

Abstract
Salmonella enterica serovars Typhimurium (S. Typhimurium) and Enteritidis (S. Enteritidis) are foodborne pathogens, and outbreaks are often associated with poultry products. Chickens are typically asymptomatic when colonized by these serovars; however, the factors contributing to this observation are uncharacterized. Whereas symptomatic mammals have a body temperature between 37°C and 39°C, chickens have a body temperature of 41°C to 42°C. Here, in vivo experiments using chicks demonstrated that numbers of viable S. Typhimurium or S. Enteritidis bacteria within the liver and spleen organ sites were ≥4 orders of magnitude lower than those within the ceca. When similar doses of S. Typhimurium or S. Enteritidis were given to C3H/HeN mice, the ratio of the intestinal concentration to the liver/spleen concentration was 1:1. In the avian host, this suggested poor survival within these tissues or a reduced capacity to traverse the host epithelial layer and reach liver/spleen sites or both. Salmonella pathogenicity island 1 (SPI-1) promotes localization to liver/spleen tissues through invasion of the epithelial cell layer. Following in vitro growth at 42°C, SPI-1 genes sipC, invF, and hilA and the SPI-1 rtsA activator were downregulated compared to expression at 37°C. Overexpression of the hilA activators fur, fliZ, and hilD was capable of inducing hilA-lacZ at 37°C but not at 42°C despite the presence of similar levels of protein at the two temperatures. In contrast, overexpression of either hilC or rtsA was capable of inducing hilA and sipC at 42°C. These data indicate that physiological parameters of the poultry host, such as body temperature, have a role in modulating expression of virulence.
INTRODUCTION
Salmonella enterica serovars Typhimurium (S. Typhimurium) and Enteritidis (S. Enteritidis) are major causes of foodborne diseases worldwide. In the United States, S. Typhimurium and S. Enteritidis accounted for the majority of confirmed cases of Salmonella outbreaks between 1970 and 2011 (1). These two are nontyphoid Salmonella (NTS) serovars that are capable of causing disease signs in a variety of animals, which contrasts with typhoid fever serovars that exclusively infect humans. Numerous food products have been associated with Salmonella outbreaks and illnesses in humans; however, poultry products are frequently implicated in outbreaks associated with NTS (www.cdc.gov/Salmonella/outbreaks.html). In 2010, a major poultry-related outbreak occurred that involved S. Enteritidis infections across 11 states and resulted in the recall of 380 million eggs (2).
S. Typhimurium invades the host epithelial cell layer and migrates to liver and spleen tissue sites through the action of a type 3 secretion system (T3SS), encoded by Salmonella pathogenicity island 1 (SPI-1). SPI-1 is a DNA segment that is approximately 40 kb in size and encodes the structural components of the secretion system, secreted and chaperone proteins, and transcription factors that activate expression of the SPI-1 genes (3–5). Three regulators that are carried within SPI-1, hilA, hilC, and hilD, and rtsA, which is carried outside SPI-1, are critical activators of the island. Collectively, the protein products encoded by genes within SPI-1 promote uptake of the pathogen by nonphagocytic cells of the host's epithelial cell layer (6–8). Oral infection with S. Typhimurium mutants lacking the entire pathogenicity island or with Δspi1 or ΔhilA mutants results in severely limited localization of the mutant to the spleen compared to wild-type strain results, but this difference is not observed when mice are inoculated through the intraperitoneal route (9). This supports the concept that SPI-1 plays an important role in traversing the intestinal epithelial layer.
Invasion of the host epithelial cell layer is a critical aspect of S. Typhimurium virulence (10). RtsA forms a complex regulatory network with HilC and HilD that ultimately activates expression of hilA, which activates components of SPI-1 (9, 11). Recently, several works have contributed to our understanding of the complex regulation of SPI-1 (12–14). In addition, the DNA binding sites of the HilD protein have been mapped and include several genes that are coregulated by HilC and RtsA (15), suggesting that reduced activation by one of the three activators may influence the activation of coregulated genes.
Although poultry are associated with Salmonella outbreaks, poultry are largely asymptomatic. In general, oral administration of S. Typhimurium or S. Enteritidis to poultry results in poor localization and colonization of systemic tissues such as the liver and spleen compared to bacterial concentration within the ceca (16–20). Although the reasons for these results are likely multifactorial, the 3 to 5°C difference in the body temperature of poultry (41°C to 42°C) compared to susceptible mammals (37°C to 39°C) may be a factor contributing to the lack of systemic localization by these serovars in poultry. Since both serovars are capable of reaching systemic tissues in other animals, we hypothesized that the body temperature of chickens exerts a regulatory effect that limits expression of SPI-1 and localization to systemic tissues.
Here, we show that S. Typhimurium or S. Enteritidis colonized the ceca of a commercial breed of chicks but localized poorly to the liver and spleen, suggesting a low level of invasion. These results contrasted with data from the murine host, which exhibited similar concentrations of S. Typhimurium or S. Enteritidis within the intestines, spleen, and liver. Therefore, the effect of temperature on the regulation of SPI-1 was evaluated in cells grown at 37°C versus 42°C. Following growth at 42°C, there was reduced expression of SPI-1 genes. Gene expression studies conducted at 42°C demonstrated reduced activation of the rtsA gene, which is directly activated by HilD (15), suggesting a reduction in the level of either HilD or HilD protein activity in response to growth at 42°C. To gain insight into the mechanism resulting in the inability to activate SPI-1 at 42°C, we utilized an inducible system to test the roles of Fur, FliZ, HilC, RtsA (STM14_5188), and HilD. Inducible expression of either HilC or RtsA protein, but not of Fur, FliZ, or HilD, was sufficient to activate hilA following growth at 42°C. As previously shown, the Lon protease inhibited activation of SPI-1; however, the lack of lon still resulted in the inability to activate SPI-1 following growth at 42°C. Our results supported the hypothesis that the body temperature of poultry caused a regulatory change in the expression of SPI-1 genes that likely contributed to the poor localization to the liver and spleen. Thus, the body temperature of poultry is a significant barrier to activation of SPI-1 and may contribute to diminished invasion of S. Typhimurium or S. Enteritidis in vivo.
MATERIALS AND METHODS
Bacterial strains, plasmids constructions, and reagents.
The bacterial strains used throughout this study and their construction are described and listed in Table S1 in the supplemental material.
S. Typhimurium and S. Enteritidis challenge of 1-day-old chicks.
A total of 50 1-day-old layer-type chicks (W-36; Hy-Line North America, West Des Moines, IA) were randomly assigned to two HEPA-filtered 934-WP animal isolators (L. H. Leathers, Inc., Athens, GA) (25 birds each). Each isolator is equipped with wire mesh racks located above plastic trays to collect waste. Chicks were provided water and feed (All Grain Start-N-Grow; Southern States, Richmond, VA) ad libitum. Isolators were temperature controlled and were maintained between 29°C and 31°C for the duration of the experiment. The Institutional Animal Care and Use Committee (IACUC) approved the animal study protocol (protocol 15-065-A).
S. Typhimurium strain NC1040 was cultivated overnight under standing conditions in Luria-Bertani broth (LB) medium (per liter, 10 g tryptone, 5 g yeast extract, and 10 g NaCl) at 37°C, concentrated by centrifugation, and washed with phosphate-buffered saline (PBS). The cell pellet was resuspended in PBS to a concentration of ∼5 × 109 CFU/ml (optical density at 600 nm [OD600] of ∼10 for S. Typhimurium and ∼20 for S. Enteritidis). Chicks were individually inoculated by oral gavage with 100 μl of the cell suspension (∼5 × 108 CFU per bird). The inoculum of strain NC1040 was quantified by serial dilution and plating to ensure the accuracy of the dose given to animals. Control birds were housed separately and given equal volumes of PBS. At the indicated days postinoculation (dpi), 5 birds from each treatment group were euthanized and the cecal contents, liver, and spleen were aseptically removed and placed in PBS–1 mM MgCl2. The weights of cecal content, liver, and spleen samples were recorded. Cecal contents were serially diluted and plated on XLT4 agar plates with 100 mM MOPS (morpholinepropanesulfonic acid) (pH 7.4) without Tergitol (BD Difco, Franklin Lakes, NJ). Liver and spleen samples were homogenized (Kimble Chase Kontes tissue grinder; VWR International, Radnor, PA), serially diluted, and plated on XLT4 agar plates. XLT4 agar plates contained kanamycin sulfate to select for strain NC1040 and 100 μg/ml rifampin to select for strain BTNC0025, and H2S-positive (black) colonies were counted. CFU counts were normalized to the weight of cecal contents or tissue.
The detection limit for this procedure was determined by spiking liver samples from control birds with strain BTNC0025. Livers were homogenized and plated on XLT4-MOPS with rifampin to determine mean levels of CFU per gram. Three separate experiments were used to determine the means ± standard deviations (SD) corresponding to the detection limit (log10 CFU per gram, 2.3 ± 1.7). When 50% or less of the tissue samples were culture negative, the mean detection limit was substituted for the zero value to determine the mean value for the samples for that population and time point (simple replacement approach).
Measurement of cloacal temperatures within chicks.
The body temperatures of a W-36 line of chicks, reared at North Carolina State University, were determined by measuring the cloacal temperature over time. Seventy-one birds were housed in two separate isolators as mentioned above, and birds were euthanized over time to maintain the appropriate space requirements for the animals within the isolators. A digital thermometer (Easy-Read Flex-Tip digital thermometer; Walgreens, Deerfield, IL) was used to measure cloacal temperatures at 1 (n = 71), 2 (n = 71), 4 (n = 61), 6 (n = 43), 8 (n = 32), 13 (n = 19), and 22 (n = 12) days posthatch. A body temperature reading was obtained following insertion of the temperature probe approximately 1 to 2 in. within the cloacal orifice. A smooth, sterile probe cover was used during measurements to prevent tissue damage.
S. Typhimurium and S. Enteritidis challenge of mice.
Sixteen female C3H/HeN mice (Ityr, Salmonella resistant) were purchased from Harlan Laboratories (Indianapolis, IN) and subjected to gavage with S. Typhimurium strain NC1040 or S. Enteritidis strain BTNC0025 as described above. Mice were housed in disposable cages (4 mice per cage), and each mouse was given ∼5 × 108 CFU of strain NC1040. In accordance with previous work (21), disease symptoms were monitored and mice were given a body condition score (BCS). At the indicated dpi, mice were euthanized and the concentration of strain NC1040 within the intestines, liver, and spleen was determined as described above. The IACUC approved the animal study protocol (protocol 15-035-B).
β-Galactosidase assays, SDS-PAGE, and immunoblotting.
For all experiments, frozen stocks (−80°C) of bacteria were inoculated into LB (Fisher) medium and incubated overnight at 37°C. Samples were then diluted at either 1:50 or 1:500 into LB medium containing 1 mM glucose and 100 mM MOPS buffered to pH 7.4 with NaOH or were left unbuffered (pH 6.0) and then were split into separate 15-ml conical tubes (Fisher) and cultured overnight at 37°C or 42°C. To induce expression of IPTG (isopropyl-β-d-thiogalactopyranoside)-controlled promoters, 0.1 or 0.01 mM IPTG was added to the growth medium. To induce expression of the hilD gene, tetracycline-HCl was added to reach a final concentration of 2.5 μg/ml. Transcriptional fusions to the lacZ gene were assayed for β-galactosidase as described previously (22). β-Galactosidase activity measured in experiments performed with the pSP417 multicopy plasmid and derivatives was determined by the use of 10 μl of sample to measure activity, whereas all other fusions used 100 μl of sample. β-Galactosidase activity was measured in stationary-phase cells following overnight growth (approximately 16 to 18 h) under the specified conditions, and, when appropriate, cells were pelleted from a portion of the sample and prepared for SDS-PAGE as described below. When expression studies used derivatives of rifampin-resistant S. Enteritidis strain BTNC0025, to avoid SPI-1 repression through metabolism of the solvent dimethyl sulfoxide (DMSO), rifampin was not added (14, 23).
Expression of FLAG-tagged proteins was determined by Western blotting, where the cell pellets were suspended in Laemmli sample buffer and samples were reduced and denatured by boiling. Samples were separated by size on 15% acrylamide gels (SDS-PAGE) and transferred to 0.2 μM nitrocellulose membranes (Bio-Rad, Hercules, CA). Immunoblotting was performed as described previously (24, 25). Briefly, membranes were stained with Ponceau S (0.1% Ponceau S [wt/vol], 1% acetic acid) to ensure equivalent loading of samples. For immunoblotting, membranes were blocked in a blocking buffer (i.e., PBS containing 0.05% Tween 20 and 1% powered nonfat milk, pH 7.4) and probed with primary antibody (monoclonal anti-FLAG M2; Sigma-Aldrich) at 1:2,000 for 2 to 3 h. Membranes were washed 2 to 3 times with the blocking buffer and probed with secondary antibody (peroxidase-conjugated goat anti-mouse antibody; Jackson ImmunoResearch Laboratories, West Grove, PA) at 1:10,000 for ∼1 h. Membranes were washed 2 to 3 times with Tris-NaCl (50 mM Tris, 200 mM NaCl, pH 7.6), and detection of horseradish peroxidase activity was determined in Tris-NaCl using 4-chloro-1-naphthol (4CN; dissolved in methanol) and H2O2 (Thermo Fisher Scientific, Waltham, MA).
qRT-PCR.
In response to growth at 42°C, changes in gene expression of target genes hilC, rtsA, and hilD and the rrsA reference gene were determined by quantitative reverse transcriptase PCR (qRT-PCR). Bacteria were grown overnight in LB medium and diluted 100-fold in 15-ml conical tubes containing 10 ml of LB-MOPS (pH 7.4) with 1 mM glucose. Cultures were incubated at 37°C or 42°C until an OD600 of ∼1 was reached (∼5.5 h after dilution). When expression studies used derivatives of the rifampin-resistant S. Enteritidis BTNC0025 strain, to avoid SPI-1 repression through metabolism of the solvent DMSO, rifampin was not added (14, 23). A 20-ml volume of cold RNAlater solution (Life Technologies, Grand Island, NY) was added to stabilize RNA. Samples were centrifuged at ∼10,000 × g, and cell pellets were resuspended in 1 ml of TRIzol reagent (Life Technologies). RNA was extracted according to manufacturer's specifications and treated with DNase I (New England BioLabs) for 1.5 h at 37°C. Then, the DNase-treated RNA was purified using the RNA cleanup protocol with an RNeasy miniprep kit (Qiagen). cDNA was synthesized as described previously (24). Briefly, 1 μl of a 10 mM deoxynucleoside triphosphate (dNTP) mixture (2.5 mM [each] dNTP), 0.5 μl of 100 μM gene-specific primer, total RNA, and H2O were added to reach a final volume of 13 μl. The sample was heated at 65°C for 5 min and then placed on ice for 1 min. Then, 5 μl of 5× first-strand synthesis buffer (Invitrogen), 1 μl of 0.1 M dithiothreitol (DTT; Invitrogen), 1 μl of RNase OUT (Invitrogen), and 1 μl of Superscript reverse transcriptase III (Invitrogen) were added. The sample was incubated at room temperature for 5 min and then at 50°C for 60 min and at 70°C for 15 min. Following cDNA synthesis, 20 or 30 μl of double-distilled water (ddH2O) was added to the sample. A control receiving no reverse transcriptase was included for each RNA sample.
qRT-PCR was performed, using RT2 SYBR green ROX quantitative PCR (qPCR) Mastermix, as follows: 10 μl of qPCR Mastermix, 0.75 μM (each) primers, 2 μl of cDNA, and ddH2O (used to adjust the reaction mixture to a 20-μl final volume) were added to a 96-well plate. An iCycler (Bio-Rad, Hercules, CA) machine was used with the following PCR parameters: 95°C for 15 min with 40 cycles of 95°C for 15 s, 50°C for 30 s, and 72°C for 30 s. The data were analyzed using Bio-Rad Optical System software, version 3.1, according to manufacturer specifications. Melting-curve analysis confirmed the presence of a single PCR product for each sample. A standard curve of rrsA DNA (102 to 108 copies per reaction mixture) was used to quantify transcripts, and expression data were normalized per the numbers of copies of rrsA, which is the R001 gene in S. Enteritidis. qPCR performed with no reverse transcriptase control, targeting rrsA, confirmed the reduction of ≥5 orders in magnitude of copy numbers in the samples, indicating that genomic DNA contamination was near the background level.
Primers are listed in Table S2 in the supplemental material and were purchased from IDT DNA Technologies (Coralville, IA) and were designed to target the hilC (STM14_3465), rtsA (STM14_5188), hilD (STM14_3474), and rrsA (STM14_4794) genes in S. Typhimurium; these genes correspond to SEN_2709, SEN_4086, SEN_2717, and the R001 gene in S. Enteritidis, respectively.
Statistical analysis.
Figures and statistical analysis were accomplished using GraphPad Prism v4.0. Throughout the study, Student's t test, with Bonferroni's correction for multiple comparisons when appropriate, was used to determine significance. In addition, an unpaired Student t test with Welch's correction was used to determine significant differences in Salmonella concentrations within livers and spleens of infected chicks compared to C3H/HeN mice. With strains that harbor derivatives of pSP417, β-galactosidase activities of some samples had a broad range (∼3 orders of magnitude). To address this, the reporter activities were log10 transformed prior to analysis.
RESULTS
S. Typhimurium and S. Enteritidis exhibit temporal localization to the liver and spleen in chicks compared the murine host.
To determine the tissue burden of S. Typhimurium in vivo, 25 1-day-old chicks of the W-36 line were subjected to gavage with ∼5 × 108 CFU/bird of the kanamycin-resistant “wild-type” strain (NC1040). At the indicated days postinoculation (dpi), 5 birds per group were euthanized and the concentration of NC1040 was enumerated from the cecal contents and tissue homogenates of the liver and spleen. At day 3, all livers were positive for NC1040 but at a much lower level than the concentrations in the ceca (Table 1). In addition, after 3 dpi, NC1040 was detected in the liver of only 1 bird (Table 1). Eleven of 25 birds were positive for NC1040 in the spleen at any time point during the study, with decreasing levels of NC1040 from 10 dpi until undetectable levels were seen at 21 dpi. All the liver-positive birds were identified before 10 days postinoculation (Table 1). In contrast, 25 of 25 birds had S. Typhimurium in the cecal content, with a mean concentration of >107 CFU/g throughout the course of the study (Table 1). Taken together, these results demonstrated that S. Typhimurium colonization of liver and spleen tissues was temporal among the birds and that, when detected, the concentrations were low (<104 CFU/g tissue), whereas ceca concentrations were >107 CFU/g and were detectable for the duration of the study.
TABLE 1.
Colonization of 1-day-old chickens following challenge with S. Typhimuriuma
| No. of days postinoculation with NC1040 | NC1040 CFU/g (no. of culture-positive birds/total no. of birds) |
||
|---|---|---|---|
| Cecum | Liver | Spleen | |
| 3 | 8.2 ± 0.3 (5/5) | 2.8 ± 0.3 (5/5) | 2.6 ± 0.3 (2/5) |
| 6 | 7.8 ± 0.2 (5/5) | 2.5 (1/5) | 2.8 ± 0.4 (4/5) |
| 10 | 8.0 ± 0.2 (5/5) | <2.3 (0/5) | 3.3 ± 0.9 (3/5) |
| 15 | 7.5 ± 1.4 (5/5) | <2.3 (0/5) | 2.7 ± 0.6 (2/5) |
| 21 | 7 ± 0.4 (5/5) | <2.3 (0/5) | <2.3 (0/5) |
W-36 chicks (1 day old) were inoculated with ∼5 × 108 CFU of the kanamycin-resistant (fnr′::ha) “wild-type” strain (NC1040). Data shown are the mean log10 CFU values per gram of cecal content or tissue homogenate from the indicated number of culture-positive birds among those sampled (shown in parentheses), with a detection limit of 2.3 log10 CFU/g. Results below the detection limit are shown as <2.3 (0/5).
To test whether these results were specific to S. Typhimurium, the experiment described above was repeated using strain BTNC0025, which is a rifampin-resistant isolate of S. Enteritidis ATCC 31194. W-36 1-day-old chicks were inoculated with ∼5 × 108 CFU of BTNC0025, and the bacterial burdens in the ceca, spleens, and livers were determined. Similarly to data obtained with S. Typhimurium strain NC1040, BTNC0025 did not localize well to these tissue sites. Throughout the study, concentrations of BTNC0025 within the spleen were undetectable (Table 2). Collectively, these data indicated that BTNC0025 exhibited poor localization to these tissue sites, which supported results obtained with S. Typhimurium. Although BTNC0025 was poorly detected within these tissue sites, the ceca was colonized to a level of >105 CFU/g within all birds until 10 dpi (Table 2). In contrast to the data obtained with NC1040, the cecal concentrations of the BTNC0025 strain declined after 10 dpi to undetectable levels at 21 dpi (Table 2), which was in agreement with earlier data (26). The results indicated that the Enteritidis strain exhibited poor localization to the livers and spleens.
TABLE 2.
Colonization of 1-day-old chickens following challenge with S. Enteritidisa
| No. of days postinoculation with BTNC0025 | BTNC0025 CFU/g (no. of culture-positive birds/total no. of birds) |
||
|---|---|---|---|
| Cecum | Liver | Spleen | |
| 1 | 7.7 ± 0.6 (5/5) | <2.3 (0/5) | <2.3 (0/5) |
| 3 | 6.4 ± 1.1 (5/5) | <2.3 (0/5) | <2.3 (0/5) |
| 6 | 6.9 ± 0.4 (5/5) | <2.3 (0/5) | <2.3 (0/5) |
| 10 | 5.1 ± 0.8 (5/5) | <2.3 (0/5) | <2.3 (0/5) |
| 15 | 5.0 ± 2.0 (4/5) | <2.3 (0/5) | <2.3 (0/5) |
| 21 | <2.3 (0/5) | <2.3 (0/5) | <2.3 (0/5) |
W-36 chicks (1 day old) were inoculated with ∼5 × 108 CFU of the rifampin-resistant “wild-type” strain (BTNC0025). Data shown are the mean log10 CFU values per gram of intestinal content or tissue homogenate from the indicated number of culture-positive birds among those sampled (shown in parentheses), with a detection limit of <2.3 log10 CFU/g.
S. Typhimurium causes typhoid-like disease signs in susceptible strains of mice; in resistant strains of mice, however, there are gastroenteritis-like symptoms, accompanied by an acute systemic infection, which can be resolved in approximately 20% to 60% of infected mice (27, 28). These resistant strains of mice develop disease signs more similar to what occurs during human infections with invasive NTS. Therefore, we used C3H/HeN mice (NRAMP+/+ TLR4+/+) to determine the phenotype of NC1040 or BTNC0025 in this host background. Oral gavage of mice with strain NC1040 (∼5 × 108 CFU) gave positive results in the intestines, livers, and spleens of all mice at 3 dpi (Table 3). From 3 to 8 dpi, the concentrations of strain NC1040 within the intestines were similar to those measured in liver and spleen tissues, i.e., an approximately 1:1 ratio, which contrasts with data from the avian host, which exhibited a difference of ∼4 orders of magnitude (Table 1). Moreover, this experiment was repeated with S. Enteritidis strain BTNC0025 and demonstrated results similar to those obtained with the S. Typhimurium strain. From 3 to 8 dpi, the concentrations of BTNC0025 within the intestines and livers/spleens were similar (Table 4).
TABLE 3.
Colonization of murine tissues following challenge with S. Typhimuriuma
| No. of days postinoculation with NC1040 | NC1040 CFU/g (no. of culture-positive mice/total no. of mice) |
||
|---|---|---|---|
| Intestine | Liver | Spleen | |
| 1 | 5.7 ± 1.3 (4/4) | 2.8 (1/4) | 2.7 ± 0.6 (2/4) |
| 3b | 5.0 ± 1.2 (3/3) | 3.8 ± 1.3 (3/3) | 4.3 ± 1.0 (3/3) |
| 4b | 5.1 ± 1.7 (3/3) | 4.4 ± 1.4 (3/3) | 4.4 ± 1.3 (3/3) |
| 8 | 5.5 ± 1.5 (4/4) | 4.4 ± 1.4 (4/4) | 5.1 ± 0.6 (4/4) |
C3H/HeN female mice (4 to 6 weeks old) were inoculated with ∼5 × 108 CFU of the kanamycin-resistant (fnr′::ha) “wild-type” strain (NC1040). Data shown are the mean log10 CFU values per gram of intestinal content or tissue homogenate from the indicated number of culture-positive mice among those sampled (shown in parentheses), with a detection limit of <2.3 log10 CFU/g.
A mouse in this group was found dead on the day of sampling.
TABLE 4.
Colonization of murine tissues following challenge with S. Enteritidisa
| No. of days postinoculation with BTNC0025 | BTNC0025 CFU/g (no. of culture-positive mice/total no. of mice) |
||
|---|---|---|---|
| Intestine | Liver | Spleen | |
| 1 | 5.3 ± 1.1 (4/4) | 2.4 ± 0.1 (3/4) | 2.5 ± 0.3 (2/4) |
| 3 | 3.6 ± 0.8 (4/4) | 5.2 ± 1.5 (4/4) | 5.5 ± 1.5 (4/4) |
| 6 | 4.1 ± 0.9 (4/4) | 5.6 ± 1.1 (4/4) | 6.9 ± 1.0 (4/4) |
| 8b | 5.1 (1/1) | 4.9 (1/1) | 5.9 (1/1) |
C3H/HeN female mice (4 to 6 weeks old) were inoculated with ∼5 × 108 CFU of the rifampin-resistant “wild-type” strain (BTNC0025). Data shown are the mean log10 CFU values per gram of intestinal content or tissue homogenate from the indicated number of culture-positive mice among those sampled (shown in parentheses), with a detection limit of <2.3 log10 CFU/g.
Three mice in this group were found dead on the day of sampling.
Expression of SPI-1 is decreased at 42°C.
The body temperature of poultry is known to increase during development. To determine the body temperatures of chickens under our conditions, we measured the internal body temperatures in a W-36 line of birds. As demonstrated previously (29–31), there was a distinct developmental increase in the body temperature of birds (Fig. 1A). At 1, 2, 4, 6, 8, 13, and 22 days posthatch, the birds had mean body temperatures of 40.3 ± 0.4°C, 40.7 ± 0.4°C, 41.3 ± 0.2°C, 41.6 ± 0.3°C, 41.5 ± 0.3°C, 41.5 ± 0.3°C, and 41.7 ± 0.3°C, respectively (Fig. 1A). At day 8, the body temperature was not significantly different from that measured at day 6 (P < 0.05). This indicated that the body temperature had stabilized between days 6 and 8 posthatch. Based on these data and earlier work regarding heat shock regulation within S. Typhimurium (32), gene regulation experiments were conducted at 42°C.
FIG 1.
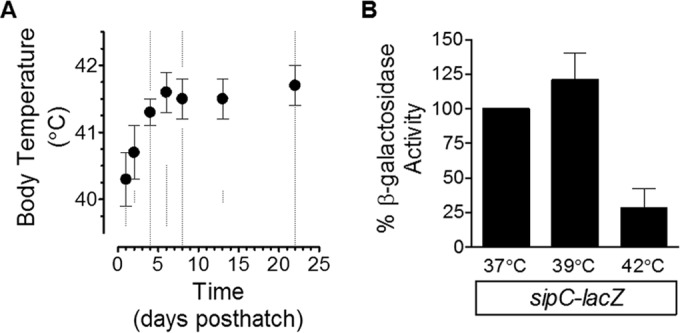
Expression of the SPI-1 gene sipC is repressed following growth at 42°C. (A) The body temperatures of a W-36 line of chickens were measured 1, 2, 4, 6, 8, 13, and 22 days posthatch (indicated by dashed lines). By day 8, the body temperature was not significantly different than at day 6 (P < 0.05 [Student's t test and Bonferroni adjustment for multiple comparisons]). (B) A sipC-lacZ fusion strain (RM5385) was used to test the impact of different temperatures on the regulation of SPI-1. Bacteria were grown overnight in LB medium under standing conditions at 37°C and diluted 50-fold in LB medium containing MOPS (pH 7.4, 100 mM) and glucose (1 mM) and incubated at different temperatures under standing conditions for approximately 16 to 18 h. β-Galactosidase activity was determined from experiments performed on three separate occasions. Data shown are the means ± SD, with reporter activity under the 37°C conditions set to 100%.
SPI-1 is an important factor that promotes invasion of the intestinal epithelium and, ultimately, systemic infection. Because infected chicks had significantly lower concentrations of NC1040 within the livers and spleens while simultaneously having significantly higher concentrations within the ceca than C3H/HeN mice, the results suggested that there may be reduced expression of SPI-1 within the avian host. Although this could have been due to a variety of host factors, the body temperature of chickens is higher than that of some other animals. Therefore, the expression of the SPI-1 genes, sipC-lacZ, was tested in response to different temperatures. Compared to expression following growth at 37°C, there was no change in expression of sipC-lacZ at 39°C, which contrasts with the data determined following growth at 42°C that resulted in reporter activity that was ∼25% of that seen at 37°C (Fig. 1B). This suggested that an environmental temperature of 42°C is a crucial factor in the regulation of SPI-1 genes.
To further test the role of 42°C in the regulation of SPI-1, expression of a hilA-lacZ fusion was monitored following growth at 37°C and 42°C. In addition, the expression of a hmpA-lacZ fusion was monitored to determine the specificity of temperature for SPI-1 gene regulation within S. Typhimurium. The influence of pH 6.0 was also tested, since this is a known environmental condition that represses expression of hilA-lacZ (33). The data showed that expression of hilA-lacZ was repressed at 42°C independently of pH, whereas that of hmpA-lacZ was not influenced by temperature (Fig. 2A). Moreover, expression of the HilA-activated invF-lacZ and sipC-lacZ reporter genes was also repressed at 42°C independently of pH (Fig. 2B). These data demonstrated that the expression of the SPI-1 activator, hilA, as well as of the HilA-activated genes invF and sipC, was transcriptionally repressed by growth at 42°C.
FIG 2.
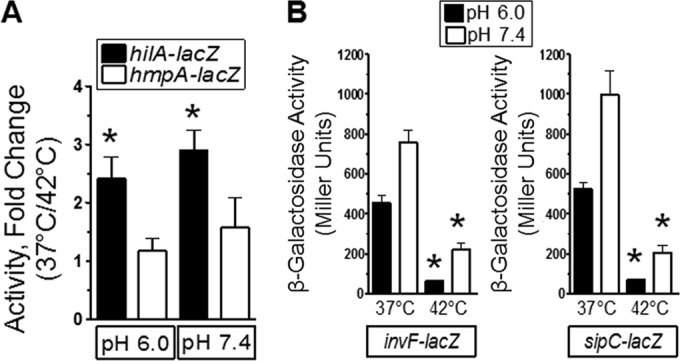
Growth at 42°C repressed SPI-1 genes independently of pH. (A) Bacteria were grown as described in the Fig. 1B legend. The pH of the medium was left unchanged or buffered to 7.4 with NaOH. Bacteria were grown overnight, and β-galactosidase activity was determined. The hmpA-lacZ strain was used as a control. Data are from 4 separate experiments, and an asterisk indicates a significant difference in the fold change measured for the hilA-lacZ strain compared to the control, the hmpA-lacZ strain. Strains used were RM5948 and AV0305. (B) Bacteria were treated as described for panel A, and the reporter activity of strains was measured. Data are from 3 separate experiments, and an asterisk indicates a significant difference in reporter activity at 42°C compared to 37°C under the same pH conditions. Strains used were CA701 and RM5385.
Effect of 42°C on transcriptional activators of SPI-1.
To understand how growth at 42°C repressed SPI-1 genes, the promoters of the three AraC/XylS-type activators, hilC, rtsA, and hilD, were cloned into the multicopy, promoterless pSP417 lacZ shuttle vector (34). These constructs were transformed into strains NC1040 and BTNC0025, and expression of each reporter fusion was determined following growth at 37°C or 42°C. Importantly, these plasmid constructs do not disrupt the chromosomal copies of hilC, rtsA, or hilD, which avoids complications in the interpretation of the data when one of these activators is mutated (9, 35). The activity seen with the empty vector, PhilC, or the PhilD fusion was not significantly altered by growth at 42°C. However, the expression of the HilD-activated PrtsA-lacZ fusion was significantly reduced in both serovars (Fig. 3A; see also Fig. S1A in the supplemental material). Thus, growth at 42°C reduced transcriptional control of sipC, invF, hilA, and rtsA and suggested that the activation of SPI-1 was diminished at 42°C.
FIG 3.
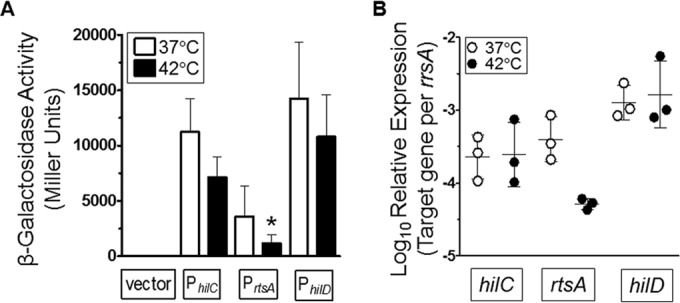
The promoters of the SPI-1 activators, hilC and rtsA, are differentially regulated by growth at 42°C. (A) The promoters of hilC, rtsA, and hilD were cloned into the pSP417 multicopy shuttle vector (empty vector) upstream of a promoterless lacZ gene. Bacteria were grown as described for Fig. 1B and diluted 1:50 into LB-MOPS medium with 1 mM glucose at pH 7.4 at 37°C or 42°C. β-Galactosidase activity was measured after overnight growth. Data are from 4 separate experiments, and a statistically significant result compared to activity at 37°C was determined. The strains used were BTNC0002 to BTNC0005. (B) Bacteria were grown in LB-MOPS medium with 1 mM glucose at pH 7.4 at 37°C or 42°C. Total RNA was extracted at an OD600 of ∼1. cDNA was generated using gene-specific primers, and expression data were normalized to the rrsA 16S rRNA gene.
Expression data from lacZ fusions were confirmed in strains NC1040 and BTNC0025 by measuring mRNA levels of hilC, rtsA, and hilD. Bacteria were grown to an OD600 of ∼1 at 37°C or 42°C, and RNA was extracted for qRT-PCR. As with the lacZ data, expression of hilD was not influenced by growth at 42°C; however, expression of rtsA was reduced ∼9-fold (Fig. 3B). Although β-galactosidase levels from the pPhilC-lacZ construct approached statistical significance under conditions of growth at 42°C, the expression of hilC was not reduced (Fig. 3B). With strain BTNC0025, expression of rtsA was reduced >5-fold following growth at 42°C (see Fig. S1 in the supplemental material). Considering the results from both serovars, the data supported the conclusion that growth at 42°C reduced expression of the rtsA gene.
Previous work from our laboratory and others demonstrated that the Fur transcription factor is required for activation of SPI-1 genes (35–38). Therefore, we determined the effect of Fur on the expression of the PhilC-lacZ, PrtsA-lacZ, and PhilD-lacZ fusions following growth at 37°C. Because growth at 42°C repressed expression of the PrtsA-lacZ fusion, identifying that Fur also regulates SPI-1 in this manner would suggest that temperature control of SPI-1 may act through Fur. However, activation of the PrtsA-lacZ and PhilD-lacZ fusions was inhibited by deletion of the fur gene (Fig. 4A). The reduction in PrtsA and PhilD activation was consistent with earlier data, which showed that Fur regulated SPI-1 by controlling hilD expression when the hilD gene was present (35, 37). Following growth at 37°C and consistent with earlier findings (35, 36), the overexpression of Fur increased expression of hilA-lacZ by ∼4-fold (Fig. 4B, right panel). Despite the enhanced expression of the hilA-lacZ fusion following induction of the Fur protein with IPTG at 37°C, induction at 42°C did not increase expression of hilA-lacZ even though the levels of the Fur protein under the two sets of conditions were strikingly similar (Fig. 4B, right panel). These data indicated that the inability to activate SPI-1 following growth at 42°C was not related to Fur.
FIG 4.

Growth at 42°C repressed expression of SPI-1 genes independently of the activator Fur. (A) The promoters of hilC, rtsA, and hilD were cloned into the pSP417 multicopy shuttle vector (empty vector) upstream of a promoterless lacZ gene. Bacteria were grown as described for Fig. 1B, and the promoter activities were determined for the NC1040 and fur::cat strains following overnight growth at 37°C. Data are from 3 separate experiments. The strains used were BTNC0002 to BTNC0005 and BTNC0006 to BTNC0009. (B) Expression of a hilA-lacZ fusion at the chromosomal att site was determined following overnight growth with or without induction of the Fur-FLAG protein. Bacteria were grown as described for Fig. 1B, except that samples were diluted 500-fold and cultivated at either 37°C or 42°C. A portion of each sample was removed to measure β-galactosidase activity, and the remainder was treated for SDS-PAGE and Western blotting to detect the FLAG epitope. Following transfer of proteins to the nitrocellulose membrane, the membrane was stained with Ponceau S to ensure that equivalent levels of protein were loaded for samples and that ∼2 × 108 cells were loaded per lane (left panel). IPTG was added to the growth medium to reach a concentration of 0.1 mM to induce Fur-FLAG. Western blotting with the anti-FLAG antibody revealed cross-reactivity to the Fur protein of the expected size, indicated by the arrowhead with the appropriate label (right panel). The β-galactosidase activity for each sample is listed below each lane (right panel). Samples shown are representative of the results of 3 separate experiments. The complete β-galactosidase activity data are listed here. For BTNC0017 (pfur-flag), the values measured at 37°C were 156 ± 33 under uninduced conditions and 657 ± 89 under induced conditions and the values measured at 42°C were 316 ± 70 under uninduced conditions and 345 ± 4 under induced conditions.
Lon and ClpPX differentially control regulation of SPI-1 independently of growth at 42°C.
Earlier work showed that the heat shock response influences regulation of SPI-1 genes (32, 39). Because the heat shock response involves activation of the two major proteases, Lon and Clp, regulation of SPI-1 genes at 42°C was studied in the presence and absence of these two factors. Consistent with earlier results, deletion of the lon gene enhanced expression of sipC-lacZ (Fig. 5A). However, the temperature-dependent expression of sipC was reduced ∼3-fold to 5-fold following growth at 42°C for both the NC1040 and lon::cat strains (Fig. 5A). The role of the Clp protease in the regulation of SPI-1 appeared to be operating at the level of activation of the pathway instead of repression. Expression of the sipC-lacZ fusion was reduced ∼3-fold upon deletion of the clpP and clpX (clpPX) genes (Fig. 5A). Furthermore, deletion of clpPX resulted in a level of inactivation similar to that seen with strain NC1040 following growth at 42°C (Fig. 5A). Thus, the clpPX heat shock genes do not appear to be the cause for reduced expression of SPI-1 at 42°C.
FIG 5.
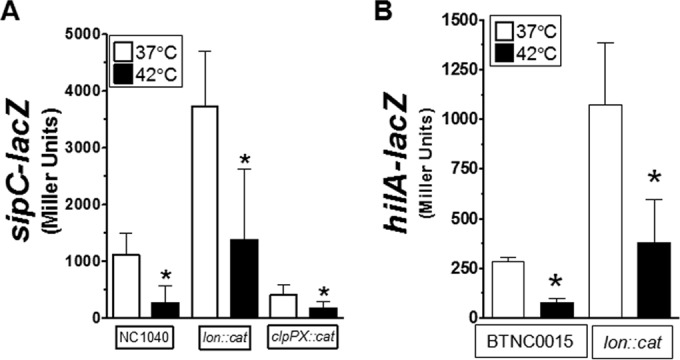
The Lon heat shock protease contributes to the basal level of SPI-1 repression but does not perturb inactivation following growth at 42°C. (A) The sipC-lacZ fusion was used to determine the contribution of lon or clpPX to the regulation of SPI-1 following growth at 42°C. Bacteria were grown as described for Fig. 1B, and reporter activity was measured after overnight growth. The strains used were BTNC0010 to BTNC0012. (B) The hilA-lacZ fusion integrated into the att within the chromosome was used to determine the influence of lon on the reduced expression following growth at 42°C. Samples were cultivated as described for Fig. 1B after overnight growth. Data are from 4 separate experiments. The strains used were BTNC0015 and BTNC0016.
Additional experiments were performed to further test the contribution of lon to the temperature-dependent regulation of SPI-1. Even though the basal expression of a hilA-lacZ fusion was ∼4-fold higher in the absence of lon, a similar level of inactivation still occurred following growth at 42°C (Fig. 5B). Thus, it is apparent that Lon does influence the basal expression of SPI-1 genes, but Lon does not appear to be responsible for inactivation of SPI-1 following growth at 42°C.
The inability to activate SPI-1 expression by growth at 42°C is overcome by overexpression of RtsA or HilC but not by overexpression of FliZ or HilD.
To determine how the overexpression of SPI-1 activators influences the regulation of hilA-lacZ following growth at 42°C, three plasmid constructs carrying fliZ, hilC, and rtsA whose protein production is under the control of the allactose analog IPTG were generated. Each activator was tagged with the FLAG epitope at the 3′ end, cloned into the pUHE21-2lacIq plasmid (40), and transformed into the hilA-lacZ att site strain (BTNC0015). To study the influence of hilD, we utilized published strain JS1180, which carries a tetracycline-inducible chromosomal copy of hilD-3×flag and the hilA-lacZ fusion integrated into the chromosomal att site (13). Along with measurement of the β-galactosidase levels in each sample, a portion was treated for SDS-PAGE and immunoblot analysis to detect expression of the FLAG-tagged proteins.
Induction of the HilD-3×FLAG protein induced expression of the hilA-lacZ fusion by ∼13-fold following growth at 37°C; however, when cells were cultivated at 42°C, induction of HilD-3×FLAG resulted in modest (only ∼2-fold) activation of the hilA-lacZ fusion (Fig. 6A). This was surprising, since similar levels of the HilD-3×FLAG protein were produced at 37°C and 42°C (Fig. 6A). This suggested that the activity of the HilD protein was regulated by growth at 42°C and not by the level of the protein.
FIG 6.

Overexpression of either the HilD or the FliZ SPI-1 activator does not activate SPI-1 at 42°C, but overexpression of either HilC or RtsA does. (A) Expression of a hilA-lacZ fusion at the chromosomal att site was determined following overnight growth with or without induction of the HilD-3×FLAG protein. Bacteria were grown as described for Fig. 1B, except that samples were diluted 500-fold and cultivated at either 37°C or 42°C. A portion of each sample was removed to measure β-galactosidase activity, and the remainder was treated for SDS-PAGE and Western blotting to detect the FLAG epitope. Following transfer of proteins to the nitrocellulose membrane, the membrane was stained with Ponceau S to ensure the equivalent levels of protein were loaded for samples and that ∼2 × 108 cells were loaded per lane (left panel). Tetracycline was added to reach a concentration of 2.5 μg/ml to induce HilD-3×FLAG. Western blotting with the anti-FLAG antibody revealed cross-reactivity to the HilD protein of the expected size, indicated by the arrowhead with the appropriate label (right panel). The β-galactosidase activity for each sample is listed below each lane (right panel). Samples shown are representative of the results of 3 separate experiments. The complete β-galactosidase activity data are listed here. For JS1180, the values measured at 37°C were 133 ± 13 under uninduced conditions and 1,716 ± 146 under induced conditions and the values measured at 42°C were 28 ± 1 under uninduced conditions and 62 ± 34 under induced conditions. (B) Expression of a hilA-lacZ fusion at the chromosomal att site was determined following overnight growth with or without induction of FliZ-FLAG. Samples were prepared as described for panel A for the Ponceau S staining (left panel), and the Western blot analysis was performed with anti-FLAG (right panel). IPTG was used at 0.1 mM to induce FliZ-FLAG. Sample data shown are representative of the results of 3 separate experiments. The complete β-galactosidase activity data for BTNC0018 (pfliZ-flag) are listed here; the values measured at 37°C were 527 ± 133 under uninduced conditions and 1,539 ± 243 under induced conditions, and the values measured at 42°C were 89 ± 36 under uninduced conditions and 177 ± 92 under induced conditions. (C) Samples were treated as described for panel A for the RtsA-FLAG or HilC-FLAG strains containing the hilA-lacZ fusion and induced with 0.1 mM IPTG. Samples data are representative, and the β-galactosidase activity for each sample is shown below each lane. The complete β-galactosidase activity data are listed here. For BTNC0019 (prtsA-flag), the values measured at 37°C were 580 ± 30 under uninduced conditions and 1,490 ± 317 under induced conditions, and the values measured at 42°C were 367 ± 66 under uninduced conditions and 2,177 ± 504 under induced conditions. For BTNC0020 (philC-flag), the values measured at 37°C were 811 ± 9 under uninduced conditions and 1,936 ± 319 under induced conditions, and the values measured at 42°C were 965 ± 113 under uninduced conditions and 3,142 ± 649 under induced conditions. (D) Strain RM5385 was transformed with prtsA-flag or philC-flag and reporter activity was determined at different temperatures. IPTG was used at 0.01 mM to induce RtsA-FLAG and HilC-FLAG. The Ponceau S (left panel) and Western blotting (right panel) are shown. The complete β-galactosidase activity data are listed here. For BTNC0023 (prtsA-flag), the values measured at 37°C were 1,966 ± 781 under uninduced conditions and 3,409 ± 103 under induced conditions, and the values measured at 42°C were 712 ± 469 under uninduced conditions and 2,666 ± 171 under induced conditions. For BTNC0024 (philC-flag), the values measured at 37°C were 2,801 ± 769 under uninduced conditions and 3,727 ± 906 under induced conditions, and the values measured at 42°C were 1,774 ± 521 under uninduced conditions and 3,122 ± 790 under induced conditions.
The FliZ regulator is known to enhance activity of the HilD protein (41) through an unknown posttranslational step; therefore, the influence of overexpression of FliZ on hilA-lacZ in response to temperature was tested. Similarly to the results seen with Fur and HilD, overexpression of FliZ increased expression of hilA-lacZ ∼4-fold at 37°C but not at 42°C even though the protein levels of FliZ were similar at the two temperatures (Fig. 6B). Collectively, these data indicated that expression of SPI-1 genes was reduced at 42°C through decreased function of the HilD protein, which resulted in reduced expression of rtsA and hilA. In addition, this implied that expression of SPI-1 genes at 42°C could be restored through HilD-independent expression of HilC or RtsA.
To test this, HilC and RtsA were placed under IPTG control to determine if expression of hilA-lacZ could be activated by either regulator at 42°C. In contrast to results seen with Fur, HilD, and FliZ, IPTG induction of either RtsA or HilC following growth at 37°C or 42°C was sufficient to enhance expression of the hilA-lacZ fusion to a similar level of activity (Fig. 6C). Moreover, overexpression of either HilC or RtsA was sufficient to activate expression of sipC-lacZ (Fig. 6D), supporting the conclusion that HilD-independent expression of HilC or RtsA is capable of restoring expression of SPI-1 genes at 42°C.
DISCUSSION
In pathogenic bacteria, temperature is a known environmental cue that alters expression of virulence factors. In the Lyme disease agent Borrelia burgdorferi, growth at different temperatures is used to model gene expression within the tick vector or the mammalian host (42–44). In addition, the infectivity of Yersinia pestis (formerly Pasteurella pestis) decreases 100-fold when cultivated at temperatures below the host body temperature (45). More recently, Elhadad et al. demonstrated that Salmonella bacteria causing typhoid fever alter gene regulation and invasion in response to temperatures consistent with a febrile response (46). After ∼6 days posthatch, the body temperature of chickens stabilized near 42°C (Fig. 1A), a temperature that negatively influences the regulation of SPI-1 genes. The data indicated that the activity of the HilD protein was inhibited during growth at 42°C. This effect was specific for HilD since overexpression of either RtsA or HilC, the other AraC/XylS type activators of hilA, was sufficient to activate SPI-1 expression at 42°C (Fig. 6C). Lon and ClpPX do not appear involved in the lack of activation of SPI-1 by growth at 42°C. Instead, the function of the HilD protein is diminished following growth at the body temperature of poultry (42°C). It is becoming evident that activation of the HilD protein through an unknown posttranslational mechanism(s) is a critical aspect in the regulation of SPI-1 (13, 14, 41). Future work is required to determine how growth at 42°C integrates into the existing understanding of the regulation of SPI-1. Regardless, it is clear from this work that activators of SPI-1 do not activate SPI-1 at 42°C, suggesting that the body temperature of the avian host appears to have adapted to limiting invasion of the liver and spleen by these two serovars.
A difference in body temperature may explain the marginal requirement of SPI-1 for S. Typhimurium and S. Enteritidis within the avian host.
Numerous studies have tested the contribution of SPI-1 genes to the cecal and systemic colonization of S. Typhimurium and S. Enteritidis in birds. Despite differences in experimental designs, Salmonella strains, and chickens, the collective data indicate a marginal contribution of SPI-1 to invasion of the liver and spleen during infection. When 1-day-old chicks were orally infected with a strain containing deletion of the SPI-1 gene spaS, Jones et al. (47) observed a delay in colonization of the liver of infected birds without a reduction in the cecal burden compared to the wild type. However, even the livers of birds infected with the wild-type strain exhibit low concentrations of S. Typhimurium by 14 days postinoculation (47). Sivula et al. (48) observed a similar phenotype when White Leghorn chicks were infected with S. Typhimurium. Moreover, mutation of the invA SPI-1 gene did not influence invasion of the cecal epithelium of birds (48). However, other work did detect a reduction in the concentration of a Δspi1 strain in the spleens of infected chickens compared to the wild-type strain (49). Conclusions from this finding are complicated by the observation that the Δspi1 strain also had a defect in cecal colonization compared to the wild-type strain (49). Thus, the reduction in localization of the Δspi1 strain to the spleen may be due to a fitness defect within the ceca. Indeed, gene expression data corresponding to colonization of the avian ceca by S. Typhimurium in vivo demonstrate that expression of hilC and other activators of SPI-1 (e.g., tdcA) is significantly downregulated (50).
Experiments performed with S. Enteritidis in chickens exhibit a trend similar to that seen with data from S. Typhimurium experiments. Rychlik et al. (51) tested the contribution of SPI-1, SPI-2, SPI-3, SPI-4, and SPI-5 to cecal and systemic colonization in chickens. Deletion of spi1 resulted in a significant reduction in liver and spleen colonization at an early time point (5 dpi) but not at a later time point (51). In addition, earlier work showed that SPI-1 marginally contributed to systemic localization (26). Moreover, deletion of the hilA SPI-1 activator reduced localization to systemic organs early during infection but not at a later time point (52). These results, in combination with our gene regulation data, suggest that SPI-1 may be important early in infection before the body temperature of birds has plateaued at ∼42°C. Although our data support the idea of a reduced ability of these two serovars to reach the livers and spleens of chicks, a thorough screening performed with additional isolates from these serovars may reveal more-invasive phenotypes. Recent work shows that S. Typhimurium isolates exhibit broad ranges of invasive phenotypes (46).
In contrast to data from the murine host, our results indicated that both S. Typhimurium and S. Enteritidis poorly localized to the spleen and liver sites within chickens despite colonization of the ceca at high concentrations. Expression of SPI-1 genes is decreased when the isolates are cultivated at the body temperature of poultry, which may explain the reduced ability of each serovar to reach the livers and spleens in chickens. In addition to contributing to invasion, expression of SPI-1 is linked to inflammation and tissue damage within the digestive tract of the animal host (53). For these two serovars, the reduced expression of SPI-1 genes at the body temperature of poultry (41°C to 42°C) may be a factor contributing to the overall lack of disease signs (i.e., liver abscesses and splenomegaly) in chickens.
Supplementary Material
ACKNOWLEDGMENTS
We thank J. M. Slauch for providing strain JS1180, E. A. Groisman for providing pUHE21-2lacIq, and T. J. Larson for providing pSP417. We thank Becca Wysocky and Rizwana Ali for assistance in the maintenance and hatching of the W-36 line of birds at North Carolina State University.
Funding was provided through USDA-NIFA 2012-68003-18621 (to H.M.H. and M.D.K.) and 2013-67012-21012 (to B.T.). R.P. was supported by a doctoral scholarship from Fundação de Amparo à Pesquisa do Estado de São Paulo—FAPESP, CNPJ 43.828.151/0001-45. The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AEM.02622-15.
REFERENCES
- 1.CDC. 2011. National Salmonella surveillance annual report. US Department of Health and Human Services, CDC, Atlanta, GA. [Google Scholar]
- 2.CDC. 2013. Surveillance for foodborne disease outbreaks—United States, 2009–2010. MMWR Morb Mortal Wkly Rep 62:41–47. [PMC free article] [PubMed] [Google Scholar]
- 3.Ellermeier JR, Slauch JM. 2007. Adaptation to the host environment: regulation of the SPI1 type III secretion system in Salmonella enterica serovar Typhimurium. Curr Opin Microbiol 10:24–29. doi: 10.1016/j.mib.2006.12.002. [DOI] [PubMed] [Google Scholar]
- 4.Eichelberg K, Galan JE. 1999. Differential regulation of Salmonella typhimurium type III secreted proteins by pathogenicity island 1 (SPI-1)-encoded transcriptional activators InvF and hilA. Infect Immun 67:4099–4105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Altier C. 2005. Genetic and environmental control of salmonella invasion. J Microbiol 43(Spec No):85–92. [PubMed] [Google Scholar]
- 6.Galán JE, Curtiss R III. 1989. Cloning and molecular characterization of genes whose products allow Salmonella typhimurium to penetrate tissue culture cells. Proc Natl Acad Sci U S A 86:6383–6387. doi: 10.1073/pnas.86.16.6383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Groisman EA, Ochman H. 1993. Cognate gene clusters govern invasion of host epithelial cells by Salmonella typhimurium and Shigella flexneri. EMBO J 12:3779–3787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jones BD, Ghori N, Falkow S. 1994. Salmonella typhimurium initiates murine infection by penetrating and destroying the specialized epithelial M cells of the Peyer's patches. J Exp Med 180:15–23. doi: 10.1084/jem.180.1.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ellermeier CD, Ellermeier JR, Slauch JM. 2005. HilD, HilC and RtsA constitute a feed forward loop that controls expression of the SPI1 type three secretion system regulator hilA in Salmonella enterica serovar Typhimurium. Mol Microbiol 57:691–705. doi: 10.1111/j.1365-2958.2005.04737.x. [DOI] [PubMed] [Google Scholar]
- 10.Penheiter KL, Mathur N, Giles D, Fahlen T, Jones BD. 1997. Non-invasive Salmonella typhimurium mutants are avirulent because of an inability to enter and destroy M cells of ileal Peyer's patches. Mol Microbiol 24:697–709. doi: 10.1046/j.1365-2958.1997.3741745.x. [DOI] [PubMed] [Google Scholar]
- 11.Bajaj V, Hwang C, Lee CA. 1995. hilA is a novel ompR/toxR family member that activates the expression of Salmonella typhimurium invasion genes. Mol Microbiol 18:715–727. doi: 10.1111/j.1365-2958.1995.mmi_18040715.x. [DOI] [PubMed] [Google Scholar]
- 12.Lawhon SD, Maurer R, Suyemoto M, Altier C. 2002. Intestinal short-chain fatty acids alter Salmonella typhimurium invasion gene expression and virulence through BarA/SirA. Mol Microbiol 46:1451–1464. doi: 10.1046/j.1365-2958.2002.03268.x. [DOI] [PubMed] [Google Scholar]
- 13.Hung CC, Garner CD, Slauch JM, Dwyer ZW, Lawhon SD, Frye JG, McClelland M, Ahmer BM, Altier C. 2013. The intestinal fatty acid propionate inhibits Salmonella invasion through the post-translational control of HilD. Mol Microbiol 87:1045–1060. doi: 10.1111/mmi.12149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Golubeva YA, Sadik AY, Ellermeier JR, Slauch JM. 2012. Integrating global regulatory input into the Salmonella pathogenicity island 1 type III secretion system. Genetics 190:79–90. doi: 10.1534/genetics.111.132779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Petrone BL, Stringer AM, Wade JT. 2014. Identification of HilD-regulated genes in Salmonella enterica serovar Typhimurium. J Bacteriol 196:1094–1101. doi: 10.1128/JB.01449-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kallapura G, Morgan MJ, Pumford NR, Bielke LR, Wolfenden AD, Faulkner OB, Latorre JD, Menconi A, Hernandez-Velasco X, Kuttappan VA, Hargis BM, Tellez G. 2014. Evaluation of the respiratory route as a viable portal of entry for Salmonella in poultry via intratracheal challenge of Salmonella Enteritidis and Salmonella Typhimurium. Poult Sci 93:340–346. doi: 10.3382/ps.2013-03602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pei Y, Parreira VR, Roland KL, Curtiss R III, Prescott JF. 2014. Assessment of attenuated Salmonella vaccine strains in controlling experimental Salmonella Typhimurium infection in chickens. Can J Vet Res 78:23–30. [PMC free article] [PubMed] [Google Scholar]
- 18.Chen CY, Tsen HY, Lin CL, Yu B, Chen CS. 2012. Oral administration of a combination of select lactic acid bacteria strains to reduce the Salmonella invasion and inflammation of broiler chicks. Poult Sci 91:2139–2147. doi: 10.3382/ps.2012-02237. [DOI] [PubMed] [Google Scholar]
- 19.Revolledo L, Ferreira CS, Ferreira AJ. 2009. Prevention of Salmonella Typhimurium colonization and organ invasion by combination treatment in broiler chicks. Poult Sci 88:734–743. doi: 10.3382/ps.2008-00410. [DOI] [PubMed] [Google Scholar]
- 20.Yang X, Brisbin J, Yu H, Wang Q, Yin F, Zhang Y, Sabour P, Sharif S, Gong J. 2014. Selected lactic acid-producing bacterial isolates with the capacity to reduce Salmonella translocation and virulence gene expression in chickens. PLoS One 9:e93022. doi: 10.1371/journal.pone.0093022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ullman-Culleré MH, Foltz CJ. 1999. Body condition scoring: a rapid and accurate method for assessing health status in mice. Lab Anim Sci 49:319–323. [PubMed] [Google Scholar]
- 22.Miller JH. 1972. Experiments in molecular genetics. Cold Spring Harbor Laboratory, Cold Spring Harbor, NY. [Google Scholar]
- 23.Antunes LC, Buckner MM, Auweter SD, Ferreira RB, Lolic P, Finlay BB. 2010. Inhibition of Salmonella host cell invasion by dimethyl sulfide. Appl Environ Microbiol 76:5300–5304. doi: 10.1128/AEM.00851-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Troxell B, Ye M, Yang Y, Carrasco SE, Lou Y, Yang XF. 2013. Manganese and zinc regulate virulence determinants in Borrelia burgdorferi. Infect Immun 81:2743–2752. doi: 10.1128/IAI.00507-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Troxell B, Xu H, Yang XF. 2012. Borrelia burgdorferi, a pathogen that lacks iron, encodes manganese-dependent superoxide dismutase essential for resistance to streptonigrin. J Biol Chem 287:19284–19293. doi: 10.1074/jbc.M112.344903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Desin TS, Lam PK, Koch B, Mickael C, Berberov E, Wisner AL, Townsend HG, Potter AA, Koster W. 2009. Salmonella enterica serovar enteritidis pathogenicity island 1 is not essential for but facilitates rapid systemic spread in chickens. Infect Immun 77:2866–2875. doi: 10.1128/IAI.00039-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Monack DM, Bouley DM, Falkow S. 2004. Salmonella typhimurium persists within macrophages in the mesenteric lymph nodes of chronically infected Nramp1+/+ mice and can be reactivated by IFNgamma neutralization. J Exp Med 199:231–241. doi: 10.1084/jem.20031319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Husain M, Jones-Carson J, Song M, McCollister BD, Bourret TJ, Vazquez-Torres A. 2010. Redox sensor SsrB Cys203 enhances Salmonella fitness against nitric oxide generated in the host immune response to oral infection. Proc Natl Acad Sci U S A 107:14396–14401. doi: 10.1073/pnas.1005299107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Scholes JC. 1940. The relationship between body temperature and genetic resistance to Salmonella pullorum in the fowl. Cornell University, Ithaca, NY. [Google Scholar]
- 30.Dunnington EA, Siegel PB. 1985. Surface and cloacal temperatures in chicks of different genetic stocks given an Escherichia coli inoculation. Avian Dis 29:177–187. doi: 10.2307/1590706. [DOI] [PubMed] [Google Scholar]
- 31.Malheiros RD, Moraes VMB, Bruno LDG, Malheiros EB, Furlan RL, Macari M. 2000. Environmental temperature and cloacal and surface temperatures of broiler chicks in first week post-hatch. J Appl Poultry Res 9:111–117. doi: 10.1093/japr/9.1.111. [DOI] [Google Scholar]
- 32.Matsui M, Takaya A, Yamamoto T. 2008. Sigma32-mediated negative regulation of Salmonella pathogenicity island 1 expression. J Bacteriol 190:6636–6645. doi: 10.1128/JB.00744-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bajaj V, Lucas RL, Hwang C, Lee CA. 1996. Co-ordinate regulation of Salmonella typhimurium invasion genes by environmental and regulatory factors is mediated by control of hilA expression. Mol Microbiol 22:703–714. doi: 10.1046/j.1365-2958.1996.d01-1718.x. [DOI] [PubMed] [Google Scholar]
- 34.Podkovyrov SM, Larson TJ. 1995. A new vector-host system for construction of lacZ transcriptional fusions where only low-level gene expression is desirable. Gene 156:151–152. doi: 10.1016/0378-1119(95)00053-9. [DOI] [PubMed] [Google Scholar]
- 35.Ellermeier JR, Slauch JM. 2008. Fur regulates expression of the Salmonella pathogenicity island 1 type III secretion system through HilD. J Bacteriol 190:476–486. doi: 10.1128/JB.00926-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Troxell B, Sikes ML, Fink RC, Vazquez-Torres A, Jones-Carson J, Hassan HM. 2011. Fur negatively regulates hns and is required for the expression of HilA and virulence in Salmonella enterica serovar Typhimurium. J Bacteriol 193:497–505. doi: 10.1128/JB.00942-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Teixidó L, Carrasco B, Alonso JC, Barbe J, Campoy S. 2011. Fur activates the expression of Salmonella enterica pathogenicity island 1 by directly interacting with the hilD operator in vivo and in vitro. PLoS One 6:e19711. doi: 10.1371/journal.pone.0019711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Thompson A, Rolfe MD, Lucchini S, Schwerk P, Hinton JC, Tedin K. 2006. The bacterial signal molecule, ppGpp, mediates the environmental regulation of both the invasion and intracellular virulence gene programs of Salmonella. J Biol Chem 281:30112–30121. doi: 10.1074/jbc.M605616200. [DOI] [PubMed] [Google Scholar]
- 39.Sirsat SA, Burkholder KM, Muthaiyan A, Dowd SE, Bhunia AK, Ricke SC. 2011. Effect of sublethal heat stress on Salmonella Typhimurium virulence. J Appl Microbiol 110:813–822. doi: 10.1111/j.1365-2672.2011.04941.x. [DOI] [PubMed] [Google Scholar]
- 40.Soncini FC, Vescovi EG, Groisman EA. 1995. Transcriptional autoregulation of the Salmonella typhimurium phoPQ operon. J Bacteriol 177:4364–4371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chubiz JE, Golubeva YA, Lin D, Miller LD, Slauch JM. 2010. FliZ regulates expression of the Salmonella pathogenicity island 1 invasion locus by controlling HilD protein activity in Salmonella enterica serovar Typhimurium. J Bacteriol 192:6261–6270. doi: 10.1128/JB.00635-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yang X, Goldberg MS, Popova TG, Schoeler GB, Wikel SK, Hagman KE, Norgard MV. 2000. Interdependence of environmental factors influencing reciprocal patterns of gene expression in virulent Borrelia burgdorferi. Mol Microbiol 37:1470–1479. doi: 10.1046/j.1365-2958.2000.02104.x. [DOI] [PubMed] [Google Scholar]
- 43.Stevenson B, Schwan TG, Rosa PA. 1995. Temperature-related differential expression of antigens in the Lyme disease spirochete, Borrelia burgdorferi. Infect Immun 63:4535–4539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Stevenson B, Bono JL, Schwan TG, Rosa P. 1998. Borrelia burgdorferi erp proteins are immunogenic in mammals infected by tick bite, and their synthesis is inducible in cultured bacteria. Infect Immun 66:2648–2654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Fukui GM, Lawton WD, Ham DA, Janssen WA, Surgalla MJ. 1960. The effect of temperature on the synthesis of virulence factors by Pasteurella pestis. Ann N Y Acad Sci 88:1146–1151. [DOI] [PubMed] [Google Scholar]
- 46.Elhadad D, McClelland M, Rahav G, Gal-Mor O. 2015. Feverlike temperature is a virulence regulatory cue controlling the motility and host cell entry of typhoidal Salmonella. J Infect Dis 212:147–156. doi: 10.1093/infdis/jiu663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Jones MA, Hulme SD, Barrow PA, Wigley P. 2007. The Salmonella pathogenicity island 1 and Salmonella pathogenicity island 2 type III secretion systems play a major role in pathogenesis of systemic disease and gastrointestinal tract colonization of Salmonella enterica serovar Typhimurium in the chicken. Avian Pathol 36:199–203. doi: 10.1080/03079450701264118. [DOI] [PubMed] [Google Scholar]
- 48.Sivula CP, Bogomolnaya LM, Andrews-Polymenis HL. 2008. A comparison of cecal colonization of Salmonella enterica serotype Typhimurium in white leghorn chicks and Salmonella-resistant mice. BMC Microbiol 8:182. doi: 10.1186/1471-2180-8-182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Dieye Y, Ameiss K, Mellata M, Curtiss R III. 2009. The Salmonella pathogenicity island (SPI) 1 contributes more than SPI2 to the colonization of the chicken by Salmonella enterica serovar Typhimurium. BMC Microbiol 9:3. doi: 10.1186/1471-2180-9-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Harvey PC, Watson M, Hulme S, Jones MA, Lovell M, Berchieri A Jr, Young J, Bumstead N, Barrow P. 2011. Salmonella enterica serovar Typhimurium colonizing the lumen of the chicken intestine grows slowly and upregulates a unique set of virulence and metabolism genes. Infect Immun 79:4105–4121. doi: 10.1128/IAI.01390-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Rychlik I, Karasova D, Sebkova A, Volf J, Sisak F, Havlickova H, Kummer V, Imre A, Szmolka A, Nagy B. 2009. Virulence potential of five major pathogenicity islands (SPI-1 to SPI-5) of Salmonella enterica serovar Enteritidis for chickens. BMC Microbiol 9:268. doi: 10.1186/1471-2180-9-268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bohez L, Ducatelle R, Pasmans F, Botteldoorn N, Haesebrouck F, Van Immerseel F. 2006. Salmonella enterica serovar Enteritidis colonization of the chicken caecum requires the HilA regulatory protein. Vet Microbiol 116:202–210. doi: 10.1016/j.vetmic.2006.03.007. [DOI] [PubMed] [Google Scholar]
- 53.Winter SE, Winter MG, Poon V, Keestra AM, Sterzenbach T, Faber F, Costa LF, Cassou F, Costa EA, Alves GE, Paixao TA, Santos RL, Baumler AJ. 2014. Salmonella enterica serovar Typhi conceals the invasion-associated type three secretion system from the innate immune system by gene regulation. PLoS Pathog 10:e1004207. doi: 10.1371/journal.ppat.1004207. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.