during the systemic challenge of TcdB. Although CSPG4 plays a more dominant role in the two human cell lines that we tested, the vis-a-vis significance of these two receptors in mouse might be different from that in human. A two-receptor working model of TcdA has also been proposed as both TcdA1-1874 and TcdA-CROPs could bind to cell surface49,50. In fact, it is not uncommon that a single bacterial toxin has more than one receptor, such as anthrax toxin17.
In summary, the identification of the TcdB receptor is an important step toward the better understanding of the underlying molecular mechanism of host intoxication by the toxin and may provide a novel therapeutic target for treating CDI.
Materials and Methods
Cell lines and medium
HeLa, HT29 and HEK293T cells were cultured in Dulbecco's Modified Eagle's Medium (DMEM; Gibco #12800-017) containing 10% fetal bovine serum (FBS; Hyclone #SN30087.02).
Antibodies and reagents
We used the following primary antibodies: rabbit monoclonal antibody against human CSPG4 (Epitomics, 6871-1); mouse monoclonal antibodies against TcdB (GeneTex, GTX41668), non-glucosylated Rac1 (BD, #610650), Rac1 (Millipore, #05-389), and Flag-tag (Sigma, A8592); rabbit polyclonal antibody against Claudin 1 (Sigma, SAB4200534); goat anti-human IgG against Fc-tag (Zsbio, ZB-2304). The HRP-conjugated goat anti-mouse IgG (H+L) (#115-035-003) and HRP-conjugated goat anti-rabbit IgG (H+L; #111-035-003) secondary antibodies were purchased from Jackson ImmunoResearch.
Plasmids
The full-length CSPG4 gene was obtained by merging all of the exons of this gene, which were cloned separately from human genomic DNA, and was subsequently cloned into the pEF6/MycHis-B (Invitrogen #V962-20) and pIRES2 vectors (a kind gift from Dr Xianwen Hu), respectively. CSPG4 (GAG*), CSPG4-N1-640-Fc and CSPG4-N401-560-Fc (with signal peptide) were cloned into the pIRES2 vector, and CSPG4ΔN30-640 was cloned into the pEF6/MycHis-B vector.
shRNAmir library construction and functional screening
The shRNAmir library plasmids in the pGIPZ lentiviral vector (OpenBiosystems, Inc.) were kindly provided by Dr Hong Zhang (University of Massachusetts Medical School). HEK293T cells (lentivirus packaging cell line) were transfected with the shRNAmir library plasmids using the PEI method, according to the manufacturer's recommendations51. The transfected cells were incubated in DMEM supplemented with 30% FBS at 37 °C for 72 h, and the lentiviruses were harvested from the supernatants. HeLa cells were infected with an MOI of 0.05, and the HeLa cell library was obtained following selection with 1 μg ml−1 puromycin; the complexity of the library was estimated to be about 2 × 106. The library of cells was challenged with 70 pg ml−1 TcdB for 8 h before the loosely attached round cells were removed by repeated pipetting. The survival, spindle-shaped cells were transferred to fresh DMEM medium. After these cells grew to about 30%-40% confluence, the cells were challenged again with TcdB toxin (70 pg ml−1) for 8 h, followed by repeated pipetting to remove the loosely attached cells. After this procedure was repeated 6 times, the cells no longer became round after TcdB exposure. Genomic DNA was isolated from these survival clones, as well as from the original library cells. Using these genomic DNAs as templates, the shRNA-coding regions were PCR amplified using a pair of specific primers: F-shRNA (5′-ACGTCGAGGTGCCCGAAGGA) and R-shRNA (5′-TACATCTGTGGCTTCACTA). The PCR products were subjected to high-throughput sequencing analysis (Illumina Hiseq 2000).
Construction of stable knockout cell lines using the TALEN and CRISPR/Cas9 techniques
The design and assembly of the two pairs of TALEN constructs used for CSPG4 gene-knockout were based on our own ULtiMATE protocol13. More specifically, the two targeting sequences for the CSPG4 locus are 5′-CTGGCCAACATAGTC-3′ for TALENL(CSPG4) and 5′-TCCAGCCCCCGGCCT-3′ for TALENR(CSPG4), with a spacer sequence (5′-GGCCTTGGCTTTGACCCT-3′). The identification and verification of the gene knockout events were based on sequencing analysis of the genomic PCR fragments of the target loci and immunoblotting analysis using antibodies specifically against CSPG4. The sgRNA-binding sequence (5′-TTGGCCAGACTTGCATCCG) was used for CSPG4 disruption with the CRISPR/Cas9 system.
Cell rounding assay
The green fluorescence of cells transfected with maxGFP was used to determine cell shape. The shape factors for circle and square are pre-determined as 1 and 0, respectively. The round cells were defined as having an area less than 500 μm2 and a shape factor above 0.95. The rounding percentage (P) at any given time (t) was calculated using the following equation:
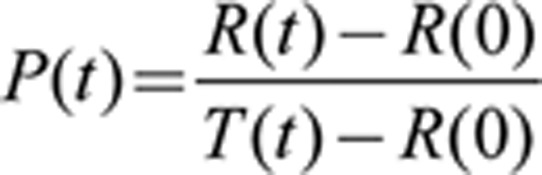 |
P(t): rounding percentage (P) at time point t;
R(t): total number of round cells at time point t;
R(0): total number of round cells at time point 0;
T(t): total number of cells at time point t.
The images were captured and analyzed using the ImageXpress Micro XLS Widefield High Content Screening System (Molecular Device).
LDH cytotoxicity assay
HeLa cells were seeded in 96-well plates for 16 h before the addition of the serially diluted toxin. LDH staining and detection were performed as described in the product instructions (CytTox96, Promega). The starting concentration of cells was 5 000 cells per well, and the cells were incubated with TcdB toxin for variable periods of time at 37 °C prior to the LDH assay. The spectrophotometer readings at 490 nm were determined using a Multi-Detection Reader (TECAN, Infinite M200). The death signal represented by the amount of LDH release was normalized to the wells with the maximum LDH activity of total lysed cells. Each data point and related error bars shown in the figures for the LDH assays represent the average results from three repeats.
Protein production and purification
TcdB protein was produced and purified as previously described19. TcdB-Alexa 488 protein was obtained using Alexa Fluor 488 Protein Labeling Kit (Life Technologies, #A-10235). CSPG4N30-640-Fc, CSPG4N401-560-Fc and ANTXR1N-Fc were produced and purified using CHO cells and HiTrap Protein G HP (GE, #17-0404-01).
Immunoblotting and Co-IP analyses
For immunoblotting analysis, the cells were treated with the toxin for different periods of time and were then lysed for immunoblotting analysis according to a standard protocol52. For the Co-IP assay, the plasmids were transfected into HeLa cells prior to treatment with the toxin, and immunoprecipitation analysis was performed according to the manufacturer's protocol. Specifically, the cells were lysed with the lysis buffer (0.5% Triton X-100, 20 mM HEPES pH 7.4, 150 NaCl, 12.5 mM β-glycerophosphate, 1.5 mM MgCl2, 10 mM NaF, 2 mM DTT, 1 mM Na3VO4, and 1 mM PMSF) at 4 °C for 45 min before being subjected to centrifugation at 20 800 g at 4 °C for 10 min. The proteins from the cell lysates or purified proteins in PBS were incubated with Ni-NTA Superflow (Qiagen, 30450), Protein A Sepharose (GE, 17-5280-02), or Anti-Flag M2 Affinity Gel (Sigma, A2220) at 4 °C overnight. After being washed three times with the lysis buffer or PBS, the proteins were subjected to immunoblotting analysis.
Immunofluorescence microscopy
HeLa cells (CSPG4−/− and CSPG4−/−/CSPG4) were grown on poly-L-lysine coverslips in 24-well plates and were treated with TcdB (10 μg ml−1) for 30 min at 4 °C. The cells were then washed 3 times with MEM, 2% FBS. Primary polyclonal mouse antibody against TcdB (1:1 000) and rabbit polyclonal antibody targeting NG2/CSPG4 (1:250, suspended in PBS, 0.1% BSA) were added for overnight incubation at 4 °C. After washing 3 times with PBS, the secondary antibodies (Alexa-Fluor 488 goat anti-mouse IgG and Alexa-Fluor 568 goat anti-rabbit IgG) were added at a dilution of 1:500 and incubated for 30 min at room temperature. After 3 times of PBS washing, the coverslips were mounted onto slides with a ProLong Gold antifade reagent, and cells were observed under confocal microscope.
TcdB binding assay
The cells were seeded in 6-well plates at a concentration of 1 × 106 cells per well. After incubation at 37 °C for 16 h, the cells were transferred to 4 °C for 15 min and then incubated with 10 μg ml−1 TcdB at 4 °C for 1 h. The cells were then washed five times with ice-cold PBS before lysis. The lysate was subjected to immunoblotting analysis.
SPR measurements
Dynamic interactions of CSPG4-N401-560 and TcdB were analyzed using Biacore T200 system (GE). Mouse anti-human IgG was amine-coupled on a CM5 sensor chip according to the manufacturer's instructions, and CSPG4-N401-560-Fc was captured at ∼500 RU. A blank flow cell with amine-coupled mouse anti-human IgG was used to subtract the buffer effect on sensograms. All SPR measurements were performed in the buffer (10 mM Hepes, 150 mM NaCl, and 0.05% Tween-20 at pH 7.4) at 25 °C with a flow rate of 30 μl/min, and the chip surface was regenerated by 3 M of MgCl2. Five concentrations of TcdB (15.625 nM, 31.25 nM, 62.5 nM, 125 nM and 250 nM) were injected for 180 s per cycle, and the dissociation was monitored for 600 s. Kinetic constants were calculated using the Biacore evaluation software.
FACS analysis
Cells were detached from the culture dishes by incubation with cell stripper (CORNING, #25-056-CI) to preserve cell surface proteins from degradation. Suspensions of cells were then mixed with 5 μg of Alexa-Fluor 488-labelled TcdB and incubated for 30 min on ice. After being washed twice with cold PBS, cells were subjected to FACS analysis. Cell surface-bound fluorescence was detected with an argon-ion laser (488 nm) and the 530-nm-band-pass filter (FL1).
TcdB in the membrane fraction
Cells were seeded in 100-mm plates at a concentration of 4 × 106 cells per plate. After incubation at 37 °C for 16 h, TcdB was added to a final concentration of 10 μg ml−1 and incubated for 30 min to allow toxin binding and endocytosis. The cell membrane fraction was extracted using the FractionPREP Cell Fraction Kit (BioVision, K270-50) and was evaluated using immunoblotting analysis.
GST pull-down assay
The pull-down assay was performed using MagneGST Pull-Down System (Promega# V8870).
Animal work
All mice used in this study were kindly provided by William Stallcup (Sanford/Burnham Medical Research Institute). Eight-week-old wild-type and NG2-knockout C57BL/6 mice were housed in dedicated pathogen-free facilities. The animal study protocol was approved by the Institutional Animal Care and Use Committee of Peking University. To assess the IL-8 induction of mice in response to TcdB intoxication, mice were challenged intraperitoneally with TcdB (10 ng per mouse). Blood samples were collected by submandibular bleeding technique the night before (0 h) and 5 h after challenge, and plasma IL-8 concentrations were measured using the ELISA kit (NeoBioscience #EMC104). For survival assay, mice were intraperitoneally injected with TcdB (20 ng per mouse), and data were analysed using Kaplan-Meier method.
Acknowledgments
We thank Hong Zhang (University of Massachusetts Medical School) for the kind gift of the shRNAmir library plasmids, William Stallcup (Sanford/Burnham Medical Research Institute) for reagents and NG2-knockout mice, Fuchou Tang (Peking University) for technical advice regarding the high-throughput sequencing analysis, Feng Shao (NIBS) for toxin-related reagents and Xianwen Hu (Beijing Institute of Biotechnology) for the pIRES2 vector and ANTXR1N-Fc protein. This work was supported by funds from the National Natural Science Foundation of China (NSFC31170126), the National Basic Research Program of China (2010CB911800), and the Peking-Tsinghua Center for Life Sciences to WW, and by grants from the National Institute of Health (R01AI088748, R01DK084509, U19AI109776, and R56AI99458) to HF.
Footnotes
(Supplementary information is linked to the online version of the paper on the Cell Research website.)
Supplementary Material
shRNAmir library screen for host genes important for TcdB toxicity.
Effect of TALEN-mediated CSPG4 knockout on the susceptibility of HeLa cells to TcdB.
GAG is irrelevant to CSPG4's function in mediating TcdB toxicity.
Effect of CSPG4 expression on the susceptibility of HT29 cells to TcdB toxicity.
CSPG4 has no effect on TcdA-mediated toxicity.
Production and purification of proteins.
Coimmunoprecipitation of TcdB-His with the indicated CSPG4 proteins in HEK293T cells transfected with indicated Flag-CSPG4 constructs.
Surface plasmon resonance (SPR) measurements for the binding constant between TcdB and CSPG4-N401-560.
References
- 1Voth DE, Ballard JD. Clostridium difficile toxins: mechanism of action and role in disease. Clin Microbiol Rev 2005; 18:247–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2Lyras D, O'Connor JR, Howarth PM, et al. Toxin B is essential for virulence of Clostridium difficile. Nature 2009; 458:1176–1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3Kuehne SA, Cartman ST, Heap JT, Kelly ML, Cockayne A, Minton NP. The role of toxin A and toxin B in Clostridium difficile infection. Nature 2010; 467:711–713. [DOI] [PubMed] [Google Scholar]
- 4Just I, Gerhard R. Large clostridial cytotoxins. Rev Physiol Biochem Pharmacol 2004; 152:23–47. [DOI] [PubMed] [Google Scholar]
- 5Just I, Selzer J, Wilm M, von Eichel-Streiber C, Mann M, Aktories K. Glucosylation of Rho proteins by Clostridium difficile toxin B. Nature 1995; 375:500–503. [DOI] [PubMed] [Google Scholar]
- 6Papatheodorou P, Zamboglou C, Genisyuerek S, Guttenberg G, Aktories K. Clostridial glucosylating toxins enter cells via clathrin-mediated endocytosis. PLoS One 2010; 5:e10673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7Florin I, Thelestam M. Internalization of Clostridium difficile cytotoxin into cultured human lung fibroblasts. Biochim Biophys Acta 1983; 763:383–392. [DOI] [PubMed] [Google Scholar]
- 8Falnes PO, Sandvig K. Penetration of protein toxins into cells. Curr Opin Cell Biol 2000; 12:407–413. [DOI] [PubMed] [Google Scholar]
- 9Greco A, Ho JG, Lin SJ, Palcic MM, Rupnik M, Ng KK. Carbohydrate recognition by Clostridium difficile toxin A. Nat Struct Mol Biol 2006; 13:460–461. [DOI] [PubMed] [Google Scholar]
- 10Na X, Kim H, Moyer MP, Pothoulakis C, LaMont JT. GP96 is a human colonocyte plasma membrane binding protein for Clostridium difficile toxin A. Infect Immun 2008; 76:2862–2871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11Stubbe H, Berdoz J, Kraehenbuhl JP, Corthesy B. Polymeric IgA is superior to monomeric IgA and IgG carrying the same variable domain in preventing Clostridium difficile toxin A damaging of T84 monolayers. J Immunol 2000; 164:1952–1960. [DOI] [PubMed] [Google Scholar]
- 12Eisenmann KM, McCarthy JB, Simpson MA, et al. Melanoma chondroitin sulphate proteoglycan regulates cell spreading through Cdc42, Ack-1 and p130cas. Nat Cell Biol 1999; 1:507–513. [DOI] [PubMed] [Google Scholar]
- 13Yang J, Yuan P, Wen D, et al. ULtiMATE System for rapid assembly of customized TAL effectors. PLoS One 2013; 8:e75649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14Campoli M, Ferrone S, Wang X. Functional and clinical relevance of chondroitin sulfate proteoglycan 4. Adv Cancer Res 2010; 109:73–121. [DOI] [PubMed] [Google Scholar]
- 15Chumbler NM, Farrow MA, Lapierre LA, et al. Clostridium difficile Toxin B causes epithelial cell necrosis through an autoprocessing-independent mechanism. PLoS Pathog 2012; 8:e1003072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16Farrow MA, Chumbler NM, Lapierre LA, et al. Clostridium difficile toxin B-induced necrosis is mediated by the host epithelial cell NADPH oxidase complex. Proc Natl Acad Sci USA 2013; 110:18674–18679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17Zhou Y, Zhu S, Cai C, et al. High-throughput screening of a CRISPR/Cas9 library for functional genomics in human cells. Nature 2014; 509:487–491. [DOI] [PubMed] [Google Scholar]
- 18Duan HF, Hu XW, Chen JL, et al. Antitumor activities of TEM8-Fc: an engineered antibody-like molecule targeting tumor endothelial marker 8. J Natl Cancer Inst 2007; 99:1551–1555. [DOI] [PubMed] [Google Scholar]
- 19Yang G, Zhou B, Wang J, et al. Expression of recombinant Clostridium difficile toxin A and B in Bacillus megaterium. BMC Microbiol 2008; 8:192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20Genisyuerek S, Papatheodorou P, Guttenberg G, Schubert R, Benz R, Aktories K. Structural determinants for membrane insertion, pore formation and translocation of Clostridium difficile toxin B. Mol Microbiol 2011; 79:1643–1654. [DOI] [PubMed] [Google Scholar]
- 21Smith FO, Rauch C, Williams DE, et al. The human homologue of rat NG2, a chondroitin sulfate proteoglycan, is not expressed on the cell surface of normal hematopoietic cells but is expressed by acute myeloid leukemia blasts from poor-prognosis patients with abnormalities of chromosome band 11q23. Blood 1996; 87:1123–1133. [PubMed] [Google Scholar]
- 22Grako KA, Ochiya T, Barritt D, Nishiyama A, Stallcup WB. PDGF (alpha)-receptor is unresponsive to PDGF-AA in aortic smooth muscle cells from the NG2 knockout mouse. J Cell Sci 1999; 112 (Part 6):905–915. [DOI] [PubMed] [Google Scholar]
- 23Steele J, Chen K, Sun X, et al. Systemic Dissemination of Clostridium difficile toxins A and B is associated with severe, fatal disease in animal models. J Infect Dis 2012; 205:384–391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24Lanis JM, Heinlen LD, James JA, Ballard JD. Clostridium difficile 027/BI/NAP1 encodes a hypertoxic and antigenically variable form of TcdB. PLoS Pathog 2013; 9:e1003523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25Linevsky JK, Pothoulakis C, Keates S, et al. IL-8 release and neutrophil activation by Clostridium difficile toxin-exposed human monocytes. Am J Physiol 1997; 273:G1333–G1340. [DOI] [PubMed] [Google Scholar]
- 26Tixier E, Lalanne F, Just I, Galmiche JP, Neunlist M. Human mucosa/submucosa interactions during intestinal inflammation: involvement of the enteric nervous system in interleukin-8 secretion. Cell Microbiol 2005; 7:1798–1810. [DOI] [PubMed] [Google Scholar]
- 27Rao K, Walk ST, Micic D, et al. Procalcitonin levels associate with severity of Clostridium difficile infection. PLoS One 2013; 8:e58265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28Bluemel C, Hausmann S, Fluhr P, et al. Epitope distance to the target cell membrane and antigen size determine the potency of T cell-mediated lysis by BiTE antibodies specific for a large melanoma surface antigen. Cancer Immunol Immunother 2010; 59:1197–1209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29Pluschke G, Vanek M, Evans A, et al. Molecular cloning of a human melanoma-associated chondroitin sulfate proteoglycan. Proc Natl Acad Sci USA 1996; 93:9710–9715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30Stallcup WB, Dahlin K, Healy P. Interaction of the NG2 chondroitin sulfate proteoglycan with type VI collagen. J Cell Biol 1990; 111:3177–3188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31Tillet E, Ruggiero F, Nishiyama A, Stallcup WB. The membrane-spanning proteoglycan NG2 binds to collagens V and VI through the central nonglobular domain of its core protein. J Biol Chem 1997; 272:10769–10776. [DOI] [PubMed] [Google Scholar]
- 32Nishiyama A, Lin XH, Giese N, Heldin CH, Stallcup WB. Interaction between NG2 proteoglycan and PDGF alpha-receptor on O2A progenitor cells is required for optimal response to PDGF. J Neurosci Res 1996; 43:315–330. [DOI] [PubMed] [Google Scholar]
- 33Barritt DS, Pearn MT, Zisch AH, et al. The multi-PDZ domain protein MUPP1 is a cytoplasmic ligand for the membrane-spanning proteoglycan NG2. J Cell Biochem 2000; 79:213–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34Goretzki L, Lombardo CR, Stallcup WB. Binding of the NG2 proteoglycan to kringle domains modulates the functional properties of angiostatin and plasmin(ogen). J Biol Chem 2000; 275:28625–28633. [DOI] [PubMed] [Google Scholar]
- 35Stegmuller J, Werner H, Nave KA, Trotter J. The proteoglycan NG2 is complexed with alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptors by the PDZ glutamate receptor interaction protein (GRIP) in glial progenitor cells. Implications for glial-neuronal signaling. J Biol Chem 2003; 278:3590–3598. [DOI] [PubMed] [Google Scholar]
- 36Wen Y, Makagiansar IT, Fukushi J, Liu FT, Fukuda MN, Stallcup WB. Molecular basis of interaction between NG2 proteoglycan and galectin-3. J Cell Biochem 2006; 98:115–127. [DOI] [PubMed] [Google Scholar]
- 37Chatterjee N, Stegmuller J, Schatzle P, et al. Interaction of syntenin-1 and the NG2 proteoglycan in migratory oligodendrocyte precursor cells. J Biol Chem 2008; 283:8310–8317. [DOI] [PubMed] [Google Scholar]
- 38Iida J, Dorchak J, Lehman JR, et al. FH535 inhibited migration and growth of breast cancer cells. PLoS One 2012; 7:e44418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39Majumdar M, Vuori K, Stallcup WB. Engagement of the NG2 proteoglycan triggers cell spreading via rac and p130cas. Cell Signal 2003; 15:79–84. [DOI] [PubMed] [Google Scholar]
- 40Yang J, Price MA, Neudauer CL, et al. Melanoma chondroitin sulfate proteoglycan enhances FAK and ERK activation by distinct mechanisms. J Cell Biol 2004; 165:881–891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41Terada N, Ohno N, Murata S, Katoh R, Stallcup WB, Ohno S. Immunohistochemical study of NG2 chondroitin sulfate proteoglycan expression in the small and large intestines. Histochem Cell Biol 2006; 126:483–490. [DOI] [PubMed] [Google Scholar]
- 42Wang X, Wang Y, Yu L, et al. CSPG4 in cancer: multiple roles. Curr Mol Med 2010; 10:419–429. [DOI] [PubMed] [Google Scholar]
- 43Hamm EE, Voth DE, Ballard JD. Identification of Clostridium difficile toxin B cardiotoxicity using a zebrafish embryo model of intoxication. Proc Natl Acad Sci USA 2006; 103:14176–14181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44Staub E, Hinzmann B, Rosenthal A. A novel repeat in the melanoma-associated chondroitin sulfate proteoglycan defines a new protein family. FEBS Lett 2002; 527:114–118. [DOI] [PubMed] [Google Scholar]
- 45Bradley KA, Mogridge J, Mourez M, Collier RJ, Young JA. Identification of the cellular receptor for anthrax toxin. Nature 2001; 414:225–229. [DOI] [PubMed] [Google Scholar]
- 46Scobie HM, Rainey GJ, Bradley KA, Young JA. Human capillary morphogenesis protein 2 functions as an anthrax toxin receptor. Proc Natl Acad Sci USA 2003; 100:5170–5174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47Cryan LM, Rogers MS. Targeting the anthrax receptors, TEM-8 and CMG-2, for anti-angiogenic therapy. Front Biosci 2011; 16:1574–1588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48Schorch B, Song S, van Diemen FR, et al. LRP1 is a receptor for Clostridium perfringens TpeL toxin indicating a two-receptor model of clostridial glycosylating toxins. Proc Natl Acad Sci USA 2014; 111:6431–6436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49Gerhard R, Frenzel E, Goy S, Olling A. Cellular uptake of Clostridium difficile TcdA and truncated TcdA lacking the receptor binding domain. J Med Microbiol 2013; 62(Part 9):1414–1422. [DOI] [PubMed] [Google Scholar]
- 50Olling A, Goy S, Hoffmann F, Tatge H, Just I, Gerhard R. The repetitive oligopeptide sequences modulate cytopathic potency but are not crucial for cellular uptake of Clostridium difficile Toxin A. PLoS One 2011; 6:e17623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51Boussif O, Lezoualc'h F, Zanta MA, et al. A versatile vector for gene and oligonucleotide transfer into cells in culture and in vivo: polyethylenimine. Proc Natl Acad Sci USA 1995; 92:7297–7301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52Harlow E, Lane D. Using Antibodies: A Laboratory Manual: Cold Spring Harbor Lab. Press, Plainview, NY 1999.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
shRNAmir library screen for host genes important for TcdB toxicity.
Effect of TALEN-mediated CSPG4 knockout on the susceptibility of HeLa cells to TcdB.
GAG is irrelevant to CSPG4's function in mediating TcdB toxicity.
Effect of CSPG4 expression on the susceptibility of HT29 cells to TcdB toxicity.
CSPG4 has no effect on TcdA-mediated toxicity.
Production and purification of proteins.
Coimmunoprecipitation of TcdB-His with the indicated CSPG4 proteins in HEK293T cells transfected with indicated Flag-CSPG4 constructs.
Surface plasmon resonance (SPR) measurements for the binding constant between TcdB and CSPG4-N401-560.