

Whole-genome sequences of Francisella tularensis isolated from 10 patients involved in a respiratory tularemia outbreak were compared with 110 global archived isolates. Surprisingly, outbreak and archived isolates were often extremely similar despite sometimes great separation in time and/or space.
Keywords: bacterial infections, tularemia, Francisella tularensis, epidemiology, genomics
Abstract
Background. The bacterium Francisella tularensis is recognized for its virulence, infectivity, genetic homogeneity, and potential as a bioterrorism agent. Outbreaks of respiratory tularemia, caused by inhalation of this bacterium, are poorly understood. Such outbreaks are exceedingly rare, and F. tularensis is seldom recovered from clinical specimens.
Methods. A localized outbreak of tularemia in Sweden was investigated. Sixty-seven humans contracted laboratory-verified respiratory tularemia. F. tularensis subspecies holarctica was isolated from the blood or pleural fluid of 10 individuals from July to September 2010. Using whole-genome sequencing and analysis of single-nucleotide polymorphisms (SNPs), outbreak isolates were compared with 110 archived global isolates.
Results. There were 757 SNPs among the genomes of the 10 outbreak isolates and the 25 most closely related archival isolates (all from Sweden/Finland). Whole genomes of outbreak isolates were >99.9% similar at the nucleotide level and clustered into 3 distinct genetic clades. Unexpectedly, high-sequence similarity grouped some outbreak and archival isolates that originated from patients from different geographic regions and up to 10 years apart. Outbreak and archival genomes frequently differed by only 1–3 of 1 585 229 examined nucleotides.
Conclusions. The outbreak was caused by diverse clones of F. tularensis that occurred concomitantly, were widespread, and apparently persisted in the environment. Multiple independent acquisitions of F. tularensis from the environment over a short time period suggest that natural outbreaks of respiratory tularemia are triggered by environmental cues. The findings additionally caution against interpreting genome sequence identity for this pathogen as proof of a direct epidemiological link.
Francisella tularensis, the causative agent of tularemia, is well known for its potential for bioterrorism, owing to high infectiousness and virulence, and ease of aerosol spread. In nature, tularemia is a zoonosis, an infection that can spread from animals to humans [1]. The pathogen has 4 closely related subspecies, of which 2 are clinically important: F. tularensis subspecies tularensis (Jellison type A) and F. tularensis subspecies holarctica (Jellison type B). Although disease presentation is similar with both varieties, type A tularemia is generally more aggressive and associated with significantly higher mortality in humans, up to 30% if untreated [1]. Two routes of infection predominate: inoculation through the skin via an arthropod bite, and inhalation of an infectious aerosol. Respiratory tularemia resulting from inhalation of F. tularensis is the clinical form that is most feared in the event of a bioterrorist attack. In a scenario with a future investigation of a suspected intentional aerosol release of F. tularensis resulting in severe human illness, it is expected that whole-genome sequencing (WGS) will be applied. Tracking transmission represents a challenge for any microorganism, and especially so for F. tularensis because of its high genomic homogeneity [2, 3].
The application of WGS in combination with more traditional epidemiology methods is highly promising for improving outbreak investigations of infectious disease. Recent reports on community- and hospital-acquired infections indicate that a combined approach may enhance outbreak detection, improve characterization of transmission events, and potentially aid in identifying the source of an outbreak, thereby providing guidance on how its effects on humans could be mitigated [4–7]. An outbreak is defined here as the occurrence of cases of disease in excess of what would normally be expected in a limited geographic area [8]. Time- and space-clustered infections may be caused by indistinguishable or closely linked pathogens as characterized by a genetic typing method (eg, WGS) [9]. Importantly, perfect or near-perfect WGS matching between causative pathogens within an outbreak is often taken as an indication of a confined and common infectious source where the pathogens share a very recent evolutionary origin [7, 10–13]. Although WGS is highly promising because it provides the full genomic information of the causative pathogen from multiple infected individuals, there is still a great need for learning how to interpret WGS data from suspected outbreaks.
In August 2010, clinicians working in Jämtland County in Sweden noted several patients diagnosed with respiratory tularemia. At the end of the summer, it became clear that this was the largest outbreak of respiratory tularemia reported worldwide since 1966–1967; this previous outbreak also occurred in Jämtland County [14]. Isolation of F. tularensis from respiratory secretions, pleural fluid, or blood is very sparsely reported even in large case series of tularemia [15–18]. However, we successfully isolated F. tularensis from a set of patients with severe respiratory tularemia, which we then analyzed using WGS. We used this rare dataset to quantify the within-outbreak genetic diversity of F. tularensis and test the hypothesis of a common environmental source causing the respiratory outbreak.
MATERIALS AND METHODS
Setting
Jämtland County in central Sweden is a sparsely populated area of 49 443 km2 with 126 000 citizens. More than one-third of the population lives in the countryside and about half in the county capital of Östersund. By obligation of the Swedish Communicable Disease and Prevention Act, all suspected and confirmed cases of tularemia are reported to the County Medical Office of Communicable Disease Control in Östersund and to the Public Health Agency of Sweden. From 2000 through 2009, 78 cases of tularemia (annual median, 5 [range, 0–32]) were notified in the county. In response to a respiratory tularemia outbreak in 2010, the County Medical Officer organized an epidemiological investigation team in collaboration with researchers at the Swedish Defence Research Agency and Umeå University to perform an outbreak investigation, including WGS of F. tularensis cultured from patients. The study was approved by the Regional Ethical Review Board in Umeå (2014-114-31M).
Data Collection
Epidemiological information was collected for each patient, including probable time of infection, date of disease onset, level of care for treatment, and likely geographical place of infection based on information electronically reported to SmiNet, a national system for communicable disease surveillance in Sweden. Missing epidemiological information was retrieved by personnel of the County Medical Office in Jämtland County using patient telephone interviews and by contacting medical practitioners. The County Medical Officer reviewed medical records and radiology reports for each patient and determined the clinical form of tularemia; no environmental sampling was performed. The case definition and the laboratory diagnostics used are detailed in the Supplementary Appendix 1.
Geographical Distribution
Using Google maps and a custom JavaScript, coordinates for the most likely place of infection were plotted on a map. In addition, the disease onset date and the level of care needed for treatment were recorded for each case, with the latter coded as (1) outpatient care with or without admission to hospital, or (2) hospital care with a minimum stay of 2 days.
Selection of Archival Reference Genomes
Archival F. tularensis reference genomes were selected based on multiple alignments of genomic sequences for the outbreak isolates and 110 genomic sequences representing maximal genetic and geographical diversity within F. tularensis subspecies holarctica as judged by high-resolution molecular typing methods applied to >400 isolates from Asia, North America, and Europe, including Sweden. Genomes that differed at ≤15 nucleotide positions to any respiratory outbreak genome were included as archival references in the final analyses.
Sequencing and Bioinformatic Analyses
Sequencing of isolates was performed at SciLifeLab, Uppsala, Sweden, using the Illumina HiSeq 2000 platform according to standard protocols to produce 100-bp pair-end read data. The resulting sequence data were assembled using ABySS version 1.3.3 [19] using standard parameters, and a filtering procedure was employed to remove genomic positions with uncertain base calls. Multiple genome alignments were performed using progressiveMauve version 2.3.1 [20]. Nucleotide distances and phylogeny were inferred using Mega 5.1 [21] (complete deletion option and the number of differences method, maximum parsimony). Details of the procedures for DNA preparation, sequencing, and bioinformatic analyses are found in the Supplementary Appendix 1.
RESULTS
Tularemia Outbreak
In total, 78 patients were diagnosed with laboratory-confirmed tularemia in Jämtland County, Sweden in 2010. Sixty-seven (54 men and 13 women) were identified as case patients with respiratory tularemia (median age, 66 [range, 24–90] years). Thirty-three case patients required >2 days in hospital. One patient died 7 days after admission, whereas 66 were alive at day 30. The case patients contracted disease over a large geographic area extending 266 km north–south and 195 km east–west. They became ill between 16 July and 27 October 2010 with a peak at the end of August and beginning of September (Figure 1). Ten F. tularensis isolates grown from blood (n = 9) or pleural fluid (n = 1) of hospitalized case patients (Table 1) were subjected to genome sequencing. Analysis of date of disease onset and place of infection revealed a complex pattern compatible with disease transmission that continued locally for several weeks (Figure 2; Video available online as Supplementary Video 1).
Figure 1.

Cases of respiratory tularemia by week of onset in Jämtland County, Sweden, 16 July–27 October 2010. Dates provided in the histogram represent Mondays of each week. Case patients from which Francisella tularensis was isolated and whole-genome sequenced are indicated in red; other laboratory-verified cases are in blue.
Table 1.
Descriptive Data for 10 Francisella tularensis Outbreak Genome Sequences
| Isolate ID | Genetic Clade | Disease Onset Date | Clinical Specimen Type | Depth of Coverage | No. of Contigs |
|---|---|---|---|---|---|
| FSC952 | B.10 | 16 July 2010 | Blood | 827 | 93 |
| FSC953 | B.7 | 11 August 2010 | Blood | 926 | 93 |
| FSC956 | B.7 | 18 August 2010 | Blood | 745 | 93 |
| FSC960 | B.7 | 28 August 2010 | Blood | 2102 | 93 |
| FSC961 | B.7 | 01 September 2010 | Blood | 1112 | 93 |
| FSC962 | B.7 | 08 September 2010 | Blood | 1280 | 93 |
| FSC965 | B.7 | 06 September 2010 | Blood | 508 | 94 |
| FSC970 | B.12 | 04 September 2010 | Pleural fluid | 931 | 93 |
| FSC987 | B.7 | 01 September 2010 | Blood | 1016 | 93 |
| FSC988 | B.10 | 22 September 2010 | Blood | 917 | 94 |
Figure 2.

This image displays the respiratory tularemia outbreak including spatial patterns. The Supplementary Video 1 displays and describes the rise and fall of the respiratory tularemia outbreak including spatial patterns over time.
Multiple Major Phylogenetic Clades Represented in the Outbreak
Our whole-genome single-nucleotide polymorphism (SNP) phylogeny revealed 3 major clusters separated by long branch lengths, and both outbreak and archival strains were assigned to each of these groups (Figures 3 and 4). These 3 groups correspond to previously identified genetic groups B.12, B.10, and B.7 (shortened versions of designations in Vogler et al [22], see Table S1 in the Supplementary Appendix 1). There were a total of 757 variable sites among the 10 outbreak and 25 archival genomes. The midpoint root of the WGS phylogeny (Figure 3) gives rise to 2 branches: a long branch of approximately 250 SNPs leading out to B.12 strains and a shorter branch of approximately 160 SNPs leading to further division. Two branches arise from this second division, with SNP lengths of 119 and 128, respectively, and ending in the clusters with B.7 or B.10 strains. This branching order is consistent with previous publications by Vogler et al [22] and Svensson et al [23]. There were no nucleotide states in the data in conflict to the inferred topology (ie, no homoplasy), indicating high quality of the data.
Figure 3.
Maximum parsimony phylogenetic tree illustrating evolutionary relationships among 10 Francisella tularensis outbreak genomes (red text), 25 closely related archival genomes (black text), and the complete genome sequence for the strain FSC200 (black text, included to support merging of subalignments). The tree is midpoint rooted and drawn to scale with branch lengths representing the number of nucleotide changes. The rooting is consistent with results obtained by the use of multiple outgroups (not shown).
Figure 4.
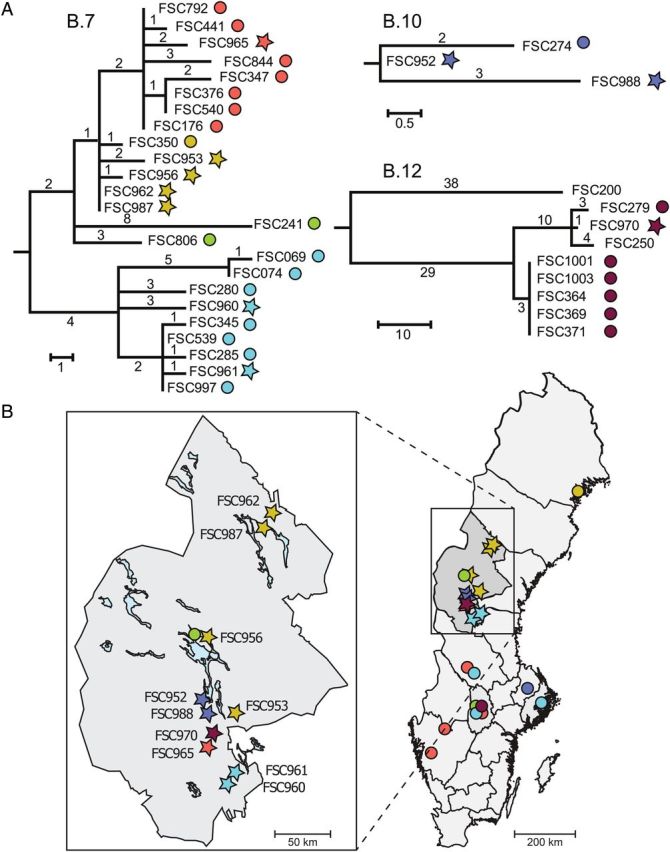
Phylogenetic relationships in relation to geographical origin of isolates. A, Detailed views of phylogenetic relationships within the genetic subclades B.7, B.10, and B.12. The outbreak genomes are labeled by stars and archival genomes by circles, and the different subgroups are color coded (red, yellow, green, turquoise, blue, and brown). The completed high-quality FSC200 genome was included for reference only. B, Geographical distribution of outbreak and archival isolates in Sweden, with Jämtland County magnified. The colored symbols correspond with the labels in (A). Arrows identify phylogenetically closely related and geographically linked taxa. The archival genome FSC250 originates in Finland outside the map.
Outbreak Strains Are Closely Related to Archival Strains Native to Sweden
Genome analysis of outbreak and archival strains found high similarity among the 2010 Swedish outbreak strains and archived strains isolated in Sweden and nearby regions. Among the 110 global archival strains evaluated, the 25 most closely related to the outbreak strains originated from Sweden and western Finland (24 and 1, respectively). The archival strains from other countries in Europe, Asia, or North America were more genetically distant (not shown). Both outbreak and archival strains from all 3 major phylogenetic groups were found in Jämtland County and other counties in Sweden (Figure 4), suggesting that these major groups occur concomitantly, are widespread, and apparently persist long-term in the environment in Sweden.
Genetic Distance Does Not Correlate With Either Spatial or Temporal Distance
In most cases, outbreak strains were more closely related to archival strains than to other outbreak strains. This occurred despite the sometimes large spatial and temporal distances between these closely related archival and outbreak strains. Indeed, there was no significant correlation between genetic distance and spatial or temporal distance (Supplementary Figure 1; Mantel r = 0.007, P = .371).
In a few cases, outbreak strains did exhibit spatial and/or temporal clustering. Two isolates from the north of the study area, FSC962 and FSC987, were indistinguishable at the genome level and represented infections contracted only 17 km and 7 days apart (Table 1 and Figure 4). Two isolates from the south, FSC961 and FSC960, differed by 6 SNPs and were from infections contracted 9 km and 4 days apart. Finally, 2 isolates from the central outbreak area, FSC952 and FSC988, differed by 3 SNPs and were from infections 12 km apart; they were separated in time by almost 4 months.
There are, however, numerous other examples of similar or closer genetic relationships between outbreak and archival strains. Within clade B.7, the previously mentioned outbreak strain FSC961 differed by just 1 SNP from archival strains FSC997 and FSC539. These archival strains were both recovered 330 km away from FSC961 and in 2002 and 2004, respectively (Figures 3 and 4). In addition, the 2 outbreak strains that were genetically indistinguishable, FSC962 and FSC987, still differed by just 1 SNP from archival strain FSC350 that was recovered in 2002, >290 km away in northern Sweden (Figure 4). Outbreak strains FSC962 and FSC987 were also closely related to 2 other outbreak strains, FSC956 and FSC953, but these strains were isolated from patients separated by at least 170 km. Finally, the outbreak strain FSC965 is most closely related to multiple archival strains even though those archival strains were isolated in different years and in different Swedish counties (Figure 4). Within clades B.10 and B.12, there is a similar lack of correlation between genetic distance and spatial and/or temporal distance (Figure 4). In the B.10 clade, the outbreak strain FSC952 is more closely related to the archival strain FSC274 that was isolated in 2000 and in a different county than it is to the outbreak strain FSC988, which was isolated very close to FSC952. In clade B.12, the sole outbreak strain, FSC970, is closely related to archival strains isolated 400 km away.
SNP Accumulation Rate Is Not Correlated With Time
No difference in SNP accumulation (ie, branch lengths) was observed between recent outbreak and older archival strains. Thus, there were no consistent time-dependent patterns for accumulations of SNPs, neither for the complete set of strains (outbreak isolates and archival isolates) nor during the outbreak (see scatterplots of number of SNPs accumulated from the most recent common ancestor of each clade in Supplementary Appendix 1, Figure 1). We found by comparing the historic FSC176 strain with the outbreak FSC962 and FSC987 strains (clade B.7) that there was no nucleotide change accumulated during 15 years. In contrast, FSC844, recovered 2 years before in the outbreak area, had 5 accumulated nucleotide changes. The differences in accumulated SNPs suggest great variations in the number of replication cycles within the same time unit for this bacterium.
DISCUSSION
In this work, our combined genomic and epidemiological approach allowed us to analyze the time course and geographical clustering of infection for all patients and, moreover, to determine in detail the genetic relationships between outbreak isolates and epidemiologically unrelated F. tularensis isolates. We show that tularemia was acquired at multiple locations and that genetic similarity only, even to the degree of single SNPs between F. tularensis genomes, did not constitute hard proof that 2 individuals had been infected at the same time and place, underscoring that WGS on its own should be regarded as supportive rather than decisive data. Importantly, the use of a large database containing archival genomes that were not part of the outbreak certified that nucleotide substitutions in whole genomes were insufficient to unequivocally distinguish all of the outbreak isolates from all of the archival isolates. As we found multiple major genetic clades and a highly interspersed phylogenetic pattern among respiratory and nonrespiratory isolates (ie, the archival set), it was also clear that the respiratory form of tularemia is not tied to specific genotypes of F. tularensis. Although both a single environmental source (containing the multiple clones) and multiple environmental sources are principally compatible with the genomic data, the wide geographic spread of locations for infection provides strong support for the latter. Taken together, our data indicate that the respiratory outbreak was caused by multiple environmental sources containing a diversity of F. tularensis clones and caution against interpreting sequence identity for this pathogen as conclusive evidence for an epidemiological link.
The nucleotide distances among outbreak isolates within each of the genetic clades were small (maximum pairwise distance of 15 SNPs) and, thus, within a range in which mutations principally could have accumulated during a phase of bacterial amplification that led to the outbreak or even during its progression. The phylogenomic analyses performed in this study, however, demonstrated a pattern incompatible with this possibility. Several outbreak isolates were in fact more closely related to archival isolates collected in quite distant geographical regions, and in different years. The presence of multiple genotypes has previously been observed in outbreaks of tularemia including a deer fly–associated outbreak in the US state of Utah [24] and in mosquito-associated outbreaks in Sweden [23, 25]. Viewed in the context of previous data, our results support that polyclonal isolate origin is common during natural tularemia outbreaks. Indeed, we found that the outbreak we investigated involved both dissemination of genetically indistinguishable isolates and unique isolates, and that the overall outbreak pattern was really an example of multiple disease clusters occurring in a single season. Such simultaneous activation of multiple F. tularensis clones suggests that tularemia outbreaks are often triggered by ecological or other environmental factors generally favorable to F. tularensis transmission and not because a specific F. tularensis clone has acquired new properties that increases its fitness, spread, and/or infectivity.
This study suggests that the rate of SNP accumulation has varied among different strains. In contrast to recent WGS studies of Staphylococcus aureus, Vibrio cholerae, and Streptococcus pneumoniae that identified a correlation between evolution and time [26–28], we showed that some F. tularensis isolates had acquired no change over a 15-year period. A possible explanation for this variation in change over time may be in the ecology of F. tularensis maintenance in the environment. Under an assumption of a constant mutation rate per replication, our results suggest a life cycle in nature where F. tularensis replicates rarely. As a consequence, this may necessitate a change in strategy for searching for the maintenance reservoir for tularemia from finding a host wherein F. tularensis replicates in between outbreaks to finding a reservoir where the bacterium resides in a dormant, or near-dormant, stage with little or no replication. A scenario also emerges wherein F. tularensis experiences replication bursts during outbreaks, which would be similar to what has recently been inferred for Yersinia pestis, the cause of plague [29].
If the probability of dispersal does not correlate with the rate of mutation, the small annual mutation rates observed in F. tularensis may theoretically explain the finding of a large geographical distribution of closely related genotypes but provides little insight into actual mechanisms involved. As F. tularensis has been associated with birds [30], it is possible that dispersal occurs through migratory bird populations. Another intriguing alternative is that aerosolized bacteria are transported by wind in the troposphere. It is generally known that microorganisms are abundant in the troposphere and can be transported vast distances, even between continents [31, 32]. The well-known propensity for aerosolization and environmental survival of F. tularensis could potentially allow for rapid, long-distance, wind-assisted dispersal [33]. In either scenario, it is likely that local replication of F. tularensis after its transport may vary considerably. In fact, low permissive conditions for replication resulting in low annual mutation rates and long-distance transports may explain the lack of time-dependent evolution as observed in this study.
This is the first study to use WGS to investigate an outbreak of tularemia, and we have used high-quality whole-genome assemblies with maximal nucleotide position coverage for determining exact phylogenetic relationships among F. tularensis isolates. In previously reported respiratory outbreaks [14, 16, 17, 34–38], F. tularensis cultures were, to the best of our knowledge, not recovered from multiple individuals and hence no molecular investigations were possible. Our conclusions on the genetic ancestry of outbreak genomes were possible only because we used an extensive set of reference genomes. An outbreak analysis using fewer reference genomes might not have revealed the complexity in correlating genetic distance with geographical location and time.
We anticipate that our results will aid the interpretation of genetic data in future outbreaks of tularemia, including when an intentional release of F. tularensis is suspected. It will also be useful for future exploration of replication rates and the natural life cycle of F. tularensis in nature. Finally, these data will stimulate discussion on the strengths and limitations of using WGS in outbreak investigations of infectious diseases spread from environmental sources to humans.
Supplementary Data
Supplementary materials are available at Clinical Infectious Diseases online (http://cid.oxfordjournals.org). Supplementary materials consist of data provided by the author that are published to benefit the reader. The posted materials are not copyedited. The contents of all supplementary data are the sole responsibility of the authors. Questions or messages regarding errors should be addressed to the author.
Notes
Acknowledgments. We thank doctors and staff at district health care centers and the department of infectious diseases (Jämtland County Council, Sweden) for cooperation in the outbreak investigation and at the clinical microbiology laboratories (Jämtland County Council and Västerbotten County Council, Sweden) for access to bacterial isolates. We also acknowledge support from Science for Life Laboratory; the national infrastructure Swedish National Infrastructure for Large-scale Sequencing; and Uppmax for providing assistance in sequencing and computational infrastructure.
Financial support. This work was supported by the US Department of Homeland Security's Science and Technology Directorate (award number HSHQDC-10-C-00139) and the Swedish Civil Contingencies Agency (grant number TA 014-2010-01) pursuant to the agreement between the US government and the Kingdom of Sweden on Cooperation in Science and Technology for Homeland Security Matters; and also the Swedish Research Council for Environment, Agricultural Sciences and Spatial Planning (Formas number 2012-1070).
Potential conflicts of interest. All authors: No reported conflicts.
All authors have submitted the ICMJE Form for Disclosure of Potential Conflicts of Interest. Conflicts that the editors consider relevant to the content of the manuscript have been disclosed.
References
- 1.Dennis DT, Inglesby TV, Henderson DA, et al. Tularemia as a biological weapon: medical and public health management. JAMA. 2001;285:2763–73. doi: 10.1001/jama.285.21.2763. [DOI] [PubMed] [Google Scholar]
- 2.Larsson P, Elfsmark D, Svensson K, et al. Molecular evolutionary consequences of niche restriction in Francisella tularensis, a facultative intracellular pathogen. PLoS Pathog. 2009;5:e1000472. doi: 10.1371/journal.ppat.1000472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Keim P, Johansson A, Wagner DM. Molecular epidemiology, evolution, and ecology of Francisella . Ann N Y Acad Sci. 2007;1105:30–66. doi: 10.1196/annals.1409.011. [DOI] [PubMed] [Google Scholar]
- 4.Köser CU, Holden MTG, Ellington MJ, et al. Rapid whole-genome sequencing for investigation of a neonatal MRSA outbreak. New Engl J Med. 2012;366:2267–75. doi: 10.1056/NEJMoa1109910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Grad YH, Lipsitch M, Feldgarden M, et al. Genomic epidemiology of the Escherichia coli O104:H4 outbreaks in Europe, 2011. Proc Natl Acad Sci U S A. 2012;109:3065–70. doi: 10.1073/pnas.1121491109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Snitkin ES, Zelazny AM, Thomas PJ, et al. Tracking a hospital outbreak of carbapenem-resistant Klebsiella pneumoniae with whole-genome sequencing. Sci Transl Med. 2012;4:148ra116. doi: 10.1126/scitranslmed.3004129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Walker TM, Ip CLC, Harrell RH, et al. Whole-genome sequencing to delineate Mycobacterium tuberculosis outbreaks: a retrospective observational study. Lancet Infect Dis. 2013;13:137–46. doi: 10.1016/S1473-3099(12)70277-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dicker RC. Chapter 2: A brief review of the basic principles of epidemiology. In: Gregg MB, editor. Field epidemiology. New York: Oxford University Press; 2008. p. 592. [Google Scholar]
- 9.Gerner-Smidt P, Hyytiä-Trees E, Rota PA. Chapter 8: Molecular epidemiology. In: Versalovic J, Carrol KC, Funke G, Landry ML, Warnock DW, editors. Manual of clinical microbiology. Washington, DC: ASM Press; 2011. p. 2552. [Google Scholar]
- 10.Hendriksen RS, Price LB, Schupp JM, et al. Population genetics of Vibrio cholerae from Nepal in 2010: evidence on the origin of the Haitian outbreak. MBio. 2011;2:e00157–11. doi: 10.1128/mBio.00157-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Biek R, O'Hare A, Wright D, et al. Whole genome sequencing reveals local transmission patterns of Mycobacterium bovis in sympatric cattle and badger populations. PLoS Pathog. 2012;8:e1003008. doi: 10.1371/journal.ppat.1003008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Reuter S, Harrison TG, Köser CU, et al. A pilot study of rapid whole-genome sequencing for the investigation of a Legionella outbreak. BMJ Open. 2013;3:e002175. doi: 10.1136/bmjopen-2012-002175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Eyre DW, Cule ML, Wilson DJ, et al. Diverse sources of C. difficile infection identified on whole-genome sequencing. N Engl J Med. 2013;369:1195–205. doi: 10.1056/NEJMoa1216064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dahlstrand S, Ringertz O, Zetterberg B. Airborne tularemia in Sweden. Scand J Infect Dis. 1971;3:7–16. doi: 10.3109/inf.1971.3.issue-1.02. [DOI] [PubMed] [Google Scholar]
- 15.Weber IB, Turabelidze G, Patrick S, Griffith KS, Kugeler KJ, Mead PS. Clinical recognition and management of tularemia in Missouri: a retrospective records review of 121 cases. Clin Infect Dis. 2012;55:1283–90. doi: 10.1093/cid/cis706. [DOI] [PubMed] [Google Scholar]
- 16.Feldman KA, Enscore RE, Lathrop SL, et al. An outbreak of primary pneumonic tularemia on Martha's Vineyard. N Engl J Med. 2001;345:1601–6. doi: 10.1056/NEJMoa011374. [DOI] [PubMed] [Google Scholar]
- 17.Syrjälä H, Kujala P, Myllylä V, Salminen A. Airborne transmission of tularemia in farmers. Scand J Infect Dis. 1985;17:371–5. doi: 10.3109/13813458509058777. [DOI] [PubMed] [Google Scholar]
- 18.Maurin M, Pelloux I, Brion JP, Del Banõ J-N, Picard A. Human tularemia in France, 2006–2010. Clin Infect Dis. 2011;53:e133–41. doi: 10.1093/cid/cir612. [DOI] [PubMed] [Google Scholar]
- 19.Simpson JT, Wong K, Jackman SD, Schein JE, Jones SJM, Birol I. ABySS: a parallel assembler for short read sequence data. Genome Res. 2009;19:1117–23. doi: 10.1101/gr.089532.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Darling AE, Mau B, Perna NT. ProgressiveMauve: multiple genome alignment with gene gain, loss and rearrangement. PLoS One. 2010;5:e11147. doi: 10.1371/journal.pone.0011147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol. 2011;28:2731–9. doi: 10.1093/molbev/msr121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Vogler AJ, Birdsell D, Price LB, et al. Phylogeography of Francisella tularensis: global expansion of a highly fit clone. J Bacteriol. 2009;191:2474–84. doi: 10.1128/JB.01786-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Svensson K, Bäck E, Eliasson H, et al. Landscape epidemiology of tularemia outbreaks in Sweden. Emerg Infect Dis. 2009;15:1937–47. doi: 10.3201/eid1512.090487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Petersen JM, Carlson JK, Dietrich G, et al. Multiple Francisella tularensis subspecies and clades, tularemia outbreak, Utah. Emerg Infect Dis. 2008;14:1928–30. doi: 10.3201/eid1412.080482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Karlsson E, Svensson K, Lindgren P, et al. The phylogeographic pattern of Francisella tularensis in Sweden indicates a Scandinavian origin of Eurosiberian tularaemia. Environ Microbiol. 2013;15:634–45. doi: 10.1111/1462-2920.12052. [DOI] [PubMed] [Google Scholar]
- 26.Harris SR, Feil EJ, Holden MTG, et al. Evolution of MRSA during hospital transmission and intercontinental spread. Science. 2010;327:469–74. doi: 10.1126/science.1182395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mutreja A, Kim DW, Thomson NR, et al. Evidence for several waves of global transmission in the seventh cholera pandemic. Nature. 2011;477:462–5. doi: 10.1038/nature10392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Croucher NJ, Harris SR, Fraser C, et al. Rapid pneumococcal evolution in response to clinical interventions. Science. 2011;331:430–4. doi: 10.1126/science.1198545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cui Y, Yu C, Yan Y, et al. Historical variations in mutation rate in an epidemic pathogen, Yersinia pestis . Proc. Natl Acad Sci U S A. 2013;110:577–82. doi: 10.1073/pnas.1205750110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lopes de Carvalho I, Zé-Zé L, Alves AS, et al. Borrelia garinii and Francisella tularensis subsp. holarctica detected in migratory shorebirds in Portugal. Eur J Wildl Res. 2012;58:857–61. [Google Scholar]
- 31.Burrows SM, Elbert W, Lawrence MG, Pöschl U. Bacteria in the global atmosphere—part 1: review and synthesis of literature data for different ecosystems. Atmos Chem Phys. 2009;9:9263–80. [Google Scholar]
- 32.Smith DJ, Timonen HJ, Jaffe DA, et al. Intercontinental dispersal of bacteria and archaea by transpacific winds. Appl Environ Microbiol. 2013;79:1134–9. doi: 10.1128/AEM.03029-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Brodie EL, DeSantis TZ, Parker JPM, Zubietta IX, Piceno YM, Andersen GL. Urban aerosols harbor diverse and dynamic bacterial populations. Proc Natl Acad Sci U S A. 2007;104:299–304. doi: 10.1073/pnas.0608255104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Puntigam F. Erkrankungen an thorakalen Formen der Tularämie bei Arbeitnehmern in Zuckerfabriken. Zeitschrift für Hyg und Infekt. 1960;147:162–8. [Google Scholar]
- 35.Teutsch SM, Martone WJ, Brink EW, et al. Pneumonic tularemia on Martha's Vineyard. N Engl J Med. 1979;301:826–8. doi: 10.1056/NEJM197910113011507. [DOI] [PubMed] [Google Scholar]
- 36.Siret V, Barataud D, Prat M, et al. An outbreak of airborne tularaemia in France, August 2004. Euro Surveill. 2006;11:58–60. [PubMed] [Google Scholar]
- 37.Allue M, Sopeña CR, Gallardo MT, et al. Tularaemia outbreak in Castilla y León, Spain, 2007: an update. Euro Surveill. 2008 13. pii=18948. [PubMed] [Google Scholar]
- 38.Hauri AM, Hofstetter I, Seibold E, et al. Investigating an airborne tularemia outbreak, Germany. Emerg Infect Dis. 2010;16:238–43. doi: 10.3201/eid1602.081727. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.