

Abstract
Background
Hepatitis C Virus (HCV) infection is associated with chronically evolving disease and development of hepatocellular carcinoma (HCC), albeit the mechanism of HCC induction by HCV is still controversial. The nucleocapsid (core) protein of HCV has been shown to be directly implicated in cellular transformation and immortalization, enhancing the effect of oncogenes and decreasing the one of tumor suppressor genes, as RB1 and its protein product pRB. With the aim of identifying novel molecular mechanisms of hepatocyte transformation by HCV, we examined the effect of HCV core protein on the expression of the whole Retinoblastoma (RB) family of tumor and growth suppressor factors, i.e. pRb, p107 and pRb2/p130.
Methods
We used a model system consisting of the HuH-7, HCV-free, human hepatocellular carcinoma cell line and of the HuH-7-CORE cells derived from the former and constitutively expressing the HCV core protein. We determined pRb, p107 and pRb2/p130 protein and mRNA amount of the respective genes RB1, RBL1 and RBL2, RBL2 promoter activity and methylation as well as DNA methyltransferase 1 (DNMT1) and 3b (DNMT3b) expression level. The effect of pRb2/p130 over-expression on the HCV core-expressing HuH-7-CORE cells was also evaluated.
Results
We found that the HCV core protein expression down-regulated pRb2/p130 protein and mRNA levels in HuH-7-CORE cells by inducing promoter hyper-methylation with the concomitant up-regulation of DNMT1 and DNMT3b expression. When pRb2/p130 expression was artificially re-established in HuH-7-CORE cells, cell cycle analysis outlined an accumulation in the G0/G1 phase, as expected.
Conclusions
HCV core appears indeed able to significantly down-regulate the expression and the function of two out of three RB family tumor and growth suppressor factors, i.e. pRb and pRb2/p130. The functional consequences at the level of cell cycle regulation, and possibly of more complex cell homeostatic processes, may represent a plausible molecular mechanism involved in liver transformation by HCV.
Keywords: Hepatitis C Virus, Hepatocellular carcinoma, HCV core protein, RBL2, pRb2/p130, Cell cycle, Viral oncogenesis, Cell transformation
Background
Hepatitis C Virus (HCV), a member of the Flaviviridae family, has a single-stranded, positive-sense RNA genome of approximately 9.6 kb in length, which encodes a large polyprotein precursor of about 3,000 amino acids. This precursor is subsequently cleaved, by a combination of host and viral proteases, into at least ten proteins, four structural proteins [core, envelope 1 (E1), envelope 2 (E2) and p7] and six non-structural proteins (NS2, NS3, NS4A, NS4B, NS5A, and NS5B) [1, 2]. Infection by the hepatotropic HCV is a leading cause of chronic liver disease, with more than 170 million chronically infected individuals worldwide [3]. Chronic HCV infection is associated with the development of chronic hepatitis, fibrosis and cirrhosis, and is a major risk factor for the development and progression to hepatocellular carcinoma (HCC) [4].
Despite the successful development of the HCV sub-genomic replicon and the establishment of the JFH1 infectious virus model, the mechanisms underlying HCV-mediated tumorigenesis are still not fully understood [5]. It is known that the HCC development associated with HCV infection is a slow progressing oncogenic process, probably requiring multiple subsequent steps of genetic and epigenetic alterations. Indeed, activation of cellular oncogenes, inactivation of tumor suppressor genes, and dysregulation of multiple signal transduction pathways have been reported as possible pro-oncogenic mechanisms of HCV associated tumor [6]. Actually, virus-encoded factors establish a set of complex interactions with various cellular proteins and are actively implicated in several cellular signal transduction pathways that affect cell survival, proliferation, migration and transformation, thus contributing to viral persistence and pathogenicity. [7]. Among the HCV proteins, the 21 kDa core protein has been shown to modulate cellular genes expression, being involved in apoptosis, signal transduction, ROS formation, lipid metabolism, transcriptional activation, transformation and immune modulation [8–13]. HCV core binds to host tumor suppressor proteins, such as p53, p73 and pRb, modulates the expression of the cyclin dependent inhibitor p21Waf1/Cip1 [14, 15], a major target of p53, and regulates the activities of cyclin/cyclin-dependent kinase complexes involved in cell-cycle control and tumor onset/progression [16]. Furthermore, HCV core protein may also influence the growth and proliferation of host cells through activation of signaling pathways such as Raf/MAPK, Wnt/β-catenin and TGF-β, all known to be activated in HCC [17]. Lately, several studies have reported the presence of HCV core mutant proteins in HCV-infected patients who developed HCC, although the functional relevance of these mutations on malignant transformation is still not clear [18].
Due to the apparent involvement of the RB1 gene and its product, pRb, in HCC onset and/or progression [19, 20], as well as the interplay with HCV infection and HCV core expression [21, 22], we sought to analyze the effect of the sole HCV core protein expression on all the Retinoblastoma (RB) family of tumor and growth suppressor factors, i.e. pRb, p107 and pRb2/p130, the protein products of the RB1, RBL1 and RBL2 genes, respectively [23–26]. These proteins are defined “pocket proteins”, due to the high homology they share at the level of the so-called pocket region, a domain fundamental for the accomplishment of their cellular effects [23, 24]. The pocket region is also the preferred target of several small DNA virus oncoproteins in order to overcome the growth suppressive properties of these endogenous factors [27]. The function of all the RB family proteins is post-translationally regulated by a complex modulation of their phosphorylation status [28]. They share overlapping functions, but possess also unique traits, often associated with cell and tissue types [24, 29, 30]. In particular, pRb2/p130 plays an important role in G0 non-proliferating cells, where it is found in its under-phosphorylated form, the one able to sequester and block its main E2F partners (E2F4 and E2F5). When cells re-enter the cell cycle, pRb2/p130 becomes phosphorylated and the release of the E2F partners reactivates the transcription of the cell growth-related genes [31, 32]. pRb2/p130 expression results altered in a number of human cancers, such as lung cancer [33, 34], endometrial [35], oral squamous cell carcinomas [36] and leukemias/lymphomas [37]. Available evidences support that pRb2/p130 may play an essential role in regulating growth and differentiation also in liver epithelial cells, and its elevated expression in HCCs, a context in which pRb is frequently down-regulated [30], has been considered as a possible protective mechanism to limit their uncontrolled growth [38]. Consistent with this, pRb2/p130 over-expression in HepG2 HCC cell lines results in G0/G1 cell cycle arrest, growth inhibition in vitro and in vivo, while its down-regulation appears thus implicated in the progression of the disease [39].
All the above observations prompted us to investigate on the effects of the HCV core protein on the RB family genes mRNA and protein expression in human HCC cell lines, with the aim to estimate the involvement of the RB family factors in the HCV core protein oncogenic mechanisms associated with HCV-induced carcinogenesis. To this end, we set up a model system composed of the HuH-7 human hepatocellular carcinoma cell line, originally established from an HCV-negative liver cancer patient [40–42], and the HuH-7-CORE cell clones, obtained from the former and capable to stably express the HCV core protein. This model appeared suitable for the investigation of cell cycle-related molecular mechanisms associated with the expression of the HCV core protein and allowed us to assess the ability of this viral protein in down-modulating the RB family proteins as well as its involvement in some molecular underlying events.
Methods
Cells transfection and plasmid construction
Human HCC cells HuH-7 were cultured at 37 °C in a 5 % CO2 atmosphere in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10 % fetal bovine serum, 100 U/ml penicillin and 100 μg/ml streptomycin and 1X minimum essential amino acids solution (MEM). Cell cultures were passed twice a week and culture medium was changed every other day. Stable cell lines, expressing the HCV core protein, HuH-7-CORE, were established by transfection with Lipofectamine 2000 (Invitrogen Life Technologies, Carlsbad, CA) of pcCAG39neo (expressing the core protein) plasmid (constructed by A. Ruggieri), followed by selection with 400 μg/ml G418 (Geneticin, Gibco Invitrogen). In order to avoid the effect of G418 selected individual clones, the experiments were performed with a pool of clones obtained after the selection in G418 and by trypsinization of multiple clones from one plate.
pcDNA3Rb2/p130 sense and antisense expression plasmids [43] were transfected into HuH-7 and HuH-7-CORE cell lines with Lipofectamine 2000. Briefly, before transfection with pcDNA3Rb2 and RBL2 (pRb2/p130) promoter constructs (see below for plasmid description), HCV core stable transfectants were cultivated in the absence of G418 for at least two passages. Cells were plated 24 h before transfection and grown to 80 % confluence in serum-free medium; cells were then incubated with Optimem medium (Invitrogen Life Technologies) under standard conditions for the first 6 h after transfection; then the medium was changed to DMEM containing 10 % fetal calf serum without antibiotics for another 48–72 h, when they were harvested for cell cycle analysis (Methods below).
pGL2bRb2P construct containing the full length Rb2/p130 promoter was obtained from PBS SK vector (kindly provided by Prof. A. Giordano) by excision, HindIII digestion, of the/HindIII digestion, of the 2,34 k b fragment, corresponding to the RBL2 promoter region. The excised fragment was inserted and ligated into the cloning site of pGL2-Basic vector (Promega, Madison, WI) that was linearized by HindIII/SacI digestion before ligation. The resulting plasmid, pGL2bRb2P, contained the RBL2 promoter region whose functional expression could be evaluated by the induced expression of Firefly Luciferase gene downstream the cloning site. pGL2bRb2P was transfected into HuH-7 and in HuH-7-CORE cell lines using Lipofectamine 2000 as above. phRL-TK vector expressing Renilla Luciferase was co-transfected as an internal control for normalization of luciferase values.
Western blot analysis
HuH-7 and HuH-7-CORE cells were harvested and dissolved in RIPA buffer (Tris-HCl 50 mM, NaCl 150 mM, 1 % sodium desoxycholate, 1 % Triton X-100, 0.1 % SDS, pH 7.5) or in pRb2/p130 buffer as previously described [43] supplemented with 1X proteinase inhibitor cocktail (Sigma Aldrich, Milan, Italy). Cell lysates (25 μg) were loaded onto a 10 % SDS-PAGE and analyzed under reduced conditions. Electrophoretically separated proteins were transferred on a PVDF membrane and probed with specific monoclonal antibody for 1.5 h at 37 °C or overnight at 4 °C. After extensive washing with TBS-T (Tris-HCl 20 mM, NaCl 137 mM, 0.5 % Tween, pH 7.6), membranes were incubated with appropriate HRP-conjugated secondary antibody. Protein bands were revealed by chemiluminescent substrates (ECL, Amersham Biosciences, Sweden). Antibodies: anti-HCV core protein mouse monoclonal antibody (Anogen Yes Biotech Laboratories. Ltd, Mississauga, Ontario, Canada) A1/3D1clone; anti-pRb2/p130 clone (BD Transduction Laboratories, Franklin Lakes, NJ); anti-pRb H-125 clone (Santa-Cruz Biotechnology, Santa Cruz, CA)¸ anti-p107 C-18 clone (Santa-Cruz); secondary antibody anti-mouse-HRP (Amersham Biosciences, Piscataway, NJ).
Gene expression analysis by qRT-PCR
For the real-time quantitative RT-PCR, total cellular RNA was extracted from the HuH-7 and HuH-7-CORE cells using the Epicentre Master Pure RNA Purification kit according to the manufacturer’s instructions (Epicentre Biotechnologies, Madison, WI). Total RNA was digested with DNase I and then subjected to reverse transcription using high capacity cDNA Reverse Transcription kit (Applied Biosystems, Branchburg, NJ). Real-time quantitative PCR was performed using SensiMix SYBR kit (Bioline, Taunton, MA). The cycling program comprises initial denaturation at 95 °C for 10 min, 40 cycles of denaturation at 95 °C for 15 sec, annealing at 59 °C for 15 sec, extension at 72 °C for 15 sec and final extension at 72 °C for 5 min. DNA amplification was performed using a 7500 Fast Real-time PCR system (Applied Biosystems).
The primer sequences were as follows:
RBL1 (p107) forward: 5’-AAGTGTGAGCCGGTTAC-3’
RBL1 (p107) reverse: 5’-AGGATTACGCACACAAGA-3’
RB1 (pRb) forward: 5’-GAGCTTGGTTAACTTGGG-3’
RB1 (pRb) reverse: 5’-CAGATTCCCCACAGTTCC-3’
RBL2 (pRb2/p130) forward: 5’-GGGTGACTGAAGTTCGTGCT-3’
RBL2 (pRb2/p130) reverse: 5’-TGTGGTTGGAGATGTTATGCTC-3’
mRNA levels of all three RB family members were normalized using TATA-box binding protein (TBP). mRNA and relative expression profiles were generated using the comparative Ct method (ΔΔCT) [44, 45].
Luciferase reporter assay
Dual luciferase Reporter Assay System (Promega) kit was used to measure the reporter luciferase activity induced by the expression of pRb2/p130 promoter in core expressing cell lines and in the control HuH-7, according to the protocol suggested by the manufacturer’s manual. Briefly, 5 × 104 cells/well were plated in a 24-well plate, 24 h before transfection with 0.6 μg of pGL2bRb2P along with 70 ng/well of Renilla Luciferase expressing plasmid, as internal control. Forty-eight h after transfection, cells were lysed directly in wells with 1X PBL buffer and cell lysates were stored in aliquots at -80 °C or processed immediately for luciferase assay. For the assay, 20 μg of cell lysate were mixed with 100 μl of luciferase substrate, and light emission was measured with the LumiCount Luminometer (Perkin Elmer, Waltham, MA Life Sciences). The luciferase activity was normalized to protein concentration, determined by BCA method (Sigma-Aldrich). The value obtained was normalized to the Renilla-Luc activity measured in the corresponding cell extracts. Each experiment was repeated at least three times prepared in triplicate.
DNA methylation analysis
Bisulfite treatment and recovery of genomic DNA samples were carried out with the EpiTect Bisulfite kit (Qiagen, Milan, Italy). Briefly, 2 μg of genomic DNA in 20 μl volume was used for each reaction and mixed with bisulfite mix (85 μl) and DNA protect buffer (35 μl). Bisulfite conversion was performed on a thermocycler as follows: 95 °C for 5 min, 60 °C for 25 min, 95 °C for 5 min, 60 °C for 85 min, 95 °C for 5 min, 60 °C for 175 min and 8 °C indefinitely. The bisulfite-treated DNA was recovered by EpiTect spin column by following the manufacturer’s instructions.
PCR, performed using pRb2/p130 primers that distinguish methylated (M) from unmethylated (U) sequences, was carried out in a 50 μl volume containing approximately 50 ng bisulfite-modified DNA, oligonucleotide primers (0.3 μM each) and 1x GoTaq Master Mix (Promega). DNA amplification was carried out with initial denaturing at 95 °C for 5 min, followed by 40 cycles of denaturing at 95 °C for 30 sec, annealing for 35 s at 55 °C and extension for 30 s at 72 °C. A final extension was added for 10 min at 72 °C after the last cycle. DNA products were electrophoresed on 2 % agarose gels and visualized by ethidium bromide staining.
Sequences to forward and reverse primers were as follows:
RBL2 (pRb2/p130)/U-For 5’-AACACAATACAAACAACAAACAAACAAACA-3’
RBL2 (pRb2/p130)/U-Rev 5’-GTTGTTTTAGGTTTTGGTTTGTGTTGTTTT-3’
RBL2 (pRb2/p130)/M-For 5’-GATACGAACGACGAACGAACGAACG-3’
RBL2 (pRb2/p130)/M-Rev 5’-TTTTAGGTTTCGGTTCGCGTCGTTTC-3’ [46].
The choice of two different experimental systems for luciferase and Methylation-Specific PCR (MSP) transcriptional assays is due to their specific technical features and responses.
DNMT1 and DNMT3b mRNA expression analysis
Real-Time quantitative PCR assessment of DNMT1 and DNMT3b mRNA expression in HuH-7 and HuH-7-CORE cell lines was performed using the ABI PRISM 7500 Sequence Detection System (Applied Biosystems). 800 ng of total RNA were in vitro reverse-transcribed and 100 ng of cDNA was subjected to Real-Time quantitative PCR, performed in triplicate using TaqMan chemistry with primers and probe sets from the Assay-on-Demand list (Hs00154749_m1 and Hs00171876_m1 - Life Technologies). 18S RNA co-amplification was used as endogenous control reference.
Cell cycle analysis
Cell cycle progression analysis of HuH-7 and HuH-7-CORE cell lines transiently expressing pRb2 and their relative controls was performed by flow cytometry, as previously described [47]. Cells were fixed and permeabilized with ice-cold ethanol for 30 min and then washed twice in PBS. DNA staining was performed by incubating cells at 37 °C in PBS containing 40 μg/ml propidium iodide and 0.4 mg/ml DNase-free RNase (type 1-A). Samples were analyzed by collecting FL2 red fluorescence in a linear scale at above 620 nm. The percentage of cells in the different phases of the cell cycle was determined by ModFIT software analysis. Apoptotic cells and debris were excluded by these analyses. Each experiment was repeated three times with duplicate samples.
Statistical analysis
Results are expressed as a mean ± Standard Deviation (S.D.). Differences between groups were analyzed using the Student’s two-tailed t test (GraphPad Prism v5). Difference was considered to be significant if P < 0.05.
Results
HCV core expression and cellular localization in a human HCC cell line
To investigate the interplay between the HCV core protein and the cellular factors involved in cell growth and replication, we engineered the HCV-negative HuH-7 human HCC cell line in order to constitutively express high levels of the HCV core protein from genotype 1b, by transfection with the plasmid pcCAG39neo (Fig. 1, panel a). Transfected cell clones stably expressing the HCV core protein were selected in G418 and named HuH-7-CORE. The expression of the HCV core protein was assessed by Western blot (Fig. 1, panel b). HCV core appeared mainly localized in the cytoplasm, displaying a punctate distribution in the peri-nuclear area (Fig. 1, panel c).
Fig. 1.
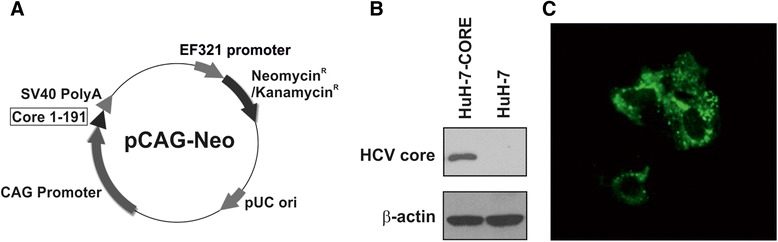
HCV core protein expression in HuH-7 stable transfectants. a Map of pcCAG39EFneo, the mammalian expression vector for cloning and expressing HCV core protein fragment (nt 1-191). b Western Blot detection of the HCV core protein constitutively expressed in HuH-7-CORE cell lysates. β-actin detection in the same blot is shown as loading control. c Immunofluorescence of the HCV core protein expression in HuH-7-CORE cell lines showing its peri-nuclear localization
Modulation of the RB family proteins expression by the HCV core protein
HCV core protein is known to down-modulate pRb expression, the product of the RB1 gene, in Rat-1 immortalized embryo fibroblasts [21] as well as its activity in other human liver and non-liver cell systems [22, 48].
In order to examine the effect of the HCV core protein on the expression of all the RB family proteins, we determined by Western blot pRb, p107 and pRb2/p130 protein expression in both HuH-7 (parental) and HuH-7-CORE (HCV core-expressing) cells (Fig. 2). Cells were assayed during the logarithmic growth phase and after that confluence was reached in the culture dish. Consistent with literature data [21], HCV core strongly inhibited pRb expression in HuH-7-CORE cells (Fig. 2, panel a). Also pRb2/p130 appeared down-regulated in HCV core-expressing cells and, interestingly, such effect regarded essentially the under-phosphorylated, fast-migrating, form of the protein, the one able to exert growth suppressive properties [24, 49]. In the case of pRb2/p130, the effect resulted by far more evident when cells reached the confluence (Fig. 2, panel a). Conversely, under the same conditions, p107 expression appeared definitely less affected by the expression of the HCV core protein (Fig. 2, panel b). α tubulin determination was employed in both panels a and b as a loading control. p107 was always detected using a replica gel, due to its apparent molecular mass, and consequent gel migration, that partially overlapped with the one of pRb and pRb2/p130.
Fig. 2.
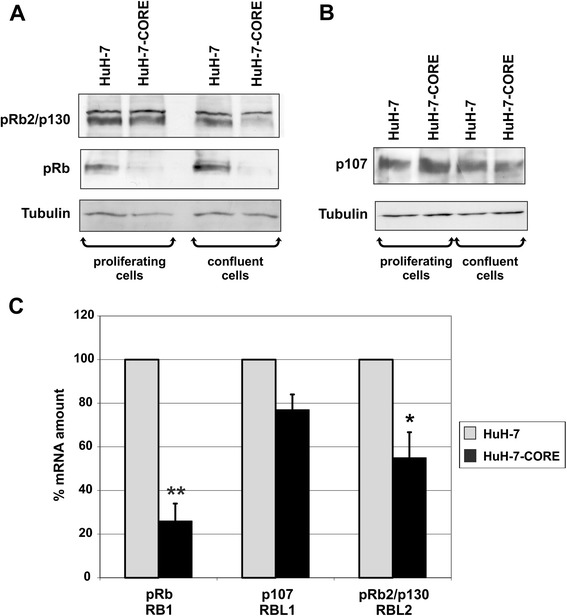
RB family proteins and mRNA modulation in HuH-7 parental and HuH-7-CORE cell lines. a and b Western blot analysis of pRb, pRb2/p130 and p107 proteins in parental HuH-7 and in core expressing HuH-7-CORE cell lines, showing a marked down-modulation of pRb and the phosphorylated form of pRb2/p130 in the HuH-7-CORE cell line. c Quantitative PCR determination of mRNA levels relative to the three RB family gene products, as indicated, in the parental HuH-7 cell line (grey bars, which refer to a 100 % relative amount) and in the HuH-7-CORE cells (black bars) constitutively expressing HCV core protein. Statistical significance: *P = <0.05; **P = <0.01
We thus determined by quantitative RT-PCR the mRNA levels related to the three RB family genes, RB1, RBL1 and RBL2. In HuH-7-CORE cells, when compared with HuH-7 cells, results showed a significant reduction in mRNA levels for RB1 and RBL2, while for RBL1 the reduction appeared less evident (Fig. 2, panel c). Such mRNA expression results were quite consistent with the determination of the respective protein levels (Fig. 2, panels a and b).
HCV core modulates RBL2 promoter activity
In search for the molecular mechanisms underlying pRb2/p130 down-regulation in HuH-7-CORE cells, we considered that HCV core is implicated, via promoter hypermethylation, in the negative modulation of several cellular genes [50–54]. Therefore, we sought to determine whether the decreased level of RBL2 mRNA and protein found in the HuH-7-CORE cells could be related to a negative modulation of the RBL2 promoter activity. We transfected the construct pGL2bRb2, containing the full length RBL2 promoter region [43], linked to a luciferase reporter gene, into both HuH-7 and HuH-7-CORE cells. Evaluation of the promoter activity indicated that RBL2 promoter was significantly down-regulated in the HuH-7-CORE cells when compared with the parental HuH-7 cells (Fig. 3, panel a), with more than three-fold decrease of the relative luciferase activity ratio (7.52 ± 1.34 vs. 23.90 ± 2.90, respectively with a statistical significance of P < 0.05).
Fig. 3.

Evaluation of the RBL2 promoter activity, its methylation status and DNMT1 and DNMT3b mRNA expression in HuH-7 parental and HuH-7-CORE cell lines. a The pGL2bRb2 construct, containing the full-length promoter region of RBL2 linked to a luciferase reporter gene, was constructed and then transfected into HuH-7 and HuH-7-CORE cell lines. Measure of Luciferase activity (Luc/Renilla ratio) showed a more than three-fold down-modulation of RBL2 promoter activity in HuH-7-CORE cells. The values plotted are the mean of three independent transfections, for each of which two different aliquots have been analyzed for Luciferase quantitation. Statistical significance: *P = <0.05. b Methylation-specific PCR (MSP) analysis covering the region abundant in CpG sequences was carried out on genomic DNA from HuH-7 and HuH-7-CORE cells. U = Unmethylated; M = Methylated. c qPCR determination of DNMT1 and DNMT3b mRNA expression shows its up-regulation in HuH-7-CORE cells with respect to the parental HuH-7 cell line, whose value has been normalized to 1. The results shown are the mean of two independent experiments performed in triplicate. Statistical significance: *P = <0.05
RBL2 epigenetic silencing by CpG methylation
DNA methylation of tumor suppressor genes has been described as one of the major epigenetic alterations in HCC [55] and such epigenetic modulations appear now also crucial for non-coding RNAs [56]. Considerably high incidence of promoter methylation in genes, like SOCS1 [57], GSTP1 [58, 59], APC [60], and CDKN2A (p16) [54] has been observed in HCV-positive HCCs [61, 62]. In addition, the HCV core protein appears able to up-regulate the levels of DNMT1 and DNMT3b and to induce promoter hypermethylation of tumor suppressor genes like E-cadherin and p16, resulting in down-regulation of their expression [51, 52, 54]. Thus, we sought to determine whether down-regulation of RBL2 gene transcription in HCV core-expressing human HCC cell lines could be associated with 5’ CpG islands hypermethylation. MSP analysis covering the region abundant in CpG sequences (Fig. 3, panel b) indicated a significant RBL2 promoter hypermethylation in HuH-7-CORE cells when compared with the parental HuH-7 cells, strongly suggesting the involvement of promoter hypermethylation as a cause of RBL2 down-regulation in HuH-7-CORE cells.
To further determine the molecular mechanisms through which HCV core affected RBL2 promoter methylation, we examined the mRNA expression of DNMT1 and DNMT3b that have been reported to positively influence HCV replication in HuH-7 cell line [63, 64]. Consistent with these previously reported results, we found an up-regulation of both DNMT1 and DNMT3b mRNA level in HuH-7-CORE cells, when compared with the parental HuH-7 cells (Fig. 3, panel c). In spite of an evident increase of transcription for both DNMT1 and DNMT3b, statistical significance was reached for DNMT3b expression only.
Forced pRb2/p130 expression in HuH-7-CORE cells
The RB pocket proteins are the main controllers of the G1/S checkpoint in the cell cycle [24]. Consequently, their down-regulation by HCV core protein facilitates the transition of the cells toward the S phase. In order to estimate the specific role of the HCV core protein on the pRb2/p130 function, we over-expressed pRb2/p130 in the HuH-7-CORE cells through transfection of the pcDNA3RB2/p130 construct. After cytofluorimetric cell cycle analysis, we found that pRb2/p130 over-expression induced a slight, but statistically significant, increase in the percentage of the cells in G1, when compared with the mock-transfected HuH-7-CORE cells (Table 1). Concomitantly, an average decrease in the percentage of the S phase cells was detected. Analysis of cell cycle in HuH-7-CORE cells transfected with the RBL2 antisense construct was included as a control. The amount of rescue elicited by pRb2/p130 over-expression in HuH-7-CORE cells can thus represent the functional role of the viral protein as a pRb2/p130 antagonist in the G1/S checkpoint dysregulation.
Table 1.
HuH-7-CORE cells: cell cycle distribution
| Go/G1 (%) | S (%) | G2/M (%) | |
|---|---|---|---|
| HuH-7-CORE (mock) | 67 ± 5.8 | 24.5 ± 5.7 | 8.7 ± 2.2 |
| HuH-7-CORE (pRb2/p130) | 70 ± 7.0 | 21 ± 6.0 | 8.7 ± 0.9 |
| HuH-7-CORE (pRb2/p130 as) | 66.7 ± 5.6 | 24.6 ± 4.9 | 8.67 ± 0.69 |
Effect of pRb2/p130 over-expression on cell cycle distribution in HuH-7-CORE cells. HuH-7-CORE cells were transfected with a mock construct or with the pRb2/p130 construct. pRb2/p130 antisense construct (pRb2/p130 as) was also included as an additional control
Discussion
HCC development in patients with chronic HCV infection is considered a multifactorial process, in which chronic liver inflammation (cirrhosis) and hepatocellular injury play an important role. However, HCC can still develop in a small proportion of non-cirrhotic patients with chronic hepatitis C infection, suggesting a direct involvement of HCV in hepatocarcinogenesis.
HCV-infected cells are forced by complex viral strategies to increase their survival ability and actively replicate, and HCV proteins are functional effectors evolved to obtain such goals. The present work enforces the notion that HCV core protein inhibits the G1/S transition of the cell cycle, demonstrates that pRb is not the only viral target to perturb this checkpoint and that a considerable role is played by targeting the sister protein pRb2/p130. Here we traced the flow of the events responsible for pRb2/p130 down-regulation by HCV core, going up to RBL2 promoter hyper-methylation, a pretty common mechanism elicited by this viral protein. Such a loss in cell cycle control at the G1/S boundary exposes infected hepatocytes to repeated unscheduled mitoses that, combined with a reduction of the apoptotic processes, increase exponentially the risk of mitotic errors, as polyploidy, and ultimately neoplastic transformation [65]. Indeed, HCV is a cancer-associated RNA virus that functionally retraces some molecular mechanisms usually considered peculiar of the small DNA viruses [66], i.e. disruption of cell cycle checkpoints, down-regulation of key oncosuppressor genes, as pRb and p53, impairment of genomic integrity and mitotic machinery [67]. All these phenomena obviously concur in raising and maintaining a transformed cell clone. It is worth of note, in this context, that HCV core protein is constantly expressed in hepatocytes during HCV chronic infection and circulating anti-core antibodies are detectable in chronically infected patients.
Conclusions
The observations we describe in the present work represent another step toward the comprehension of the molecular mechanisms of HCV-induced liver carcinogenesis. The RB family proteins are key regulators in pivotal cellular processes, e.g. cell cycle, apoptosis, genomic stability, senescence [23, 68] and the impairment of pRb functions by the HCV core protein has been widely described. Now we demonstrated that the pRb cognate protein pRb2/p130, which shares several functions with pRb, but shows also peculiar features [24], is likewise down regulated by HCV core and that this phenomenon is due, at least partially, to an increase in DNMT1 and DNMT3b activity, with consequent RBL2 promoter hypermethylation and pRb2/p130 protein down-regulation. Now, another negative cell cycle regulator appears strongly impaired in its function, thus strengthening the possibility of a neoplastic drift.
Being HCV core a key factor in sustaining HCV infection and subsequent host cell transformation, a better knowledge at the molecular level of the effects of this viral protein on the cell machinery can help in combating HCV infection and HCV-related hepatocarcinoma as well.
Acknowledgements
The authors thank Prof. Antonio Giordano for the pRb2/p130 (RBL2) promoter construct, Prof Y. Matsuura for the HCV Core protein plasmids and for helpful discussion and Dr. T. Harada for helpful suggestions to establish cell lines stably expressing the HCV core protein. This work was financed in part by intramural ISS funds.
Abbreviations
- DMEM
Dulbecco’s modified Eagle’s Medium
- DNMT1
DNA methyltransferase 1
- DNMT3b
DNA methyltransferase 3b
- HCC
hepatocellular carcinoma
- HCV
Hepatitis C Virus
- MSP
Methylation-Specific PCR
- PVDF
polyvinylidene difluoride
- RB
retinoblastoma
- ROS
reactive oxygen species
- TBS
tris-buffered saline
Footnotes
Anna Maria Mileo and Stefano Mattarocci contributed equally to this work.
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
SM: contributed to experimental design and to acquisition WB and PCR data. AMM: contributed to the research planning and to qRT-PCR data analysis and interpretation. PM: contributed to cell cycle analysis and interpretation and to related statistical analysis. SA: contributed to establish cell culture model, to interpretation of data and to statistical analysis. CA: contributed to qRT-PCR data acquisition and statistical analysis. SC: contributed to qRT-PCR data acquisition, plasmid construction and statistical analysis. RS: contributed to the interpretation of data, helped to draft the manuscript. MGP: contributed to conceiving the study and revised the manuscript critically for the intellectual content. AR: involved in design the study, coordination of the whole project, manuscript drafting. All authors read and approved the final manuscript.
References
- 1.Suzuki R, Suzuki T, Ishii K, Matsuura Y, Miyamura T. Processing and functions of Hepatitis C virus proteins. Intervirology. 1999;42(2-3):145–52. doi: 10.1159/000024973. [DOI] [PubMed] [Google Scholar]
- 2.Lohmann V, Koch JO, Bartenschlager R. Processing pathways of the hepatitis C virus proteins. J Hepatol. 1996;24(2 Suppl):11–9. [PubMed] [Google Scholar]
- 3.Global Burden Of Hepatitis CWG. Global burden of disease (GBD) for hepatitis C. J Clin Pharmacol. 2004;44(1):20–9. doi: 10.1177/0091270003258669. [DOI] [PubMed] [Google Scholar]
- 4.de Oliveria Andrade LJ, D’Oliveira A, Melo RC, De Souza EC, Costa Silva CA, Parana R. Association between hepatitis C and hepatocellular carcinoma. J Glob Infect Dis. 2009;1(1):33–7. doi: 10.4103/0974-777X.52979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wakita T, Pietschmann T, Kato T, Date T, Miyamoto M, Zhao Z, et al. Production of infectious hepatitis C virus in tissue culture from a cloned viral genome. Nat Med. 2005;11(7):791–6. doi: 10.1038/nm1268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Levrero M. Viral hepatitis and liver cancer: the case of hepatitis C. Oncogene. 2006;25(27):3834–47. doi: 10.1038/sj.onc.1209562. [DOI] [PubMed] [Google Scholar]
- 7.Banerjee A, Ray RB, Ray R. Oncogenic potential of hepatitis C virus proteins. Viruses. 2010;2(9):2108–33. doi: 10.3390/v2092108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hino K, Hara Y, Nishina S. Mitochondrial reactive oxygen species as a mystery voice in hepatitis C. Hepatol Res. 2014;44(2):123–32. doi: 10.1111/hepr.12247. [DOI] [PubMed] [Google Scholar]
- 9.Koike K. Hepatitis C, virus contributes to hepatocarcinogenesis by modulating metabolic and intracellular signaling pathways. J Gastroenterol Hepatol. 2007;22(Suppl 1):S108–11. doi: 10.1111/j.1440-1746.2006.04669.x. [DOI] [PubMed] [Google Scholar]
- 10.Liang TJ, Heller T. Pathogenesis of hepatitis C-associated hepatocellular carcinoma. Gastroenterology. 2004;127(5 Suppl 1):S62–71. doi: 10.1053/j.gastro.2004.09.017. [DOI] [PubMed] [Google Scholar]
- 11.Moriya K, Nakagawa K, Santa T, Shintani Y, Fujie H, Miyoshi H, et al. Oxidative stress in the absence of inflammation in a mouse model for hepatitis C virus-associated hepatocarcinogenesis. Cancer Res. 2001;61(11):4365–70. [PubMed] [Google Scholar]
- 12.Reed KE, Rice CM. Overview of hepatitis C virus genome structure, polyprotein processing, and protein properties. Curr Top Microbiol Immunol. 2000;242:55–84. doi: 10.1007/978-3-642-59605-6_4. [DOI] [PubMed] [Google Scholar]
- 13.Jahan S, Ashfaq UA, Khaliq S, Samreen B, Afzal N. Dual behavior of HCV Core gene in regulation of apoptosis is important in progression of HCC. Infect Genet Evol. 2012;12(2):236–9. doi: 10.1016/j.meegid.2012.01.006. [DOI] [PubMed] [Google Scholar]
- 14.Chang ML, Chen TH, Chang MY, Yeh CT. Cell cycle perturbation in the hepatocytes of HCV core transgenic mice following common bile duct ligation is associated with enhanced p21 expression. J Med Virol. 2009;81(3):467–72. doi: 10.1002/jmv.21403. [DOI] [PubMed] [Google Scholar]
- 15.Ray RB, Steele R, Meyer K, Ray R. Hepatitis C virus core protein represses p21WAF1/Cip1/Sid1 promoter activity. Gene. 1998;208(2):331–6. doi: 10.1016/S0378-1119(98)00030-4. [DOI] [PubMed] [Google Scholar]
- 16.Ohkawa K, Ishida H, Nakanishi F, Hosui A, Ueda K, Takehara T, et al. Hepatitis C virus core functions as a suppressor of cyclin-dependent kinase-activating kinase and impairs cell cycle progression. JBiolChem. 2004;279(12):11719–26. doi: 10.1074/jbc.M308560200. [DOI] [PubMed] [Google Scholar]
- 17.Liu J, Wang Z, Tang J, Tang R, Shan X, Zhang W, et al. Hepatitis C virus core protein activates Wnt/beta-catenin signaling through multiple regulation of upstream molecules in the SMMC-7721 cell line. Arch Virol. 2011;156(6):1013–23. doi: 10.1007/s00705-011-0943-x. [DOI] [PubMed] [Google Scholar]
- 18.Battaglia S, Benzoubir N, Nobilet S, Charneau P, Samuel D, Zignego AL, et al. Liver cancer-derived hepatitis C virus core proteins shift TGF-beta responses from tumor suppression to epithelial-mesenchymal transition. PLoS One. 2009;4(2) doi: 10.1371/journal.pone.0004355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhang X, Xu H-J, Murakami Y, Sachse R, Yashima K, Hirohashi S, et al. Deletions of chromosome 13q, mutations in retinoblastoma 1, and retinoblastoma protein state in human hepatocellular carcinoma. Cancer Res. 1994;54:4177–82. [PubMed] [Google Scholar]
- 20.Murakami Y, Hayashi K, Hirohashi S, Sekiya T. Aberrations of the tumor suppressor p53 and retinoblastoma genes in human hepatocellular carcinomas. Cancer Res. 1991;51:5520–5. [PubMed] [Google Scholar]
- 21.Cho J, Baek W, Yang S, Chang J, Sung YC, Suh M. HCV core protein modulates Rb pathway through pRb down-regulation and E2F-1 up-regulation. BiochimBiophysActa. 2001;1538(1):59–66. doi: 10.1016/s0167-4889(00)00137-3. [DOI] [PubMed] [Google Scholar]
- 22.Hassan M, Ghozlan H, Abdel-Kader O. Activation of RB/E2F signaling pathway is required for the modulation of hepatitis C virus core protein-induced cell growth in liver and non-liver cells. Cell Signal. 2004;16(12):1375–85. doi: 10.1016/j.cellsig.2004.04.005. [DOI] [PubMed] [Google Scholar]
- 23.Paggi MG, Baldi A, Bonetto F, Giordano A. Retinoblastoma protein family in cell cycle and cancer: A review. J Cell Biochem. 1996;62(3):418–30. doi: 10.1002/(SICI)1097-4644(199609)62:3<418::AID-JCB12>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- 24.Paggi MG, Giordano A. Who Is the Boss in the Retinoblastoma Family? The Point of View of Rb2/p130, the Little Brother. Cancer Res. 2001;61(12):4651–4. [PubMed] [Google Scholar]
- 25.Henley SA, Dick FA. The retinoblastoma family of proteins and their regulatory functions in the mammalian cell division cycle. Cell Div. 2012;7(1):10. doi: 10.1186/1747-1028-7-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Indovina P, Pentimalli F, Casini N, Vocca I, Giordano A. RB1 dual role in proliferation and apoptosis: cell fate control and implications for cancer therapy. Oncotarget. 2015;6(20):17873–90. doi: 10.18632/oncotarget.4286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Felsani A, Mileo AM, Paggi MG. Retinoblastoma family proteins as key targets of the small DNA virus oncoproteins. Oncogene. 2006;25(38):5277–85. doi: 10.1038/sj.onc.1209621. [DOI] [PubMed] [Google Scholar]
- 28.Cobrinik D. Pocket proteins and cell cycle control. Oncogene. 2005;24(17):2796–809. doi: 10.1038/sj.onc.1208619. [DOI] [PubMed] [Google Scholar]
- 29.Dean JL, McClendon AK, Stengel KR, Knudsen ES. Modeling the effect of the RB tumor suppressor on disease progression: dependence on oncogene network and cellular context. Oncogene. 2010;68(1):68–80. doi: 10.1038/onc.2009.313. [DOI] [PubMed] [Google Scholar]
- 30.Reed CA, Mayhew CN, McClendon AK, Knudsen ES. Unique impact of RB loss on hepatic proliferation: tumorigenic stresses uncover distinct pathways of cell cycle control. J Biol Chem. 2010;285(2):1089–96. doi: 10.1074/jbc.M109.043380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Vairo G, Livingston DM, Ginsberg D. Functional interaction between E2F-4 and p130: Evidence for distinct mechanisms underlying growth suppression by different retinoblastoma protein family members. Genes Dev. 1995;9:869–81. doi: 10.1101/gad.9.7.869. [DOI] [PubMed] [Google Scholar]
- 32.Hijmans EM, Voorhoeve PM, Beijersbergen RL, Veer LJ V ’t, Bernards R. E2F-5, a new E2F family member that interacts with p130 in vivo. Mol Cell Biol. 1995;15:3082–9. doi: 10.1128/MCB.15.6.3082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Helin K, Holm K, Niebuhr A, Eiberg H, Tommerup N, Hougaard S, et al. Loss of the retinoblastoma protein-related p130 protein in small cell lung carcinoma. Proc Natl Acad Sci U S A. 1997;94(13):6933–8. doi: 10.1073/pnas.94.13.6933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ho VM, Schaffer BE, Karnezis AN, Park KS, Sage J. The retinoblastoma gene Rb and its family member p130 suppress lung adenocarcinoma induced by oncogenic K-Ras. Oncogene. 2009;28(10):1393–9. doi: 10.1038/onc.2008.491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.D’Andrilli G, Bovicelli A, Paggi MG, Giordano A. New insights in endometrial carcinogenesis. J Cell Physiol. 2012;227(7):2842–6. doi: 10.1002/jcp.24016. [DOI] [PubMed] [Google Scholar]
- 36.Tanaka N, Ogi K, Odajima T, Dehari H, Yamada S, Sonoda T, et al. pRb2/p130 Protein expression is correlated with clinicopathologic findings in patients with oral squamous cell carcinoma. Cancer. 2001;92(8):2117–25. doi: 10.1002/1097-0142(20011015)92:8<2117::AID-CNCR1553>3.0.CO;2-O. [DOI] [PubMed] [Google Scholar]
- 37.Takeuchi S, Takeuchi N, Tsukasaki K, Fermin AC, De VS, Seo H, et al. Mutations in the retinoblastoma-related gene RB2/p130 in adult T-cell leukaemia/lymphoma. LeukLymphoma. 2003;44(4):699–701. doi: 10.1080/1042819031000063480. [DOI] [PubMed] [Google Scholar]
- 38.Huynh H, Chow PK, Soo KC. AZD6244 and doxorubicin induce growth suppression and apoptosis in mouse models of hepatocellular carcinoma. Mol Cancer Ther. 2007;6(9):2468–76. doi: 10.1158/1535-7163.MCT-07-0162. [DOI] [PubMed] [Google Scholar]
- 39.Huynh H. Overexpression of tumour suppressor retinoblastoma 2 protein (pRb2/p130) in hepatocellular carcinoma. Carcinogenesis. 2004;25(8):1485–94. doi: 10.1093/carcin/bgh154. [DOI] [PubMed] [Google Scholar]
- 40.Nakabayashi H, Taketa K, Miyano K, Yamane T, Sato J. Growth of human hepatoma cells lines with differentiated functions in chemically defined medium. Cancer Res. 1982;42(9):3858–63. [PubMed] [Google Scholar]
- 41.Nakabayashi H, Taketa K, Yamane T, Miyazaki M, Miyano K, Sato J. Phenotypical stability of a human hepatoma cell line, HuH-7, in long-term culture with chemically defined medium. Gan. 1984;75(2):151–8. [PubMed] [Google Scholar]
- 42.Vecchi C, Montosi G, Pietrangelo A. Huh-7: a human “hemochromatotic” cell line. Hepatology. 2010;51(2):654–9. doi: 10.1002/hep.23410. [DOI] [PubMed] [Google Scholar]
- 43.Paggi MG, Bonetto F, Severino A, Baldi A, Battista T, Bucci F, et al. The retinoblastoma-related Rb2/p130 gene is an effector downstream of AP-2 during neural differentiation. Oncogene. 2001;20(20):2570–8. doi: 10.1038/sj.onc.1204356. [DOI] [PubMed] [Google Scholar]
- 44.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25(4):402–8. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 45.Abbruzzese C, Mattarocci S, Pizzuti L, Mileo AM, Visca P, Antoniani B, et al. Determination of SGK1 mRNA in non-small cell lung cancer samples underlines high expression in squamous cell carcinomas. J Exp Clin Cancer Res. 2012;31(1):4. doi: 10.1186/1756-9966-31-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cinti C, Macaluso M, Giordano A. Tumor-specific exon 1 mutations could be the ‘hit event’ predisposing Rb2/p130 gene to epigenetic silencing in lung cancer. Oncogene. 2005;24(38):5821–6. doi: 10.1038/sj.onc.1208880. [DOI] [PubMed] [Google Scholar]
- 47.Conti L, Rainaldi G, Matarrese P, Varano B, Rivabene R, Columba S, et al. The HIV-1 vpr protein acts as a negative regulator of apoptosis in a human lymphoblastoid T cell line: possible implications for the pathogenesis of AIDS. J Exp Med. 1998;187(3):403–13. doi: 10.1084/jem.187.3.403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Munakata T, Liang Y, Kim S, McGivern DR, Huibregtse J, Nomoto A, et al. Hepatitis C virus induces E6AP-dependent degradation of the retinoblastoma protein. PLoSPathog. 2007;3(9):1335–47. doi: 10.1371/journal.ppat.0030139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Baldi A, De Luca A, Claudio PP, Baldi F, Giordano GG, Tommasino M, et al. The Rb2/p130 gene product is a nuclear protein whose phosphorylation is cell cycle regulated. J Cell Biochem. 1995;59(3):402–8. doi: 10.1002/jcb.240590311. [DOI] [PubMed] [Google Scholar]
- 50.Ray RB, Ray R. Hepatitis C virus core protein: intriguing properties and functional relevance. FEMS Microbiol Lett. 2001;202(2):149–56. doi: 10.1111/j.1574-6968.2001.tb10796.x. [DOI] [PubMed] [Google Scholar]
- 51.Park J, Jang KL. Hepatitis C virus represses E-cadherin expression via DNA methylation to induce epithelial to mesenchymal transition in human hepatocytes. Biochem Biophys Res Commun. 2014;446(2):561–7. doi: 10.1016/j.bbrc.2014.03.009. [DOI] [PubMed] [Google Scholar]
- 52.Ripoli M, Barbano R, Balsamo T, Piccoli C, Brunetti V, Coco M, et al. Hypermethylated levels of E-cadherin promoter in Huh-7 cells expressing the HCV core protein. Virus Res. 2011;160(1-2):74–81. doi: 10.1016/j.virusres.2011.05.014. [DOI] [PubMed] [Google Scholar]
- 53.Lee H, Woo YJ, Kim SS, Kim SH, Park BJ, Choi D, et al. Hepatitis C virus Core protein overcomes all-trans retinoic acid-induced cell growth arrest by inhibiting retinoic acid receptor-beta2 expression via DNA methylation. Cancer Lett. 2013;335(2):372–9. doi: 10.1016/j.canlet.2013.02.057. [DOI] [PubMed] [Google Scholar]
- 54.Lim JS, Park SH, Jang KL. Hepatitis C virus Core protein overcomes stress-induced premature senescence by down-regulating p16 expression via DNA methylation. Cancer Lett. 2012;321(2):154–61. doi: 10.1016/j.canlet.2012.01.044. [DOI] [PubMed] [Google Scholar]
- 55.Mah WC, Lee CG. DNA methylation: potential biomarker in Hepatocellular Carcinoma. Biomark Res. 2014;2(1):5. doi: 10.1186/2050-7771-2-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Liu X, Chen X, Yu X, Tao Y, Bode AM, Dong Z, et al. Regulation of microRNAs by epigenetics and their interplay involved in cancer. J Exp Clin Cancer Res. 2013;32:96. doi: 10.1186/1756-9966-32-96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Yoshikawa H, Matsubara K, Qian GS, Jackson P, Groopman JD, Manning JE, et al. SOCS-1, a negative regulator of the JAK/STAT pathway, is silenced by methylation in human hepatocellular carcinoma and shows growth-suppression activity. NatGenet. 2001;28(1):29–35. doi: 10.1038/ng0501-29. [DOI] [PubMed] [Google Scholar]
- 58.Tchou JC, Lin X, Freije D, Isaacs WB, Brooks JD, Rashid A, et al. GSTP1 CpG island DNA hypermethylation in hepatocellular carcinomas. Int J Oncol. 2000;16(4):663–76. doi: 10.3892/ijo.16.4.663. [DOI] [PubMed] [Google Scholar]
- 59.Jain S, Chen S, Chang KC, Lin YJ, Hu CT, Boldbaatar B, et al. Impact of the location of CpG methylation within the GSTP1 gene on its specificity as a DNA marker for hepatocellular carcinoma. PLoSONE. 2012;7(4):e35789. doi: 10.1371/journal.pone.0035789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Xu B, Nie Y, Liu X, Feng S, Yang Z, Wang Z, et al. Quantitative analysis of APC promoter methylation in hepatocellular carcinoma and its prognostic implications. Oncol Lett. 2014;7(5):1683–8. doi: 10.3892/ol.2014.1951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zekri AR, Bahnasy AA, Shoeab FE, Mohamed WS, El-Dahshan DH, Ali FT, et al. Methylation of multiple genes in hepatitis C virus associated hepatocellular carcinoma. J Adv Res. 2014;5(1):27–40. doi: 10.1016/j.jare.2012.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Formeister EJ, Tsuchiya M, Fujii H, Shpyleva S, Pogribny IP, Rusyn I. Comparative analysis of promoter methylation and gene expression endpoints between tumorous and non-tumorous tissues from HCV-positive patients with hepatocellular carcinoma. Mutat Res. 2010;692(1-2):26–33. doi: 10.1016/j.mrfmmm.2010.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Benegiamo G, Vinciguerra M, Mazzoccoli G, Piepoli A, Andriulli A, Pazienza V. DNA methyltransferases 1 and 3b expression in Huh-7 cells expressing HCV core protein of different genotypes. Dig Dis Sci. 2012;57(6):1598–603. doi: 10.1007/s10620-012-2160-1. [DOI] [PubMed] [Google Scholar]
- 64.Chen C, Pan D, Deng AM, Huang F, Sun BL, Yang RG. DNA methyltransferases 1 and 3B are required for hepatitis C virus infection in cell culture. Virology. 2013;44(1):57–65. doi: 10.1016/j.virol.2013.03.005. [DOI] [PubMed] [Google Scholar]
- 65.Machida K, Liu JC, McNamara G, Levine A, Duan L, Lai MM. Hepatitis C virus causes uncoupling of mitotic checkpoint and chromosomal polyploidy through the Rb pathway. J Virol. 2009;83(23):12590–600. doi: 10.1128/JVI.02643-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Bellacchio E, Paggi MG. Understanding the targeting of the RB family proteins by viral oncoproteins to defeat their oncogenic machinery. J Cell Physiol. 2013;228(2):285–91. doi: 10.1002/jcp.24137. [DOI] [PubMed] [Google Scholar]
- 67.McGivern DR, Lemon SM. Tumor suppressors, chromosomal instability, and hepatitis C virus-associated liver cancer. Annu Rev Pathol. 2009;4:399–415. doi: 10.1146/annurev.pathol.4.110807.092202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Dick FA, Rubin SM. Molecular mechanisms underlying RB protein function. Nat Rev Mol Cell Biol. 2013;14(5):297–306. doi: 10.1038/nrm3567. [DOI] [PMC free article] [PubMed] [Google Scholar]