

Abstract
We performed genetic analysis of Bartonella isolates from rodent populations from Heixiazi Island in northeast China. Animals were captured at four sites representing grassland and brushwood habitats in 2011 and examined for the prevalence and genetic diversity of Bartonella species, their relationship to their hosts, and geographic distribution. A high prevalence (57.7%) and a high diversity (14 unique genotypes which belonged to 8 clades) of Bartonella spp. were detected from 71 rodents comprising 5 species and 4 genera from 3 rodent families. Forty-one Bartonella isolates were recovered and identified, including B. taylorii, B. japonica, B. coopersplainsensis, B. grahamii, B. washoensis subsp. cynomysii, B. doshiae, and two novel Bartonella species, by sequencing of four genes (gltA, the 16S rRNA gene, ftsZ, and rpoB). The isolates of B. taylorii and B. grahamii were the most prevalent and exhibited genetic difference from isolates identified elsewhere. Several isolates clustered with strains from Japan and far-eastern Russia; strains isolated from the same host typically were found within the same cluster. Species descriptions are provided for Bartonella heixiaziensis sp. nov. and B. fuyuanensis sp. nov.
INTRODUCTION
Significant numbers of zoonotic Bartonella species have been recognized in the past 25 years. Before 1991, B. bacilliformis was the only member of the Bartonella genus. At present, the genus Bartonella contains over 30 formally described species of Bartonella and many unnamed Bartonella spp. isolated from a variety of mammal species, most often from rodents, ruminants, and carnivores (1). Some Bartonella spp. are long-known etiological agents of human diseases (Carrion's disease, cat scratch disease, and trench fever) and some specific syndromes (verruga peruana, bacillary angiomatosis, endocarditis, pericarditis, and neuroretinitis) associated with these agents. Rodents are the largest source of Bartonella spp. There are more than 40 species of rodents known to be associated with 26 different species of Bartonella, seven of which have been implicated as occasional causes of human infections (1). People become infected with rodent-borne Bartonella incidentally, when they are exposed to habitats and ectoparasites of wild rodents harboring various Bartonella species.
Previous field and clinical studies in mainland China demonstrated circulation of a variety of different Bartonella spp. associated with peridomestic, feral, and exotic animals across the country (2–5) and low levels of human Bartonella infections, diagnosed primary as cat scratch disease due to B. henselae, in heavily populated areas (6). Heixiazi Island has a beautiful natural landscape and plentiful wildlife; it is expected to become a tourist attraction because of its special history and geographical position. This island was previously unpopulated and isolated from mainland northeast Asia before its recent opening to tourists. Thus, in the present study, to evaluate the potential risk for transmission of rodent-borne Bartonella on Heixiazi Island after opening it up to tourists, we conducted a baseline survey of the occurrence and prevalence of Bartonella infections in rodents on Heixiazi Island, China. The genetic diversity of Bartonella isolates and their relationship to hosts and geographic distributions were examined and evaluated in the context of current knowledge about Bartonella species which are associated with rodents.
MATERIALS AND METHODS
Ethics statement.
This study was performed after review and approval by the Ethics Committee of the National Institute for Communicable Disease Control and Prevention of the Chinese CDC. All animals were treated according to the approved protocols and the guidelines for the Laboratory Animal Use and Care from the Chinese CDC and the Rules for the Implementation of Laboratory Animal Medicine (1998) from the Ministry of Health, China.
Sampling sites and tissue collections.
Rodents were captured using the trap-at-night method in May 2011. Six hundred traps baited with peanuts were set up in four sites of Heixiazi Island consisting of grassland and brushwood, the predominant habitats on the island. The trapped animals were morphologically identified to species. Collection time, site, habitat, species, gender, weight, head-body length, and tail length were recorded. Liver and spleen were collected into 1.5-ml Nunc CryoTubes and stored in liquid nitrogen prior to processing and laboratory testing.
Isolation and cultivation of Bartonella organisms, phenotypic characterization, and electron microscopy.
Approximate 20 mg of organ samples was disrupted using a Dounce-Potter homogenizer in 0.6 ml of tryptic soy broth (TSB) (BD), and suspensions were inoculated onto tryptic soy agar (TSA) plates (BD) supplemented with 5% defibrinated sheep blood. Plates were kept in a 5% CO2 humidified atmosphere at 35°C for 30 days and were checked daily for bacterial growth starting at day 3 after inoculation. Small, round, gray-white colonies were morphologically identified as Bartonella and passaged onto fresh plates.
Material from a single colony was suspended in 100 μl of deionized water, heated for 10 min at 95°C, and cleared by centrifugation for 5 min at 6,000 × g at 4°C. The supernatant was used as a source of DNA template for PCR. The presence of Bartonella DNA was first determined by PCR detecting a portion of the citrate synthase gene (gltA) with the primers BhCS.781p and BhCS.1137n (Table 1) as described elsewhere (7). Bacterial isolates identified as Bartonella were cloned by limiting dilution, and established cultures were preserved in brain heart infusion (BHI) broth (BD) supplemented with 30% glycerol at −70°C. Isolates were passaged into flasks containing insect Schneider's medium and propagated according to previously established procedures to observe their growth characteristics (8, 9).
TABLE 1.
Oligonucleotide primers used for PCR
| Target gene | Primer | Primer direction | Primer sequence (5′–3′) | Product length (bp) |
|---|---|---|---|---|
| gltA | BhCS.781p | Forward | GGG GAC CAG CTC ATG GTG G | 379 |
| BhCS.1137n | Reverse | AAT GCA AAA AGA ACA GTA AAC A | ||
| 16S rRNA gene | fDl | Forward | AGA GTT TGA TCC TGG CTC AG | 1,400–1,500 |
| rDl | Reverse | AAG GAG GTG ATC CAG CC | ||
| ftsZ | Bfp1 | Forward | ATT AAT CTG CAY CGG CCA GA | 896 |
| Bfp2 | Reverse | ACV GAD ACA CGA ATA ACA CC | ||
| rpoB | 1400F | Forward | CGC ATT GGC TTA CTT CGT ATG | 866 |
| 2300R | Reverse | GTA GAC TGA TTA AAC GCT G |
To prepare samples for electron microscopy, bacterial cells were suspended in deionized water, spread onto a water surface, absorbed onto Formvar stabilized with carbon support film, stained for 2 min in 1% uranyl acetate, and then air dried. Samples were examined with a transmission electron microscope (Hitachi, HT7700) at 80 kV with negative staining.
Biochemical testing was performed using commercially available reagents (Rapid ID 32 A; bioMérieux) according to the manufacturer's instructions. Fatty acid methyl esters were obtained by saponification, methylation, and extraction according to the MIS operation manual (Midi Inc., 2002) and then analyzed using Agilent Hp6890 as Chromatograph and ChemStation v A5.05.
PCR amplification and sequencing of Bartonella genes.
DNA was extracted from bacterial isolates using the DNeasy tissue kit (Qiagen, Hilden, Germany) according to the manufacturer's instructions. For all identified isolates, four gene fragments were amplified and their sequences were obtained: gltA, the 16S rRNA gene (10), cell division protein gene ftsZ (11), and RNA polymerase beta-subunit gene rpoB (12). Nucleotide primers used in this study are listed in Table 1.
PCR amplification was performed using Pfu DNA polymerase (TransGen Biotech, Beijing, China) and 20 to 50 ng of genomic DNA in a final volume of 50 μl containing 0.2 μM each primer and 25 μl 2× TransTaq-T PCR SuperMix. The PCR amplification conditions were as follows: an initial step of 94°C for 2 min; 30 amplification cycles, each consisting of 94°C for 30 s and 53°C for 30 s; an elongation step of 72°C for 2 min; and a final incubation at 72°C for 5 min. Amplified products were analyzed by electrophoresis on 1% agarose gels supplemented with 0.005% of GoldView (SBS Genetech, Beijing, China) and visualized under UV light. PCR products of the expected length were then purified and sequenced on both strands by Tsingke Biotechnology (Beijing, China).
Phylogenetic analysis.
Individual gene sequences and concatenated sequences assembled for gltA (326 bp), 16S rRNA gene (1245 bp), ftsZ (767 bp), and rpoB (771 bp) fragments were aligned using ClustalW, and neighbor-joining analysis was performed using MEGA6 (13) with the Kimura 2-parameter model. A phylogeny was constructed based on the concatenated sequences using the maximum-likelihood method and HKY85 model implemented in PhyML software (14). Homologous sequences of Brucella abortus were used as an outgroup. Bootstrap analysis was carried out with 1,000 resamplings. Homologous sequences for reference Bartonella species were downloaded from the NCBI GenBank, and their accession numbers are listed in Table S1 in the supplemental material.
Recombination analysis.
The multiple-alignment sequences (MAS) were scanned for intragenic and intergenic recombination using RDPv.3.44 (15), GENECONV, Bootscan, MaxChi, Chimaera, and SiScan as implemented in RDP with default settings. A phylogenetic tree was also inferred using ClonalFrame v1.1 (16) with the number of Markov chain Monte Carlo (MCMC) iterations set to 50,000, following a burn-in period of 50,000 iterations.
The Neighbor-Net implemented in the software SplitsTree 4.13 (17) with 1,000 bootstrap replicates was used to create the EqualAngle phylogenic network for the concatenated sequences. The pairwise homoplasy index (PHI) in SplitsTree 4.13 was used to test the role of past recombination events in generating allelic variation.
Population diversity and distribution analysis.
Nucleotide diversity indices (π) and the polymorphic level (haplotype diversity [Hd]) were calculated using DNAsp v5.10 (18). The aligned segment of gltA was used to construct a phylogenetic network with the postprocessing of maximum parsimony (MP) calculation (19) using the median joining method (20) after the preprocessing of star contraction (21) implemented in the program Network 4.6.1.2. The parameters of characters were set at the default values (epsilon and weight values = 0 and 10). Homologous sequences for the Bartonella strains were downloaded from the NCBI GenBank, and their accession numbers are listed in Tables S1, S2, S3, and S4 in the supplemental material.
Statistical analysis.
SPSS software (version 19.0) was used for statistical analysis. Chi-square and Fisher's exact tests were applied for assessing the differences of infection rates in the tissues tested. A P value of <0.05 was considered statistically significant.
Nucleotide sequence accession numbers.
Sequences of the Bartonella gene fragments generated in this study were deposited in the NCBI GenBank database under accession numbers KJ175028 to KJ175068 for gltA, KJ361602 to KJ361642 for the 16S rRNA gene, KJ361684 to KJ361724 for ftsZ, and KJ361725 to KJ361765 for rpoB.
RESULTS
Study site description.
Heixiazi Island (134°24′E, 48°22′N) (also called Fuyuan Delta or Bolshoi Ussuriysky Island), located at the Chinese-Russian border, is a natural sedimentary island at the confluence of Heilongjiang and Wusulijiang Rivers (called the Ussuri and Amur Rivers in Russia) (Fig. 1). Heixiazi Island serves as a natural border between China and Russia; it is 327 km2 with an average altitude of about 40 m. China restored sovereign control over half of Heixiazi Island (171 km2) from Russia on 14 October 2008. The Chinese section of the island is part of Fuyuan County, Heilongjiang Province. The landscape consists mainly of wetlands and lush growth of elms, poplars, willows oak trees, sedges, and grasses.
FIG 1.
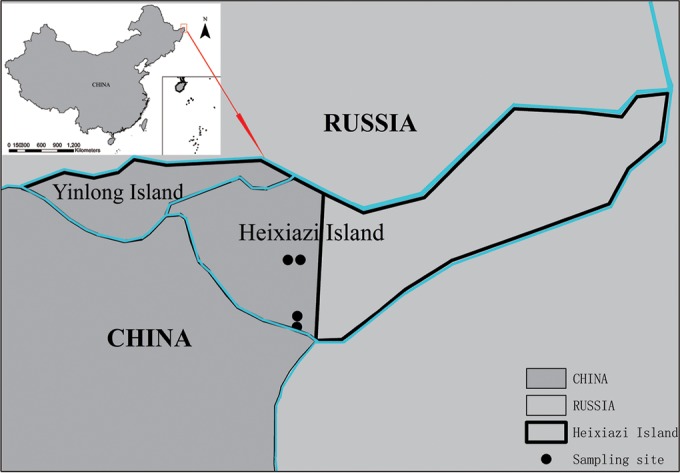
Distribution of sampling sites. The map shows the locations of the geographic sites where the rodents were collected on Heixiazi Island, China, during 2011. Map created using ArcGis v10.2 (ESRI, Redland, CA).
Animal trapping.
A total of 71 small mammals belonging to 6 species and 4 genera from 3 rodent families were captured in 4 sampling sites. Forty-three animals were trapped in woodland habitats and 28 animals in grassland habitats. The northern red-backed vole Myodes rutilus was the most prevalent animal species and accounted for 64.8% (n = 46) of the rodents captured. Striped field mouse Apodemus agrarius (n = 12), reed vole Microtus fortis (n = 5), Maximowicz's vole Microtus maximowiczii (n = 1), Korean field mouse Apodemus peninsulae (n = 6), and Siberian chipmunk Eutamias sibiricus (n = 1) were also collected and accounted for 16.9%, 7.0%, 1.4%, 8.5%, and 1.4%, respectively, of all trapped animals.
Bartonella prevalence and identification.
The prevalence of Bartonella in rodents was 57.7% (41/71) but there were no significant variations of infection rate (P > 0.05, Fisher's exact test) between the two species belonging to Apodemus spp. In total, 66 Bartonella isolates were established from tissue samples of 71 rodents, including 34 (48.6%) isolates from 70 liver tissues and 32 (47.8%) isolates from 67 spleen tissues. There were 25 paired isolates recovered from the spleens and livers, respectively, of 25 rodents. The other 16 isolates were obtained from livers (9 isolates) or spleens (7 isolates) of different rodents (Table 2; see Table S5 in the supplemental material). The prevalence of Bartonella infection in livers did not differ from the prevalence of Bartonella infection in spleens (P > 0.05, chi-square test). Bartonella growth could be observed as early as day 4 after inoculation of the tissue homogenate onto agar plates. Colony morphology and growth rate were consistent with the characteristics of Bartonella isolates described in the literature (22–24).
TABLE 2.
Bartonella infection rates in liver and spleen tissues of rodents collected on Heixiazi Island, Chinaa
| Rodent species | Liver |
Spleen |
Liver and spleen coisolation | ||
|---|---|---|---|---|---|
| n | No. (%) positive | n | No. (%) positive | ||
| Microtus fortis | 5 | 1 (20.0) | 5 | 1 (20.0) | 1 |
| Microtus maximowiczii | 1 | 1 (100.0) | 1 | 1 (100.0) | 1 |
| Myodes rutilus | 46 | 25 (54.3) | 43 | 22 (51.2) | 19 |
| Apodemus agrarius | 11 | 4 (36.4) | 11 | 5 (45.5) | 2 |
| Eutamias sibiricus | 1 | 0 | 1 | 1 (100) | 0 |
| Apodemus peninsulae | 6 | 3 (50) | 6 | 2 (33.3) | 2 |
| Total | 70 | 34 (48.6) | 67 | 32 (47.8) | 25 |
There were no significant differences in the rates of Bartonella isolate recovery from liver and spleen (P > 0.05).
Genetic identification and phylogenetic relationships of rodent Bartonella isolates from Heixiazi Island.
Based on sequence analysis of the 326-nucleotide (nt) gltA fragment, 41 newly established isolates of Bartonella belonged to six known Bartonella species and two potentially novel species. The isolates of B. taylorii (11 isolates), B. japonica (1 isolate), B. coopersplainsensis (3 isolates), B. grahamii (5 isolates), B. washoensis subsp. cynomysii (1 isolate), and B. doshiae (12 isolates) were obtained from voles, field mice, and chipmunks from Heixiazi Island. Seven isolates from voles (one from M. maximowiczii and 6 from M. rutilus) and one isolate from a mouse (Apodemus agrarius) had unique genotypes, and the representative strains were CR90HXZ and AA137HXZ. The isolates CR90HXZ and AA137HX, each shared less than 95% nucleotide sequence similarity in their gltA fragment with homologous genes of the nearest species of Bartonella, B. vinsonii subsp. arupensis and B. grahamii, respectively.
To further explore the relationship of the new Bartonella isolates from Chinese rodents with recognized species of Bartonella, we performed a phylogenetic analysis based on the alignment of the individual gene fragments of the gltA, 16S rRNA, ftsZ, and rpoB genes (see Fig. S1 in the supplemental material). Furthermore, 3,110-bp fragments of concatenated sequences of the same four conserved genes of 41 Bartonella isolates obtained in this study and homologous sequences of 26 Bartonella type strains retrieved from GenBank were analyzed. The same topology trees were produced for each individual gene fragment and for the concatenated sequence fragments using both the neighbor-joining (Fig. 2) and maximum-likelihood methods (see Fig. S2 in the supplemental material). Overall, the 41 isolates exhibited 14 distinct genotypes which were predicted using DnaSP v5.10.1; these 14 genotypes were clustered into 8 clades (I to VIII). The largest number of isolates was associated with clades formed by six from all recognized species of Bartonella, including B. taylorii (clade I), B. japonica (clade II), B. coopersplainsensis (clade III), B. grahamii (clade V), B. washoensis subsp. cynomysii (clade VI), and B. doshiae (clade VIII) (Fig. 2). Eleven Chinese isolates clustered with B. taylorii exhibited two distinct genotypes: one genotype comprised 8 isolates from M. rutilis (Myodes genotype) and a second genotype consisted of 3 isolates from 2 A. peninsulae animals and one A. agrarius animal (Apodemus genotype). Nucleotide sequences of another isolate from A. peninsulae (AP73HXZ) were identical to those of homologous gene fragments of B. japonica recovered from A. argenteus in Japan (AB242289). Furthermore, sequences of three isolates from A. agrarius (AA95HXZ, AA86HXZ, and AA123HXZ) were identical to each other and had the most similarity with the sequences of B. coopersplainsensis obtained from Rattus leucopus in Australia (EU111803). The B. grahamii clade (clade V) included 5 genetically distinct Chinese isolates recovered from A. agrarius (2 isolates), M. rutilus (2 isolates), and M. fortis (1 isolate). A single isolate, ES132HXZ from E. sibiricus, was grouped with B. washoensis subsp. cynomysii in cluster VI. Twelve isolates from M. rutilus had the same sequences and were most related to B. doshiae. Seven isolates from 6 M. rutilus and one from M. maximowiczii were identical to each other and were included in cluster VII, which represented a unique lineage within the trees evaluated. The nucleotide sequences of these isolates exhibited 4.6% nucleotide divergence from the homologous sequences of its closest neighbor, B. washoensis subsp. cynomysii. One isolate from A. agrarius formed cluster IV and had 4.5% genetic divergence from its nearest neighbor, B. rattimassiliensis. Based on these genetic differences, Bartonella isolates from M. rutilus and M. maximowiczii and an isolate from A. agrarius closest to B. rattimassiliensis represent novel species.
FIG 2.
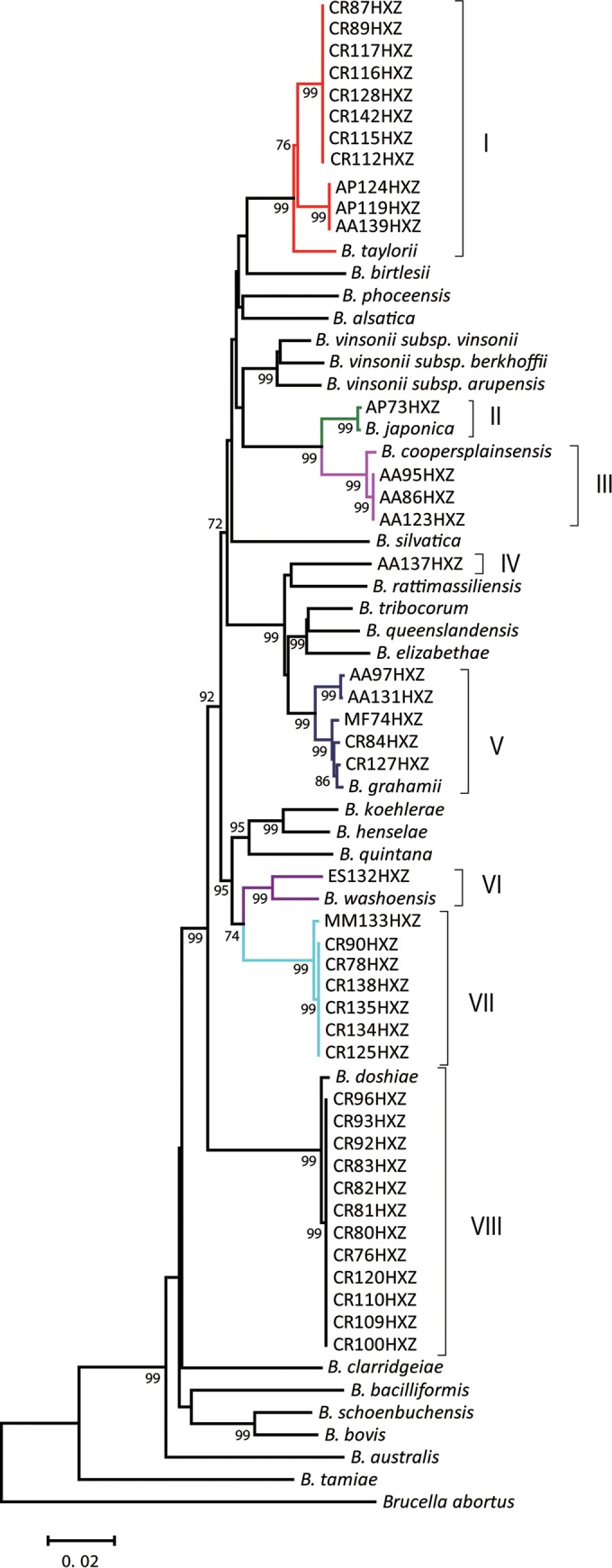
Phylogenetic relationships of the Bartonella type strains and the Bartonella isolates from Heixiazi Island based on the concatenation of fragments of gltA, the 16S rRNA gene, ftsZ, and rpoB. The phylogenetic tree was inferred using the neighbor-joining method based on the Kimura 2-parameter model in MEGA6 (13). The percentages of replicate trees (>70%) in which the associated taxa clustered together in the bootstrap test (1,000 replicates) are shown next to the branches. The analysis involved 3,110 concatenated nucleotides from 68 isolates. Roman numerals I to VIII on the right side of the tree correspond to different Bartonella genetic groups. Homologous sequences of Brucella abortus were used as an outgroup. All sequences were trimmed to the same size, and gaps were excluded from the analysis.
Scanning of multiple sequence alignments for intragenic and intergenic mosaicism and effects of recombination on phylogenetic relationships.
Individual DNA alignments of gltA, the 16S rRNA gene, ftsZ, and rpoB were analyzed with RDPv.3.44 programs to detect putative interspecies recombinant sequences. Potential mosaic regions were considered likely recombinants only if they were detected by at least two programs. There were no significant interspecies recombination events detected in the individual DNA alignments. Accordingly, there was no interference from recombination for the phylogenetic trees constructed based on the four individual gene fragments.
To include more informative sites, the concatenated sequences of four genes were analyzed for occurrence of possible interspecies recombination events using the same method. Three significant recombinations were detected, which affected mostly the two different genotypes of B. taylorii. The recombination breakpoint position was located between nt 153 and 392 of the concatenated sequences of the Myodes-type isolates of B. taylorii, while the minor parental sequence originated from CR84HXZ of B. grahamii. These parts of the concatenated fragment were from the hypervariable 3′ end of the gltA fragment (nt 153 to 326) and 16S rRNA gene fragment (nt 1 to 67). The topology of the phylogenetic tree was not changed upon exclusion of those nucleotides from the analysis. Furthermore, the split network also supported the same relationship among the isolates, and this was not reflected in the recombination breakpoint pattern. The fit, quite high at 98.2%, indicates that the data are very tree-like, further suggesting that the bootstrap split tree (Fig. 3) was congruent with the clustering in the phylogenetic binary tree (Fig. 2).
FIG 3.

Neighbor-Net tree of associations of Bartonella isolates obtained from different rodents inhabiting Heixiazi Island, China, and the Bartonella type strains based on concatenated conserved genes (gltA, 16S rRNA gene, ftsZ, and rpoB) representing a total of 3,110 nucleotides using SplitsTree4. The numbers in parentheses represent the number of isolates at each branch. The percentages of replicate trees in which the associated taxa clustered together in the bootstrap test (1,000 replicates) are shown next to the branches; only bootstrap values of >70% are indicated. Different colors refer to individual Bartonella genetic groups. Recombination is illustrated by the reticulation structures in the center among Bartonella strains. Bartonella tamiae was used as an outgroup. The fit parameter values were 98.2% in all cases, indicating that a substantial fraction of the phylogenetic signals could be depicted on the graph. The bar in the upper left corner of the network graph indicates the scale of branches.
For assessing the relative contributions of recombination and mutation in the delineation of various Bartonella genotypes from a common ancestor, we calculated the ratio of recombination to mutation value (r/m and ρ/θ ratio) using ClonalFrame v1.1 (21). The inferred outputs were estimated at 0.83286 (r/m) (95% confidence interval [CI95] = 0.507585 to 1.269109) and 0.036913 (ρ/θ) (CI95 = 0.021735 to 0.058028), suggesting that nucleotide changes in these conserved genes occur more frequently by de novo mutation than by recombination.
Genetic polymorphism of rodent-adapted Bartonella species.
Overall, the extent of the intraspecies variation within the different Bartonella clades was different and closely linked to their host associations based on the 4 conserved gene fragments. Twelve B. doshiae isolates from M. rutilus had an identical genotype. Similarly, three isolates of B. coopersplainsensis from Apodemus spp. exhibited the same genotype. In contrast, B. taylorii, B. grahamii, and B. washoensis-like isolates from Heixiazi Island exhibited noticeable intraspecies genetic diversity. Therefore, we examined the nucleotide polymorphisms of the 4 conserved genes in these isolates recovered from Heixiazi Island compared to the levels of inter- and intraspecies genetic variation among isolates of B. taylorii, B. grahamii and B. washoensis circulating in different parts of the world, using sequences from GenBank. The level of nucleotide polymorphism differed between two groups, and the nucleotide diversities varied with the different alleles (Table 3). The 16S rRNA gene exhibited the lowest genetic diversity in each population of Bartonella isolates analyzed and had the lowest calculated genetic distance (D) and nucleotide diversity (π) values. The nucleotide diversity and genetic distance for the gltA, ftsZ, and rpoB gene fragments varied among the B. taylorii, B. grahamii, and B. washoensis-like isolates from Heixiazi Island. Analysis of the D and π parameters calculated for all Bartonella isolates from Heixiazi Island suggested that the highest polymorphism was within gltA and rpoB, similar to the conclusions derived when similar parameters were calculated for sequences of known Bartonella species. Our observations suggest that these two genes have more significant impact for defining species of Bartonella than the other two genes.
TABLE 3.
Nucleotide polymorphism and nucleotide diversity of rodent-associated isolates from Heixiazi Island (China) and type strains of Bartonella speciesa
| Bartonella sp. (no. of isolates) | Gene | Size (bp) | D | S | PI | π (mean ± SD) | Hd (mean ± SD) | k |
|---|---|---|---|---|---|---|---|---|
| B. taylorii (11) | gltA | 326 | 0.033 | 0 | 23 | 0.03079 ± 0.01050 | 0.436 ± 0.133 | 10.036 |
| 16S rRNA gene | 1,247 | 0.002 | 0 | 5 | 0.00175 ± 0.00078 | 0.436 ± 0.133 | 2.182 | |
| ftsZ | 766 | 0.006 | 0 | 10 | 0.00570 ± 0.00217 | 0.436 ± 0.133 | 4.364 | |
| rpoB | 771 | 0.009 | 0 | 15 | 0.00847 ± 0.00303 | 0.436 ± 0.133 | 6.545 | |
| Concatenate | 3,110 | 0.008 | 0 | 53 | 0.00744 ± 0.00239 | 0.436 ± 0.133 | 23.127 | |
| B. grahamii (5) | gltA | 326 | 0.007 | 1 | 6 | 0.01227 ± 0.00359 | 0.700 ± 0.218 | 4.000 |
| 16S rRNA gene | 1,245 | 0.000 | 0 | 1 | 0.00048 ± 0.00014 | 0.600 ± 0.175 | 0.600 | |
| ftsZ | 766 | 0.011 | 4 | 11 | 0.01069 ± 0.00260 | 1.000 ± 0.126 | 8.200 | |
| rpoB | 771 | 0.009 | 12 | 22 | 0.02335 ± 0.00480 | 1.000 ± 0.126 | 18.000 | |
| Concatenate | 3,108 | 0.010 | 17 | 40 | 0.00991 ± 0.00219 | 1.000 ± 0.126 | 30.800 | |
| B. heixiaziensis (7) | gltA | 326 | 0.002 | 2 | 0 | 0.00175 ± 0.00120 | 0.286 ± 0.196 | 0.571 |
| 16S rRNA gene | 1,245 | 0.000 | 0 | 0 | 0.00000 | 0.000 | 0.000 | |
| ftsZ | 766 | 0.002 | 5 | 0 | 0.00186 ± 0.00128 | 0.286 ± 0.196 | 1.429 | |
| rpoB | 771 | 0.000 | 1 | 0 | 0.00037 ± 0.00026 | 0.286 ± 0.039 | 0.286 | |
| Concatenate | 3,107 | 0.001 | 8 | 0 | 0.00074 ± 0.00051 | 0.286 ± 0.196 | 2.286 | |
| All HXZ isolates (41) | gltA | 326 | 0.110 | 15 | 78 | 0.09707 ± 0.00390 | 0.856 ± 0.033 | 31.645 |
| 16S rRNA gene | 1,245 | 0.006 | 6 | 26 | 0.00567 ± 0.00085 | 0.848 ± 0.032 | 7.040 | |
| ftsZ | 766 | 0.080 | 12 | 143 | 0.07373 ± 0.00327 | 0.860 ± 0.034 | 56.554 | |
| rpoB | 771 | 0.073 | 21 | 172 | 0.09479 ± 0.00339 | 0.860 ± 0.034 | 73.083 | |
| Concatenate | 3,106 | 0.057 | 54 | 419 | 0.05419 ± 0.00223 | 0.860 ± 0.034 | 168.322 | |
| All type strains (26) | gltA | 326 | 0.145 | 49 | 104 | 0.12614 ± 0.00836 | 1.000 ± 0.011 | 41.123 |
| 16S rRNA gene | 1,236 | 0.015 | 52 | 43 | 0.01515 ± 0.00202 | 1.000 ± 0.011 | 18.723 | |
| ftsZ | 765 | 0.121 | 80 | 191 | 0.10947 ± 0.00837 | 1.000 ± 0.011 | 83.745 | |
| rpoB | 771 | 0.132 | 48 | 224 | 0.11947 ± 0.00560 | 1.000 ± 0.011 | 92.108 | |
| Concatenate | 3,097 | 0.081 | 230 | 560 | 0.07553 ± 0.00494 | 1.000 ± 0.011 | 233.609 |
HXZ, Heixiazi Island; D, genetic distance; S, singleton variable sites; PI, parsimony-informative sites; π, nucleotide diversity; Hd, haplotype diversity; k, average number of nucleotide differences.
Isolates identified as B. taylorii belong to two genotypes which exhibit estimated genetic distances of 0.008: the Apodemus-associated-genotype and the Myodes-associated genotype. The Apodemus-associated B. taylorii isolates were from two sympatric species of wood mice, A. agrarius and A. peninsulae. These isolates had identical nucleotide sequences for all four gene fragments sequenced, but these sequences were different from the sequences of the homologous genes of M. rutilus-associated B. taylorii isolates. No indel was found within the four gene fragments sequenced for all B. taylorii isolates from Heixiazi Island examined here; however, 53 polymorphic and parsimony informative sites were identified within 3,110 nt of concatenated gltA-16S rRNA gene-ftsZ-rpoB sequences of 11 B. taylorii isolates from the island. In particular, gltA, the 16S rRNA gene, ftsZ and rpoB contained 23, 5, 10 and 15 polymorphic sites, respectively, which accounted for 0.74%, 0.16%, 0.32%, and 0.48%, respectively, of the entire 3,110-nt concatenated fragment. The haplotype diversity (Hd), π value, and average number of nucleotide differences (k) are estimated to be 0.436, 0.00744, and 23.127, respectively. In comparison, analysis of the 258-nt sequence of gltA from the 108 entries listed as B. taylorii sequences in NCBI GenBank detected 63 polymorphic sites. Among them, there were 13 single nucleotide polymorphism (SNP) sites and 49 parsimony-informative sites; accordingly, the calculated π value per site was 0.03881, and the k value was 10.013. Furthermore, it was estimated that 53 haplotypes exist among previously identified isolates of B. taylorii; their predicted Hd value was 0.971.
Five Heixiazi Island isolates, including two isolates from M. rutilus (CR84HXZ and CR127HXZ), one isolate from M. fortis (MF74HXZ), and two isolates from A. agrarius (AA97HXZ and AA131HXZ) were clustered with B. grahamii, but all had distinct genotypes with an overall genetic distance of 0.011. Sequences of four conserved genes compared for these isolates of B. grahamii had 57 polymorphic sites, no indels were present. There were 17 SNP and 40 parsimony-informative sites; they were found in gltA, the 16S rRNA gene, ftsZ and rpoB and accounted for 0.22% (7 nt), 0.03% (1 nt), 0.48% (15 nt), and 1.09% (34 nt), respectively, of the entire 3,109-nt concatenated fragment. The Hd, π, and k values were estimated to be 1.000, 0.00991, and 30.8, respectively. In comparison, analysis of the 307-nt sequence of gltA of the 64 entries identified as B. grahamii sequences found in GenBank detected 54 polymorphic sites. Among them, there were 27 SNP and were 27 parsimony-informative sites; the estimated π value per site was 0.02241, and the k value was 6.880. It was estimated that there are at least 36 haplotypes among previously identified isolates of B. grahamii; the predicted Hd value among known B. grahamii isolates was estimated to be 0.961.
Seven Bartonella isolates from Heixiazi Island, including six isolates from M. rutilus and one isolate from M. maximowiczii, exhibited two distinct genetic types, and both were clustered with B. washoensis as a nearest neighbor. Indel and parsimony-informative sites were not found in sequences generated for these isolates. The 16S rRNA gene sequences were conserved among all seven isolates. Eight SNP sites were found in the gltA, ftsZ, and rpoB sequences, which accounted for 0.06% (2 nt), 0.16% (5 nt), and 0.03% (1 nt) of the entire 3,107-nt concatenated fragment, respectively. The Hd, π, and k values among the two genotypes were 0.286, 0.00074, and 2.286, respectively. In comparison, analysis of the 306-nt sequence of gltA of the 82 of B. washoensis sequences found in GenBank detected 56 polymorphic sites. Among them, there were 17 SNP and 39 parsimony-informative sites; the estimated π value per site was 0.02984, and the k value was 9.132. Overall, there were 53 haplotypes among previously identified isolates of B. washoensis, and their predicted Hd is 0.9584.
Relationship of Bartonella, hosts, and geographic distributions.
Since isolates of B. taylorii, B. grahamii, and B. washoensis are known for their ubiquitous distribution, the phylogenetic network of these Bartonella species was analyzed in the context of their geographic relationship and their host associations in order to define their influence on the Bartonella population genetic structure. Our analysis revealed that Bartonella isolates from Asia, Europe, and North America do not exhibit any obvious geographic clustering pattern; however, they could be easily grouped based on their host associations (Fig. 4).
FIG 4.

Median-joining networks of gltA genotypes established based on maximum-parsimony calculation for Bartonella isolates from Heixiazi Island and all other isolates of homologous species whose nucleotide sequences are available in the NCBI GenBank database. Genotypes are colored according to their geographical origins and host species. The smallest colored circle corresponds to a single isolate. Isolates from Heixiazi Island are indicated by a secondary dashed-line circle. A1, network of B. taylorii according to its geographic distribution; A2, network of B. taylorii according to its host species associations; B1, network of B. grahamii according to its geographic distribution; B2, network of B. grahamii according to its host species associations; C1, network of B. washoensis according to its geographic distribution; C2, network of B. washoensis according to its host species associations.
The genetically different isolates of B. taylorii from voles and mice in Heixiazi Island belong to two different clades of Bartonella; one includes B. taylorii from Japan, and the other includes B. taylorii isolates from France, Greece, Japan, Lithuania, Poland, the Russian Far East, and Thailand (Fig. 4A1). This grouping of B. taylorii isolates matched the grouping based on their host associations (Fig. 4A2). Three Apodemus genotype isolates (AA139HXZ, AP119HXZ, and AP124HXZ) from Heixiazi Island were in the same cluster group as B. taylorii from Europe and the Apodemus- and Myodes-associated B. taylorii isolates from Japan. The eight Myodes genotype isolates (CR87HXZ, CR89HXZ, CR112HXZ, CR115HXZ, CR116HXZ, CR117HXZ, CR128HXZ, and CR142HXZ) grouped with the isolates from Asia and Europe, which have different animal species as hosts. Four isolates of B. taylorii from Ctenophthalmus lushuiensis, one isolate from Eothenomys miletus trapped in Yunnan Province, China, and an isolate from Japanese Microtus motebelli clustered separately from the Heixiazi Island isolates (Fig. 4A1 and A2).
Five Heixiazi Island isolates of B. grahamii formed two clusters; one included 2 isolates (AA131HXZ and AA97HXZ) from A. agrarius and isolates from Apodemus spp. trapped in Japan and the Russian Far East (Fig. 4B1). The second cluster consisted of isolates originating from a variety of different hosts, including one from M. fortis (MF74HXZ) and two from M. rutilus (CR84HXZ and CR127HXZ) trapped on Heixiazi Island and four Bartonella strains isolated from a human, A. agrarius, and M. ochrogaster in Finland, the Russian Far East, and North America (Fig. 4B2).
Previously characterized isolates of B. washoensis associate with various species of squirrels in Asia and North America (25). The B. washoensis isolates from Hebei, Zhejiang Province, and Heixiazi Island in China belong to three groups, one of which also includes the Heixiazi Island isolates from E. sibiricus (Fig. 4C1). The other two clusters include 17 isolates of B. washoensis from Spermophilus dauricus, one isolate from A. chevrieri, and one isolate from a tick, Haemaphysalis longicornis (Fig. 4C2). B. washoensis isolates from North America exhibited a similar clustering pattern based on their host associations (Fig. 4C2).
Host specificity of Bartonella isolates.
Overall, 28 Bartonella isolates of 4 different species were obtained from M. rutilus (Table 4). There were 12 isolates of B. doshiae, 8 of B. taylorii, 6 of B. washoensis, and 2 of B. grahamii. Eleven isolates of B. taylorii were established from M. rutilus (8 isolates; 72.7%), A. peninsulae (2 isolates, 18.2%), and A. agrarius (1 isolate, 9.1%). The vole was the predominant host of B. taylorii. Genetic variation among individual isolates of B. taylorii from Heixiazi Island separated the Apodemus genotype and the Myodes rutilus genotype. The majority of B. washoensis-like isolates originated from M. rutilus and M. fortis voles. B. grahamii isolates were recovered from M. rutilus, A. agrarius, and M. fortis. Except for E. sibiricus, five species of rodents trapped on the island harbored more than one species of Bartonella. Only one strain of B. washoensis was isolated from E. sibiricus from Heixiazi Island. A new genotype (VII) of Bartonella spp., which was isolated from 6 M. rutilus animals and 1 M. maximowiczii animal, appears to be predominantly associated with voles.
TABLE 4.
Rodent species from which the Bartonella isolates were established and their habitats on Heixiazi Island, China
| Host | Nearest known Bartonella sp. | Similarity for 3,109 bp (%) | No. of isolates |
Constituent ratio (%) | ||
|---|---|---|---|---|---|---|
| Grassland | Woodland | Total | ||||
| Apodemus agrarius | B. coopersplainsensis | 99.6 | 2 | 1 | 3 | 42.9 |
| B. grahamii | 98.4 | 2 | 0 | 2 | 28.6 | |
| B. rattimassiliensis | 95.5 | 1 | 0 | 1 | 14.3 | |
| B. taylorii | 97.6 | 1 | 0 | 1 | 14.3 | |
| Apodemus peninsulae | B. taylorii | 97.6 | 0 | 2 | 2 | 66.7 |
| B. japonica | 99.9 | 1 | 0 | 1 | 33.3 | |
| Myodes rutilus | B. taylorii | 97.8 | 4 | 4 | 8 | 28.6 |
| B. doshiae | 99.5 | 1 | 11 | 12 | 42.9 | |
| B. grahamii | 99.7 | 2 | 0 | 2 | 7.1 | |
| B. washoensis subsp. cynomysii | 95.4 | 5 | 1 | 6 | 21.4 | |
| Eutamias sibiricus | B. washoensis subsp. cynomysii | 97.1 | 1 | 0 | 1 | 100.0 |
| Microtus fortis | B. grahamii | 99.5 | 0 | 1 | 1 | 50.0 |
| Microtus maximowiczii | B. washoensis subsp. cynomysii | 95.4 | 1 | 0 | 1 | 50.0 |
Characterization of novel Bartonella isolates.
Light microscopy examination of the two isolates CR90HXZ and AA137HXZ revealed short, straight, or slightly curved Gram-negative bacilli. Electron microscopy with negative staining of isolate AA137HXZ detected small rods with unipolar surface pili; in contrast, pili were not observed for isolate CR90HXZ (Fig. 5).
FIG 5.
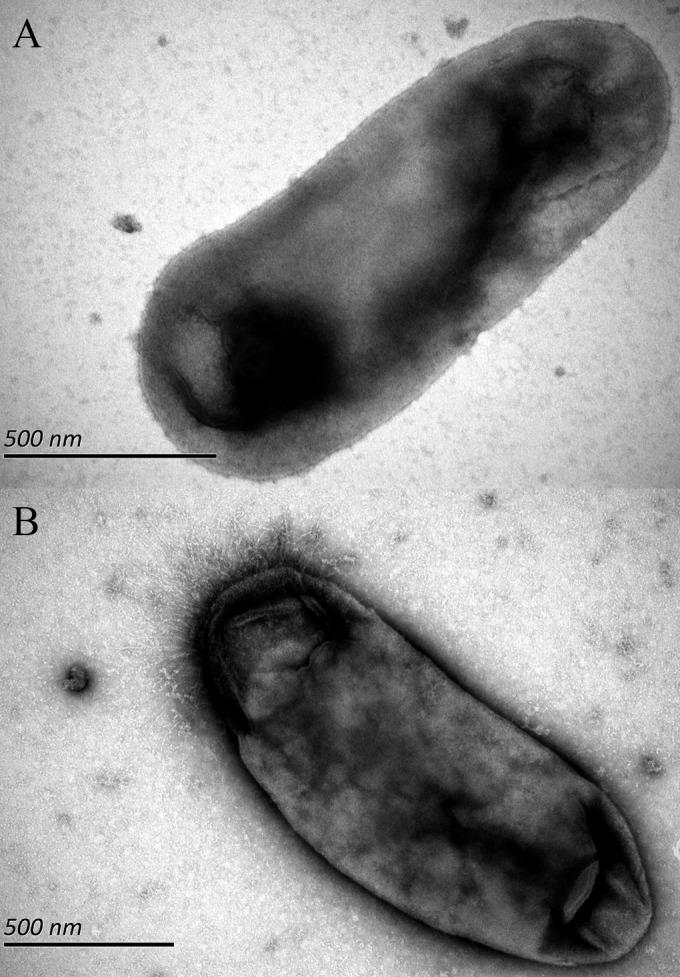
Electron micrographs of the two novel Bartonella strains, CR90HXZ without pili (A) and AA137HXZ showing the presence of polar pili (B). Bars, 0.5 μm.
Isolates CR90HXZ and AA137HXZ were catalase and oxidase negative and had no urease activity, similar to the case for other Bartonella isolates. Tests for α-glucosidase, α-l-fucosidase, α-l-arabinofuranosidase, mannose, raffinose, and nitrate reduction and spot indole tests using both stains were negative. Amino acidarylamidase activity was observed with the following amino acids: arginine (Arg), phenylalanine (Phe), leucine (Leu), tyrosine (Tyr), alanine (Ala), glycine (Gly), histidine (His), and serine (Ser).
To further confirm that these novel isolates were typical of Bartonella species, their fatty acid profiles were determined. The common cellular fatty acid proportions of C18:1ω7c, C18:0, C16:0, and C18:1ω7c were, respectively, 62.39%, 18.7%, 10.82%, and 1.01% for isolate CR90HXZ and 56.29%, 12.66%, 22.79%, and 2.16% for isolate AA137HXZ. C18:1ω9c and C20:1ω7c accounted for, respectively, 1.71% and 2.06% in isolate CR90HXZ; C17:0 accounted for 4.02% of total fatty acids in isolate AA137HXZ, while C18:1ω9c and C20:1ω7c were absent.
DISCUSSION
On Heixiazi Island, six species of rodents harbored eight Bartonella spp., including six known Bartonella species and two potentially novel Bartonella species. In particular, Apodemus rodents harbored the largest variety of Bartonella spp. (B. coopersplainsensis, B. grahamii, B. taylorii, B. japonica, and a novel species close to B. rattimassiliensis), M. rutilus harbored B. taylorii, B. doshiae, B. grahamii, and a novel species closely related to B. washoensis, M. maximowiczii harbored B. grahamii and the same novel species, and one E. sibiricus animal and one M. fortis animal harbored B. washoensis and B. grahamii, respectively. Since rodent species captured in our study widely inhabit the Sanjiang Plain (26), it is very likely that circulation of the same Bartonella spp. may be expected in areas surrounding the river and associated mainland territories.
Livers and spleens of infected animals had similar burdens of Bartonella as determined by the isolation results. These and previous observations suggest that these organs may act as depositories of harbored Bartonella, although they may be damaged as a result of a long-term Bartonella persistence in these tissues (27). These interactions underlie the primary etiology of Bartonella-caused hepatosplenomegaly and tissue inflammation in humans and some animals, including dogs (28–30). Nevertheless, the spleen is regarded as an organ for retaining and filtering infected erythrocytes rather than to be a sanctuary for persisting Bartonella, as suggested by observations of chronic B. birtlesii infection in an in vivo mouse model (31); however, the precise mechanisms of Bartonella persistence in animals are not well understood.
Previous studies suggested that genes of rodent-borne Bartonella exhibit more frequent recombination events than those of the species typically associated with humans and cats (32, 33). Since any recombination event affecting housekeeping genes will compromise the accuracy of the phylogenetic trees (34), we examined both fragments of the individual genes and multigene concatenated sequences for occurrence of recombination events. No recombination events were found upon analyzing sequences of the four individual gene fragments, suggesting that these genes are suitable for the phylogenetic analysis of binary trees. Detection of recombination events in the multigene concatenated sequences is suggestive of an artifact due to concatenation or an indication of recombination in the regions adjacent to the primer annealing sites and external to the PCR fragments (15). Since the topology of the trees remained the same upon exclusion of these putative recombined portions of the fragments, it is unlikely that the phylogenetic relationship within this group of Bartonella species is affected by their inclusion in the concatenated fragments. Furthermore, the split-tree analysis also confirmed that the topology of the binary tree tends to be a tree, not a network. Although the multigene concatenation did not influence the results of phylogenetic tree construction in the present study, considering the illusion of recombination, owing to artificial connection of the multiple locus genes, could cause an inaccurate phylogenetic estimation of taxonomic relationships.
The extent of the genetic divergence established for different Bartonella populations matched the diversity of their associated hosts present on Heixiazi Island. Genetic diversity estimated for B. grahamii on Heixiazi Island is greater than the genetic diversity established for B. taylorii from the same area (1 and 0.436); however, our findings are in contrast to the results of comparative analysis of B. grahamii and B. taylorii isolates of European and American origin (35). Furthermore, the information available for gltA fragment suggests that the genetic diversity of B. taylorii isolates (Hd = 0.971, π = 0.03881) is slightly greater than that of B. grahamii isolates (Hd = 0.961, π = 0.02241). Clear genetic diversity of gltA was demonstrated for B. grahamii upon comparison of 36 strains from Asia (Hd = 0.943, π = 0.02154) and 27 strains from Europe and North America (Hd = 0.889, π = 0.01427). The nucleotide diversity of gltA is consistent with the result of Berglund et al., who analyzed 26 strains from 11 species of wild rodents in seven countries (32, 36). Therefore, our analysis corroborates previous speculation that the European and American lineages of Bartonella strains evolved as an expansion of the genetically diverse Asian clade. Further in-depth spatial and temporal sampling with evenly distributed statistical sampling will be required to demonstrate the ecological significance and value of these observations. Multigene locus analysis targeting genes with different levels of genetic conservation at the population, species, and genus levels or whole-genome level comparisons would be ideal for such an approach, but this will require substantial computational effort and multicenter cooperation to assemble data for a representative collection of isolates.
Previous investigations of Bartonella infections of small mammals and their ectoparasites were conducted in southeast and southwest China, including Zhejiang (37), Fujian (38), and Yunnan (22, 23, 39, 40) Provinces. In the present study, we determined the prevalence of Bartonella in wild rodents under the undisturbed natural ecological conditions on an unpopulated sedimentary island at the Chinese-Russian border in northeast China. Analysis of the geographical distribution pattern of Bartonella isolates from Heixiazi Island compared to that of Bartonella from other geographic regions determined that the occurrence of various genotypes of least three species of Bartonella (B. grahamii, B. taylorii, and B. washoensis) was associated with the geographic locations of their animal hosts. The impacts of host availability at each particular geographic location were different among different species of Bartonella. Furthermore, distribution patterns of different genotypes in a given population were indeed related to the host species and appeared to have a more critical role than the geographical factors. This is confirmed by an observation that the isolates of B. taylorii and B. grahamii on Heixiazi Island were not present in the same phylogenetic clusters but instead were clustered together with the strains detected in or isolated from the same animal hosts in various geographic areas. Another example was the isolates of B. grahamii from North America which did not belong to the same cluster because of their different host origins. Obviously, the internal distribution pattern of genotypes originated from same hosts was impacted by geographic locations. For instance, it was easy to observe that all presently known isolates of B. grahamii from Apodemus spp. are split into two clades; one clade includes isolates from Asia, and another clade includes isolates from Europe and North America. Based on these observations, we speculate that hosts and environmental factors may have different effects on genetic variations observed among isolates of Bartonella spp. at the strain and species levels. Consequently, to identify the effects of geographical factors on Bartonella isolates in a given population, analysis of intra- and interspecies variations should be performed for isolates of the same genus or species of rodent hosts collected in different geographic locations.
Since Bartonella spp. are facultative intracellular pathogens, the reservoir animal hosts can be viewed as an ecological niche which captures the range of environmental conditions enabling isolated persistence of these species (41). In theory, the variation of intracellular pathogens is low, and it likely to be the first impacted by availability of hosts inhabiting specific niches within certain geographic parameters; in contrast, it may be determined by the particular effects of the infecting parasite on its vertebrate host. Therefore, for Bartonella sp. diversity, host diversity appears to play a more important role than geographic factors. This hypothesis will need further evaluation and will require statistically justified and supported sampling to ensure representative collection of the isolates.
To fulfill the rules of the International Code of Nomenclature of Bacteria (42), we provide the following descriptions of the novel Bartonella species characterized in this study.
Description of Bartonella heixiaziensis sp. nov.
Bartonella heixiaziensis (Hei.xia.zi.en.sis. N.L. gen. n. from Heixiazi Island [Bolshoi Ussuriysky Island], a sedimentary island at the confluence of the Ussuri and Amur Rivers in northeast China's Heilongjiang Province where the voles [Myodes rutilis and Microtus maximowiczii] from which the first strains were isolated were trapped).
Following inoculation on blood TSA, small, off-white, translucent, smooth, round colonies about 1 to 2 mm in diameter appeared at day 5 of incubation at 35°C in a 5% CO2 humidified atmosphere. Best growth was observed on blood-enriched solid medium or Schneider's insect medium (liquid) with 10% fetal bovine serum in a moist atmosphere containing 5% CO2. Electron microscopic examination revealed small Gram-negative bacilli without flagella or pili and approximately 1 to 2 μm long and 0.5 to 0.8 μm wide (Fig. 5A). Bacteria were oxidase and catalase negative, and exhibited no urease activity. Tests for α-glucosidase, α-l-fucosidase, α-l-arabino-furanosidase, mannose, raffinose, nitrate reduction, and spot indole were negative. Amino acidarylamidase activity was observed with the following amino acids: arginine (Arg), phenylalanine (Phe), leucine (Leu), tyrosine (Tyr), alanine (Ala), glycine (Gly), histidine (His) and, serine (Ser). This new species is distinguished from other Bartonella species by 16S rRNA gene (KJ361623), gltA (KJ175047), ftsZ (KJ361705), and rpoB (KJ361744) nucleotide sequences. The type strain CR90HXZ was isolated from the blood of a red-backed vole (Myodes rutilus) collected in May 2011; it was deposited in the Leibniz Institute DSMZ-German Collection of Microorganisms and Cell Cultures as DSM 100695 and in the China General Microbiological Culture Collection Center as CGMCC1.15048. Its closest known relative is B. washoensis as determined by molecular comparison of the four gene fragments (43).
Description of Bartonella fuyuanensis sp. nov.
Bartonella fuyuanensis (fu.yuan.en.sis. N.L. gen. n. from Fuyuan Delta, which is the ancient name for Heixiazi Island in northeast China's Heilongjiang Province, where the animals were trapped).
Following inoculation on blood TSA, small, off-white, translucent, smooth, round colonies about 1 to 2 mm in diameter appeared at day 5 of incubation at 35°C in a 5% CO2 humidified atmosphere. Best growth was observed on blood-enriched solid medium or Schneider's insect medium (liquid) with 10% fetal bovine serum in a moist atmosphere containing 5% CO2. Electron microscopic examination revealed small Gram-negative bacilli, approximately 1 to 2 μm long by 0.5 to 0.8 μm wide with polar pili (Fig. 2B). Bacteria were oxidase and catalase negative and exhibited no urease activity. Tests for α-glucosidase, α-l-fucosidase, α-l-arabinofuranosidase, mannose, raffinose, nitrate reduction, and spot indole were negative. Amino acidarylamidase activity was observed with the following amino acids: arginine (Arg), phenylalanine (Phe), leucine (Leu), tyrosine (Tyr), alanine(Ala), glycine (Gly), histidine (His), and serine (Ser). This species is distinguished from other Bartonella species by 16S rRNA gene (KJ361607), gltA (KJ175033), ftsZ (KJ361689), and rpoB (KJ361730) nucleotide sequences. The type strain AA137HXZ was isolated from the blood of a wild striped field mouse (Apodemus agrarius) collected in May 2011; it is deposited in the Leibniz Institute DSMZ-German Collection of Microorganisms and Cell Cultures as DSM 100694 and in the China General Microbiological Culture Collection Center as CGMCC1.15047. Its closest known relative is B. rattimassiliensis as determined by molecular comparison of the four gene fragments (43).
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by National Critical Project for Science and Technology on Infectious Disease grant 2012ZX10004219 and Special Funds for Public Welfare Projects grant 201310072, People's Republic of China.
We declare that no competing interests exist.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AEM.02041-15.
REFERENCES
- 1.Buffet JP, Kosoy M, Vayssier-Taussat M. 2013. Natural history of Bartonella-infecting rodents in light of new knowledge on genomics, diversity and evolution. Future Microbiol 8:1117–1128. doi: 10.2217/fmb.13.77. [DOI] [PubMed] [Google Scholar]
- 2.Huang R, Liu Q, Li G, Li D, Song X, Birtles RJ, Zhao F. 2011. Bartonella quintana infections in captive monkeys, China. Emerg Infect Dis 17:1707–1709. doi: 10.3201/eid1709.110133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Li H, Bai JY, Wang LY, Zeng L, Shi YS, Qiu ZL, Ye HH, Zhang XF, Lu QB, Kosoy M, Liu W, Cao WC. 2013. Genetic diversity of Bartonella quintana in macaques suggests zoonotic origin of trench fever. Mol Ecol 22:2118–2127. doi: 10.1111/mec.12261. [DOI] [PubMed] [Google Scholar]
- 4.Sun J, Fu G, Lin J, Song X, Lu L, Liu Q. 2010. Seroprevalence of Bartonella in eastern China and analysis of risk factors. BMC Infect Dis 10:121. doi: 10.1186/1471-2334-10-121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yuan C, Zhu C, Wu Y, Pan X, Hua X. 2011. Bacteriological and molecular identification of Bartonella species in cats from different regions of China. PLoS Negl Trop Dis 5:e1301. doi: 10.1371/journal.pntd.0001301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Liu Q, Eremeeva ME, Li D. 2012. Bartonella and Bartonella infections in China: from the clinic to the laboratory. Comp Immunol Microbiol Infect Dis 35:93–102. doi: 10.1016/j.cimid.2012.01.002. [DOI] [PubMed] [Google Scholar]
- 7.Norman AF, Regnery R, Jameson P, Greene C, Krause DC. 1995. Differentiation of Bartonella-like isolates at the species level by PCR-restriction fragment length polymorphism in the citrate synthase gene. J Clin Microbiol 33:1797–1803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Li DM, Miao ZG, Song XP, Wang J, Liu QY. 2012. Optimization of liquid growth conditions and determination of growth curves for Bartonella species. Microbiol China 39:1695–1702. [Google Scholar]
- 9.Riess T, Dietrich F, Schmidt KV, Kaiser PO, Schwarz H, Schäfer A, Kempf VA. 2008. Analysis of a novel insect cell culture medium-based growth medium for Bartonella species. Appl Environ Microbiol 74:5224–5227. doi: 10.1128/AEM.00621-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Daly JS, Worthington MG, Brenner DJ, Moss CW, Hollis DG, Weyant RS, Steigerwalt AG, Weaver RE, Daneshvar MI, O'Connor SP. 1993. Rochalimaea elizabethae sp. nov. isolated from a patient with endocarditis. J Clin Microbiol 31:872–881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zeaiter Z, Liang Z, Raoult D. 2002. Genetic classification and differentiation of Bartonella species based on comparison of partial ftsZ gene sequences. J Clin Microbiol 40:3641–3647. doi: 10.1128/JCM.40.10.3641-3647.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Renesto P, Gouvernet J, Drancourt M, Roux V, Raoult D. 2001. Use of rpoB gene analysis for detection and identification of Bartonella species. J Clin Microbiol 39:430–437. doi: 10.1128/JCM.39.2.430-437.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tamura K, Stecher G, Peterson D, Filipski A, Kumar S. 2013. MEGA6: Molecular Evolutionary Genetics Analysis version 6.0. Mol Biol Evol 30:2725–2729. doi: 10.1093/molbev/mst197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Guindon S, Dufayard JF, Lefort V, Anisimova M, Hordijk W, Gascuel O. 2010. New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst Biol 59:307–321. doi: 10.1093/sysbio/syq010. [DOI] [PubMed] [Google Scholar]
- 15.Martin DP, Lemey P, Lott M, Moulton V, Posada D, Lefeuvre P. 2010. RDP3: a flexible and fast computer program for analyzing recombination. Bioinformatics 26:2462–2463. doi: 10.1093/bioinformatics/btq467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Didelot X, Falush D. 2007. Inference of bacterial microevolution using multilocus sequence data. Genetics 175:1251–1266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Huson DH, Bryant D. 2006. Application of phylogenetic networks in evolutionary studies. Mol Biol Evol 23:254–267. [DOI] [PubMed] [Google Scholar]
- 18.Librado P, Rozas J. 2009. DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics 25:1451–1452. doi: 10.1093/bioinformatics/btp187. [DOI] [PubMed] [Google Scholar]
- 19.Polzin T, Daneshmand SV. 2001. On Steiner trees and minimum spanning trees in hypergraphs. Oper Res Lett 31:12–20. [Google Scholar]
- 20.Bandelt HJ, Forster P, Rohl A. 1999. Median-joining networks for inferring intraspecific phylogenies. Mol Biol Evol 16:37–48. doi: 10.1093/oxfordjournals.molbev.a026036. [DOI] [PubMed] [Google Scholar]
- 21.Forster P, Röhl A, Lünnemann P, Brinkmann C, Zerjal T, Tyler-Smith C, Brinkmann B. 2000. A short tandem repeat-based phylogeny for the human Y chromosome. Am J Hum Genet 67:182–196. doi: 10.1086/302953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li DM, Yu DZ, Liu QY, Gong ZD. 2004. Study on the prevalence of Bartonella species in rodent hosts from different environmental areas in Yunnan. Zhonghua Liu Xing Bing Xue Za Zhi 25:934–937. [PubMed] [Google Scholar]
- 23.Bai HM, Yang FL, Yang H, Zhang Q. 2005. Study on Bartonella species in rodents in western Yunnan, China. Zhonghua Liu Xing Bing Xue Za Zhi 26:868–870. [PubMed] [Google Scholar]
- 24.Song XP, Liu QY, Lu L, Zhao W, Li G-C, Li D-M, Sun JM. 2010. Isolation and sequence analysis of Bartonella in small mammals in Hainan province. Chin J Vect Biol Control 21:131–133. [Google Scholar]
- 25.Inoue K, Kabeya H, Hagiya K, Kosoy MY, Une Y, Yoshikawa Y, Maruyama S. 2011. Multi-locus sequence analysis reveals host specific association between Bartonella washoensis and squirrels. Vet Microbiol 148:60–65. doi: 10.1016/j.vetmic.2010.08.007. [DOI] [PubMed] [Google Scholar]
- 26.Zhang XL, Zhao ZJ, Li M, Guo XM, Yang J, Qu Y-M, Li Y-J, Wen ZQ, Hu K-X, Sun XH. 2011. Survey of rodent diversity in different habitats in the central border area of China and Russia. Chin J Hyg Insect Equip 17:34–36. [Google Scholar]
- 27.Chiaraviglio L, Duong S, Brown DA, Birtles RJ, Kirby JE. 2010. An immunocompromised murine model of chronic Bartonella infection. Am J Pathol 176:2753–2763. doi: 10.2353/ajpath.2010.090862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Liston TE, Je K. 1996. Granulomatous hepatitis and necrotizing splenitis due to Bartonella henselae in a patient with cancer: case report and review of hepatosplenic manifestations of Bartonella infection. Clin Infect Dis 22:951–957. doi: 10.1093/clinids/22.6.951. [DOI] [PubMed] [Google Scholar]
- 29.Klotz SA. 2011. Cat-scratch disease. Am Fam Physician 83:152–155. [PubMed] [Google Scholar]
- 30.Hutchins RG, Breitschwerdt EB, Cullen JM, Bissett SA, Gookin JL. 2012. Limited yield of diagnoses of intrahepatic infectious causes of canine granulomatous hepatitis from archival liver tissue. J Vet Diagn Invest 24:888–894. doi: 10.1177/1040638712453583. [DOI] [PubMed] [Google Scholar]
- 31.Deng HK, Le Rhun D, Lecuelle B, Le Naour E, Vayssier-Taussat M. 2012. Role of the spleen in Bartonella spp. infection. FEMS Immunol Med Microbiol 64:143–145. doi: 10.1111/j.1574-695X.2011.00908.x. [DOI] [PubMed] [Google Scholar]
- 32.Berglund EC, Ellegaard K, Granberg F, Xie Z, Maruyama S, Kosoy MY, Birtles RJ, Sg A. 2010. Rapid diversification by recombination in Bartonella grahamii from wild rodents in Asia contrasts with low levels of genomic divergence in Northern Europe and America. Mol Ecol 19:2241–2255. doi: 10.1111/j.1365-294X.2010.04646.x. [DOI] [PubMed] [Google Scholar]
- 33.Paziewska A, Siński E. 2011. Recombination within and between species of the alpha Proteobacterium Bartonella infecting rodents. Microb Ecol 61:134–145. doi: 10.1007/s00248-010-9735-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Posada D, Ka C. 2002. The effect of recombination on the accuracy of phylogeny estimation. J Mol Evol 54:396–402. doi: 10.1007/s00239-001-0034-9. [DOI] [PubMed] [Google Scholar]
- 35.Buffet JP, Pisanu B, Brisse S, Roussel S, Félix B, Halos L, Chapuis JL, Vayssier-Taussat M. 2013. Deciphering Bartonella diversity, recombination, and host specificity in a rodent community. PLoS One 8:e68956. doi: 10.1371/journal.pone.0068956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Birtles RJ, Harrison TG, Molyneux DH. 1994. Grahamella in small woodland mammals in the U.K.: isolation, prevalence and host specificity. Ann Trop Med Parasitol 88:317–327. [DOI] [PubMed] [Google Scholar]
- 37.Liu Q, Sun J, Lu L, Fu G, Ding G, Song X, Meng F, Wu H, Yang T, Ren Z. 2010. Detection of Bartonella species in small mammals from Zhejiang Province, China. J Wildl Dis 46:179–185. doi: 10.7589/0090-3558-46.1.179. [DOI] [PubMed] [Google Scholar]
- 38.Ye X, Li GW, Yao ML, Luo W, Su LQ. 2009. Study on the prevalence and genotypes of Bartonella species in rodent hosts from Fujian coastal region. Zhong Hua Liu Xing Bing Xue Za Zhi 30:989–992. [PubMed] [Google Scholar]
- 39.Ying B, Kosoy MY, Maupin GO, Tsuchiya KR, Gage KL. 2002. Genetic and ecologic characteristics of Bartonella communities in rodents in southern China. Am J Trop Med Hyg 66:622–627. [DOI] [PubMed] [Google Scholar]
- 40.Li DM, Liu QY, Yu DZ, Zhang JZ, Gong ZD, Song XP. 2007. Phylogenetic analysis of Bartonella detected in rodent fleas in Yunnan, China. J Wildl Dis 43:609–617. doi: 10.7589/0090-3558-43.4.609. [DOI] [PubMed] [Google Scholar]
- 41.Casadevall A. 2008. Evolution of intracellular pathogens. Annu Rev Microbiol 62:19–33. doi: 10.1146/annurev.micro.61.080706.093305. [DOI] [PubMed] [Google Scholar]
- 42.Lapage SP, Sneath PHA, Lessel EF, Skerman VBD, Seeliger HPR, Clark WA (ed). 1992. International code of nomenclature of bacteria: bacteriological code, 1990 revision. ASM Press, Washington, DC. [PubMed] [Google Scholar]
- 43.La Scola B, Zeaiter Z, Khamis A, Raoult D. 2003. Gene-sequence-based criteria for species definition in bacteriology: the Bartonella paradigm. Trends Microbiol 11:318–321. doi: 10.1016/S0966-842X(03)00143-4. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.