

Abstract
Pompe disease (glycogen storage disease type II; acid maltase deficiency) is a devastating myopathy resulting from acid α-glucosidase (GAA) deficiency in striated and smooth muscle. Despite the availability of enzyme replacement therapy (ERT) with recombinant human GAA (rhGAA), the limitations of ERT have prompted the preclinical development of gene therapy. Gene therapy has the advantage of continuously producing GAA, in contrast to ERT, which requires frequent injections of rhGAA. An adeno-associated viral (AAV) vector containing a muscle-specific promoter, AAV-MHCK7hGAApA, achieved high GAA expression in heart and skeletal muscle in mice with Pompe disease. However, elevated GAA activity was not sufficient to completely clear accumulated glycogen in skeletal muscle. The process of glycogen clearance from lysosomes might require improved trafficking of GAA to the lysosomes in skeletal muscle, previously achieved with the β2-agonist clenbuterol that enhanced glycogen clearance in skeletal muscle without increasing GAA activity. Glycogen clearance was clearly enhanced by treatment with a nondepleting anti-CD4 monoclonal antibody (anti-CD4 mAb) along with muscle-specific GAA expression in cardiac muscle, but that treatment was not effective in skeletal muscle. Furthermore, anti-CD4 mAb treatment along with clenbuterol achieved synergistic therapeutic efficacy in both cardiac and skeletal muscle. This triple therapy increased both muscle strength and weight gain. Overall, triple therapy to enhance GAA trafficking and to suppress immune responses significantly improved the efficacy of muscle-targeted gene therapy in murine Pompe disease.
Introduction
Pompe disease (glycogen storage disease type II; acid maltase deficiency) is a lysosomal storage disorder caused by a deficiency of lysosomal acid α-glucosidase (GAA) activity that results in the progressive intralysosomal accumulation of glycogen. The most severely affected tissues are cardiac and skeletal muscle. Symptoms of Pompe disease include hypotonia, muscle weakness, cardiomyopathy, and respiratory failure. Enzyme replacement therapy (ERT) using recombinant human GAA (rhGAA) was approved in 2006, which is currently the only U.S. Food and Drug Administration (FDA)-approved therapy for Pompe disease. Since approval the limitations of ERT in Pompe disease have become obvious. Even in patients with a good response to ERT, residual motor weakness (neck flexor weakness, dorsiflexor weakness, myopathic facies, ptosis, and strabismus) has been observed.1–3 Thus, the correction of neuromuscular involvement has not been possible in Pompe disease, despite adherence to standard-of-care ERT.
Patients with Pompe disease without any residual GAA are deemed cross-reacting immune material (CRIM) negative. CRIM-negative patients have been shown to be poor ERT responders, who form high sustained anti-rhGAA IgG antibody titers (HSAT). Patients with HSAT have demonstrated greatly increased mortality, in comparison with CRIM-positive patients, who usually form no or only low-titer antibodies.4 The poor outcome of CRIM-negative infants occurs despite beginning ERT early in life. Furthermore, suppressing the formation of anti-rhGAA antibodies by immunosuppression significantly prolongs the survival of CRIM-negative infants.5,6 The relevance of antibody formation to therapeutic efficacy in Pompe disease has been emphasized by the poor response of CRIM-negative patients to ERT, which correlates with the onset of HSAT.4,7
To overcome the obstacle posed by HSAT, we have focused on developing tools for the suppression of immune responses as well as induction of immune tolerance against introduced GAA. A single administration of a nondepleting anti-CD4 monoclonal antibody (mAb) before administration of an AAV2/9 vector encoding GAA significantly reduced formation of anti-GAA IgGs, including IgG1, IgG2a, IgG2b, IgG2c, and IgG3.8 Anti-CD4 mAb along with a vector containing a constitutive promoter, AAV2/9-CBhGAApA, significantly increased GAA activity in muscle, resulting in a significant reduction of glycogen accumulation in the heart and to a lesser extent in skeletal muscle.
ERT depends on the uptake of rhGAA at the surface of the plasma membrane and trafficking to lysosomes, which is mediated by the cation-independent mannose 6-phosphate receptor (CI-MPR). The paucity of CI-MPR in mammalian adult muscle has underscored the concept that CI-MPR expression is one of the factors limiting the efficacy of ERT in Pompe disease.9,10 We demonstrated that increased CI-MPR expression improved efficacy from ERT in GAA-knockout (GAA-KO) mice, confirming the relevance of CI-MPR expression in therapy for Pompe disease.11 Using GAA-KO mice, we showed that clenbuterol, a selective β2-adrenergic receptor agonist, increased the expression of CI-MPR in muscle, and increased the efficacy of either ERT or gene therapy in murine Pompe disease.11–13 The underlying mechanism of the therapeutic action of clenbuterol might be related to insulin-like growth factor (IGF)-1-mediated muscle hypertrophy, which has been correlated with increased CI-MPR (also known as IGF-2 receptor) expression.14
In the present study, we evaluated a combination of anti-CD4 mAb and clenbuterol to induce immune tolerance and to increase CI-MPR-mediated uptake and trafficking of GAA, respectively. A single injection of anti-CD4 mAb blocked anti-GAA antibody formation, which was provoked by ubiquitous GAA expression with an AAV vector in GAA-KO mice.8 Simultaneous clenbuterol administration could induce CI-MPR expression in striated muscle.13 The AAV vector containing a muscle-specific promoter, AAV2/8-MHCK7hGAApA, expressed GAA in both cardiac and skeletal muscle.15 Combined anti-CD4 mAb and clenbuterol with AAV2/8-MHCK7hGAApA, termed triple therapy, revealed a synergistic therapeutic effect on biochemical correction in cardiac and skeletal muscle, as well as improved muscle strength, after AAV vector administration in GAA-KO mice.
Materials and Methods
Preparation of AAV vectors
AAV2/8-MHCK7hGAApA is composed of the MHCK7 (α-myosin heavy chain enhancer/muscle creatine kinase enhancer-promoter) regulatory cassette, human GAA cDNA, and a human growth hormone polyadenylation sequence, flanked by the AAV2 terminal repeat.16 The vector was produced in HEK 293 cells and purified by centrifugation through a cesium chloride gradient. Briefly, 293 cells were transfected with an AAV vector plasmid, the AAV8 packaging plasmid,17 and pAdHelper (Stratagene, La Jolla, CA). After 48 hr cells were harvested and freeze–thawed three times. AAV vectors were isolated by sucrose cushion pelleting followed by two cesium chloride gradient centrifugation steps. AAV stocks were dialyzed against three changes of phosphate-buffered saline with 5% sorbitol added to the third dialysis, and aliquots were stored at −80°C until use. The numbers of vector particles were determined by Southern blot analysis after digestion with DNase I and extraction of the digested DNA fragments. Viral vectors were handled under Biohazard Safety Level 2 guidelines published by the National Institutes of Health (Bethesda, MD).
Animal studies
All animal procedures were performed under the guidelines of the Duke University (Durham, NC) Institutional Animal Care and Use Committee. Two-month-old GAA-KO mice were administered 200 μl of anti-CD4 antibody (0.1 mg/ml) via intravenous injection on day −1. The mice were injected with 1.0 × 1011 vector particles (VP) of AAV2/8-MHCK7hGAApA via the tail vein on day 1. Clenbuterol (6 μg/ml) was continuously provided in the drinking water from day 1. Rotarod and wirehang testing were performed on day 0, week 6, and week 12. The mice were killed 13 weeks after AAV vector injection for collection of tissues. GAA activity and glycogen content from the collected tissues were analyzed as described.18
Statistical analysis
Data analysis was performed with GraphPad Prism 5 (GraphPad Software, San Diego, CA). Multiple comparisons were performed by two-way analysis of variance with Bonferroni's comparison test. The reference group was triple therapy (anti-CD4 mAb plus clenbuterol plus AAV2/8-MHCK7hGAApA), except for CI-MPR quantification, which used both untreated and triple-therapy groups as the reference (see Fig. 3). The statistical significance of comparisons is indicated as follows: *p < 0.05, **p < 0.01, or ***p < 0.001.
Figure 3.

Effects of therapy on cation-independent mannose 6-phosphate receptor (CI-MPR) expression in GAA-KO mice. Expression of GAA with AAV2/8-MHCK7hGAApA induced CI-MPR expression in quadriceps and gastrocnemius. Western blotting was performed on three mice from each group, and signal intensity was quantified with Quantity One (Bio-Rad, Hercules, CA) for quadriceps, gastrocnemius, heart, and liver. CI-MPR was normalized to the signal for glyceraldehyde-3-phosphate dehydrogenase (GAPDH). Statistical analysis was performed by two-way analysis of variance with Bonferroni's comparison test, using both the untreated and combination therapy groups as the reference in separate analyses (*p < 0.05, **p < 0.01, ***p < 0.001). Shown are means and standard deviation. Clen, clenbuterol.
Results
Different patterns between cardiac muscle and skeletal muscle in GAA activity and glycogen clearance
Triple therapy was evaluated in 2-month-old GAA-KO mice given clenbuterol (6 μg/ml in drinking water) and a single treatment with anti-CD4 mAb (20 μg/mouse) administered 2 days before administering the muscle-specific GAA expression vector at a low dose (1.0 × 1011 VP). Triple therapy increased GAA activity in the heart of GAA-KO mice at 13 weeks, in comparison with each of the other treatments (p < 0.001; Fig. 1A). However, GAA activity was not significantly elevated in skeletal muscle by any of the treatments (Fig. 1A).
Figure 1.
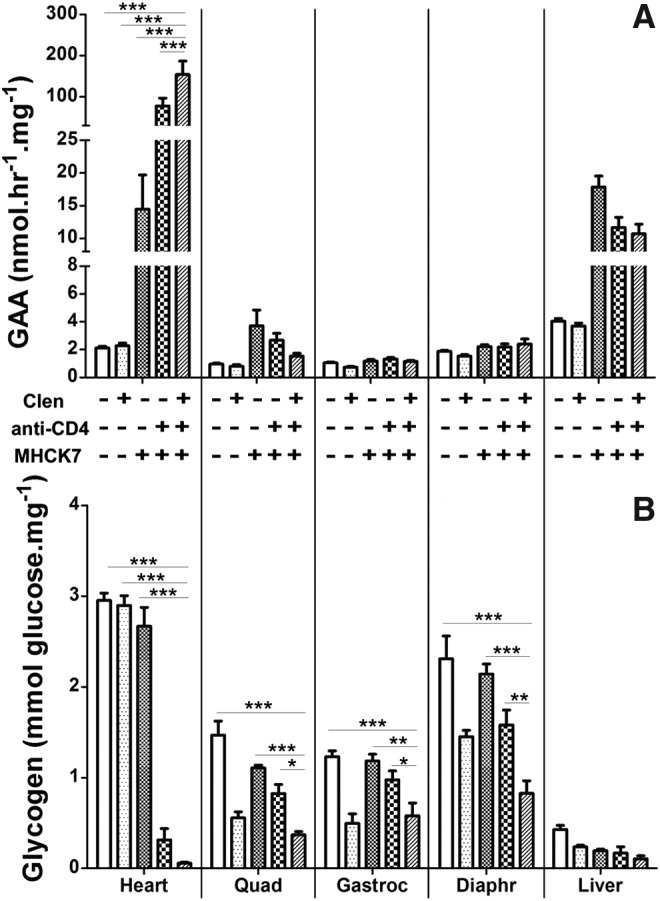
Biochemical correction by muscle-targeted gene therapy with clenbuterol and anti-CD4 monoclonal antibody (mAb). (A) Acid α-glucosidase (GAA) activity and (B) glycogen content were analyzed to demonstrate biochemical correction of Pompe disease by combinations of clenbuterol (Clen), anti-CD4 mAb, and AAV2/8-MHCK7hGAApA (MHCK7) in GAA-knockout (GAA-KO) mice. Group size was as follows: no-treatment control (n = 6), clenbuterol only (n = 6), MHCK7 only (n = 4), MHCK7 with anti-CD4 mAb (n = 7), triple therapy with MHCK7, clenbuterol, and anti-CD4 mAb (n = 7). Statistical analysis was performed by two-way analysis of variance with Bonferroni's comparison test, using the triple-therapy group as the reference (*p < 0.05, **p < 0.01, ***p < 0.001). Shown are means and standard deviation. Quad, quadriceps; Gastroc, gastrocnemius; Diaphr, diaphragm.
The administration of AAV2/8-MHCK7hGAApA has reduced accumulated lysosomal glycogen in Pompe disease; however, a low dose of the vector was not sufficient to remove accumulated glycogen in the heart and quadriceps, possibly because of inefficient trafficking of GAA to lysosomes.15 We observed the same phenomenon in the current study; however, the greatest clearance of the accumulated glycogen in heart was observed by adding anti-CD4 mAb treatment to suppress anti-GAA formation. A synergistic effect on glycogen clearance was demonstrated after triple therapy in the heart, diaphragm, and quadriceps, which reduced glycogen content to the lowest concentration observed in each of these muscles (Fig. 1B).
Therapeutic benefit from anti-CD4 mAb is comparable to AAV-mediated liver-specific expression of GAA
One of critical challenges in Pompe gene therapy is antibody formation against introduced GAA regardless of the source of GAA, either ERT or AAV-based expression vectors. In our previous studies immune tolerance was induced to GAA in mice by a liver-specific GAA expression cassette, AAV2/8-LSPhGAApA.19–21 Once immune tolerance was induced by AAV2/8-LSPhGAApA, GAA-KO mice did not form antibodies in response to an immune challenge with rhGAA. Therefore, we compared the immune-tolerogenic effects of the anti-CD4 mAb and AAV2/8-LSPhGAApA, because a single injection of anti-CD4 mAb was sufficient to prevent immune responses against AAV-MHCK7hGAApA.21 In this experiment, two AAV vectors were injected at a modest dose: 1 × 1011 VP of AAV2/8-MHCK7hGAApA and 5 × 1010 VP of AAV2/8-LSPhGAApA. The dual vector-injected mice had increased GAA activity in the heart that correlated with significantly reduced glycogen content (Fig. 2). Similarly, anti-CD4 mAb and AAV2/8-MHCK7hGAApA administration increased GAA activity in the heart. Only in the liver did dual vector administration significantly increase GAA activity, in comparison with anti-CD4 plus AAV2/8-MHCK7hGAApA (Fig. 2A). The result was attributed to administration of the liver-specific GAA expression vector that produced additional GAA activity in liver. However, administration of anti-CD4 mAb and AAV2/8-MHCK7hGAApA achieved a comparable therapeutic effect regarding glycogen clearance in all muscles, in comparison with dual vector administration, except for the diaphragm, where dual vector treatment was more effective (Fig. 2B). Overall, the addition of anti-CD4 mAb was almost as effective as administration of the additional liver-expressing vector regarding biochemical correction.
Figure 2.

Comparison of biochemical correction after immune tolerance induction with anti-CD4 mAb or liver-specific expression of GAA. Effects of anti-CD4 mAb and the liver-targeted AAV vector, AAV2/8-LSPGAApA (LSP), were compared. Biochemical correction was compared regarding (A) GAA activity and (B) glycogen content in striated muscle, including cardiac muscle and skeletal muscle. Group size was as follows: no-treatment control (n = 6), LSP with MHCK7 (n = 7), triple therapy with MHCK7, clenbuterol (Clen), and anti-CD4 mAb (n = 7), wild type (n = 5). *p < 0.05, ***p < 0.001. Shown are means and standard deviation. Quad, quadriceps; Gastroc, gastrocnemius.
CI-MPR expression is increased by GAA expression
Receptor-mediated uptake of GAA, like other lysosomal enzymes, occurs mainly through a receptor-mediated endocytosis mechanism using CI-MPR,22,23 and clenbuterol treatment during ERT or gene therapy increased CI-MPR expression in the skeletal muscle of GAA-KO mice with positive therapeutic effects.11,13 In this study, clenbuterol significantly increased the expression of CI-MPR in skeletal muscle (Fig. 3), including the quadriceps (p < 0.01) and gastrocnemius (p < 0.05). Surprisingly, the expression of CI-MPR was much higher after AAV-MHCK7hGAApA administration, either alone or in combination with anti-CD4 mAb (Fig. 3).
Effects of biochemical correction on body weight and muscle strength
Over the course of this 13-week experiment, the GAA-KO mice treated with triple therapy gained significantly more body weight, in comparison with untreated mice (Fig. 4A). Triple therapy also increased weight gain, in comparison with anti-CD4 plus AAV2/8-MHCK7hGAApA (p < 0.05) or AAV2/8-MHCK7hGAApA alone (p < 0.05; Fig. 4A). The induction of immune tolerance was suggested by the lack of anti-GAA antibody formation in mice treated with anti-CD4, in contrast to treatment with AAV2/8-MHCK7hGAApA alone, which provoked anti-GAA (Fig. 4B). Triple therapy increased muscle strength as evaluated by the wirehang test at week 12, but not Rotarod latency, in comparison with AAV2/8-MHCK7hGAApA alone (Fig. 4C and D). Wirehang testing has previously detected changes in muscle strength with more sensitivity than Rotarod testing.13 Treatment with clenbuterol alone increased wirehang latency, in comparison with untreated mice (Fig. 4D). Similarly, the addition of clenbuterol during triple therapy increased wirehang latency, in comparison with anti-CD4 mAb plus AAV2/8-MHCK7hGAApA (Fig. 4D). Anti-CD4 mAb administration increased wirehang latency, in comparison with AAV2/8-MHCK7hGAApA alone (Fig. 4D). In summary, additive effects from clenbuterol and anti-CD4 mAb administration were observed in vector-treated mice regarding biochemical correction (Fig. 1) and muscle function (Fig. 4), consistent with a synergistic effect from this triple therapy.
Figure 4.

Synergistic effects from clenbuterol (Clen) and anti-CD4 mAb on weight gain and muscle function. The effect of each treatment on (A) body weight, (B) anti-GAA, (C) Rotarod latency, and (D) wirehang latency was evaluated. *p < 0.05, ***p < 0.001. Shown are means and standard deviation. OD, optical density.
Discussion
Pompe disease is a neuromuscular disorder caused by the deficiency of lysosomal enzyme GAA, currently treated by ERT. However, ERT induces severe immune responses to rhGAA that reduce therapeutic efficacy, especially in infantile-onset patients. The formation of HSAT blocked the uptake of GAA and provoked fatal hypersensitivity reactions in GAA-KO mice with Pompe disease.20 Although Nayak and colleagues demonstrated that immune reactions were mediated by a helper T cell type 2 response associated with high-titer IgG1 during ERT,24 we observed alterations in many subclasses of immunoglobulins, implying a more complex response to AAV vector-mediated GAA expression in GAA-KO mice,8 Therefore, we have developed coreceptor blockade to eliminate CD4+ T lymphocyte help, and combined this immune tolerance induction with β2-agonists to enhance CI-MPR-mediated uptake of GAA during gene therapy in Pompe disease.
Strategies to overcome the obstacles of immune responses and inefficient uptake of GAA in skeletal muscle were developed independently to enhance Pompe therapy. Induction of immune tolerance was achieved by two different approaches: one approach was focused on the suppression of immune responses through liver-specific expression of GAA25 and the second acted through blockade of helper T lymphocyte activation with anti-CD4 mAb.8 At present the need for the induction of immune tolerance is greatest for patients with infantile-onset Pompe disease. In the meantime, the need to induce CI-MPR expression for improvement of GAA uptake in skeletal muscle is greatest for patients with late-onset Pompe disease. However, it is conceivable that infantile-onset patients will require enhancement of GAA uptake in skeletal muscle to address residual neuromuscular defects despite ERT.26 Therefore, the current triple therapy was evaluated to provide a more comprehensive therapy for Pompe disease. Surprisingly, we found a synergistic therapeutic effect from treatment with clenbuterol and anti-CD4 mAb in the context of muscle-directed gene therapy, resulting in higher biochemical efficacy that was comparable to the result produced by the liver-specific GAA expression method using AAV2/8-LSPhGAApA. The latter represents a well-established strategy for immune suppression and widespread correction of skeletal muscle in GAA-KO mice. Our group has confirmed the induction of immune tolerance using AAV vector-mediated liver-specific GAA expression, but that method has not been translated to clinical use. Therefore, the development of alternative methods for immune tolerance induction including blockade with nondepleting anti-CD4 mAb is justified.
In the current study, we found that glycogen clearance was dependent not only on increasing GAA activity, but also on β-adrenergic receptor-mediated endocytosis and trafficking to the lysosomes. We previously found that receptor-mediated uptake of GAA could be enhanced via increased CI-MPR expression, using β2-agonists including clenbuterol and albuterol.11 However, a muscle-specific GAA expression vector, AAV2/8-MHCK7hGAApA, did not clear accumulated glycogen from the heart and skeletal muscles despite a large increase in the observed GAA activity.15 We observed this phenomenon again in the current study (Fig. 1). GAA activity was dramatically elevated by treatment with vector alone, but the elevated GAA activity was not sufficient to remove accumulated glycogen in muscle. Moreover, the efficiency of glycogen clearance was clearly increased by treatment with clenbuterol in skeletal muscle, including quadriceps, gastrocnemius, and diaphragm. As previously demonstrated, clenbuterol alone reduced glycogen content in skeletal muscle in the absence of GAA replacement by AAV vector administration in GAA-KO mice.15 Treatment with a β2-agonist was well tolerated and efficacious in an open label clinical trial in combination with ERT,27 and this adjunctive therapy would be feasible in the context of gene therapy. Furthermore, the reduction of glycogen in cardiac muscle was dependent on immune tolerance induction with anti-CD4 mAb. Therefore, immunosuppression improved efficacy in cardiac muscle, whereas CI-MPR induction was effective in skeletal muscle.
In previous studies using GAA-KO mice, clenbuterol increased CI-MPR expression in a skeletal muscle (gastrocnemius) during gene therapy with a liver-specific GAA expression vector, AAV2/8-LSPhGAApA.13 The current study demonstrated increased CI-MPR expression in quadriceps and gastrocnemius by a muscle-specific GAA expression vector, AAV2/8-MHCK7hGAApA (Fig. 3). Furthermore, the ability of muscle-specific GAA expression to increase CI-MPR expression was unprecedented, and possibly resulted from the effect of biochemical correction on CI-MPR recycling. Thus, the prevalence of CI-MPR was modulated by both β2-agonist and AAV vector administration.
Liver-specific expression of GAA sustained immune tolerance to subsequent administration of rhGAA,28 and we observed a similar beneficial effect from the current strategy of anti-CD4 mAb pretreatment combined with muscle-specific expression of GAA. In this study, we also demonstrated that immune suppression by anti-CD4 mAb could increase GAA activity in heart and skeletal muscle, using a muscle-expressing AAV vector (Fig. 1), which was similar to the effect of anti-CD4 mAb accompanied by ubiquitous expression of GAA.8 Thus, the prevention of anti-GAA formation by any method might enhance the efficacy of GAA activity in heart and skeletal muscles, regardless of enzyme sources, either transgene-mediated from gene therapy or from ERT.
Multiple approaches for the induction of immune tolerance to a therapeutic protein (human factor IX) have been demonstrated in models for hemophilia, each of which might be adapted to the treatment of Pompe disease, including (1) liver-specific expression of therapeutic genes,29,30 (2) neonatal exposure to the protein,31,32 (3) expression of the therapeutic gene in precursor forms of immunologic cells33 or in tolerogenic B cells,34 or (4) blockade of coreceptors.35 Coreceptor blockade has advantages over transgene-mediated approaches, at least regarding lack of toxicity from chromosomal integration or vector-directed immune responses.36 Furthermore, coreceptor blockade by treatment with a nondepleting anti-CD4 mAb has achieved efficacy from low dosages of vector,8 which could avoid T cell responses directed toward AAV capsid proteins.37
Triple therapy with anti-CD4, clenbuterol, and AAV2/8-MHCK7hGAApA enhanced biochemical correction, which correlated with changes in body weight and muscle strength of GAA-KO mice. Clenbuterol treatment previously increased weight gain by GAA KO mice, in comparison with untreated GAA-KO mice, over the course of a 4-week experiment.13 In this experiment body weight increased after 3 months of exposure to clenbuterol in GAA-KO mice (Fig. 4A). In addition, we also observed a synergistic effect on weight gain from triple therapy. Similarly, synergistic effects on muscle function were observed, as demonstrated by the increased latency in the wirehang test (Fig. 4D). In this study, a combination therapy using anti-CD4 mAb coreceptor blockade and clenbuterol for activation of β-adrenergic receptors clearly enhanced muscle-targeted gene therapy in murine Pompe disease.
Acknowledgments
This work was supported by grant 241701 from the Muscular Dystrophy Association (USA) and by a grant from Genethon. The AAV packaging plasmids were provided courtesy of Dr. James M. Wilson (University of Pennsylvania, Philadelphia, PA). GAA-KO mice were provided courtesy of Dr. Nina Raben (National Institute of Arthritis and Musculoskeletal and Skin Diseases). Nondepleting anti-CD4 mAb (YTS177) was provided by Tolerx (Cambridge, MA).
Author Disclosure
Dr. Koeberl has been awarded a patent for the use of β2-agonists in the treatment of Pompe disease.
References
- 1.Nicolino M, Byrne B, Wraith JE, et al. . Clinical outcomes after long-term treatment with alglucosidase alfa in infants and children with advanced Pompe disease. Genet Med 2009;11:210–219 [DOI] [PubMed] [Google Scholar]
- 2.Jones HN, Muller CW, Lin M, et al. . Oropharyngeal dysphagia in infants and children with infantile Pompe disease. Dysphagia 2009;25:277–283 [DOI] [PubMed] [Google Scholar]
- 3.Yanovitch TL, Banugaria SG, Proia AD, et al. . Clinical and histologic ocular findings in pompe disease. J Pediatr Ophthalmol Strabismus 2010;47:34–40 [DOI] [PubMed] [Google Scholar]
- 4.Banugaria SG, Prater SN, Ng YK, et al. . The impact of antibodies on clinical outcomes in diseases treated with therapeutic protein: lessons learned from infantile Pompe disease. Genet Med 2011;13:729–736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Messinger YH, Mendelsohn NJ, Rhead W, et al. . Successful immune tolerance induction to enzyme replacement therapy in CRIM-negative infantile Pompe disease. Genet Med 2012;14:135–142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Banugaria SG, Prater SN, Patel TT, et al. . Algorithm for the early diagnosis and treatment of patients with cross reactive immunologic material-negative classic infantile pompe disease: a step towards improving the efficacy of ERT. PLoS One 2013;8:e67052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kishnani PS, Goldenberg PC, DeArmey SL, et al. . Cross-reactive immunologic material status affects treatment outcomes in Pompe disease infants. Mol Genet Metab 2010;99:26–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Han SO, Li S, Brooks ED, et al. . Enhanced efficacy from gene therapy in Pompe disease using coreceptor blockade. Hum Gene Ther 2015;26:26–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Raben N, Danon M, Gilbert AL, et al. . Enzyme replacement therapy in the mouse model of Pompe disease. Mol Genet Metab 2003;80:159–169 [DOI] [PubMed] [Google Scholar]
- 10.Raben N, Fukuda T, Gilbert AL, et al. . Replacing acid α-glucosidase in Pompe disease: recombinant and transgenic enzymes are equipotent but neither completely clears glycogen from type II muscle fibers. Mol Ther 2005;11:48–56 [DOI] [PubMed] [Google Scholar]
- 11.Koeberl DD, Li S, Dai J, et al. . β2-Agonists enhance the efficacy of simultaneous enzyme replacement therapy in murine Pompe disease. Mol Genet Metab 2012;105:221–227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Koeberl DD, Luo X, Sun B, et al. . Enhanced efficacy of enzyme replacement therapy in Pompe disease through mannose-6-phosphate receptor expression in skeletal muscle. Mol Genet Metab 2011;103:107–112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Li S, Sun B, Nilsson MI, et al. . Adjunctive β2-agonists reverse neuromuscular involvement in murine Pompe disease. FASEB J 2013;27:34–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Matsumoto T, Akutsu S, Wakana N, et al. . The expressions of insulin-like growth factors their receptors and binding proteins are related to the mechanism regulating masseter muscle mass in the rat. Arch Oral Biol 2006;51:603–611 [DOI] [PubMed] [Google Scholar]
- 15.Farah BL, Madden L, Li S, et al. . Adjunctive β2-agonist treatment reduces glycogen independently of receptor-mediated acid α-glucosidase uptake in the limb muscles of mice with Pompe disease. FASEB J 2014;28:2272–2280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sun B, Young SP, Li P, et al. . Correction of multiple striated muscles in murine Pompe disease through adeno-associated virus-mediated gene therapy. Mol Ther 2008;16:1366–-1371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gao GP, Alvira MR, Wang L, et al. . Novel adeno-associated viruses from rhesus monkeys as vectors for human gene therapy. Proc Natl Acad Sci U S A 2002;99:11854–11859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sun B, Zhang H, Franco LM, et al. . Correction of glycogen storage disease type II by an adeno-associated virus vector containing a muscle-specific promoter. Mol Ther 2005;11:889–898 [DOI] [PubMed] [Google Scholar]
- 19.Franco LM, Sun B, Yang X, et al. . Evasion of immune responses to introduced human acid α-glucosidase by liver-restricted expression in glycogen storage disease type II. Mol Ther 2005;12:876–884 [DOI] [PubMed] [Google Scholar]
- 20.Sun B, Kulis MD, Young SP, et al. . Immunomodulatory gene therapy prevents antibody formation and lethal hypersensitivity reactions in murine Pompe disease. Mol Ther 2010;18:353–360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhang P, Sun B, Osada T, et al. . Immunodominant liver-specific expression suppresses transgene-directed immune responses in murine pompe disease. Hum Gene Ther 2012;23:460–472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cardone M, Porto C, Tarallo A, et al. . Abnormal mannose-6-phosphate receptor trafficking impairs recombinant α-glucosidase uptake in Pompe disease fibroblasts. Pathogenetics 2008;1:6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhu Y, Jiang JL, Gumlaw NK, et al. . Glycoengineered acid α-glucosidase with improved efficacy at correcting the metabolic aberrations and motor function deficits in a mouse model of Pompe disease. Mol Ther 2009;17:954–963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nayak S, Sivakumar R, Cao O, et al. . Mapping the T helper cell response to acid α-glucosidase in Pompe mice. Mol Genet Metab 2012;106:189–195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sun B, Bird A, Young SP, et al. . Enhanced response to enzyme replacement therapy in Pompe disease after the induction of immune tolerance. Am J Hum Genet 2007;81:1042–1049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Prater SN, Banugaria SG, DeArmey SM, et al. . The emerging phenotype of long-term survivors with infantile Pompe disease. Genet Med 2012;14:800–810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Koeberl DD, Austin S, Case LE, et al. . Adjunctive albuterol enhances the response to enzyme replacement therapy in late-onset Pompe disease. FASEB J 2014;28:2171–2176 [DOI] [PubMed] [Google Scholar]
- 28.Wang S, Young S, Bali D, et al. . Assessment of toxicity and biodistribution of recombinant AAV2/8 vector-mediated immunomodulatory gene therapy in mice with Pompe disease. Mol Ther Methods Clin Dev 2014;1:14018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mingozzi F, Liu YL, Dobrzynski E, et al. . Induction of immune tolerance to coagulation factor IX antigen by in vivo hepatic gene transfer. J Clin Invest 2003;111:1347–1356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hoffman BE, Dobrzynski E, Wang L, et al. . Muscle as a target for supplementary factor IX gene transfer. Hum Gene Ther 2007;18:603–613 [DOI] [PubMed] [Google Scholar]
- 31.Ohlfest JR, Frandsen JL, Fritz S, et al. . Phenotypic correction and long-term expression of factor VIII in hemophilic mice by immunotolerization and nonviral gene transfer using the Sleeping Beauty transposon system. Blood 2005;105:2691–2698 [DOI] [PubMed] [Google Scholar]
- 32.Hu C, Cela RG, Suzuki M, et al. . Neonatal helper-dependent adenoviral vector gene therapy mediates correction of hemophilia A and tolerance to human factor VIII. Proc Natl Acad Sci U S A 2011;108:2082–2087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Doering CB, Gangadharan B, Dukart HZ, et al. . Hematopoietic stem cells encoding porcine factor VIII induce pro-coagulant activity in hemophilia A mice with pre-existing factor VIII immunity. Mol Ther 2007;15:1093–1099 [DOI] [PubMed] [Google Scholar]
- 34.Wang X, Moghimi B, Zolotukhin I, et al. . Immune tolerance induction to factor IX through B cell gene transfer: TLR9 signaling delineates between tolerogenic and immunogenic B cells. Mol Ther 2014;22:1139–1150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Peng B, Ye P, Rawlings DJ, et al. . Anti-CD3 antibodies modulate anti-factor VIII immune responses in hemophilia A mice after factor VIII plasmid-mediated gene therapy. Blood 2009;114:4373–4382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chuah MK, Nair N, and VandenDriessche T. Recent progress in gene therapy for hemophilia. Hum Gene Ther 2012;23:557–565 [DOI] [PubMed] [Google Scholar]
- 37.Manno CS, Pierce GF, Arruda VR, et al. . Successful transduction of liver in hemophilia by AAV-Factor IX and limitations imposed by the host immune response. Nat Med 2006;12:342–347 [DOI] [PubMed] [Google Scholar]