

Abstract
Sphingobium sp. strain SYK-6 is able to degrade various lignin-derived biaryls, including a phenylcoumaran-type compound, dehydrodiconiferyl alcohol (DCA). In SYK-6 cells, the alcohol group of the B-ring side chain of DCA is initially oxidized to the carboxyl group to generate 3-(2-(4-hydroxy-3-methoxyphenyl)-3-(hydroxymethyl)-7-methoxy-2,3-dihydrobenzofuran-5-yl) acrylic acid (DCA-C). Next, the alcohol group of the A-ring side chain of DCA-C is oxidized to the carboxyl group, and then the resulting metabolite is catabolized through vanillin and 5-formylferulate. In this study, the genes involved in the conversion of DCA-C were identified and characterized. The DCA-C oxidation activities in SYK-6 were enhanced in the presence of flavin adenine dinucleotide and an artificial electron acceptor and were induced ca. 1.6-fold when the cells were grown with DCA. Based on these observations, SLG_09480 (phcC) and SLG_09500 (phcD), encoding glucose-methanol-choline oxidoreductase family proteins, were presumed to encode DCA-C oxidases. Analyses of phcC and phcD mutants indicated that PhcC and PhcD are essential for the conversion of (+)-DCA-C and (−)-DCA-C, respectively. When phcC and phcD were expressed in SYK-6 and Escherichia coli, the gene products were mainly observed in their membrane fractions. The membrane fractions of E. coli that expressed phcC and phcD catalyzed the specific conversion of DCA-C into the corresponding carboxyl derivatives. In the oxidation of DCA-C, PhcC and PhcD effectively utilized ubiquinone derivatives as electron acceptors. Furthermore, the transcription of a putative cytochrome c gene was significantly induced in SYK-6 grown with DCA. The DCA-C oxidation catalyzed by membrane-associated PhcC and PhcD appears to be coupled to the respiratory chain.
INTRODUCTION
Lignin, one of the major components of plant cell walls, is a complex phenolic polymer resulting from the oxidative combinatorial coupling of 4-hydroxycinnamyl alcohols (1). Although lignin has various intermolecular linkages between phenylpropane units and contains a number of asymmetric carbons, it is considered to be optically inactive, implying the racemic nature of the lignin backbone (2–4). In nature, lignin is initially decomposed by phenol oxidases such as lignin peroxidase, manganese peroxidase, versatile peroxidase, and laccase secreted by white rot fungi (5–7). Recently, dye-decolorizing peroxidases (Dyp) of Rhodococcus (8) and Amycolatopsis (9) and small laccase of Streptomyces (10) were characterized, and these enzymes have been implicated as being involved in lignin degradation. In addition, bacteria play key roles in the degradation and mineralization of low-molecular-weight aromatic compounds derived from lignin (11, 12). Since fragmented oligomers from lignin consist of stereoisomers that contain various types of intermolecular linkages between phenylpropane units, catabolic enzymes necessary for the conversion of such stereoisomers must have evolved in bacteria to fully utilize structurally and stereochemically complicated lignin-derived aromatics as a source of carbon and energy. In fact, we found the involvement of three stereospecific dehydrogenases (Cα-dehydrogenases) (13) and three enantioselective glutathione S-transferases (β-etherases) (14, 15), respectively, of Sphingobium sp. strain SYK-6 in the conversion of four different stereoisomers of guaiacylglycerol-β-guaiacyl ether into enantiomers of α-(2-methoxyphenoxy)-β-hydroxypropiovanillone (MPHPV) and the ether cleavage of enantiomers of MPHPV. Therefore, detailed analyses of the enzyme genes in each catabolic pathway of lignin-derived aromatics are essential to fully understand bacterial lignin degradation. Furthermore, an understanding of these catabolic systems will establish a solid foundation for the challenges encountered in lignin valorization, including the production of value-added chemicals from lignin (16–18).
An alphaproteobacterium, Sphingobium sp. strain SYK-6, is the best-characterized bacterial degrader of lignin-derived aromatic compounds (19). The catabolic pathways and catabolic genes in this strain for β-aryl ether (13–15), biphenyl (20–22), pinoresinol (23), ferulate (24, 25), vanillin (26, 27), and syringate (28) have been extensively characterized. However, the catabolic genes for a phenylcoumaran compound, dehydrodiconiferyl alcohol (DCA), remain largely unknown. The phenylcoumaran linkage accounts for 3 to 10% of the total intermolecular linkages in lignin (29), and DCA is known as one of the major dilignols generated in lignifying xylem (30, 31). An outline of the catabolic pathway of DCA in Sphingomonas paucimobilis TMY1009 was first proposed by Habu et al. (32). We later found a similar catabolic route for DCA in SYK-6, and provided a detailed characterization (Fig. 1) (33). In SYK-6 cells, the alcohol group of the B-ring side chain of DCA was first oxidized to the carboxyl group to generate 3-(2-(4-hydroxy-3-methoxyphenyl)-3-(hydroxymethyl)-7-methoxy-2,3-dihydrobenzofuran-5-yl) acrylic acid (DCA-C) via an aldehyde derivative, 3-(2-(4-hydroxy-3-methoxyphenyl)-3-(hydroxymethyl)-7-methoxy-2,3-dihydrobenzofuran-5-yl) acrylaldehyde (DCA-L). Then the resulting DCA-C was converted to 5-(2-carboxyvinyl)-2-(4-hydroxy-3-methoxyphenyl)-7-methoxy-2,3-dihydrobenzofuran-3-carboxylic acid (DCA-CC) by the two-step oxidations of the alcohol group of the A-ring side chain of DCA-C. DCA-CC was decarboxylated to produce 3-(4-hydroxy-3-(4-hydroxy-3-methoxystyryl)-5-methoxyphenyl) acrylic acid (DCA-S), and this lignostilbene was further cleaved between Cα and Cβ of the A-ring side chain to form vanillin and 5-formylferulate. Our previous study also suggested that the multiple genes encoding quinohemoprotein alcohol dehydrogenases (quinohemoprotein ADH) and aryl ADH, as well as four aldehyde dehydrogenase (ALDH) genes, are involved in the oxidation of DCA and DCA-L, respectively (33). In addition, Kamoda et al. reported that four isozymes of different lignostilbene α,β-dioxygenases of TMY1009 had the ability to cleave the interphenyl α, β double bond of DCA-S (34, 35). Nevertheless, no reports on the genes and enzymes involved in the conversions of DCA-C to DCA-S have been documented to date. Interestingly, quinohemoprotein ADH and aryl ADH, which were able to oxidize the alcohol group of the B-ring side chain of DCA, showed little or no activity in the oxidation of the alcohol group of the A-ring side chain of DCA-C (33). These facts suggested that other types of enzymes participate in the oxidation of DCA-C in SYK-6.
FIG 1.

(A) Proposed catabolic pathway of DCA in Sphingobium sp. strain SYK-6. Enzymes: ADHs, alcohol dehydrogenases; ALDHs, aldehyde dehydrogenases; PhcC and PhcD, DCA-C oxidases. (B) Gene organization of SLG_09480 (phcC) and SLG_09500 (phcD) involved in the oxidation of DCA-C. Arrows indicate the genes from SLG_09470 to SLG_09520. The thick line under the map indicates the location of amplified RT-PCR product shown in panel C. (C) Agarose gel electrophoresis of RT-PCR products (a phcC-phcD intergenic region) amplified with primers shown in Materials and Methods using the following templates: total DNA of SYK-6 (lane 1), reverse transcripts of total RNA of SYK-6 (lane 2), and total RNA of SYK-6 (lane 3). M, molecular size markers.
In this study, on the basis of the cofactor requirements and induction profiles of the enzyme activities for the conversion of DCA-C in SYK-6, we identified two genes encoding glucose-methanol-choline (GMC) oxidoreductase family proteins. Gene disruption experiments and characterization of the gene products uncovered the actual roles of these genes in the catabolism of DCA.
MATERIALS AND METHODS
Bacterial strains, plasmids, and culture conditions.
The strains and plasmids used in this study are listed in Table 1. Sphingobium sp. strain SYK-6 and its mutants were grown in lysogeny broth (LB; 10 g/liter of Bacto tryptone, 5 g/liter of yeast extract, and 5 g/liter of NaCl) and Wx minimal medium (25) containing SEMP (10 mM sucrose, 10 mM glutamate, 0.13 mM methionine, and 10 mM proline) and 2 mM DCA or DCA-C at 30°C. When necessary, 50 mg of kanamycin (Km)/liter was added to the cultures. Escherichia coli strains were grown in LB at 37°C. For cultures of cells carrying antibiotic resistance markers, the media for E. coli transformants were supplemented with 100 mg of ampicillin (Ap)/liter, 25 mg of Km/liter, or 12.5 mg of tetracycline (Tc)/liter.
TABLE 1.
Strains and plasmids used in this study
| Strain or plasmid | Relevant characteristic(s)a | Reference or source |
|---|---|---|
| Sphingobium sp. strains | ||
| SYK-6 | Wild type; Nalr Smr | 68 |
| SME110 | SYK-6 derivative; phcC::kan; Nalr Smr Kmr | This study |
| SME111 | SYK-6 derivative; phcD::kan; Nalr Smr Kmr | This study |
| SME112 | SYK-6 derivative; phcC-phcD::kan; Nalr Smr Kmr | This study |
| E. coli strains | ||
| BL21(DE3) | F− ompT hsdSB(rB− mB−) gal dcm (DE3); T7 RNA polymerase gene under the control of the lacUV5 promoter | 69 |
| HB101 | recA13 supE44 hsd20 ara-14 proA2 lacY1 galK2 rpsL20 xyl-5 mtl-1 | 70 |
| NEB 10-beta | araD139 Δ(ara-leu)7697 fhuA lacX74 galK (ϕ80 ΔlacZ M15) recA1 endA1 nupG rpsL (Smr) Δ(mrr-hsdRMS-mcrBC) | New England Biolabs |
| Plasmids | ||
| pT7Blue | Cloning vector; Apr | Novagen |
| pBluescript II KS(+) | Cloning vector; Apr | 71 |
| pET-16b | Expression vector; T7 promoter; Apr | Novagen |
| pJB866 | RK2 broad-host-range expression vector; Tcr Pm xylS | 72 |
| pJB861 | RK2 broad-host-range expression vector; Kmr Pm xylS | 72 |
| pK19mobsacB | oriT sacB; Kmr | 73 |
| pIK03 | pBluescript II KS(+) with a 1.3-kb EcoRV fragment carrying kan of pUC4K; Apr Kmr | 28 |
| pKS09480-500 | pBluescript II KS(+) with a 3.9-kb PCR-amplicon carrying phcC and phcD | This study |
| pKS09480 | pBluescript II KS(+) with a 2.4-kb BglII-BamHI fragment carrying phcC from pKS09480-500 | This study |
| pKS09500 | pBluescript II KS(+) with a 2.7-kb XhoI-SacI fragment carrying phcD from pKS09480-500 | This study |
| pKS09480K | pKS09480 with a 1.3-kb EcoRV fragment carrying kan from pIK03 into HincII site of phcC | This study |
| pKS09500K | pKS09500 with a 1.3-kb EcoRV fragment carrying kan from pIK03 into HincII site of phcD | This study |
| pKmb09480K | pK19mobsacB with a 3.7-kb KpnI-BamHI fragment carrying disrupted phcC of pKS09480K | This study |
| pKmb09500K | pK19mobsacB with a 4.0-kb KpnI-SacI fragment carrying disrupted phcD of pKS09500K | This study |
| pT09480-500 | pT7Blue with a 3.9-kb PCR-amplicon carrying phcC and phcD | This study |
| pT09480-500K | pT09480-500 with a 1.3-kb EcoRV fragment carrying kan from pIK03 into HincII site of phcC and phcD | This study |
| pKmb09480-500K | pK19mobsacB with a 3.0-kb SphI-BamHI fragment carrying disrupted phcC and phcD of pT09480-500K | This study |
| pT09480 | pT7Blue with a 1.7-kb PCR-amplicon carrying phcC | This study |
| pT09500 | pT7Blue with a 1.8-kb PCR-amplicon carrying phcD | This study |
| pT09480Xba | pT7Blue with a 1.8-kb XbaI-BamHI fragment carrying phcC from pT09480 | This study |
| pT09500Xba | pT7Blue with a 1.9-kb XbaI-BamHI fragment carrying phcD from pT09500 | This study |
| pET09480 | pET-16b with a 1.7-kb NdeI-BamHI fragment carrying phcC from pT09480 | This study |
| pET09500 | pET-16b with a 1.8-kb NdeI-BamHI fragment carrying phcD from pT09500 | This study |
| pJBVI09480 | pJB866 with a 1.8-kb HindIII-BamHI fragment carrying phcC from pT09480Xba | This study |
| pJBVI09500 | pJB866 with a 1.9-kb HindIII-BamHI fragment carrying phcD from pT09500Xba | This study |
| pJBI09480 | pJB861 with a 1.8-kb KpnI-BamHI fragment carrying phcC from pJBVI09480 | This study |
| pJBI09500 | pJB861 with a 1.9-kb KpnI-BamHI fragment carrying phcD from pJBVI09500 | This study |
Kmr, Nalr, Smr, Apr, and Tcr, resistance to kanamycin, nalidixic acid, streptomycin, ampicillin, and tetracycline, respectively.
Preparation of substrates.
DCA, DCA-L, DCA-C, DCA-CC, and DCA-S were prepared as described previously (33). Guaiacylglycerol-β-guaiacyl ether (GGE), coniferyl alcohol, and vanillyl alcohol were purchased from Tokyo Chemical Industry Co., Ltd., or Wako Pure Chemical Industries, Ltd. 2-(4-Hydroxy-3-methoxyphenyl)-5-(3-hydroxyprop-1-enyl)-7-methoxy-2,3-dihydrobenzofuran-3-carboxylic acid (DCA-AC) was prepared by selective reduction of DCA-CC. To a stirred solution of DCA-CC diethyl ester (221 mg; 0.5 mmol; prepared by oxidative coupling of ethyl ferulate) and imidazole (68 mg; 1.0 mmol) in tetrahydrofuran (8 ml), tert-butyldimethylsilyl chloride (TBSCl; 106 mg; 0.7 mmol) was added. After being stirred at room temperature for 12 h, the reaction mixture was concentrated in vacuo, and the residue was purified by a silica gel column chromatography (hexane-ethyl acetate, 5/1) to give TBS-protected DCA-CC diethyl ester (compound A; 248.3 mg; 89%) as a white solid. To a stirred solution of compound A (240 mg; 0.43 mmol) in tetrahydrofuran (20 ml) at −10°C, diisobutylaluminum hydride (1.04 M in hexane; 1.0 ml; 1.08 mmol) was slowly added. After being stirred at 0°C for 30 min, a saturated aqueous solution of NH4Cl was added. The reaction was warmed to room temperature and diluted with ethyl acetate. A saturated aqueous solution of Rochelle's salt was added, and the biphasic mixture was vigorously stirred for 6 h. The organic layer was separated and concentrated in vacuo, and the crude product was purified by silica gel column chromatography (hexane-ethyl acetate, 3/2) to give TBS-protected DCA-AC ethyl ester (compound B; 170.9 mg; 85%) as a colorless oil. To a stirred solution of compound B (55.9 mg; 0.109 mmol) in dioxane (1.0 ml), 2 N NaOH (0.5 ml) was added dropwise at room temperature. After being stirred for 1.5 h, 1 N HCl (2.0 ml) was added, and the reaction mixture was extracted by ethyl acetate. The organic layer was concentrated in vacuo, and the residue was purified by silica gel column chromatography (ethyl acetate-methanol, 10/1) to give DCA-AC (36.6 mg; 91%) as a white solid. 1H nuclear magnetic resonance (NMR) (400 MHz, acetone-d6): δ (ppm) 7.72 (1H, bs), 7.09 (1H, d, J = 2.0 Hz), 7.08 (1H, bs), 7.03 (1H, bs), 6.92 (1H, dd, J = 8.1, 2.0 Hz), 6.84 (1H, d, J = 8.1 Hz), 6.56 (1H, d, J = 15.6 Hz), 6.28 (1H, dt, J = 15.6, 5.4 Hz), 5.99 (1H, d, J = 8.1 Hz), 4.37 (1H, d, J = 8.1 Hz), 4.21 (1H, dd, J = 5.4, 1.0 Hz), 3.88 (3H, s), 3.85 (3H, s). 13C NMR (100 MHz, acetone-d6): δ (ppm) 172.2, 148.5, 148.4, 147.8, 145.5, 132.7, 132.4, 130.1, 129.0, 127.2, 120.0, 116.0, 115.8, 112.0, 110.6, 63.3, 56.4, 56.3, 56.2.
Enzyme assays.
Cells of SYK-6 grown in LB were washed with Wx medium, resuspended in Wx-SEMP to an optical density at 600 nm (OD600) of 0.2, and grown at 30°C. When the OD600 of the culture reached 0.5, DCA (2 mM) was added to the culture and then the culture was incubated for 2 h. Cells were collected by centrifugation (5,000 × g for 10 min at 4°C) and washed twice with 50 mM Tris-HCl buffer (pH 7.5; buffer A). The cells were resuspended in the same buffer and sonicated by an ultrasonic disintegrator (UD201; Tomy Seiko Co.) (23). After the cell lysate was centrifuged at 19,000 × g for 15 min at 4°C, the resulting supernatant was used as the cell extract. The cell extract was further centrifuged at 120,000 × g for 1 h at 4°C. The resultant pellet and supernatant were used for the precipitated fraction (defined as the membrane fraction) and the soluble fraction, respectively. Protein concentrations were determined by Lowry's assay using a commercially available detection kit (DC protein assay; Bio-Rad Laboratories). Cell extracts of SYK-6 (300 μg of protein/ml, 500 μg/ml, 50 μg/ml, and 200 μg/ml) were incubated with 100 μM substrate (DCA, DCA-C, DCA-CC, and DCA-S) in the presence and absence of 300 μM NAD+, 300 μM pyrroloquinoline quinone (PQQ) plus 300 μM 1-methoxy-5-methylphenazinium methylsulfate (PMS), or 300 μM flavin adenine dinucleotide (FAD) plus 300 μM PMS at 30°C. The reactions were stopped by the addition of methanol (final concentration, 50%) after 10, 30, 5, and 10 min of incubation with DCA, DCA-C, DCA-CC, and DCA-S, respectively. Precipitated proteins were removed by centrifugation at 19,000 × g for 15 min. The resulting supernatant diluted with 50% acetonitrile (final concentration, 17%) was analyzed by a high-performance liquid chromatography (HPLC; Acquity ultraperformance liquid chromatography [UPLC]) system (Waters) coupled with an Acquity TQ detector (Waters) using a TSKgel ODS-140HTP column (2.1 by 100 mm; Tosoh) as described previously (36). The mobile phase of the HPLC system was a mixture of water (75%) and acetonitrile (25%) containing formic acid (0.1%) at a flow rate of 0.3 ml/min. In the electrospray ionization-mass spectrometry (ESI-MS) analysis, MS spectra were obtained using the negative-ion mode according to the settings described for a previous study (36). DCA, DCA-C, DCA-CC, and DCA-S were detected at 277, 327, 324, and 323 nm, respectively. One unit of enzyme activity was defined as the amount that degrades 1 μmol of substrate/min.
Microarray preparation.
An array of 5,542 specific oligonucleotides (5,447 45-mer and 95 35-mer; melting temperature, 72 ± 5°C) was designed based on the Sphingobium sp. SYK-6 genome sequence (GenBank accession no. AP012222 and AP012223). The oligonucleotides were designed using sequences from the 4,063 putative genes and synthesized by Sigma-Genosys (Sigma-Aldrich) and printed onto glass slides by KakenGeneqs. As a positive control, all of the oligonucleotides were mixed and printed at the corners of each subgrid.
RNA isolation and cDNA synthesis for microarray analysis.
Cells of SYK-6 were grown in Wx-SEMP until the OD600 of the culture reached 0.5. Cultures were further incubated with 2 mM DCA for 2 h, with 2 mM vanillate for 6 h, or without any substrates for 2 h. Total RNA was isolated using Isogen II reagent (Nippon Gene) according to the manufacturer's instructions. Purified RNA was then treated with RNase-free DNase I (Roche) to remove any contaminated DNA. RNA samples isolated from three independent cultures were used for cDNA synthesis. Total RNA (6 μg) was labeled using an indirect method in which aminoallyl-dUTPs were incorporated by reverse transcription using a mixture of two kinds of random hexamers, normal GC content (Invitrogen) and high GC content (70%) (Sigma), 5-(3-aminoallyl)-dUTP (Ambion), and PrimeScript II reverse transcriptase (TaKaRa Bio Inc.). The RNA template was then degraded through incubation with 0.2 N NaOH and 0.1 M EDTA, followed by a neutralization using 1 M HEPES (pH 7.5). For a control of microarray hybridization, genomic DNA (gDNA) was prepared. gDNA was isolated from SYK-6 and fragmented by sonication to an average size of 1,000 bp. Resultant gDNA (4 μg) was labeled using 5-(3-aminoallyl)-dUTP and Klenow fragment (Roche). Cy3 and Cy5 dyes were coupled to the aminoallyl-dUTP in the cDNA and gDNA, respectively, in the presence of 0.1 M sodium bicarbonate (pH 9.0). The unlabeled dyes were removed using the QIAquick PCR purification system (Qiagen).
Microarray hybridization and data analysis.
We performed duplicate competitive hybridization experiments using equal amounts of Cy3- and Cy5-labeled probes. Hybridizations were performed in a GeneTac HybStation instrument (Genomic Solutions). Hybridized arrays were immediately scanned using a GenePix 4000B scanner (Axon Instruments), and the spot intensities were quantified using Imagene 6.1 (BioDiscovery). To clarify the expression pattern of Wx-SEMP plus DCA–Wx-SEMP and Wx-SEMP plus vanillate–Wx-SEMP, in silico analysis was conducted by the LIMMA (linear model for microarray analysis) loess (subgrid) method using ArrayPipe 2.0. Average normalized expression ratios (treatment/control) were calculated for each gene and tested for significant variation between treatments (analysis of variance [ANOVA]).
Sequence analysis.
Sequence analysis was performed with the MacVector program (MacVector, Inc.). Sequence similarity searches were carried out using the BLAST program (37). Pairwise and multiple alignments were performed with the EMBOSS alignment tool (38) and the ClustalW2 program (39), respectively. Phylogenetic trees were generated using the FigTree program (http://tree.bio.ed.ac.uk/software/figtree/).
Construction of mutants.
A DNA fragment carrying phcC and phcD was amplified by PCR using total DNA of SYK-6 as a template and primers 5′-GCGGGATCCACGGCGGGAAGAGGG-3′ and 5′-ATCTTCACCGGGCCGGCGCCGTAGC-3′. A 3.9-kb HindIII-BamHI fragment of the PCR amplicon was ligated in pBluescript II KS(+) and pT7Blue to obtain pKS09480-500 and pT09480-500, respectively. The 2.4-kb BglII (blunt-ended)-BamHI fragment carrying phcC from pKS09480-500 was cloned into the XhoI (blunt-ended) and BamHI sites of pBluescript II KS(+) to generate pKS09480. The 1.3-kb EcoRV fragment carrying the Km resistance gene (kan) of pIK03 was inserted into the HincII site in phcC of pKS09480. The 3.7-kb KpnI-BamHI fragment of the resultant plasmid, pKS09480K, was ligated into the SalI-BamHI sites of pK19mobsacB, and then pKmb09480K was obtained. The 2.7-kb XhoI-SacI fragment of pKS09480-500 carrying phcD was cloned into the same sites of pBluescript II KS(+) to create pKS09500. The 1.3-kb EcoRV fragment carrying kan was inserted into the HincII site in phcD of pKS09500. The 4.0-kb KpnI-SacI fragment of the resultant plasmid, pKS09500K, was blunted and inserted into the SmaI site of pK19mobsacB, and pKmb09500K was obtained. The 1.3-kb EcoRV fragment carrying kan was ligated to the HincII sites in phcC and phcD of pT09480-500. The 3.0-kb SphI-BamHI fragment of the resultant plasmid, pT09480-500K, was ligated to the same sites of pK19mobsacB to obtain pKmb09480-500K.
The resulting plasmids were independently introduced into SYK-6 cells by electroporation, and candidate mutants were isolated as described previously (40). The disruption of each gene was examined by Southern hybridization analysis. Total DNA of candidates for SME110, SME111, and SME112 were digested with HindIII-EcoRI, PstI-SalI, and XhoI, respectively. A 1.7-kb NdeI-BamHI fragment carrying phcC, a 1.8-kb NdeI-BamHI fragment carrying phcD, a 3.9-kb HindIII-BamHI fragment carrying phcC and phcD, and a 1.3-kb EcoRV fragment carrying kan were labeled with the digoxigenin system (Roche) and used as probes.
Characterization of mutants.
Cells of SYK-6, SME110, SME111, and SME112 were grown in LB. After 12 h of incubation, cells were collected by centrifugation, washed twice with buffer A, and resuspended in 1 ml of the same buffer. After the addition of 100 μM substrate (DCA or DCA-C), resting cells (OD600 of 0.5 or 1.0) were incubated at 30°C with shaking. Portions of the cultures were collected at various sampling time points. The reactions were stopped by centrifugation, and the supernatants were filtered and analyzed by HPLC using a TSKgel ODS-140HTP column and Chiralcel OD-RH column (4.6 by 150 mm; DAICEL). The same mobile phase as described above was used, and the flow rates were 0.3 and 0.7 ml/min, respectively. To determine the optical rotation of the racemic DCA-C preparation, the sample was separated by an HPLC system (Shimadzu) using a Chiralcel OD-RH column. The same mobile phase as described above was used, and the flow rate was 0.8 ml/min. Optical rotations were measured with a laser polarimeter (APL4; PDR-Separations) at 426 nm.
Reverse transcription-PCR (RT-PCR).
Total RNA was prepared from SYK-6 cells grown with DCA according to the method described above. cDNA was synthesized by the method described previously (41) except for using PrimeScript II reverse transcriptase. PCR was performed with the resultant cDNA with primers 5′-GCACATCGTCATAGCTCCA-3′ and 5′-GGCATCTTCCTCAACCTGT-3′ and Q5 Hot Start high-fidelity DNA polymerase (New England BioLabs Inc.). A control PCR was performed with reverse transcriptase-negative samples to verify the absence of genomic DNA contamination. The resultant PCR products were subjected to agarose gel electrophoresis.
Expression of phcC and phcD in SYK-6 and E. coli.
A 1.7-kb fragment carrying phcC and a 1.8-kb fragment carrying phcD were amplified by PCR using total DNA of SYK-6 as a template and primer sets as follows: for phcC amplification, 5′-AACTCTGACATATGGCCAGCAAAACG-3′ and 5′-GCGGGATCCACGGCGGGAAGAGGG-3′, and for phcD amplification, 5′-CCGAGAGCATATGAATCATCATGAACTCC-3′ and 5′-GAAGGATCCGATATAGCGCTCGAAATCC-3′. The PCR-amplified fragments were ligated in pT7Blue, and the nucleotide sequences of the inserts were determined. The 1.7-kb NdeI-BamHI fragment carrying phcC and 1.8-kb NdeI-BamHI fragment carrying phcD of the resulting plasmids were cloned into pET-16b to obtain pET09480 and pET09500. pJBI09480 and pJBI09500 were created using the DNA fragments carrying phcC and phcD derived from pET09480 and pET09500 through the generation of plasmids pT09480Xba, pT09500Xba, pJBVI09480, and pJBVI09500.
E. coli BL21(DE3) cells harboring pET09480 or pET09500 were grown in LB at 30°C, and the expression of the genes was induced for 24 h at 16°C by adding 100 μM isopropyl-β-d-thiogalactopyranoside when the OD600 of the culture reached 0.5. SYK-6 cells harboring pJBI09480 and pJBI09500 were grown in LB at 30°C, and the expression of the genes was induced for 12 h at 30°C by adding 1 mM m-toluate when the OD600 of the culture reached 0.5. Cells of the E. coli and SYK-6 transformants were then harvested by centrifugation and washed with buffer A. The cells resuspended in the same buffer were sonicated, and the cell lysates were obtained. After the cell lysates were centrifuged at 19,000 × g for 15 min at 4°C, the resulting supernatants were used as the cell extracts. The cell extracts were further centrifuged at 120,000 × g for 1 h at 4°C. The resultant pellet and supernatant were used for the precipitated fraction (membrane fraction) and the soluble fraction. The expression of the genes was confirmed using SDS-12% polyacrylamide gel electrophoresis (PAGE). Protein bands in gels were stained with Coomassie brilliant blue.
Cellular localizations of PhcC and PhcD in SYK-6.
Cellular localizations of PhcC and PhcD were determined by measuring the activities for the conversion of DCA in the membrane and soluble fractions prepared from the SYK-6 cells harboring pJBI09480 and pJBI09500 grown in LB. A 1-ml assay mixture containing buffer A, 300 μM FAD, 300 μM PMS, and 100 μM DCA-C was mixed with cell extracts (100 μg of protein), the soluble fraction containing PhcC or PhcD (200 μg and 500 μg of protein), or the membrane fraction containing PhcC or PhcD (20 μg of protein). Reactions were carried out at 30°C and then stopped by the addition of methanol (final concentration, 50%) after 3 min (for the reaction with PhcC) and 5 min (for the reaction with PhcD) of incubation. Precipitated proteins were then removed by centrifugation at 19,000 × g for 15 min, and the supernatants diluted with 50% acetonitrile (final concentration, 17%) were analyzed by HPLC.
Enantiospecificities of PhcC and PhcD.
The membrane fractions prepared from E. coli cells harboring pET09480 and pET09500 grown in LB were employed for the enzyme characterization of PhcC and PhcD. To determine the enantiospecificities, the membrane fractions containing PhcC or PhcD (10 or 100 μg/ml of protein) were incubated in a reaction mixture containing buffer A, 200 μM DCA-C, 300 μM FAD, and 300 μM PMS. After incubation for 10 min, the reaction was terminated by the addition of methanol (final concentration, 50%). To isolate the remaining DCA-C in the reactions, the supernatants were extracted with ethyl acetate, and the compounds were separated by thin-layer chromatography using Silica Gel 60 F254 (Merck Millipore) with a solvent mixture of 71.4% benzene, 21.4% ethyl acetate, and 7.2% acetic acid. Compounds were visualized under UV light at 254 nm. DCA-C has an Rf of 0.41 in this system and was extracted with ethyl acetate. The resultant compound was analyzed by chiral HPLC analysis under the conditions described above.
Identification of the reaction products.
In the presence of 300 μM FAD and 300 μM PMS, 200 μM DCA-C was incubated with the membrane fractions containing PhcC (10 μg of protein/ml) or PhcD (100 μg of protein/ml) for 30 min at 30°C. The supernatants were analyzed by HPLC and ESI-MS under the conditions described above.
Substrate preferences.
The activities of PhcC and PhcD for the oxidation of DCA, DCA-L, DCA-C, GGE, coniferyl alcohol, and vanillyl alcohol were determined by measuring the decrease in the amount of substrates by HPLC analysis. The enzyme reactions were carried out in a 1-ml reaction mixture containing buffer A, 300 μM FAD, 300 μM PMS, 100 μM substrate, and the membrane fractions containing PhcC or PhcD (5 or 50 μg of protein) at 30°C for 3 min (for the reaction with PhcC) and 5 min (for the reaction with PhcD). The decrease in the substrates (DCA, DCA-L, and DCA-C) and the generation of the reaction products were determined by HPLC and ESI-MS analysis under the conditions described above. In analyzing the activities for GGE, coniferyl alcohol, and vanillyl alcohol, mobile phases consisting of a mixture of water and acetonitrile (80:20, 85:15, and 95:5) containing formic acid (0.1%) were used. DCA, DCA-L, DCA-C, GGE, coniferyl alcohol, and vanillyl alcohol were detected at 277, 346, 327, 277, 263, and 279 nm, respectively.
Optimum pH and temperatures.
The optimal pH was determined in a 1-ml reaction mixture containing 300 μM FAD, 300 μM PMS, 100 μM DCA-C, and the membrane fractions containing PhcC or PhcD (5 or 50 μg of protein) at pH ranges from 6.0 to 10.0 using 50 mM GTA buffer (50 mM 3,3-dimethylglutarate, 50 mM Tris, and 50 mM 2-amino-2-methyl-1,3-propanediol; pH 6.0 to 9.0) and 50 mM CHES [2-(N-cyclohexylamino)ethanesulfonic acid] buffer (pH 8.6 to 10) at 30°C. The optimal temperatures for PhcC and PhcD were determined in a 1-ml reaction mixture containing 50 mM GTA buffer (pH 9.0 for PhcC and pH 7.5 for PhcD), 300 μM FAD, 300 μM PMS, 100 μM DCA-C, and the membrane fractions containing PhcC or PhcD (5 or 50 μg of protein) at temperature ranges from 10 to 50°C.
Identification of the flavin cofactor.
In order to identify the flavin cofactor in PhcC, a 100-μl solution of the membrane fraction containing PhcC (485 μg of protein) was incubated in boiling water for 10 min. Precipitated proteins were removed by centrifugation at 19,000 × g for 15 min. The resulting supernatant was analyzed using HPLC with an Acquity UPLC BEH C18 column (2.1 by 100 mm; Waters). The mobile phase of the HPLC system was a mixture of water (90%) and acetonitrile (10%) containing formic acid (0.1%) at a flow rate of 0.5 ml/min.
Effects of ubiquinone derivatives on the enzyme activities.
To investigate the role of ubiquinone as an electron acceptor of PhcC and PhcD, the activities of these enzymes for the conversion of DCA-C in the presence of water-soluble short-chain ubiquinone (coenzyme Q10 [CoQ10]) analogs, 2,3-dimethoxy-5-methyl-1,4-benzoquinone (CoQ0) and 2,3-dimethoxy-5-methyl-6-(3-methyl-2-butenyl)-1,4-benzoquinone (CoQ1), were determined. The enzyme reactions were carried out in a 1-ml reaction mixture containing GTA buffer (pH 9.0 or 7.5), 200 μM DCA-C, 300 μM FAD, 300 μM CoQ0 or CoQ1, and the membrane fractions containing PhcC or PhcD (5 or 50 μg of protein) at 30°C for 3 min (for the reaction with PhcC) and 5 min (for the reaction with PhcD). The decrease in the substrates was determined by HPLC analysis.
Microarray data accession number.
Details of the microarray design, transcriptomic experimental design, and transcriptomic data have been deposited in the NCBI Gene Expression Omnibus (http://www.ncbi.nlm.nih.gov/geo/) and are accessible through GEO series accession number GSE71789.
RESULTS
Cofactor requirements, cellular localizations, and induction profiles of DCA catabolic enzymes.
In order to characterize the enzymes involved in the catabolism of DCA in Sphingobium sp. SYK-6, localizations and induction profiles of the enzyme activities in SYK-6 toward DCA, DCA-C, DCA-CC, and DCA-S were examined (Table 2). The effects of the addition of NAD+, PQQ plus PMS, and FAD plus PMS on the enzyme activities to convert DCA and DCA-C were also investigated (Table 2).
TABLE 2.
Enzyme activities of cell extract, soluble fraction, and membrane fraction of SYK-6 toward DCA and its derivatives
| Substrate | Fraction | Cofactor(s) | Activitya (mU) |
||
|---|---|---|---|---|---|
| SEMP | SEMP + DCA | SEMP + DCA-C | |||
| DCA | Cell extract | None | 21 ± 7 | 36 ± 3 | 46 ± 10 |
| NAD+ | 160 ± 2 | 170 ± 2 | 140 ± 1 | ||
| PQQ + PMS | 61 ± 2 | 59 ± 0.6 | 56 ± 0.2 | ||
| FAD + PMS | 43 ± 0.8 | 73 ± 3 | 74 ± 4 | ||
| Soluble | None | 5.7 ± 3 | 11 ± 6 | —c | |
| NAD+ | 110 ± 2 | 120 ± 2 | — | ||
| PQQ + PMS | 20 ± 0.4 | 16 ± 2 | — | ||
| FAD + PMS | 15 ± 3 | 16 ± 1 | — | ||
| Membrane | None | 6.8 ± 0.4 | 12 ± 0.4 | — | |
| NAD+ | 6.3 ± 0.1 | 13 ± 0.7 | — | ||
| PQQ + PMS | 18 ± 0.2 | 18 ± 0.1 | — | ||
| FAD + PMS | 9.4 ± 0.3 | 21 ± 0.8 | — | ||
| DCA-C | Cell extract | None | 3.5 ± 0.4 | 14 ± 0.8 | 6.0 ± 0.6 |
| NAD+ | 2.4 ± 1 | 14 ± 0.2 | 6.4 ± 1 | ||
| PQQ + PMS | 3.2 ± 0.5 | 16 ± 0.3 | 9.1 ± 2 | ||
| FAD + PMS | 19 ± 0.6 | 32 ± 0.4 | 27 ± 0.7 | ||
| Soluble | None | NDb | 1.7 ± 0.2 | — | |
| NAD+ | ND | 1.4 ± 0.5 | — | ||
| PQQ + PMS | 1.3 ± 0.3 | 3.0 ± 0.1 | — | ||
| FAD + PMS | 5.3 ± 0.6 | 6.9 ± 0.2 | — | ||
| Membrane | None | 0.8 ± 0.04 | 3.0 ± 0.1 | — | |
| NAD+ | 0.7 ± 0.05 | 3.1 ± 0.08 | — | ||
| PQQ + PMS | 0.8 ± 0.1 | 3.2 ± 0.05 | — | ||
| FAD + PMS | 2.4 ± 0.02 | 3.8 ± 0.07 | — | ||
| DCA-CC | Cell extract | None | 1,300 ± 30 | 2,400 ± 70 | 2,200 ± 100 |
| Soluble | None | 1,300 ± 20 | 2,100 ± 10 | — | |
| Membrane | None | ND | ND | — | |
| DCA-S | Cell extract | None | 180 ± 40 | 240 ± 20 | — |
| Soluble | None | 130 ± 8 | 190 ± 20 | — | |
| Membrane | None | ND | 4.1 ± 2 | — | |
Cell extracts of SYK-6 (10 mg of protein) were fractionated into the soluble (7.5 mg of protein) and membrane (0.94 mg of protein) fractions. Enzyme activities in the cell extract, soluble fraction, and membrane fraction are shown as milliunits/10 mg of protein, milliunits/7.5 mg protein, and milliunits/0.94 mg of protein, respectively. The data are averages ± standard deviations from three independent experiments.
ND, not detected.
—, not tested.
The extract of SYK-6 cells grown with DCA exhibited the highest specific activities for the conversion of DCA when NAD+ was added to the reaction mixture. In contrast, the specific activities in the presence of PQQ plus PMS or FAD plus PMS were ca. 35 to 43% of the activity obtained with NAD+. To examine the localization of the enzyme, cell extracts of SYK-6 were fractionated into the soluble fraction and the precipitated fraction (defined as the membrane fraction) by ultracentrifugation. The amount of proteins in the membrane fraction was estimated to account for ca. 9.4% (wt/wt) of the total cellular proteins. During the fractionation, a considerable portion of membrane proteins seemed to be trapped in an intermediate layer which appeared after ultracentrifugation. To examine the localization of the DCA-converting enzyme, the activities of the soluble (7.5 mg of protein) and membrane fractions (0.94 mg of protein) derived from the cell extract (10 mg of protein) were compared. Major activity was observed in the soluble fraction in the presence of NAD+, suggesting that the DCA-converting enzymes are mainly localized in the cytoplasm. In addition, since the activities of extracts of the SYK-6 cells grown with or without DCA were almost the same, the genes encoding DCA-converting enzymes seem to be constitutively expressed. In our previous study, we suggested the involvement of multiple quinohemoprotein ADHs and aryl ADHs in the DCA oxidation (33). However, since quinohemoprotein ADHs are usually localized in the membrane or periplasm (42), aryl ADHs appear to play an important role in the oxidation of DCA.
Enzyme activities of the SYK-6 cell extract for the conversion of DCA-C were significantly enhanced in the presence of FAD plus PMS (Table 2), as shown in our previous report (33). The activities for the conversion of DCA-C in the presence of FAD plus PMS were observed in both the soluble and membrane fractions (Table 2). Considering the possible loss of some membrane proteins during the preparation of the membrane fraction, the DCA-C-converting enzyme(s) appears to be almost equally distributed to the cytoplasm and membrane. When using the cells grown with DCA, the activities for the conversion of DCA-C in the presence of FAD plus PMS in the soluble and membrane fractions were ca. 1.3- and 1.6-fold higher than those from the cells grown without DCA. These results indicated that the activities for the conversion of DCA-C were induced at a low level in the SYK-6 cells during growth on DCA, which is in agreement with our previous results (33). In addition, similar induction was also seen in the cells grown with DCA-C.
The activities of SYK-6 converting DCA-CC and DCA-S were only detected in the soluble fraction, suggesting the cytoplasmic localization of the DCA-CC-converting enzyme(s) and DCA-S-converting enzyme(s). When the cells were grown with DCA, the activities for the conversion of DCA-CC and DCA-S were stimulated ca. 1.6- and 1.5-fold, respectively.
Search for the genes involved in the oxidation of DCA-C using microarray analysis.
Because the DCA-C oxidation in the SYK-6 cells was enhanced in the presence of FAD and induced at a low level in the cells grown with DCA, we searched for the DCA-C oxidase genes based on the induction profiles of the whole SYK-6 genes analyzed by DNA microarray analysis and domain architectures deduced from the nucleotide sequences. These analyses found SLG_09480 and SLG_09500, which showed similarities to the GMC oxidoreductase family proteins. The transcriptions of SLG_09480 and SLG_09500 were induced 2.3-fold (P value < 0.081; ANOVA) and 2.2-fold (P value < 0.013), respectively, during the growth of SYK-6 with DCA, while the transcriptions of these genes were not induced when vanillate was added to the culture. SLG_09480 and SLG_09500, which encode proteins of 541 and 576 amino acids, respectively, showed 42% amino acid sequence identity with each other and exhibited 38% and 37% identities with choline dehydrogenase of E. coli K-12 (43). SLG_09490, located between SLG_09480 and SLG_09500, encodes a hypothetical protein of 126 amino acids containing a SnoaL2 domain, whose function in the catabolism of DCA was not deduced (Fig. 1B).
To examine whether SLG_09480 and SLG_09500 form a transcription unit, RT-PCR analysis was performed using total RNA prepared from the SYK-6 cells grown in the presence of DCA. A specific amplification of the region between SLG_09480 and SLG_09500 was observed (Fig. 1B and C), indicating that the genes SLG_09480, SLG_09490, and SLG_09500 form an operon.
In the vicinity of SLG_09480 and SLG_09500, SLG_09520 and SLG_09440, which encode a putative MarR-type transcriptional regulator and a putative lignostilbene α,β-dioxygenase, respectively, were found. In addition, the SLG_09420 product was previously characterized as an aryl ADH catalyzing the oxidation of DCA (33). On the other hand, it was shown that the gene products of ALDH-encoding SLG_09400 and SLG_09510 showed no activity toward DCA-L (33). The involvement of SLG_09520 and SLG_09440 in the DCA catabolism should be examined in a future study.
Roles of SLG_09480 and SLG_09500 in the conversion of DCA-C.
In order to examine whether SLG_09480 and SLG_09500 are indeed involved in the conversion of DCA-C in SYK-6, an SLG_09480 mutant (SME110), an SLG_09500 mutant (SME111), and an SLG_09480 SLG_09500 double mutant (SME112) were created. The ability of SME110 and SME111 to convert DCA-C was assessed using resting cells. The wild-type strain completely converted 100 μM DCA-C within 2.5 h. On the other hand, the conversion rates of these mutants for DCA-C were significantly decreased, and approximately a half molar concentration of DCA-C remained after being incubated for 5 h. In addition, SME112 was no longer able to convert DCA-C (Fig. 2A). When DCA was used as a substrate, the rates of conversion between the wild type and the above-mentioned three mutants were almost identical (Fig. 2B). However, SME110 and SME111 accumulated DCA-C at a concentration half-molar to that of the added DCA. In the case of SME112, an accumulation of DCA-C at a concentration approximately equimolar to the added DCA was observed (Fig. 2B). These results indicated that both SLG_09480 and SLG_09500 are essential for the catabolism of DCA-C; thus, we designated these genes phcC and phcD. The results also suggested that PhcC and PhcD converted one of the different enantiomers probably included in the DCA-C preparation.
FIG 2.
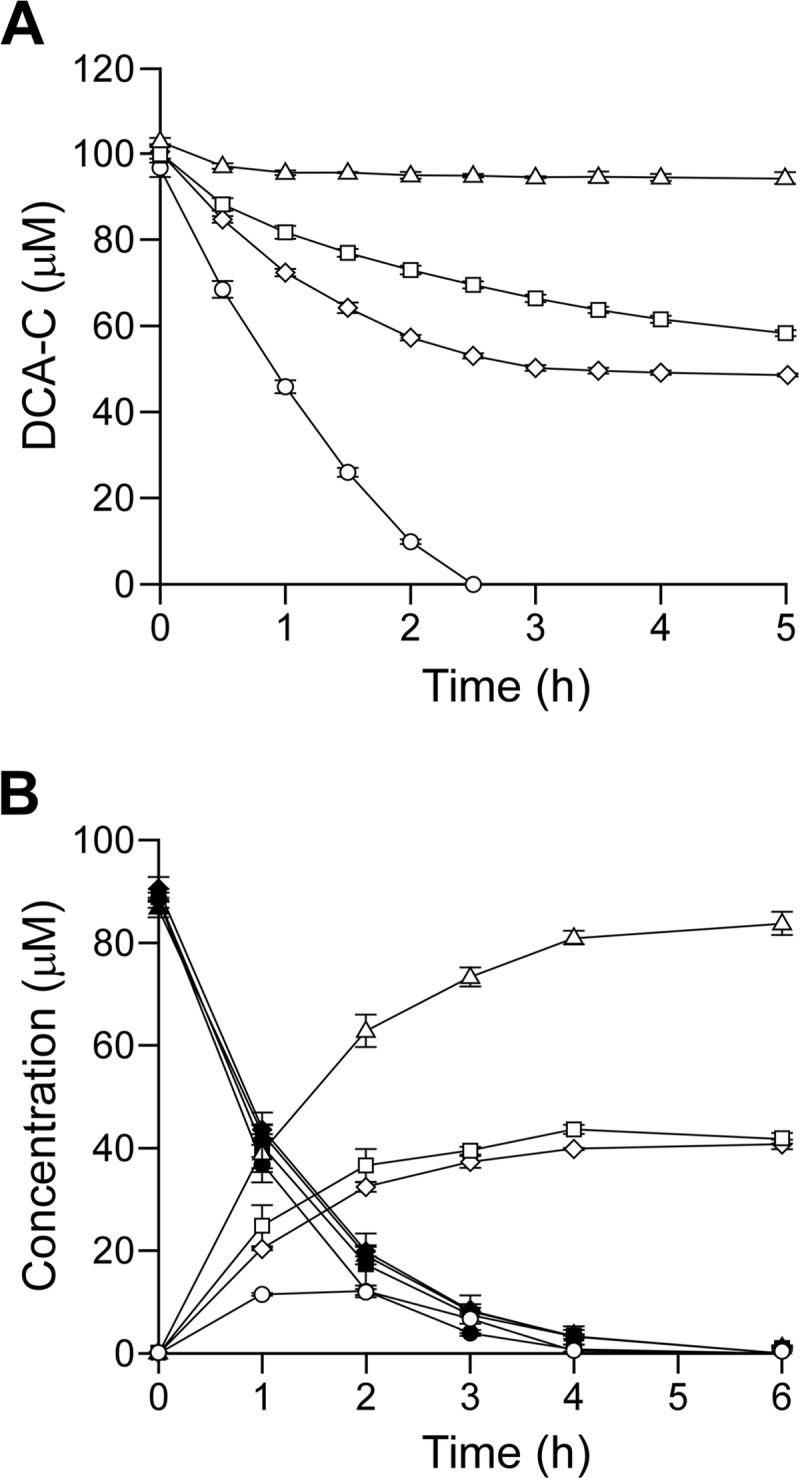
Conversion of DCA and DCA-C by SME110, SME111, and SME112. (A) Conversions of 100 μM DCA-C by resting cells (OD600 of 1.0) of SYK-6 (circles), SME110 (squares), SME111 (diamonds), and SME112 (triangles). (B) Conversions of 100 μM DCA by resting cells (OD600 of 0.5) of SYK-6 (circles), SME110 (squares), SME111 (diamonds), and SME112 (triangles). Open and closed symbols indicate the concentrations of DCA-C and DCA, respectively. The data are the averages ± standard deviations (error bars) of at least three measurements.
In order to examine the existence of enantiomers in the DCA-C preparation, DCA-C was analyzed by chiral HPLC, and separated into two peaks with retention times at 10.8 min and 12.2 min (Fig. 3A). From the results of the optical-rotation analysis, the former and latter peaks were identified to be (+)-DCA-C and (−)-DCA-C, respectively (Fig. 3B). The racemic DCA-C preparation consists of equal amounts of (+)-DCA-C (ca. 49.4%) and (−)-DCA-C (ca. 50.6%). The resting cells of SME110 and SME111 grown in LB were incubated with 100 μM DCA-C for 5 h, and the reaction mixtures were analyzed by chiral HPLC. In the reaction mixture of SME110, (−)-DCA-C almost completely disappeared while (+)-DCA-C remained (Fig. 4B). On the other hand, (+)-DCA-C was almost completely transformed, while (−)-DCA-C remained in the reaction mixture of SME111 (Fig. 4C). All these results strongly suggest that PhcC and PhcD specifically converted (+)-DCA-C and (−)-DCA-C, respectively.
FIG 3.
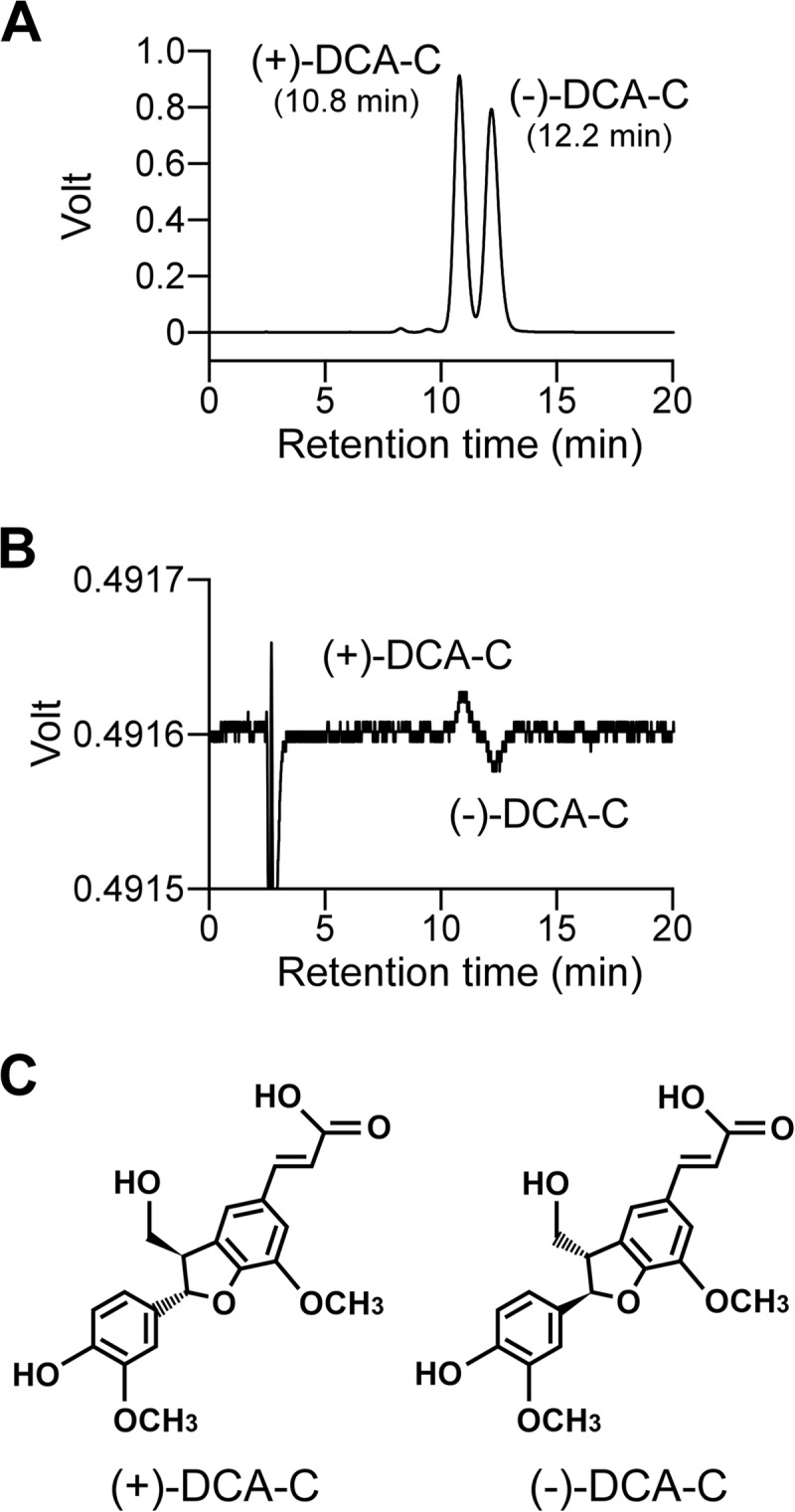
Chiral-HPLC separation of the racemic DCA-C. Chromatograms with UV detection (A) and optical-rotation detection are shown (B). Deduced stereochemistries of (+)-DCA-C and (−)-DCA-C based on the absolute configurations of (+)-(2S,3R)-DCA and (−)-(2R,3S)-DCA are shown in panel C (53).
FIG 4.
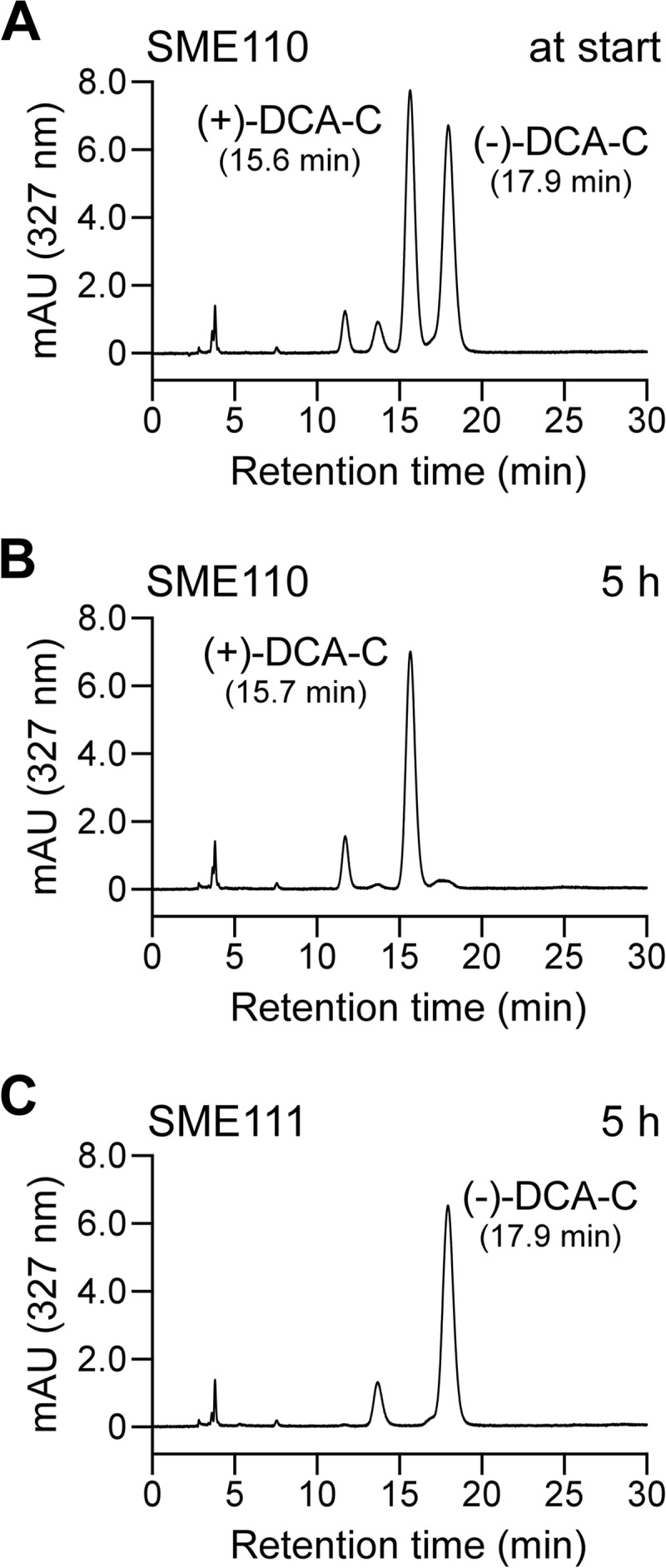
Chiral-HPLC analysis of the conversion of DCA-C by SME110 and SME111. DCA-C (200 μM) was incubated with resting cells of SME110 and SME111 (OD600 of 1.0). Portions of the reaction mixtures were collected at the start of incubation (incubation with SME110) (A) and after 5 h of incubation with SME110 (B) and SME111 (C) and analyzed by chiral HPLC.
Cellular localizations of PhcC and PhcD.
In order to determine the cellular localizations of PhcC and PhcD, each of phcC and phcD was overexpressed in SYK-6 cells using pJB861 as a vector. The activities for the conversion of DCA-C of the extracts prepared from the SYK-6 cells carrying phcC and phcD expression plasmids (pJBI09480 and pJBI09500) grown in LB were increased 95-fold (180 ± 4 mU/mg of protein) and 26-fold (49 ± 4 mU/mg of protein), respectively, compared to that of the wild type. The extracts of the SYK-6 cells harboring pJBI09480 and pJBI09500 were fractionated to the soluble and membrane fractions. SDS-PAGE of the fractions of the SYK-6 cells harboring pJBI09480 showed the production of PhcC in both the soluble and membrane fractions (Fig. 5A). On the other hand, PhcD was observed in the membrane fraction of the SYK-6 cells harboring pJBI09500. The activities for the conversion of DCA-C in the membrane fractions containing PhcC and PhcD (1.1 and 1.2 mg of protein) were estimated to be 340 ± 40 and 330 ± 60 mU. In the soluble fractions containing PhcC and PhcD (6.8 and 6.6 mg of protein), activity levels of 162% and 11% of those found in the corresponding membrane fractions were observed. These results suggest that PhcC and PhcD were localized to both the cytoplasm and membrane, although most parts of PhcD were localized to the membrane.
FIG 5.

Expression of phcC and phcD in Sphingobium sp. SYK-6 and E. coli. Proteins (10 μg) were separated on SDS-12% polyacrylamide gels and stained with Coomassie brilliant blue. (A) Lanes: 1, 4, and 7, SYK-6 harboring pJB861 (vector); 2, 5, and 8, SYK-6 harboring pJBI09480 (phcC); 3, 6, and 9, SYK-6 harboring pJBI09500 (phcD); 1 to 3, cell extracts; 4 to 6, soluble fractions; 7 to 9, membrane fractions. (B) Lanes: 1, 4, and 7, E. coli BL21(DE3) harboring pET-16b (vector); 2, 5, and 8, E. coli BL21(DE3) harboring pET09480 (phcC); 3, 6, and 9, E. coli BL21(DE3) harboring pET09500 (phcD), 1 to 3, cell extracts; 4 to 6, soluble fractions; 7 to 9, membrane fractions; M, molecular mass markers.
Identification of the reaction products of PhcC and PhcD.
For characterization of PhcC and PhcD, phcC and phcD fused with a His tag at their 5′ termini were expressed in E. coli. SDS-PAGE of the precipitated fractions (membrane fractions) prepared from each E. coli strain harboring pET09480 and pET09500 showed the expression of phcC and phcD; however, the expression of phcD was substantially lower than that of phcC (Fig. 5B). The membrane fractions were treated with six different detergents, including n-dodecyl-β-d-maltoside to solubilize PhcC and PhcD, and then these enzymes were purified by Ni affinity chromatography. However, purified PhcD lost its activity, and the specific activity of purified PhcC was decreased to approximately 12% (260 ± 20 mU/mg) of that obtained with the membrane fraction. Moreover, His tag-fused phcD was expressed in SYK-6 cells harboring pJBI09500, which carried phcD. However, an active PhcD was not obtained after the purification. Because of these results, enzyme properties of PhcC and PhcD were investigated using the membrane fractions prepared from the cell extracts of E. coli harboring pET09480 and pET09500.
When the membrane fractions containing PhcC or PhcD were incubated with 200 μM DCA-C in the presence of FAD plus PMS, these fractions exhibited specific activities of 2,100 ± 300 mU/mg and 180 ± 20 mU/mg, respectively. HPLC analysis of the reaction mixtures obtained after 30 min of incubation indicated that DCA-C was converted into DCA-CC with a retention time of 3.1 min as a major product (Fig. 6). A small amount of DCA-CL with a retention time of 2.0 min was also observed. These results indicate that PhcC and PhcD have the ability to oxidize the alcohol group at the γ-carbon of the A-ring side chain of DCA-C.
FIG 6.

Conversion of DCA-C by membrane fractions containing PhcC and PhcD. DCA-C (100 μM) was incubated with membrane fractions containing PhcC (10 μg of protein/ml) or PhcD (100 μg of protein/ml). Portions of the reaction mixtures were collected at the start of incubation (incubation with PhcC) (A) and after 30 min of incubation with PhcC (B) and PhcD (C) and analyzed by HPLC. Negative-ion ESI-MS spectra of compounds I (D) and II (E), which were identified as DCA-CL and DCA-CC, are shown (F).
Enantiospecificity of PhcC and PhcD.
The membrane fractions, including PhcC or PhcD were incubated with 200 μM DCA-C for 10 min in the presence of FAD plus PMS. Because the retention times of DCA-C and the reaction product, DCA-CC, were similar when the chiral column was used for the analysis, DCA-C remaining after the reaction was isolated by thin-layer chromatography. Chiral-HPLC analyses indicated that the DCA-C remaining after the reactions catalyzed by PhcC and PhcD were (−)-DCA-C and (+)-DCA-C, respectively (Fig. 7). These results corresponded to the results of the analysis of the mutants, indicating the enantiospecificity of PhcC and PhcD, respectively, toward (+)-DCA-C and (−)-DCA-C.
FIG 7.
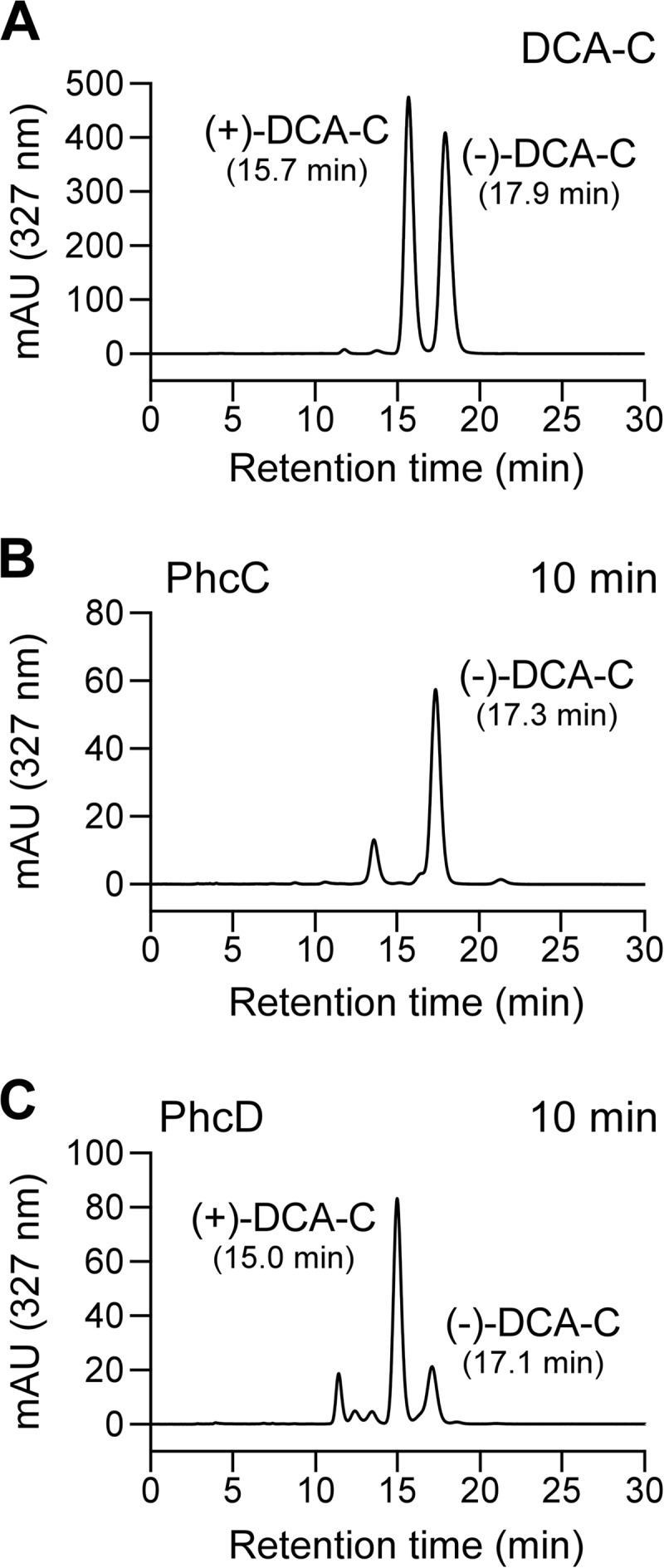
Stereospecificities of PhcC and PhcD for the oxidation of DCA-C enantiomers. (A) Chiral-HPLC profiles of the racemic DCA-C. The membrane fractions containing PhcC (B) or PhcD (C) were incubated with 200 μM DCA-C in the presence of 300 μM FAD and 300 μM PMS for 10 min. The remaining DCA-C in the reaction mixtures was separated by thin-layer chromatography and then analyzed by chiral HPLC.
Other enzyme properties of PhcC and PhcD.
The optimum pHs of PhcC and PhcD for the conversion of DCA-C were estimated to be 9.0 and 7.0 to 8.0, respectively. The optimum temperatures of PhcC and PhcD were determined to be 30°C and 40°C. HPLC analysis of the supernatant obtained by the heat treatment of the membrane fraction containing PhcC indicated that this enzyme contained FAD as a prosthetic group (data not shown).
The membrane fractions containing PhcC or PhcD were incubated with 200 μM DCA, DCA-L, GGE, coniferyl alcohol, and vanillyl alcohol, and the reaction mixtures were analyzed by HPLC. PhcC and PhcD displayed specific activities of 2,200 ± 100 mU/mg and 230 ± 8 mU/mg toward DCA, which were equivalent to the activities of them toward DCA-C. Liquid chromatography-mass spectrometry (LC-MS) analysis showed that the generation of DCA-AC (compound III) from DCA, indicating that the alcohol group at the γ-carbon of the A-ring side chain of DCA was oxidized to the carboxyl group (Fig. 8A to C and G to I). Furthermore, PhcC and PhcD were also able to oxidize DCA-L, and their activities were 67% and 47% of that toward DCA-C. The oxidation of the alcohol group at the γ-carbon of the A-ring side chain of DCA-L by PhcC was verified by LC-MS analysis (Fig. 8D to F and I). PhcC had only weak activity for coniferyl alcohol (ca. 4% of the specific activity for DCA-C). GGE and vanillyl alcohol were not substrates for PhcC and PhcD.
FIG 8.

Conversion of DCA and DCA-L by the membrane fraction containing PhcC. DCA and DCA-L (100 μM), respectively, were incubated with the membrane fraction containing PhcC (10 μg of protein/ml) in the presence of 300 μM FAD and 300 μM PMS. Portions of the reaction mixtures containing DCA and DCA-L were collected at the start (A and D) and after 10 min of incubation (B and E). The reaction products were analyzed by LC-MS, and negative-ion ESI-MS spectra of compounds III and IV are shown in panels C and F. An HPLC chromatogram of authentic DCA-AC and its negative-ion ESI-MS spectrum are shown in panels G and H, respectively. (I) Chemical structures of DCA-AC and compound IV [2-(4-hydroxy-3-methoxyphenyl)-7-methoxy-5-(3-oxoprop-1-enyl)-2,3-dihydrobenzofuran-3-carboxylic acid; DCA-LC].
Based on the observations that significant portions of PhcC and PhcD were localized to the membrane, we hypothesized that these enzymes could utilize ubiquinone (CoQ10) as an electron acceptor in the oxidation of DCA-C. Therefore, the DCA-C oxidation activities of PhcC and PhcD were measured using CoQ0 and CoQ1 as electron acceptors instead of CoQ10 because of their better solubilities. The membrane fractions containing PhcC or PhcD were incubated with 200 μM DCA-C in the presence of FAD plus CoQ0 or CoQ1, and the reaction mixtures were analyzed by HPLC. PhcC showed specific activities of 2,000 ± 200 mU/mg and 1,900 ± 600 mU/mg in the presence of CoQ0 and CoQ1, respectively. PhcD also exhibited specific activities of 180 ± 10 mU/mg and 160 ± 10 mU/mg in the presence of CoQ0 and CoQ1. These values were almost equivalent to the activities obtained using PMS as an electron acceptor (PhcC, 2,100 ± 300 mU/mg; PhcD, 180 ± 20 mU/mg).
DISCUSSION
In this study, we found that the gene products of phcC (SLG_09480) and phcD (SLG_09500), which belong to the GMC oxidoreductase family, have activities for the oxidation of DCA-C. Enzymes in this family, including glucose oxidase (EC 1.1.3.4), cholesterol oxidase (EC 1.1.3.6), pyranose oxidase (EC 1.1.3.10), methanol oxidase (EC 1.1.3.13), aryl alcohol oxidase (AAO; EC 1.1.3.7), and choline dehydrogenase (EC 1.1.99.1), oxidize primary and secondary alcohols and contain FAD as a prosthetic group (44, 45). An FAD-binding domain and a substrate-binding domain are conserved in the N-terminal and C-terminal regions, respectively, among the enzymes in this family (46). These domains are also conserved in PhcC and PhcD, and the presence of FAD in PhcC was experimentally confirmed. In the SYK-6 genome, six putative GMC oxidoreductase family enzyme genes, including phcC and phcD were found. A phylogenetic analysis suggested that all of these SYK-6 gene products are related to choline dehydrogenases (Fig. 9).
FIG 9.

Phylogenetic tree of PhcC and PhcD with known and putative GMC oxidoreductase family enzymes. The scale corresponds to a genetic distance of 0.06 substitutions per position. GMC oxidoreductase family enzymes include the following, with locus tags or GenBank accession numbers in parentheses: PhcC (SLG_09480) and PhcD (SLG_09500), DCA-C oxidases of Sphingobium sp. SYK-6; BetA_Ec, choline dehydrogenase of E. coli K-12 (P17444) (43); BetA_Cs, choline dehydrogenase of Chromohalobacter salexigens DSM 3043 (Q9L4K0) (54); BetA_Rm, choline dehydrogenase of Rhizobium meliloti 1021 (P54223) (55); AlkJ_Pp, alcohol dehydrogenase of Pseudomonas putida GPo1 (Q9WWW2) (48); PegA_St, polyethylene glycol dehydrogenase of Sphingopyxis terrae (Q93I49) (49); Pno_Ml, pyridoxine 4-oxidase of Microbacterium luteolum YK-1 (Q9AJD6) (56); BetA_Mm, 5-hydroxymethylfurfural oxidase of Methylovorus sp. MP688 (E4QP00) (57); Gox_An, glucose oxidase of Aspergillus niger NRLL-3 (P13006) (58); Gox_Pa, glucose oxidase of Penicillium amagasakiense (P81156) (46); P2ox_Pc, pyranose 2-oxidase of Phanerochaete chrysosporium BKM-F-1767 (Q6QWR1) (59); P2ox_Tv, pyranose 2-oxidase of Trametes versicolor (P79076) (60); P2ox_Pg, pyranose 2-oxidase of Phlebiopsis gigantea DSM 13218 (Q6UG02) (61); P2ox_Tm, pyranose 2-oxidase of Tricholoma matsutake (Q8J2V8) (62); ChoB_Bs, cholesterol oxidase of Brevibacterium sterolicum (P22637) (63); ChoA_Ss, cholesterol oxidase of Streptomyces sp. SA-COO (P12676) (64); ChoD_Mt, cholesterol oxidase of Mycobacterium tuberculosis H37Rv (P9WMV9) (65); Mox_Pa, methanol oxidase of Pichia angusta (P04841) (66); and Aod1_Cb, alcohol oxidase of Candida boidinii S2 (Q00922) (67). Putative GMC oxidoreductase family enzymes include the following: SLG_11400, SLG_12830, SLG_20170, and SLG_27980 of Sphingobium sp. SYK-6; WYH_02651, WYH_02653, and WYH_02869 of Altererythrobacter atlanticus 26DY36; TS85_05630 and TS85_05635 of Sphingomonas sp. WHSC-8; L288_14545 of Sphingobium quisquiliarum P25; NSU_1606 of Novosphingobium pentaromativorans US6-1; Caul_3245 of Caulobacter sp. K31; M529_19015 of Sphingobium ummariense RL-3; and YP76_13265 of Sphingobium chungbukense DJ77.
The DCA-C preparation contains almost equal amounts of (+)-DCA-C and (−)-DCA-C (Fig. 3), and PhcC and PhcD were found to be enantioselective DCA-C oxidases that are specific for (+)-DCA-C and (−)-DCA-C, respectively (Fig. 4 and 7). Based on the fact that the phcC phcD double mutant (SME112) lost the activity for the conversion of DCA-C, and an equimolar amount of DCA-C was accumulated from DCA, it was concluded that DCA is indeed catabolized via DCA-C and that phcC and phcD are essential for the conversion of (+)-DCA-C and (−)-DCA-C, respectively (Fig. 2). To date, we have demonstrated the involvement of three Cα-dehydrogenases (LigD, LigL, and LigN) and three glutathione S-transferases (LigF, LigE, and LigP) in the stereoselective oxidation of four stereoisomers of GGE and enantioselective β-ether cleavage of MPHPV enantiomers (13–15). As shown in this study, it is apparent that SYK-6 has evolved the enzyme systems to completely degrade the stereoisomers of not only β-aryl ether but also a phenylcoumaran-type lignin-derived biaryl. In this respect, it is noteworthy that phcC and phcD constitute an operon for the specific conversion of DCA-C and that their expression is induced during DCA catabolism. To utilize both enantiomers of DCA, either phcC or phcD might have arisen through gene duplication in SYK-6 or its ancestor. To our knowledge, this study is the first showing a set of enantiospecific oxidases belonging to the GMC oxidoreductase family of proteins. Interestingly, a gene set similar to phcC and phcD was found in Altererythrobacter atlanticus 26DY36 (WYH_02651 and WYH_02653) and Sphingomonas sp. WHSC-8 (TS85_05630 and TS85_05635) (Fig. 9), implying the involvement of the gene products of these strains in enzyme reactions similar to the stereospecific oxidation of DCA-C.
In SYK-6 cells, the activities for the conversion of DCA-C were observed in both the cytoplasm and membrane fractions (Table 2). Corresponding to this result, PhcC and PhcD produced in SYK-6 cells harboring phcC- and phcD-containing plasmids were localized to both the cytoplasm and membrane. Similarly, a GMC oxidoreductase family protein, polyethylene glycol dehydrogenase (PEGDH) from Sphingopyxis terrae, was suggested to be localized to both the soluble and membrane fractions (47). Another GMC oxidoreductase family protein, AlkJ, encoded on the alkane-metabolizing alk operon from Pseudomonas putida strain GPo1, was shown to be localized to the membrane (48). Since there is no predicted signal sequence or hydrophobic transmembrane spanner in PhcC, PhcD, PEGDH, and AlkJ, these enzymes are thought to be peripheral membrane proteins. Interestingly, it was reported that PEGDH and AlkJ were able to utilize ubiquinone (CoQ10) or its derivatives (CoQ0 and CoQ1) as electron acceptors for the oxidation of their substrates (48, 49). Furthermore, electron transport from AlkJ to cytochrome c in the presence of CoQ1 was observed. Therefore, the electrons that are removed from the substrate by AlkJ and PEGDH are thought to be transferred to the respiratory chain (48, 49). Meanwhile, we demonstrated that PhcC and PhcD were able to utilize CoQ0 and CoQ1 as electron acceptor and that the activities for the conversion of DCA-C were similar to that obtained in the presence of PMS. Furthermore, we found that SLG_38090, which encodes a putative cytochrome c family protein, was highly upregulated in the SYK-6 cells grown with DCA (ca. 5.9-fold) compared to the cells grown with vanillate. All these results strongly suggest that electrons removed from the substrates during the oxidation of DCA-C catalyzed by PhcC and PhcD were transferred to cytochrome c via ubiquinone. Although PhcC and PhcD in the cytoplasm may be able to oxidize DCA-C using molecular oxygen as an electron acceptor, the reaction efficiency seems to be very low, and the resulting H2O2 must be toxic to the cells. Therefore, PhcC and PhcD in the cytoplasm appear to not play a major role in the catabolism of DCA-C in vivo.
The membrane fractions containing PhcC and PhcD converted DCA-C into DCA-CC, and only a trace amount of DCA-CL was accumulated (Fig. 6). This result suggests that both enzymes were capable of oxidizing DCA-C to carboxylic acid derivatives. AAOs belonging to the GMC oxidoreductase family have activities for the oxidation of a variety of aromatic alcohols to the corresponding aromatic aldehydes, while AAOs exhibit activities for the oxidation of some aromatic aldehydes. For example, the activity of AAO from Pleurotus eringii for the oxidation of benzaldehyde was less than 1% of that for the oxidation of benzyl alcohol. In contrast, the activity of this enzyme for the oxidation of 4-nitrobenzaldehyde was ca. 50% of that for the oxidation of 4-nitorobenzyl alcohol (50). Generation of aromatic carboxylic acids from aromatic alcohols by the reaction catalyzed by AAO is thought to be the result of the oxidation of gem-diols formed by the hydration of aromatic aldehydes (51). A similar reaction is known in choline oxidase from Arthrobacter globiformis, which oxidizes choline to glycine betaine via betaine aldehyde (52). These facts suggest that PhcC and PhcD have the ability to convert DCA-C into DCA-CC through similar reactions, described above. However, since our results were obtained using the membrane fractions containing PhcC and PhcD, further experiments are necessary to verify this hypothesis using the purified enzymes. In a previous study, we observed a significant accumulation of DCA-CL when DCA-C was incubated with an ultrafiltrate of the cell extract of SYK-6 (>3 kDa) (33). After the addition of NAD+ into the same reaction mixture, DCA-CL was converted to vanillate and ferulate. These results suggest that cytoplasmic NAD+-dependent ALDHs are also involved in the oxidation of DCA-CL into DCA-CC.
Interestingly, PhcC and PhcD were able to oxidize the γ-carbon at the A-ring side chain of DCA, and their activities were equivalent to those for the oxidation of DCA-C. These results might suggest the presence of an alternative pathway in which the γ-carbon at the A-ring side chain of DCA is initially oxidized. However, after the incubation of DCA with the cell extract of SYK-6, both DCA-L and DCA-C were observed, whereas a DCA derivative, the γ-carbon at the A-ring side chain of which is carboxylic acid (DCA-AC), was not detected (data not shown). Furthermore, there was no difference in the conversion rates for DCA between the phcC phcD double mutant (SME112) and the wild-type strain (Fig. 2B). These results indicated that phcC and phcD are not involved in the conversion of DCA.
ACKNOWLEDGMENTS
This work was supported in part by the Development of Preparatory Basic Bioenergy Technology grant from The New Energy and Industrial Technology Development Organization (NEDO) of Japan and the Advanced Low Carbon Technology Research and Development Program grant from The Japan Science and Technology Agency (JST).
We thank DAICEL Corporation for performing the chiral-HPLC and optical-rotation analyses of DCA and DCA-C.
REFERENCES
- 1.Vanholme R, Demedts B, Morreel K, Ralph J, Boerjan W. 2010. Lignin biosynthesis and structure. Plant Physiol 153:895–905. doi: 10.1104/pp.110.155119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ralph J, Peng J, Lu F, Hatfield RD, Helm RF. 1999. Are lignins optically active? J Agric Food Chem 47:2991–2996. doi: 10.1021/jf9901136. [DOI] [PubMed] [Google Scholar]
- 3.Akiyama T, Magara K, Matsumoto Y, Meshitsuka G, Ishizu A, Lundquist K. 2000. Proof of the presence of racemic forms of arylglycerol-β-aryl ether structure in lignin: studies on the stereo structure of lignin by ozonation. J Wood Sci 46:414–415. doi: 10.1007/BF00776407. [DOI] [Google Scholar]
- 4.Akiyama T, Sugimoto T, Matsumoto Y, Meshitsuka G. 2002. Erythro/threo ratio of β-O-4 structures as an important structural characteristic of lignin. I. Improvement of ozonation method for the quantitative analysis of lignin side-chain structure. J Wood Sci 48:210–215. [Google Scholar]
- 5.Martínez AT, Speranza M, Ruiz-Dueñas FJ, Ferreira P, Camarero S, Guillén F, Martínez MJ, Gutiérrez A, del Río JC. 2005. Biodegradation of lignocellulosics: microbial, chemical, and enzymatic aspects of the fungal attack of lignin. Int Microbiol 8:195–204. [PubMed] [Google Scholar]
- 6.Lundell TK, Mäkelä MR, Hildén K. 2010. Lignin-modifying enzymes in filamentous basidiomycetes—ecological, functional and phylogenetic review. J Basic Microbiol 50:5–20. doi: 10.1002/jobm.200900338. [DOI] [PubMed] [Google Scholar]
- 7.Pollegioni L, Tonin F, Rosini E. 2015. Lignin-degrading enzymes. FEBS J 282:1190–1213. doi: 10.1111/febs.13224. [DOI] [PubMed] [Google Scholar]
- 8.Ahmad M, Roberts JN, Hardiman EM, Singh R, Eltis LD, Bugg TD. 2011. Identification of DypB from Rhodococcus jostii RHA1 as a lignin peroxidase. Biochemistry 50:5096–5107. doi: 10.1021/bi101892z. [DOI] [PubMed] [Google Scholar]
- 9.Brown ME, Barros T, Chang MC. 2012. Identification and characterization of a multifunctional dye peroxidase from a lignin-reactive bacterium. ACS Chem Biol 7:2074–2081. doi: 10.1021/cb300383y. [DOI] [PubMed] [Google Scholar]
- 10.Majumdar S, Lukk T, Solbiati JO, Bauer S, Nair SK, Cronan JE, Gerlt JA. 2014. Roles of small laccases from Streptomyces in lignin degradation. Biochemistry 53:4047–4058. doi: 10.1021/bi500285t. [DOI] [PubMed] [Google Scholar]
- 11.Bugg TD, Ahmad M, Hardiman EM, Rahmanpour R. 2011. Pathways for degradation of lignin in bacteria and fungi. Nat Prod Rep 28:1883–1896. doi: 10.1039/c1np00042j. [DOI] [PubMed] [Google Scholar]
- 12.Bugg TD, Ahmad M, Hardiman EM, Singh R. 2011. The emerging role for bacteria in lignin degradation and bio-product formation. Curr Opin Biotechnol 22:394–400. doi: 10.1016/j.copbio.2010.10.009. [DOI] [PubMed] [Google Scholar]
- 13.Sato Y, Moriuchi H, Hishiyama S, Otsuka Y, Oshima K, Kasai D, Nakamura M, Ohara S, Katayama Y, Fukuda M, Masai E. 2009. Identification of three alcohol dehydrogenase genes involved in the stereospecific catabolism of arylglycerol-β-aryl ether by Sphingobium sp. strain SYK-6. Appl Environ Microbiol 75:5195–5201. doi: 10.1128/AEM.00880-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Masai E, Ichimura A, Sato Y, Miyauchi K, Katayama Y, Fukuda M. 2003. Roles of the enantioselective glutathione S-transferases in cleavage of β-aryl ether. J Bacteriol 185:1768–1775. doi: 10.1128/JB.185.6.1768-1775.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tanamura K, Abe T, Kamimura N, Kasai D, Hishiyama S, Otsuka Y, Nakamura M, Kajita S, Katayama Y, Fukuda M, Masai E. 2011. Characterization of the third glutathione S-transferase gene involved in enantioselective cleavage of the β-aryl ether by Sphingobium sp. strain SYK-6. Biosci Biotechnol Biochem 75:2404–2407. doi: 10.1271/bbb.110525. [DOI] [PubMed] [Google Scholar]
- 16.Linger JG, Vardon DR, Guarnieri MT, Karp EM, Hunsinger GB, Franden MA, Johnson CW, Chupka G, Strathmann TJ, Pienkos PT, Beckham GT. 2014. Lignin valorization through integrated biological funneling and chemical catalysis. Proc Natl Acad Sci U S A 111:12013–12018. doi: 10.1073/pnas.1410657111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Otsuka Y, Nakamura M, Shigehara K, Sugimura K, Masai E, Ohara S, Katayama Y. 2006. Efficient production of 2-pyrone 4,6-dicarboxylic acid as a novel polymer-based material from protocatechuate by microbial function. Appl Microbiol Biotechnol 71:608–614. doi: 10.1007/s00253-005-0203-7. [DOI] [PubMed] [Google Scholar]
- 18.Hishida M, Shikinaka K, Katayama Y, Kajita S, Masai E, Nakamura M, Otsuka Y, Ohara S, Shigehara K. 2009. Polyesters of 2-pyrone-4,6-dicarboxylic acid (PDC) as bio-based plastics exhibiting strong adhering properties. Polym J 41:297–302. doi: 10.1295/polymj.PJ2008291. [DOI] [Google Scholar]
- 19.Masai E, Katayama Y, Fukuda M. 2007. Genetic and biochemical investigations on bacterial catabolic pathways for lignin-derived aromatic compounds. Biosci Biotechnol Biochem 71:1–15. doi: 10.1271/bbb.60437. [DOI] [PubMed] [Google Scholar]
- 20.Yoshikata T, Suzuki K, Kamimura N, Namiki M, Hishiyama S, Araki T, Kasai D, Otsuka Y, Nakamura M, Fukuda M, Katayama Y, Masai E. 2014. Three-component O-demethylase system essential for catabolism of a lignin-derived biphenyl compound in Sphingobium sp. strain SYK-6. Appl Environ Microbiol 80:7142–7153. doi: 10.1128/AEM.02236-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Peng X, Egashira T, Hanashiro K, Masai E, Nishikawa S, Katayama Y, Kimbara K, Fukuda M. 1998. Cloning of a Sphingomonas paucimobilis SYK-6 gene encoding a novel oxygenase that cleaves lignin-related biphenyl and characterization of the enzyme. Appl Environ Microbiol 64:2520–2527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Peng X, Masai E, Kasai D, Miyauchi K, Katayama Y, Fukuda M. 2005. A second 5-carboxyvanillate decarboxylase gene, ligW2, is important for lignin-related biphenyl catabolism in Sphingomonas paucimobilis SYK-6. Appl Environ Microbiol 71:5014–5021. doi: 10.1128/AEM.71.9.5014-5021.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fukuhara Y, Kamimura N, Nakajima M, Hishiyama S, Hara H, Kasai D, Tsuji Y, Narita-Yamada S, Nakamura S, Katano Y, Fujita N, Katayama Y, Fukuda M, Kajita S, Masai E. 2013. Discovery of pinoresinol reductase genes in sphingomonads. Enzyme Microb Technol 52:38–43. doi: 10.1016/j.enzmictec.2012.10.004. [DOI] [PubMed] [Google Scholar]
- 24.Masai E, Harada K, Peng X, Kitayama H, Katayama Y, Fukuda M. 2002. Cloning and characterization of the ferulic acid catabolic genes of Sphingomonas paucimobilis SYK-6. Appl Environ Microbiol 68:4416–4424. doi: 10.1128/AEM.68.9.4416-4424.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kasai D, Kamimura N, Tani K, Umeda S, Abe T, Fukuda M, Masai E. 2012. Characterization of FerC, a MarR-type transcriptional regulator, involved in transcriptional regulation of the ferulate catabolic operon in Sphingobium sp. strain SYK-6. FEMS Microbiol Lett 332:68–75. doi: 10.1111/j.1574-6968.2012.02576.x. [DOI] [PubMed] [Google Scholar]
- 26.Masai E, Yamamoto Y, Inoue T, Takamura K, Hara H, Kasai D, Katayama Y, Fukuda M. 2007. Characterization of ligV essential for catabolism of vanillin by Sphingomonas paucimobilis SYK-6. Biosci Biotechnol Biochem 71:2487–2492. doi: 10.1271/bbb.70267. [DOI] [PubMed] [Google Scholar]
- 27.Abe T, Masai E, Miyauchi K, Katayama Y, Fukuda M. 2005. A tetrahydrofolate-dependent O-demethylase, LigM, is crucial for catabolism of vanillate and syringate in Sphingomonas paucimobilis SYK-6. J Bacteriol 187:2030–2037. doi: 10.1128/JB.187.6.2030-2037.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Masai E, Sasaki M, Minakawa Y, Abe T, Sonoki T, Miyauchi K, Katayama Y, Fukuda M. 2004. A novel tetrahydrofolate-dependent O-demethylase gene is essential for growth of Sphingomonas paucimobilis SYK-6 with syringate. J Bacteriol 186:2757–2765. doi: 10.1128/JB.186.9.2757-2765.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Capanema EA, Balakshin MY, Kadla JF. 2005. Quantitative characterization of a hardwood milled wood lignin by nuclear magnetic resonance spectroscopy. J Agric Food Chem 53:9639–9649. doi: 10.1021/jf0515330. [DOI] [PubMed] [Google Scholar]
- 30.Morreel K, Ralph J, Kim H, Lu F, Goeminne G, Ralph S, Messens E, Boerjan W. 2004. Profiling of oligolignols reveals monolignol coupling conditions in lignifying poplar xylem. Plant Physiol 136:3537–3549. doi: 10.1104/pp.104.049304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Morreel K, Dima O, Kim H, Lu F, Niculaes C, Vanholme R, Dauwe R, Goeminne G, Inzé D, Messens E, Ralph J, Boerjan W. 2010. Mass spectrometry-based sequencing of lignin oligomers. Plant Physiol 153:1464–1478. doi: 10.1104/pp.110.156489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Habu N, Samejima M, Yoshimoto T. 1988. Metabolic pathway of dehydrodiconiferyl alcohol by Pseudomonas sp. TMY1009. Mokuzai Gakkaishi 34:1026–1034. [Google Scholar]
- 33.Takahashi K, Kamimura N, Hishiyama S, Hara H, Kasai D, Katayama Y, Fukuda M, Kajita S, Masai E. 2014. Characterization of the catabolic pathway for a phenylcoumaran-type lignin-derived biaryl in Sphingobium sp. strain SYK-6. Biodegradation 25:735–745. doi: 10.1007/s10532-014-9695-0. [DOI] [PubMed] [Google Scholar]
- 34.Kamoda S, Saburi Y. 1993. Structural and enzymatical comparison of lignostilbene-α,β-dioxygenase isozymes, I, II, and III, from Pseudomonas paucimobilis TMY1009. Biosci Biotechnol Biochem 57:931–934. doi: 10.1271/bbb.57.931. [DOI] [PubMed] [Google Scholar]
- 35.Kamoda S, Terada T, Saburi Y. 1997. Purification and some properties of lignostilbene-α,β-dioxygenase isozyme IV from Pseudomonas paucimobilis TMY1009. Biosci Biotechnol Biochem 61:1575–1576. doi: 10.1271/bbb.61.1575. [DOI] [PubMed] [Google Scholar]
- 36.Fukuhara Y, Inakazu K, Kodama N, Kamimura N, Kasai D, Katayama Y, Fukuda M, Masai E. 2010. Characterization of the isophthalate degradation genes of Comamonas sp. strain E6. Appl Environ Microbiol 76:519–527. doi: 10.1128/AEM.01270-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Johnson M, Zaretskaya I, Raytselis Y, Merezhuk Y, McGinnis S, Madden TL. 2008. NCBI BLAST: a better web interface. Nucleic Acids Res 36:W5–W9. doi: 10.1093/nar/gkn201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rice P, Longden I, Bleasby A. 2000. EMBOSS: the European molecular biology open software suite. Trends Genet 16:276–277. doi: 10.1016/S0168-9525(00)02024-2. [DOI] [PubMed] [Google Scholar]
- 39.Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, McWilliam H, Valentin F, Wallace IM, Wilm A, Lopez R, Thompson JD, Gibson TJ, Higgins DG. 2007. Clustal W and Clustal X version 2.0. Bioinformatics 23:2947–2948. doi: 10.1093/bioinformatics/btm404. [DOI] [PubMed] [Google Scholar]
- 40.Masai E, Shinohara S, Hara H, Nishikawa S, Katayama Y, Fukuda M. 1999. Genetic and biochemical characterization of a 2-pyrone-4,6-dicarboxylic acid hydrolase involved in the protocatechuate 4,5-cleavage pathway of Sphingomonas paucimobilis SYK-6. J Bacteriol 181:55–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kamimura N, Aoyama T, Yoshida R, Takahashi K, Kasai D, Abe T, Mase K, Katayama Y, Fukuda M, Masai E. 2010. Characterization of the protocatechuate 4,5-cleavage pathway operon in Comamonas sp. strain E6 and discovery of a novel pathway gene. Appl Environ Microbiol 76:8093–8101. doi: 10.1128/AEM.01863-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Toyama H, Mathews FS, Adachi O, Matsushita K. 2004. Quinohemoprotein alcohol dehydrogenases: structure, function, and physiology. Arch Biochem Biophys 428:10–21. doi: 10.1016/j.abb.2004.03.037. [DOI] [PubMed] [Google Scholar]
- 43.Lamark T, Kaasen I, Eshoo MW, Falkenberg P, McDougall J, Strøm AR. 1991. DNA sequence and analysis of the bet genes encoding the osmoregulatory choline-glycine betaine pathway of Escherichia coli. Mol Microbiol 5:1049–1064. doi: 10.1111/j.1365-2958.1991.tb01877.x. [DOI] [PubMed] [Google Scholar]
- 44.Romero E, Gadda G. 2014. Alcohol oxidation by flavoenzymes. Biomol Concepts 5:299–318. [DOI] [PubMed] [Google Scholar]
- 45.Dijkman WP, de Gonzalo G, Mattevi A, Fraaije MW. 2013. Flavoprotein oxidases: classification and applications. Appl Microbiol Biotechnol 97:5177–5188. doi: 10.1007/s00253-013-4925-7. [DOI] [PubMed] [Google Scholar]
- 46.Kiess M, Hecht HJ, Kalisz HM. 1998. Glucose oxidase from Penicillium amagasakiense. Primary structure and comparison with other glucose-methanol-choline (GMC) oxidoreductases. Eur J Biochem 252:90–99. doi: 10.1046/j.1432-1327.1998.2520090.x. [DOI] [PubMed] [Google Scholar]
- 47.Kawai F, Kimura T, Fukaya M, Tani Y, Ogata K, Ueno T, Fukami H. 1978. Bacterial oxidation of polyethylene glycol. Appl Environ Microbiol 35:679–684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kirmair L, Skerra A. 2014. Biochemical analysis of recombinant AlkJ from Pseudomonas putida reveals a membrane-associated, flavin adenine dinucleotide-dependent dehydrogenase suitable for the biosynthetic production of aliphatic aldehydes. Appl Environ Microbiol 80:2468–2477. doi: 10.1128/AEM.04297-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ohta T, Kawabata T, Nishikawa K, Tani A, Kimbara K, Kawai F. 2006. Analysis of amino acid residues involved in catalysis of polyethylene glycol dehydrogenase from Sphingopyxis terrae, using three-dimensional molecular modeling-based kinetic characterization of mutants. Appl Environ Microbiol 72:4388–4396. doi: 10.1128/AEM.02174-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Guillén F, Martínez AT, Martínez MJ. 1992. Substrate specificity and properties of the aryl-alcohol oxidase from the ligninolytic fungus Pleurotus eryngii. Eur J Biochem 209:603–611. doi: 10.1111/j.1432-1033.1992.tb17326.x. [DOI] [PubMed] [Google Scholar]
- 51.Ferreira P, Hernández-Ortega A, Herguedas B, Rencoret J, Gutiérrez A, Martínez MJ, Jiménez-Barbero J, Medina M, Martínez AT. 2010. Kinetic and chemical characterization of aldehyde oxidation by fungal aryl-alcohol oxidase. Biochem J 425:585–593. doi: 10.1042/BJ20091499. [DOI] [PubMed] [Google Scholar]
- 52.Fan F, Germann MW, Gadda G. 2006. Mechanistic studies of choline oxidase with betaine aldehyde and its isosteric analogue 3,3-dimethylbutyraldehyde. Biochemistry 45:1979–1986. doi: 10.1021/bi0517537. [DOI] [PubMed] [Google Scholar]
- 53.Hirai N, Okamoto M, Udagawa H, Yamamuro M, Kato M, Koshimizu K. 1994. Absolute configuration of dehydrodiconiferyl alcohol. Biosci Biotechnol Biochem 58:1679–1684. doi: 10.1271/bbb.58.1679. [DOI] [Google Scholar]
- 54.Cánovas D, Vargas C, Kneip S, Morón MJ, Ventosa A, Bremer E, Nieto JJ. 2000. Genes for the synthesis of the osmoprotectant glycine betaine from choline in the moderately halophilic bacterium Halomonas elongata DSM 3043. Microbiology 146:455–463. doi: 10.1099/00221287-146-2-455. [DOI] [PubMed] [Google Scholar]
- 55.Pocard JA, Vincent N, Boncompagni E, Smith LT, Poggi MC, Le Rudulier D. 1997. Molecular characterization of the bet genes encoding glycine betaine synthesis in Sinorhizobium meliloti 102F34. Microbiology 143:1369–1379. doi: 10.1099/00221287-143-4-1369. [DOI] [PubMed] [Google Scholar]
- 56.Kaneda Y, Ohnishi K, Yagi T. 2002. Purification, molecular cloning, and characterization of pyridoxine 4-oxidase from Microbacterium luteolum. Biosci Biotechnol Biochem 66:1022–1031. doi: 10.1271/bbb.66.1022. [DOI] [PubMed] [Google Scholar]
- 57.Dijkman WP, Fraaije MW. 2014. Discovery and characterization of a 5-hydroxymethylfurfural oxidase from Methylovorus sp. strain MP688. Appl Environ Microbiol 80:1082–1090. doi: 10.1128/AEM.03740-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Frederick KR, Tung J, Emerick RS, Masiarz FR, Chamberlain SH, Vasavada A, Rosenberg S, Chakraborty S, Schopfer LM, Massey V. 1990. Glucose oxidase from Aspergillus niger. Cloning, gene sequence, secretion from Saccharomyces cerevisiae and kinetic analysis of a yeast-derived enzyme. J Biol Chem 265:3793–3802. [PubMed] [Google Scholar]
- 59.de Koker TH, Mozuch MD, Cullen D, Gaskell J, Kersten PJ. 2004. Isolation and purification of pyranose 2-oxidase from Phanerochaete chrysosporium and characterization of gene structure and regulation. Appl Environ Microbiol 70:5794–5800. doi: 10.1128/AEM.70.10.5794-5800.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Nishimura I, Okada K, Koyama Y. 1996. Cloning and expression of pyranose oxidase cDNA from Coriolus versicolor in Escherichia coli. J Biotechnol 52:11–20. doi: 10.1016/S0168-1656(96)01618-5. [DOI] [PubMed] [Google Scholar]
- 61.Bastian S, Rekowski MJ, Witte K, Heckmann-Pohl DM, Giffhorn F. 2005. Engineering of pyranose 2-oxidase from Peniophora gigantea towards improved thermostability and catalytic efficiency. Appl Microbiol Biotechnol 67:654–663. doi: 10.1007/s00253-004-1813-1. [DOI] [PubMed] [Google Scholar]
- 62.Takakura Y, Kuwata S. 2003. Purification, characterization, and molecular cloning of a pyranose oxidase from the fruit body of the basidiomycete, Tricholoma matsutake. Biosci Biotechnol Biochem 67:2598–2607. doi: 10.1271/bbb.67.2598. [DOI] [PubMed] [Google Scholar]
- 63.Vrielink A, Lloyd LF, Blow DM. 1991. Crystal structure of cholesterol oxidase from Brevibacterium sterolicum refined at 1.8 Å resolution. J Mol Biol 219:533–554. doi: 10.1016/0022-2836(91)90192-9. [DOI] [PubMed] [Google Scholar]
- 64.Yue QK, Kass IJ, Sampson NS, Vrielink A. 1999. Crystal structure determination of cholesterol oxidase from Streptomyces and structural characterization of key active site mutants. Biochemistry 38:4277–4286. doi: 10.1021/bi982497j. [DOI] [PubMed] [Google Scholar]
- 65.Brzostek A, Dziadek B, Rumijowska-Galewicz A, Pawelczyk J, Dziadek J. 2007. Cholesterol oxidase is required for virulence of Mycobacterium tuberculosis. FEMS Microbiol Lett 275:106–112. doi: 10.1111/j.1574-6968.2007.00865.x. [DOI] [PubMed] [Google Scholar]
- 66.Evers ME, Titorenko V, Harder W, van der Klei I, Veenhuis M. 1996. Flavin adenine dinucleotide binding is the crucial step in alcohol oxidase assembly in the yeast Hansenula polymorpha. Yeast 12:917–923. [DOI] [PubMed] [Google Scholar]
- 67.Stewart MQ, Esposito RD, Gowani J, Goodman JM. 2001. Alcohol oxidase and dihydroxyacetone synthase, the abundant peroxisomal proteins of methylotrophic yeasts, assemble in different cellular compartments. J Cell Sci 114:2863–2868. [DOI] [PubMed] [Google Scholar]
- 68.Katayama Y, Nishikawa S, Nakamura M, Yano K, Yamasaki M, Morohoshi N, Haraguchi T. 1987. Cloning and expression of Pseudomonas paucimobilis SYK-6 genes involved in the degradation of vanillate and protocatechuate in P. putida. Mokuzai Gakkaishi 33:77–79. [Google Scholar]
- 69.Studier FW, Moffatt BA. 1986. Use of bacteriophage T7 RNA polymerase to direct selective high-level expression of cloned genes. J Mol Biol 189:113–130. doi: 10.1016/0022-2836(86)90385-2. [DOI] [PubMed] [Google Scholar]
- 70.Bolivar F, Backman K. 1979. Plasmids of Escherichia coli as cloning vectors. Methods Enzymol 68:245–267. doi: 10.1016/0076-6879(79)68018-7. [DOI] [PubMed] [Google Scholar]
- 71.Short JM, Fernandez JM, Sorge JA, Huse WD. 1988. λ ZAP: a bacteriophage λ expression vector with in vivo excision properties. Nucleic Acids Res 16:7583–7600. doi: 10.1093/nar/16.15.7583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Blatny JM, Brautaset T, Winther-Larsen HC, Karunakaran P, Valla S. 1997. Improved broad-host-range RK2 vectors useful for high and low regulated gene expression levels in gram-negative bacteria. Plasmid 38:35–51. doi: 10.1006/plas.1997.1294. [DOI] [PubMed] [Google Scholar]
- 73.Schäfer A, Tauch A, Jäger W, Kalinowski J, Thierbach G, Pühler A. 1994. Small mobilizable multipurpose cloning vectors derived from the Escherichia coli plasmids pK18 and pK19: selection of defined deletions in the chromosome of Corynebacterium glutamicum. Gene 145:69–73. doi: 10.1016/0378-1119(94)90324-7. [DOI] [PubMed] [Google Scholar]