

Abstract
Meat and meat products are important sources of human intestinal infections. We report the isolation of Helicobacter pullorum strains from chicken meat. Bacteria were isolated from 4 of the 17 analyzed fresh chicken meat samples, using a membrane filter method. MIC determination revealed that the four strains showed acquired resistance to ciprofloxacin; one was also resistant to erythromycin, and another one was resistant to tetracycline. Whole-genome sequencing of the four strains and comparative genomics revealed important genetic traits within the H. pullorum species, such as 18 highly polymorphic genes (including a putative new cytotoxin gene), plasmids, prophages, and a complete type VI secretion system (T6SS). The T6SS was found in three out of the four isolates, suggesting that it may play a role in H. pullorum pathogenicity and diversity. This study suggests that the emerging pathogen H. pullorum can be transmitted to humans by chicken meat consumption/contact and constitutes an important contribution toward a better knowledge of the genetic diversity within the H. pullorum species. In addition, some genetic traits found in the four strains provide relevant clues to how this species may promote adaptation and virulence.
INTRODUCTION
Infectious diarrhea is a major cause of morbidity and mortality throughout the world, particularly in children (1). Besides the most commonly associated pathogens, such as Salmonella enterica, Campylobacter spp., and Shiga toxin-producing Escherichia coli, infectious intestinal pathogens also include Helicobacter species. Among these, Helicobacter pullorum has been detected in poultry as well as in human samples (2, 3).
H. pullorum is a Gram-negative, gently curved rod, with an unsheathed monopolar flagellum (4). This enterohepatic Helicobacter species was initially isolated from the liver, duodenum, and cecum of asymptomatic poultry, but it has also been associated with enteritis and vibrionic hepatitis in broiler chickens and laying hens (4). In poultry, H. pullorum was found to colonize the cecum at high concentrations, as well as to be present on poultry carcasses, possibly due to contamination during slaughtering. Therefore, the potential role of this bacterium as an emerging foodborne human pathogen needs to be considered (3, 5–7). In humans, H. pullorum is considered an emergent agent implicated in several digestive pathologies, such as gastroenteritis (2) and chronic inflammatory conditions of the intestine (8, 9) and liver (for a review, see reference 10 and references cited therein). However, the lack of available detection tools and the difficulties in cultivating this fastidious organism result in an underdetection of the pathogen and underdiagnosis of the associated infection. Despite its zoonotic potential, there is no proof to date that one can acquire H. pullorum infection by the handling and consumption of raw or undercooked broiler chicken meat.
In the present work, we report the isolation of H. pullorum from chicken meat and present antimicrobial susceptibility data and genomic features of four isolates, supporting the view that the foodborne bacterium H. pullorum exhibits traits that may play a role in adaptation and virulence.
MATERIALS AND METHODS
Food samples, isolation, and identification.
Meat samples were collected from 15 different producers, in the context of the Portuguese Inspection Plan for Surveillance and Monitoring of Foodstuffs, comprising seven regions, distributed as follows: North (one producer, one region), Centre (six producers, two regions), Lisbon Metropolitan Area (six producers, two regions), and South (two producers, two regions). The samples were processed according to the ISO 10272-1:2006 protocol for plating, which specifies the use of an alternative plating medium (11). For each meat sample, 25 g of fresh meat was homogenized in a stomacher, and 225 ml of Bolton broth (Bolton base medium CM0983, selective supplement SR0183E, and lysed horse blood SR0048C; Oxoid, Hampshire, United Kingdom) was added, for selective preenrichment. The samples were incubated at 37 ± 1°C for 4 to 6 h, followed by 44 ± 4 h at 41.5 ± 1°C, under microaerophilic conditions with hydrogen (Anoxomat; MART Microbiology BV, Drachten, The Netherlands). Then, 100 μl of the mixture was applied onto a 0.65-μm cellulose filter membrane (Advantec MFS, CA, USA) and placed directly on Columbia agar with 5% sheep blood (bioMérieux, Marcy l'Etoile, France). This plate was incubated aerobically at room temperature for 15 min; the membrane was then removed, and the plate was incubated under a hydrogen-enriched microaerophilic atmosphere for 44 ± 4 h at 41.5 ± 1°C.
Five colonies from each colony type were chosen from each plate, tested by Gram stain and oxidase test, and then subcultured on blood agar plates if they were Gram negative, oxidase positive, and spiral shaped. Colonies that were not characteristic of Campylobacter spp. were identified by 16S rRNA gene amplification followed by Sanger sequencing, as previously described (12), using the universal forward 27F (5′-AGAGTTTGATCMTGGCTCAG-3′) and reverse 1492R (5′-TACGGYTACCTTGTTACGACTT-3′) primers (13).
Antibiotic susceptibility testing.
MICs of three antimicrobial agents, namely, ciprofloxacin, erythromycin, and tetracycline, were determined using Etest strips. Briefly, bacterial suspensions (adjusted to an opacity equivalent to an 0.5 McFarland opacity standard) were inoculated on Mueller-Hinton agar supplemented with 5% defibrinated horse blood (Oxoid, Basingstoke, Hants, United Kingdom) and 20 mg/liter β-NAD (Sigma-Aldrich, St. Louis, MO) and incubated under a microaerobic hydrogen-enriched atmosphere, at 41 ± 1°C for 48 h. Besides the isolates collected in this study, six additional H. pullorum clinical strains, presumably susceptible to ciprofloxacin, were included. These strains had been isolated from stool samples of patients suffering from gastroenteritis and are designated as follows: 2007-1794, 2008-547, 2010-1828, 2012-216H, 2014-133H, and 2014-1734 (the isolation year is used as the first number in the strain identifications).
Whole-genome sequencing (WGS).
Total DNA was extracted using the QIAmp DNA minikit (Qiagen) according to the manufacturer's instructions. The yield and quality of the purified DNA were assessed with a Qubit assay (Quant-iT double-stranded DNA [dsDNA] assay kit, broad range; Life Technologies, MA, USA) and agarose gel (0.7%) electrophoresis. High-quality DNA samples were then selected to prepare sequencing libraries using the Nextera XT DNA sample preparation kit. Library samples were subjected to cluster generation and paired-end sequencing (2 by 250 bp) on a MiSeq sequencer (Illumina Inc., San Diego, CA, USA), according to the manufacturer's instructions. The number of passing filter reads obtained per sample ranged from 1.0 to 1.9 million. The FastQC (http://www.bioinformatics.babraham.ac.uk/projects/fastqc/) and FASTX (http://hannonlab.cshl.edu/fastx_toolkit/) software tools were applied to evaluate and improve the quality of the raw sequence data, respectively. Subsequently, high-quality reads were assembled de novo using Velvet (version 1.2.10) (14) (several assemblies using different k-mer sizes were run), where the best assembly was assumed as the one with the best cumulative rank for N50, the number of contigs/scaffolds, and the length of the largest contig/scaffold. The obtained mean depth of coverage per sample ranged from 135- to 195-fold. The final contigs/scaffolds were visually inspected (using Tablet 1.14.04.10) (15) and properly corrected prior to annotation using the NCBI Prokaryotic Genomes Annotation Pipeline 2.3.
Comparative genomic analyses.
With the exception of the four genomes sequenced in the present study, only the draft genome sequence of the human MIT 98-5489 H. pullorum isolate (16) is currently available (GenBank accession no. ABQU00000000.1). Therefore, in order to achieve a wide picture of the genetic diversity within the H. pullorum species, this genome was included in all comparative genomic analyses. First, the 5 draft sequences were aligned using the progressive algorithm of MAUVE software (version 2.3.1) (17), and a core alignment was extracted by keeping and concatenating regions where genomes aligned over at least 500 bp. This core genome alignment, encompassing about 70 to 83% of each genome sequence, was subsequently applied to identify genomic regions with high single nucleotide polymorphism (SNP) density using DnaSP v5 software (18), with a window and step sizes of 1,000 bp. MEGA5 software (19) (http://www.megasoftware.net) was also used to estimate overall genetic distances among the H. pullorum genomes. Individual alignments of highly polymorphic genes and genes with SNPs likely to be associated with antibiotic resistance were further analyzed using MEGA5 to confirm annotation, homology, and polymorphism. Identification of positional homologous coding sequences (CDSs) was facilitated by using MAUVE, assuming a minimum of 60% nucleotide identity and a minimum of 70% length coverage. DnaSP and SWAAP (version 1.0.3) software analyses were further applied to evaluate the variability within relevant CDSs. The major differences in the accessory genome, particularly in regions showing strain specificity, were also studied. The identification of putative plasmids was performed by checking contigs (minimum size, 1,000 bp) with high depth of coverage and evidence of circularity. The plasmid copy numbers were estimated by comparing the mean plasmid sequence coverage to that of the remaining contigs/scaffolds. Large insertions and deletions (indels) were identified following both the inspection of the positional homolog in MAUVE and nucmer analyses using MUMmer 3.23 (minimum homology of 75% and a stretch window of 100 bp) (http://www.tigr.org/software/mummer) (20). For confirmation purposes, reads from the H. pullorum isolates collected in this study were additionally mapped using Bowtie2 (version 2.1.0 [http://bowtie-bio.sourceforge.net/bowtie2/index.shtml]) (21) against all genomic regions that displayed strain specificity or were absent in particular strains. BLASTn (http://blast.be-md.ncbi.nlm.nih.gov/Blast.cgi) and PHAST (http://phast.wishartlab.com/) analyses (22) were additionally applied over these regions to check homology and to identify, annotate, and graphically display prophage sequences, respectively.
Nucleotide sequence accession numbers.
Newly determined sequence data were deposited in GenBank under accession numbers JNOB00000000, JNOC00000000, JNUR00000000, and JNOA00000000.
RESULTS
H. pullorum isolation, identification, and antibiotic susceptibility.
We isolated H. pullorum from four fresh raw chicken meat samples out of 17 analyzed. These four samples originated from three different producers, two located in the Centre region and one in the South of Portugal. Three strains were isolated from refrigerated chicken carcass meat, and the fourth was isolated from seasoned-meat preparations intended to be cooked before consumption (Table 1). The small gray-white colonies grown were different from Campylobacter species. Subculturing and Gram staining revealed small curved Gram-negative bacilli identified as H. pullorum by 16S rRNA gene sequencing. WGS was carried out for further analysis of comparative genomics. The draft genome sequence of the H. pullorum MIT 98-5489 strain (16) isolated in Canada from the stools of a patient suffering from gastroenteritis (corresponding to strain H436 in reference 23) was included.
TABLE 1.
Origin, genome characteristics, and antimicrobial susceptibility of the four Helicobacter pullorum strains isolated from chicken meat and comparison with the human clinical isolate H436 (MIT 98-5489)
| Isolate name | Origin | Genomic data |
Antibiotic resistance-associated SNPb |
Antimicrobial susceptibility (MIC, mg/liter) |
|||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Accession no. | Genome size (Mb) | GC content (%) | No. of putative plasmids | Fluoroquinolone; GyrA, Thr84Ile mutation | Macrolide; 23S rRNA gene,c AAA2055-2057GAA mutation | Tetracycline; 16S rRNA gene,c transversion AGA925-927TGA | Ciprofloxacin | Erythromycin | Tetracycline | ||
| 229313/12d | Refrigerated chicken carcass meat | JNOB00000000 | 1.69 | 34.56 | 0 | Yes | No | No | >32 | 0.25 | 0.25 |
| 229334/12 | Seasoned-meat preparations | JNOC00000000 | 2.13 | 33.92 | 0 | Yes | No | No | >32 | 0.125 | 0.5 |
| 229336/12 | Refrigerated chicken carcass meat | JNUR00000000 | 1.81 | 34.20 | 1 | No | Yes | No | >32 | >256 | 0.38 |
| 229254/12 | Refrigerated chicken carcass meat | JNOA00000000 | 1.81 | 34.23 | 2 | Yes | No | Yes | >32 | 0.25 | >256 |
| MIT 98-5489a | Human, stool, gastroenteritis | ABQU00000000.1 | 1.95 | 34.15 | 0 | Yes | Yes | Yes | NAe | NA | NA |
H. pullorum strain MIT 98-5489 was isolated in Canada from the stools of a patient suffering from gastroenteritis and sequenced by Shen et al. (16).
Numbering according to the annotated genomes.
All three operons of the 23S and 16S rRNA genes harbored the SNPs.
A complete 36.4-kb prophage is integrated in the bacterial genome of this strain.
NA, not available.
The MIC values of the H. pullorum isolates collected in this study are shown in Table 1. For all four isolates, the MIC of ciprofloxacin was >32 mg/liter, and for three of those, the functional ACA→ATA substitution previously reported was detected in gyrA (24). This mutation was also detected in the human MIT 98-5489 strain (16). Missense mutations in gyrA reported to confer resistance to ciprofloxacin in other enterohepatic Helicobacter species were not detected in any of the four strains (16). The six additional H. pullorum strains isolated from patients exhibited a low range of MICs of ciprofloxacin (0.023 to 0.047 mg/liter), and none of these strains harbored the ACA→ATA transition in gyrA. In addition, no distinctive mutations were found in gyrB when comparing the group with low MIC values of ciprofloxacin (GenBank accession numbers of gyrB: KR014216, KR014217, KR014218, KR014219, KR014220, and KR014221) with the group of the four strains isolated in this study presenting with high MIC values. Neither the topoisomerases IV encoded by parC and parE nor the plasmid-mediated quinolone resistance gene, qnr, was found in the four genomes of the meat isolates and in the human strain MIT 98-5489.
For erythromycin, only strain 229336/12 had a high MIC (>256 mg/liter), and the transition A2075G in the 23S rRNA gene associated with macrolide resistance was detected in this strain (25). Concerning tetracycline, only strain 229254/12 presented a MIC of >256 mg/liter, and it presented a single transversion, A926T, in the 16S rRNA gene. Both these mutations were detected in the strain MIT 98-5489 but in none of the meat isolates presenting low MICs of erythromycin and tetracycline (Table 1) (16).
Core genome analysis.
Draft H. pullorum genomes were extrapolated/estimated to vary from 1.7 Mb to 2.1 Mb, with a G+C content of ∼34% (Table 1). By comparing the mean depth of coverage obtained for the rRNA genes with the one calculated for single-copy genes (e.g., gyrA), we confirmed that H. pullorum harbors three copies of the ribosomal operon (26).
The core genomes were highly similar (>98%), with a mean number of 22,031.5 (standard error [SE], ±91.9) nucleotide differences, although marked by highly polymorphic regions (named A to R in Fig. 1 and Table 2). Eighteen polymorphic genes were found to belong to different functional categories, like metabolic enzymes, outer membrane proteins (OMP), and restriction modification systems.
FIG 1.

Analysis of polymorphism in the Helicobacter pullorum core genome. The graph shows the SNP density throughout a core genome alignment encompassing about 70 to 83% of each draft genome sequence under evaluation. An SNP density threshold of 100/1,000 was defined to highlight the most polymorphic regions among the H. pullorum strains studied. Genes found to be highly polymorphic are labeled with letters and listed in Table 2. Nonlabeled peaks refer to artifacts associated with the random junction of noncontiguous blocks (see Materials and Methods for details).
TABLE 2.
Highly polymorphic genes in Helicobacter pullorum strains isolated from chicken meat
| Label | Locus taga | Gene product |
p-distance |
|
|---|---|---|---|---|
| ntd | aae | |||
| A | HPU229254_06545 | Dihydroxy acid dehydratase (ilv) | 0.081 | 0.050 |
| B | HPU229254_07315 | Putative outer membrane protein | 0.052 | 0.052 |
| C | HPU229254_07240b | Outer membrane protein | 0.110 | 0.133 |
| D | HPU229254_09065 | Antibiotic resistance protein (pyridoxamine 5′-phosphate oxidase related) | 0.112 | 0.108 |
| E | HPU229254_01025 | TonB-dependent receptor | 0.103 | 0.092 |
| F | HPU229254_01370 | Outer membrane phospholipase A family protein/porin | 0.099 | 0.102 |
| G | HPU229254_01245 | Putative maltose O-acetyltransferase | 0.088 | 0.076 |
| H | HPU229254_03825 | 1-Deoxy-d-xylulose 5-phosphate reductoisomerase (dxr) | 0.107 | 0.097 |
| I | HPU229254_03755c | Cytotoxin | 0.208 | 0.280 |
| J | HPU229254_03030 | TonB-dependent receptor | 0.156 | 0.164 |
| K | HPU229254_04335 | Modification methylase | 0.078 | 0.062 |
| L | HPU229254_02485 | Arginase | 0.068 | 0.057 |
| M | HPU229254_03115 | Transporter/outer membrane efflux protein | 0.179 | 0.186 |
| N | HPU229254_03120 | Transporter/putative efflux system membrane fusion protein | 0.096 | 0.093 |
| O | HPU229254_04745 | Asparagine synthase | 0.031 | 0.023 |
| P | HPU229254_05270 | Homoserine O-succinyltransferase (MetA) | 0.057 | 0.034 |
| Q | HPU229254_05315 | Hypothetical protein | 0.163 | 0.183 |
| R | HPU229254_06390 | Type II restriction modification enzyme/restriction endonuclease | 0.132 | 0.176 |
The locus designations are based on a draft genome annotation of the 229254/12 strain (GenBank accession no. JNOA00000000).
The corresponding gene names for the strains 229313/12, 229334/12, 229336/12, and MIT 98-5489 are HPU229313_06420, HPU229334_02340, HPU229336_08255, and HPMG_00190, respectively.
The corresponding gene names for the strains 229313/12, 229334/12, 229336/12, and MIT 98-5489 are HPU229313_03315, HPU229334_07720, HPU229336_02900, and HPMG_01076, respectively.
nt, nucleotide.
aa, amino acid.
A more detailed SNP analysis of the identified polymorphic OMP-encoding gene (label C, Fig. 1 and Table 2), varying in length from 1,407 to 1,488 bp, showed a particular pattern of variability, with 7 localized variable regions interspersed with highly conserved sequences throughout the gene (Fig. 2A), similarly to the Campylobacter major protein (cmp) gene encoding the major outer membrane protein (MOMP) of Campylobacter jejuni (27). The high density of nonsynonymous substitutions observed in the variable regions of this OMP-encoding gene (Fig. 2B) suggests that they are likely to correspond to surface-exposed regions of the OMP.
FIG 2.
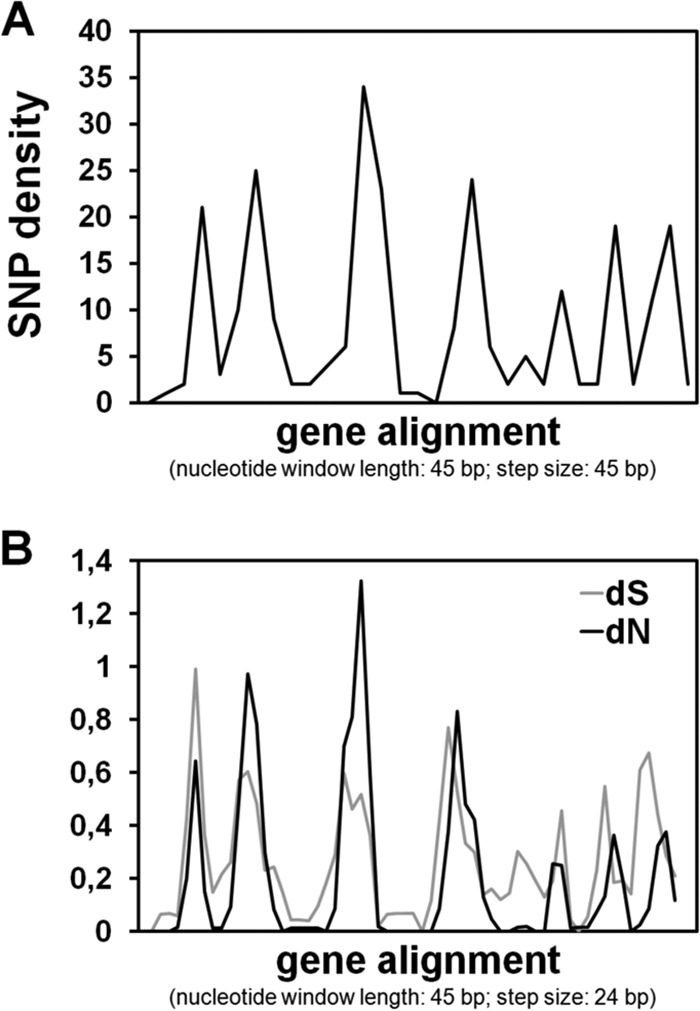
Sequence variation within the gene encoding the outer membrane protein HPU229254_07240. (A) SNP density (calculated through DnaSP) based on the nucleotide alignment of the sequences of the five H. pullorum strains under study. (B) Analysis of dN (number of nonsynonymous substitutions per nonsynonymous site) and dS (number of synonymous substitutions per synonymous site) over the same alignment (using SWAAP). Step size was adjusted according to software specificities and to improve visualization.
Other genes were highly polymorphic (Fig. 1 and Table 2), such as a gene encoding a protein belonging to the pyridoxamine 5′-phosphate oxidase-related family (label D), as well as a gene coding for a putative cytotoxin (label I) which was highly variable in length (from 2,649 to 2,817 bp) among the four H. pullorum isolates and the strain MIT 98-5489. This cytotoxin presented the highest variability at both nucleotide and protein levels (p-distances of 0.208 and 0.280, respectively), which was mostly concentrated within the 5′ end and the middle region of the gene. In silico analysis of functional domains showed that the conserved 3′ region shows features of an autotransporter β-domain (residues 592 to 882, according to the numbering of the HPMG_01076 gene of strain MIT 98-5489), with a putative signal peptide including a cleavage site (data not shown). The cytotoxin may belong to a gene family since a total of four putative paralog genes were found in the genome of all four H. pullorum strains studied, as well as in strain MIT 98-5489.
BLAST searches against the recently described genomes of other Helicobacter species (16) revealed the presence of protein homologs to both the Campylobacter-like MOMP and the cytotoxin for the species Helicobacter canadensis (strain NCTC 13241; MIT 98-5491) (blastp; query cover, 100%; identity, >76%; E value, 0, for both proteins; GenBank accession numbers WP_006656911.1 and WP_006656715.1, respectively) and “Helicobacter winghamensis” (strain ATCC BAA-430) (blastp; query cover, >98%; identity, >42%; E value, <1e−168, for both proteins; GenBank accession numbers WP_006801784.1 and WP_006802875, respectively).
Accessory genome analysis.
The analysis of the accessory genome of H. pullorum showed several genomic regions which were highly variable in length and with G+C contents deviating from the genome average (minimum, 29.42%; maximum, 35.26%) (Table 3). These include (i) two putative cryptic plasmids present in strain 229254/12 and one putative conjugative plasmid present in strain 229336/12, (ii) several strain-specific regions with contiguous CDSs coding for hypothetical proteins, (iii) a complete 36.4-kb prophage integrated in the bacterial genome of strain 229313/12, and (iv) a putative type VI secretion system (T6SS), present in all strains except for 229313/12.
TABLE 3.
Most relevant genetic differences in accessory genome of Helicobacter pullorum
| Isolate | Contig | Contig position (bp) | Predicted length (bp) | % G+C | Observation |
|---|---|---|---|---|---|
| 229254/12 | 55 | 5,149 | 30.73 | Putative plasmid (probably more than 10 copies per chromosome) | |
| 229254/12 | 88 | 4,149 | 33.21 | Putative plasmid (probably more than 10 copies per chromosome) | |
| 229313/12 | 7 | 94358–130742 | 36,385 | 35.26 | PHAST result: intact prophage |
| 229313/12 | 29 | 30609–41976 | 11,368 | 29.42 | Remnant prophage? BLAST results: Campylobacter jejuni putative prophages; no significant PHAST result |
| 229313/12 | 48 | 9,097 | 30.89 | Predicted annotation includes several hypothetical proteins | |
| 229334/12 | 58 | 1–7656 | 7,656 | 32.84 | Predicted annotation includes several hypothetical proteins |
| 229334/12 | 102 | 7,140 | 33.33 | Predicted annotation includes several hypothetical proteins | |
| 229334/12 | 103 | 13,867 | 29.96 | Predicted annotation includes several hypothetical proteins | |
| 229334/12 | 107 | 15,702 | 31.75 | Predicted annotation includes several hypothetical proteins | |
| 229334/12 | 111 | 9,151 | 31.56 | Predicted annotation includes several hypothetical proteins | |
| 229334/12 | 114 | 8,140 | 32.94 | Predicted annotation includes several hypothetical proteins | |
| 229336/12 | 46 | 24,483 | 29.48 | Putative conjugative plasmid (probably fewer than two copies per chromosome); BLAST results: Campylobacter jejuni putative plasmids and integrative conjugative elements | |
| All but 229313/12 | 1a | 25,000–27,000 | 35.12 | Type VI secretion system locus (Fig. 3) |
The T6SS genetic cluster is located in the contig “1” for all strains, except for strain 229334/12, where it is fragmented in more than one contig (contigs 1, 2, and 3).
The prophage sequence in strain 229313/12 revealed a G+C content of 35.54% and 47 predicted CDSs, representing 33,266 bp. The sequence showed homology with phage sequences found in several bacterial genera, including a predicted integrase, a transposase, and tail proteins. The last are intercalated with sequences coding for putative proteins of bacterial origin of related species, such as C. jejuni, Arcobacter butzleri, and Helicobacter hepaticus, and other DNA-associated open reading frames (ORFs) whose functions could not be predicted. The strain 229313/12 also showed another exclusive sequence of 11 kbp, most likely corresponding to a remnant prophage, with homology to C. jejuni prophages.
The T6SS genetic cluster was well conserved in three out of the four chicken meat isolates and strain MIT 98-5489, comprising 13 ORFs that encode T6SS predicted proteins, including the T6SS hallmark genes: hemolysin-coregulated protein (hcp) and valine glycine repeat G (vgrG) genes (Fig. 3 and Table 4). The H. pullorum T6SS genetic cluster was quite similar in organization to the C. jejuni T6SS, except that in the latter, the 13 T6SS genes are contiguous, whereas in H. pullorum, the T6SS genes are organized in three blocks (two of which are inverted, related to the C. jejuni gene organization) which are intercalated with genes encoding mostly proteins with no assigned function. The central block contains most of the T6SS genes. Among the extreme blocks, one is comprised of the genes fha, vasK, and hcp whereas the other (the noninverted block) corresponds to the vgrG gene (Fig. 3). It is important to note that all 13 T6SS-associated proteins display a high level of similarity between the two species (Table 4).
FIG 3.

Genetic organization of the T6SS cluster in Helicobacter pullorum. The T6SS gene order and content are displayed for Helicobacter pullorum strains and also for Campylobacter jejuni strain 414 (top row), for comparative purposes. Genes are plotted as arrows (size proportional to gene length) whose direction indicates the chromosomal strand. Genes encoding proteins related to the T6SS are in black. Genes located in regions displaying variability in the genetic content among Helicobacter pullorum strains (highlighted by gray dashed lines) are in gray. The gene organization of the T6SS for strain 229334/12 is not shown because assembly issues yielded the T6SS genes in more than one contig. However, the draft genome of this strain carries all of the genes related to the T6SS.
TABLE 4.
Type VI secretion system in Helicobacter pullorum strains isolated from chicken meata
| Label | Locus tag for strain: |
Predicted protein/putative domains | BLASTp against C. jejuni 414b |
||||
|---|---|---|---|---|---|---|---|
| MIT 98-5489 | 229254/12 | 229336/12 | Cover (%) | Identity (%) | E value | ||
| A | HPMG_00002 | HPU229254_00020 | HPU229336_00010 | Flagellar hook-associated protein (FlgK) | |||
| B | HPMG_00003 | HPU229254_00030 | HPU229336_00020 | Predicted T6SS component/FHA domain | 98 | 52 | 3e−93 |
| C | HPMG_00004 | HPU229254_00035 | HPU229336_00025 | T6S protein IcmF/VasK | 64 | 64 | 0 |
| D | HPMG_00005 | HPU229254_00040 | HPU229336_00030 | T6S effector Hcp1 family/T6SS component Hcp (secreted cytotoxin) | 100 | 82 | 8e−111 |
| E | HPMG_00006 | HPU229254_00045 | —c | Acetyltransferase (GNAT) family | |||
| F | HPMG_00007 | HPU229254_00050 | HPU229336_00045 | Helicase/NYN domain | |||
| G | HPMG_00008 | HPU229254_00055 | Helicase | ||||
| H | HPMG_00009 | HPU229254_00060 | HPU229336_00055 | Hypothetical protein | |||
| I | HPMG_00010 | HPU229254_00070 | HPU229336_00060 | T6S protein ImpH/VasB | 100 | 61 | 6e−132 |
| J | HPMG_00011 | HPU229254_00075 | HPU229336_00065 | T6S protein ImpG/VasA | 100 | 63 | 0 |
| K | HPMG_00012 | HPU229254_00080 | HPU229336_00070 | T6SS lysozyme-like protein | 98 | 61 | 1e−23 |
| L | HPMG_00013 | HPU229254_00085 | HPU229336_00075 | T6S protein ImpC | 100 | 83 | 0 |
| M | HPMG_00014 | HPU229254_00090 | HPU229336_00080 | T6S protein ImpB | 83 | 77 | 7e−72 |
| N | HPMG_00015d | HPU229254_00095 | HPU229336_00085 | T6S ImpA-related N-terminal protein | 74 | 41 | 5e−72 |
| O | HPMG_00016 | HPU229254_00100 | HPU229336_00090 | T6S lipoprotein VasD/SciN | 80 | 69 | 2e−58 |
| P | HPMG_00017 | HPU229254_00105 | HPU229336_00095 | T6S protein ImpJ/VasE | 99 | 67 | 0 |
| Q | HPMG_00018 | HPU229254_00110 | HPU229336_00100 | T6S protein ImpK/VasF; OmpA/MotB domain | 81 | 62 | 6e−84 |
| R | HPMG_00019 | HPU229254_00115 | HPU229336_00105 | Hypothetical protein/restriction endonuclease-like superfamily | |||
| S | HPMG_00020 | —e | HPU229336_00110 | Hypothetical protein/phosphatidylinositol-4-phosphate 5-kinase | |||
| T | HPMG_00021 | —e | HPU229336_00115 | Hypothetical protein | |||
| U | HPMG_00022 | Hypothetical protein | |||||
| V | HPMG_00023 | Hypothetical protein | |||||
| W | HPMG_00024 | HPU229254_00125 | HPU229336_00120 | T6SS VgrG protein/phage late control gene D protein | 92 | 47 | 0 |
| X | HPMG_00025 | HPU229254_00130 | HPU229336_00125 | Hypothetical protein | |||
| Y | HPMG_00026 | HPU229254_00135 | HPU229336_00130 | Hypothetical protein/predicted ATP-dependent endonuclease of the OLD family | |||
| Z | HPMG_00027 | HPU229254_00140 | HPU229336_00135 | Hypothetical protein | |||
T6SS genes are highlighted in bold.
BLASTp result (nonredundant protein sequence, blastp) of the T6SS proteins from Helicobacter pullorum against the genome of Campylobacter jejuni subsp. jejuni 414 (GenBank accession number NZ_CM000855) described in reference 41.
The 229336/12 strain has two putative ORFs (HPU229336_00035 and HPU229336_00040) in this region.
This region has annotation issues in the genome of the strain MIT 98-5489.
The 229254/12 strain has a putative gene fusion (HPU229254_00120) encompassing the ORFs HPMG_00020 and HPMG_00021.
DISCUSSION
The present study reports the isolation of H. pullorum from fresh raw chicken meat. Although H. pullorum is present in poultry, representing a risk to human health, and has been isolated in cases of human intestinal and liver disease, it has never been identified or cultured on poultry meat before. The finding of H. pullorum in four out of 17 chicken meat samples for human consumption suggests that this organism may pose a risk of zoonotic foodborne transmission, as is known to occur with Campylobacter sp., which is presently recognized as a major human enteropathogen worldwide. In support of this, a few studies using PCR reported high (although variable) frequencies of H. pullorum, ranging from 33.6% (28) to 100% (6, 26) in poultry cecal samples and 37% in chicken meat products (7). Presently, the difficulty in culturing this fastidious microorganism and the use of inappropriate culture media and conditions, in clinical, veterinary, and food laboratories, have hampered the correct establishment of H. pullorum as a foodborne pathogen. In this study, we demonstrate the usefulness of a membrane filter method for isolating H. pullorum from meat samples, as was previously described for poultry cecal samples (6). The application of this strategy may also be highly valuable for the accurate and sensitive detection/isolation of Campylobacter and Helicobacter species in those laboratory settings.
The four H. pullorum isolates collected in this study were tested for antibiotic susceptibility to the antibiotics commonly used to treat Campylobacter and Helicobacter infections. According to previous data on in vitro susceptibility of H. pullorum to antimicrobial agents, we may assume that all strains were resistant to fluoroquinolones; one isolate was also resistant to erythromycin, which is the first-choice antibiotic against Campylobacterales infections; and another was resistant to tetracycline (6, 24, 29). The MIC values of the resistant isolates were all high, suggesting a high level of resistance to those three antibiotics. Although to date there are no studies on antibiotic susceptibility comprising a large number of H. pullorum isolates, high resistance to these antibiotics has been described in poultry isolates of one study from Italy including nine isolates (24) and of one study from Egypt including 20 isolates (30). In contrast, lower MIC values were described for 15 isolates in another study from Italy (6) and in a study from Belgium comprising 23 strains from poultry and two human isolates (29). Resistance to ciprofloxacin, erythromycin, and tetracycline corresponded to SNPs previously reported in gyrA, 23S rRNA genes, and 16S rRNA genes, respectively (16, 24). For the ciprofloxacin-resistant strain for which no mutations in gyrA and gyrB could be assigned, we cannot exclude the putative involvement of efflux pumps in such resistance phenotypes, in line with what has been demonstrated for Campylobacter (31).
The present study also allowed the release of four H. pullorum genomes in addition to the single genome available to date (16), contributing to a better knowledge of the genome make-up of this emerging foodborne pathogen. Among the most polymorphic genes found in the H. pullorum core genome (Table 2), including some genes encoding proteins with unknown function, we highlight a gene belonging to the pyridoxamine 5′-phosphate oxidase-related family. Although members of this family (e.g., Nim proteins) have been associated with resistance to 5-nitroimidazole, the molecular basis of this resistance involves several bacterial players, so any correlation between the multiple polymorphisms found and susceptibility to nitroimidazoles would be speculative (32). Another interesting example stands for a length-variable OMP-encoding gene, whose polymorphism pattern, relying on 7 variable domains intercalated with conserved regions, resembles that of the Campylobacter major protein (cmp) gene of C. jejuni. This gene encodes a major OMP that displays features of Gram-negative bacterial porins and has important antigenic properties, making it a potential target for vaccine development (27). In addition, polymorphisms in cmp seem to reflect phylogenetic relations between C. jejuni strains, suggesting that a sequence variation of cmp might be exploited for molecular typing or phylogenetic analysis of C. jejuni. The identification of a cmp ortholog in the H. pullorum genome suggests that this gene may also code for a porin and may be involved in antigenic variability, constituting a promising research target.
The most polymorphic gene identified in the H. pullorum genomes codes for a putative cytotoxin, displaying high genetic variability within the 5′ and middle regions of the gene. This gene pattern shares similarities with the vacA gene, encoding the vacuolating cytotoxin of the gastric pathogen Helicobacter pylori, for which the signal, intermediate, and middle genotypes are strong markers of severe gastric disease (33). The well-conserved 3′ region of the H. pullorum cytotoxin shows features of an autotransporter β-domain, which is characteristic of secreted proteins (34). Accordingly, the cytotoxin also showed a putative signal peptide with a cleavage site, corroborating the hypothesis that it is a secreted protein. This scenario again parallels the vacuolating cytotoxin of H. pylori, whose gene structure, protein secretion, and proteolytic processing are characteristic of autotransporter proteins secreted by Gram-negative bacteria (35). Finally, three additional paralogs of the cytotoxin were found in the four genomes of chicken meat H. pullorum isolates as well as in the human strain MIT 98-5489, once more mirroring the scenario of the vacA gene. Altogether, these data strongly suggest that H. pullorum is likely to possess an additional cytotoxin besides the cytolethal distending toxin, which was present in the four meat isolates and is believed to be ubiquitous in H. pullorum (36). The genetic heterogeneity and role of this additional cytotoxin in the virulence of H. pullorum deserve to be further explored. Homologs of both these genetic traits, the Campylobacter-like MOMP and the cytotoxin, were also present in the genomes of two strains from other enterohepatic Helicobacter species, namely, H. canadensis and H. winghamensis. Similar to H. pullorum, both these species are considered emerging human pathogens with diverse animal reservoirs and also lack a flagellar sheath and the gamma-glutamyl transpeptidase (4, 16, 37, 38). Therefore, the contributions of the Campylobacter-like MOMP and the putative cytotoxin to the host adaptation and to the virulence of these pathogens warrant further investigation. In contrast, neither H. canadensis nor H. winghamensis displays the virulence-associated T6SS (16), which was found to be carried by three of the four H. pullorum isolates in this study, and also in sheathed-flagellum enterohepatic Helicobacter species, i.e., Helicobacter cinaedi and Helicobacter bilis. The T6SS is used by Gram-negative bacteria to deliver effector proteins into both eukaryotic and prokaryotic neighboring cells to mediate virulence and competition, respectively (39). The presently characterized T6SS consists of 13 noncontiguous genes, including the hallmark hcp and vgrG genes, and closely resembles the T6SS genes of C. jejuni at both gene organization and content levels (Fig. 3) (40). The existence of a putative functional T6SS in H. pullorum was recently proposed by Sirianni et al., who correlated the mechanism of action of this cluster with the transcellular invasion of Caco-2 intestinal epithelial cells by this pathogen (41). In C. jejuni, the prevalence of the T6SS varies greatly among isolates and appears to depend on the geographic origin of the strain, being more prevalent among strains from Asia (from 60.6% to 71.5%) than those from Western countries (<10%) (40, 42). In addition, the T6SS is associated with more severe forms of C. jejuni infection, such as bloody diarrhea and bacteremia, supporting the contribution of the system to the virulence of this pathogen (40, 42). Therefore, the presence of a complete and most likely functional T6SS in three out of the four H. pullorum isolates suggests that this system is common among this species, constituting an important virulence mechanism.
Globally, the comparative genomic analysis has shown some extent of plasticity in the H. pullorum genome (Table 3), and it is likely that other genomic features found in the accessory genome also contribute to adaptation and virulence, as in the case of a prophage. It is well known that phages integrated into the genomes of their hosts influence both bacterial behavior and virulence (43). In C. jejuni, prophages promote genomic rearrangements and resistance to DNA transformation (44), as well as in H. pylori, for which the prevalence of prophages has been underestimated until recently (45). Considering that one isolate revealed a complete prophage and also sequences from another putative remnant prophage, we hypothesize that this type of genetic element may play a role in H. pullorum adaptation and pathogenicity. Also, since phages are important drivers of microbial evolution, this finding should be important for further understanding and prediction of genetic diversity in H. pullorum.
In conclusion, our data indicate that the emerging pathogen H. pullorum can be transmitted to humans by chicken meat consumption/contact, suggesting the need to expand already-existent surveillance strategies at food level. In addition, this study has greatly enhanced the knowledge of genetic diversity within the H. pullorum species, with some highlighted genetic traits, such as OMPs, cytotoxins, prophages, and the T6SS, providing clues to how this species promotes adaptation and virulence.
ACKNOWLEDGMENT
This work was funded by the Portuguese National Institute of Health grant for Research and Development.
REFERENCES
- 1.Aboutaleba N, Kuijper E, van Dissela J. 2014. Emerging infectious colitis. Curr Opin Gastroenterol 30:106–115. doi: 10.1097/MOG.0000000000000030. [DOI] [PubMed] [Google Scholar]
- 2.Steinbrueckner B, Haerter G, Pelz K, Weiner S, Rump JA, Deissler W, Bereswill S, Kist M. 1997. Isolation of Helicobacter pullorum from patients with enteritis. Scand J Infect Dis 29:315–318. doi: 10.3109/00365549709019053. [DOI] [PubMed] [Google Scholar]
- 3.Ceelen L, Decostere A, Verschraegen G, Ducatelle R, Haesebrouck F. 2005. Prevalence of Helicobacter pullorum among patients with gastrointestinal disease and clinically healthy persons. J Clin Microbiol 43:2984–2986. doi: 10.1128/JCM.43.6.2984-2986.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Stanley J, Linton D, Burnens A, Dewhirst FE, On SL, Porter A, Owen RJ, Costas M. 1994. Helicobacter pullorum sp. nov.—genotype and phenotype of a new species isolated from poultry and from human patients with gastroenteritis. Microbiology 140:3441–3449. doi: 10.1099/13500872-140-12-3441. [DOI] [PubMed] [Google Scholar]
- 5.Atabay H, Corry J, On S. 1998. Identification of unusual Campylobacter-like isolates from poultry products as Helicobacter pullorum. J Appl Microbiol 84:1017–1024. doi: 10.1046/j.1365-2672.1998.00438.x. [DOI] [PubMed] [Google Scholar]
- 6.Zanoni R, Rossi M, Giacomucci D, Sanguinetti V, Manfreda G. 2007. Occurrence and antibiotic susceptibility of Helicobacter pullorum from broiler chickens and commercial laying hens in Italy. Int J Food Microbiol 116:168–173. doi: 10.1016/j.ijfoodmicro.2006.12.007. [DOI] [PubMed] [Google Scholar]
- 7.González A, Piqueres P, Moreno Y, Cañigral I, Owen RJ, Hernández J, Ferrús MA. 2008. A novel real-time PCR assay for the detection of Helicobacter pullorum-like organisms in chicken products. Int Microbiol 11:203–208. [PubMed] [Google Scholar]
- 8.Laharie D, Asencio C, Asselineau J, Bulois P, Bourreille A, Moreau J, Bonjean P, Lamarque D, Pariente A, Soulé J-C, Charachon A, Coffin B, Perez P, Mégraud F, Zerbib F. 2009. Association between enterohepatic Helicobacter species and Crohn's disease: a prospective cross-sectional study. Aliment Pharmacol Ther 30:283–293. doi: 10.1111/j.1365-2036.2009.04034.x. [DOI] [PubMed] [Google Scholar]
- 9.Bohr U, Glasbrenner B, Primus A, Zagoura A, Wex T, Malfertheiner P. 2004. Identification of enterohepatic Helicobacter species in patients suffering from inflammatory bowel disease. J Clin Microbiol 42:2766–2768. doi: 10.1128/JCM.42.6.2766-2768.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pellicano R, Ménard A, Rizzetto M, Mégraud F. 2008. Helicobacters and liver diseases. Association or causation? Lancet Infect Dis 8:254–260. doi: 10.1016/S1473-3099(08)70066-5. [DOI] [PubMed] [Google Scholar]
- 11.International Organization for Standardization. 2006. ISO 10272-1:2006. Microbiology of food and animal feeding stuffs. Horizontal method for detection and enumeration of Campylobacter spp. Part 1: detection method. International Organization for Standardization, Geneva, Switzerland. [Google Scholar]
- 12.Ferreira S, Júlio C, Queiroz J, Domingues F, Oleastro M. 2014. Molecular diagnosis of Arcobacter and Campylobacter in diarrhoeal samples among Portuguese patients. Diagn Microbiol Infect Dis 78:220–225. doi: 10.1016/j.diagmicrobio.2013.11.021. [DOI] [PubMed] [Google Scholar]
- 13.Suzuki M, Giovannoni S. 1996. Bias caused by template annealing in the amplification of mixtures of 16S rRNA genes by PCR. Appl Environ Microbiol 62:625–630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zerbino D, Birney E. 2008. Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Res 18:821–829. doi: 10.1101/gr.074492.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Milne I, Stephen G, Bayer M, Cock PJ, Pritchard L, Cardle L, Shaw PD, Marshall D. 2013. Using Tablet for visual exploration of second generation sequencing data. Brief Bioinform 14:193–202. doi: 10.1093/bib/bbs012. [DOI] [PubMed] [Google Scholar]
- 16.Shen Z, Sheh A, Young S, Abouelliel A, Ward DV, Earl AM, Fox JG. 2014. Draft genome sequences of six enterohepatic Helicobacter species isolated from humans and one from rhesus macaques. Genome Announc 2(5):00857-14. doi: 10.1128/genomeA.00857-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Darling A, Mau B, Perna N. 2010. progressiveMauve: multiple genome alignment with gene gain, loss and rearrangement. PLoS One 5:e11147. doi: 10.1371/journal.pone.0011147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Librado P, Rozas J. 2009. DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics 25:1451–1452. doi: 10.1093/bioinformatics/btp187. [DOI] [PubMed] [Google Scholar]
- 19.Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S. 2011. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol 28:2731–2739. doi: 10.1093/molbev/msr121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kurtz S, Phillippy A, Delcher A, Smoot M, Shumway M, Antonescu C, Salzberg SL. 2004. Versatile and open software for comparing large genomes. Genome Biol 5:R12. doi: 10.1186/gb-2004-5-2-r12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Langmead B, Salzberg S. 2012. Fast gapped-read alignment with Bowtie 2. Nat Methods 9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhou Y, Liang Y, Lynch K, Dennis J, Wishart D. 2011. PHAST: a fast phage search tool. Nucleic Acids Res 39:W347–W352. doi: 10.1093/nar/gkr485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gibson J, Ferrus M, Woodward D, Xerry J, Owen R. 1999. Genetic diversity in Helicobacter pullorum from human and poultry sources identified by an amplified fragment length polymorphism technique and pulsed-field gel electrophoresis. J Appl Microbiol 87:602–610. doi: 10.1046/j.1365-2672.1999.00858.x. [DOI] [PubMed] [Google Scholar]
- 24.Pasquali F, Rossi M, Manfreda G, Zanoni R. 2007. Complete nucleotide sequence of the gyrA gene of Helicobacter pullorum and identification of a point mutation leading to ciprofloxacin resistance in poultry isolates. Int J Antimicrob Agents 30:222–228. doi: 10.1016/j.ijantimicag.2007.04.017. [DOI] [PubMed] [Google Scholar]
- 25.Kuijper E, Stevens S, Imamura T, De Wever B, Claas E. 2003. Genotypic identification of erythromycin-resistant campylobacter isolates as Helicobacter species and analysis of resistance mechanism. J Clin Microbiol 41:3732–3736. doi: 10.1128/JCM.41.8.3732-3736.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Manfreda G, Parisi A, Lucchi A, Zanoni R, De Cesare A. 2011. Prevalence of Helicobacter pullorum in conventional, organic, and free-range broilers and typing of isolates. Appl Environ Microbiol 77:479–484. doi: 10.1128/AEM.01712-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhang Q, Meitzler J, Huang S, Morishita T. 2000. Sequence polymorphism, predicted secondary structures, and surface-exposed conformational epitopes of Campylobacter major outer membrane protein. Infect Immun 68:5679–5689. doi: 10.1128/IAI.68.10.5679-5689.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ceelen L, Decostere A, Van den Bulck K, On SLW, Baele M, Ducatelle R, Haesebrouck F. 2006. Helicobacter pullorum in chickens, Belgium. Emerg Infect Dis 12:263–267. doi: 10.3201/eid1202.050847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ceelen L, Decostere A, Devriese L, Ducatelle R, Haesebrouck F. 2005. In vitro susceptibility of Helicobacter pullorum strains to different antimicrobial agents. Microb Drug Resist 11:122–126. doi: 10.1089/mdr.2005.11.122. [DOI] [PubMed] [Google Scholar]
- 30.Mohamed M, Ibrahim R, Shahata M, El-Refaie E. 2010. Helicobacter pullorum among poultry in Assiut-Egypt: genetic characterization, virulence and MIC. Int J Poult Sci 9:521–526. doi: 10.3923/ijps.2010.521.526. [DOI] [Google Scholar]
- 31.Ge B, McDermott P, White D, Meng J. 2005. Role of efflux pumps and topoisomerase mutations in fluoroquinolone resistance in Campylobacter jejuni and Campylobacter coli. Antimicrob Agents Chemother 49:3347–3354. doi: 10.1128/AAC.49.8.3347-3354.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Löfmark S, Edlund C, Nord CE. 2010. Metronidazole is still the drug of choice for treatment of anaerobic infections. Clin Infect Dis 50(Suppl 1):S16–S23. doi: 10.1086/647939. [DOI] [PubMed] [Google Scholar]
- 33.Memon A, Hussein N, Miendje Deyi V, Burette A, Atherton J. 2014. Vacuolating cytotoxin genotypes are strong markers of gastric cancer and duodenal ulcer-associated Helicobacter pylori strains: a matched case-control study. J Clin Microbiol 52:2984–2989. doi: 10.1128/JCM.00551-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Grijpstra J, Arenas J, Rutten L, Tommassen J. 2013. Autotransporter secretion: varying on a theme. Res Microbiol 164:562–582. doi: 10.1016/j.resmic.2013.03.010. [DOI] [PubMed] [Google Scholar]
- 35.Cover T, Blanke S. 2005. Helicobacter pylori VacA, a paradigm for toxin multifunctionality. Nat Rev Microbiol 3:320–332. doi: 10.1038/nrmicro1095. [DOI] [PubMed] [Google Scholar]
- 36.Varon C, Mocan I, Mihi B, Péré-Védrenne C, Aboubacar A, Moraté C, Oleastro M, Doignon F, Laharie D, Mégraud F, Ménard A. 2014. Helicobacter pullorum cytolethal distending toxin targets vinculin and cortactin and triggers formation of lamellipodia in intestinal epithelial cells. J Infect Dis 209:588–599. doi: 10.1093/infdis/jit539. [DOI] [PubMed] [Google Scholar]
- 37.Fox J, Chien C, Dewhirst F, Paster B, Shen Z, Melito P, Woodward D, Rodgers F. 2000. Helicobacter canadensis sp. nov. isolated from humans with diarrhea as an example of an emerging pathogen. J Clin Microbiol 38:2546–2549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Melito P, Munro C, Chipman P, Woodward D, Booth T, Rodgers F. 2001. Helicobacter winghamensis sp. nov., a novel Helicobacter sp. isolated from patients with gastroenteritis. J Clin Microbiol 39:2412–2417. doi: 10.1128/JCM.39.7.2412-2417.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ho B, Dong T, Mekalanos J. 2014. A view to a kill: the bacterial type 6 secretion system. Cell Host Microbe 15:9–21. doi: 10.1016/j.chom.2013.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bleumink-Pluym N, van Alphen L, Bouwman L, Wösten M, van Putten J. 2013. Identification of a functional type VI secretion system in Campylobacter jejuni conferring capsule polysaccharide sensitive cytotoxicity. PLoS Pathog 9:e1003393. doi: 10.1371/journal.ppat.1003393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sirianni A, Kaakoush N, Raftery M, Mitchell H. 2013. The pathogenic potential of Helicobacter pullorum: possible role for the type VI secretion system. Helicobacter 18:102–111. doi: 10.1111/hel.12009. [DOI] [PubMed] [Google Scholar]
- 42.Harrison JW, Dung TTN, Siddiqui F, Korbrisate S, Bukhari H, Tra MPV, Hoang NVM, Carrique-Mas J, Bryant J, Campbell JI, Studholme DJ, Wren BW, Baker S, Titball RW, Champion OL. 2014. Identification of possible virulence marker from Campylobacter jejuni isolates. Emerg Infect Dis 20:1026–1029. doi: 10.3201/eid2006.130635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bondy-Denomy J, Davidson A. 2014. When a virus is not a parasite: the beneficial effects of prophages on bacterial fitness. J Microbiol 52:235–242. doi: 10.1007/s12275-014-4083-3. [DOI] [PubMed] [Google Scholar]
- 44.Clark C, Chong P, McCorrister S, Mabon P, Walker M, Westmacott G. 2014. DNA sequence heterogeneity of Campylobacter jejuni CJIE4 prophages and expression of prophage genes. PLoS One 9:e95349. doi: 10.1371/journal.pone.0095349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lehours P, Vale F, Bjursell M, Melefors O, Advani R, Glavas S, Guegueniat J, Gontier E, Lacomme S, Alves Matos A, Menard A, Mégraud F, Engstrand L, Andersson AF. 2011. Genome sequencing reveals a phage in Helicobacter pylori. mBio 2:e00239-11. doi: 10.1128/mBio.00239-11. [DOI] [PMC free article] [PubMed] [Google Scholar]