

Abstract
Aim:
To investigate the effects of phorbol 12-myristate 13-acetate (PMA), a PKC activator, on P-glycoprotein-mediated efflux of digoxin in two cell transport models.
Methods:
Caco-2 cells, wild MDCKII cells (MDCKII-WT) and MDCKII cells transfected stably with human MDR1-gene encoding P-gp (MDCKII-MDR1) were examined. Cell viability was evaluated with MTT assay. Bidirectional transport of digoxin was evaluated in these cells. Intracellular ATP level was measured using ATP assay. P-gp ATPase activity was analyzed using a Pgp-GloTM assay.
Results:
PMA (10 μmol/L) did not reduce the viability of the 3 types of cells. In Caco-2 and MDCKII-MDR1 cell monolayers, PMA (1, 10 and 100 nmol/L) dose-dependently inhibited the basolateral to apical transport of digoxin, but did not change the apical to basolateral transport. In addition, PMA did not affect both the basolateral to apical and apical to basolateral transport of digoxin in MDCKII-WT cell monolayer. In agreement with the above results, PMA dose-dependently reduced intracellular ATP level and stimulated P-gp ATPase activity in both Caco-2 and MDCKII-MDR1 cells. Verapamil (a positive control, 100 μmol/L) caused similar inhibition on digoxin efflux as PMA did, whereas 4α-PMA (a negative control, 100 nmol/L) had no effect.
Conclusion:
PMA significantly inhibited P-gp-mediated efflux of digoxin in both Caco-2 and MDCKII-MDR1 cell monolayers via PKC activation.
Keywords: drug-drug interaction, membrane transporter, P-glycoprotein, MDR1, Caco-2 cells, digoxin, phorbol ester, PKC, verapamil
Introduction
In recent years, the issue of potential drug-drug interactions (DDI) occurring when pharmaceuticals are co-administered has become a subject of increasing recognition. The high frequency of DDIs is largely a result of the tendency of the general populace to use complementary medicine. In addition to the cytochrome enzymes, membrane transporters are known to play a crucial role in the modulation of the pharmacokinetic profile of drugs and DDIs1. Orally administered drugs must be adequately and consistently absorbed to achieve successful therapy. Oral bioavailability may be influenced by the dissolution of drug in the gastrointestinal fluids, first pass metabolism, gastrointestinal membrane permeability, and active excretion2.
Drugs absorption can be decreased by first pass metabolism in the intestine by cytochrome enzymes and efflux transporters3. One of the major barriers limiting oral drug delivery is the active efflux of drugs from the enterocytes back into the intestinal lumen by P-glycoprotein (P-gp). P-gp is an ATP-binding cassette protein that is responsible for secreting drugs and xenobiotics out of the cell4. P-gp is a plasma membrane-bound drug efflux protein that is expressed on several barrier epithelia, including the intestine, renal epithelial cells and brain capillary endothelial cells5. In the small intestine, P-gp is predominantly located in the apical membrane of the intestinal epithelial cells, consistent with its vital role in extruding drugs and xenobiotics back into the intestinal lumen6,7. The absorption and efflux of drug substrates of P-gp may be inhibited when other pharmaceuticals are co-administered. In addition, the concentration of inhibitory agents can be much higher in the intestine than in the systemic circulation after oral administration, which leads to the poor bioavailability of certain drugs and DDIs in the intestine rather than in the liver upon the oral administration of the inhibitory agent8.
In the clinic, a number of significant DDIs involving P-gp substrates have been reported. DDIs with the cardiac glycoside digoxin, a P-gp drug substrate most commonly used for treating congestive heart failure, have been observed due to patients concomitantly taking erythromycin, verapamil (VER), ketoconazole, and cyclosporine, among other drugs9,10. Although different approaches have been developed and proposed to study drug uptake in the intestine and the other functions of differentiated intestinal cells, a human colon carcinoma cell (Caco-2) monolayer is a widely accepted model for studying intestinal absorption and secretion of drug molecules in vitro11,12.
Caco-2 cells, derived from human adenocarcinoma colon cells, can be prepared on a porous filter as a polarized monolayer capable of functions similar to intestinal enterocytes13,14. The cells are connected by tight junctions to prevent paracellular substance flux, and they express distinct intestinal transport proteins. Several reports have demonstrated the possibility of predicting the oral absorption of drugs in humans based on permeability data obtained from Caco-2 monolayers11,15. Madin-Darby canine kidney (MDCK) cells, a dog renal epithelia cell line, can differentiate into columnar epithelia and form tight junctions in a shorter time than Caco-2 cells when grown onto Transwells16. MDCKII-MDR1 cells were generated by stably transfecting the human multi-drug-resistance (MDR) 1-gene encoding P-gp into MDCK cells. An expression level of P-gp, similar to that in Caco-2 cells, has been reported in MDCKII-MDR1 cells17. In the present study, the possible impact of PKC on P-gp was verified in the corresponding parental MDCKII-wild type (WT) cell line, which has a much lower P-gp expression.
Phorbol 12-myristate 13-acetate (PMA) is a phorbol diester and a potent tumor promoter that is often applied in biomedical research to activate the signal transduction enzyme protein kinase C (PKC). PMA can activate PKC in vivo and in vitro by binding to PKC, resulting in a variety of cellular effects18,19. There is evidence that PKC plays a crucial role in cell signal transduction, modulation of tumor formation and development, cell proliferation and differentiation, oncogene activation, and cellular response cells to growth factors20,21. Previous studies have reported that the multidrug resistance of tumor cells was closely related to the PKC signaling transduction system22. PKC activity in P-gp overexpressing cancer cells was apparently higher than strains sensitive to doxorubicin and vincristine, suggesting that PKC plays a positive role in multidrug resistance23,24. However, activation of PKC may downregulate P-gp gene expression and activity through the pregnane X receptor (PXR) pathway25,26. Clearly, the roles of the PKC signaling pathway on the modulation of P-gp activity and P-gp-mediated transport of drugs are not well understood, and the underlying mechanisms are still largely unknown.
The purpose of this study was to investigate the effect of PKC activation on the P-gp-mediated transport of digoxin using the Caco-2 and MDCKII-MDR1 cell transport models. In addition, the effect of PMA on the intracellular ATP levels and the activity of P-gp ATPase were studied.
Materials and methods
Chemicals
Phorbol 12-myristate 13-acetate, 4α-PMA, digoxin, verapamil, and 3-(4,5-dimethylthiazol-2-thiazolyl)-2,5-diphenyltetrazolium bromide (MTT) were purchased from Sigma-Aldrich (St Louis, MO, USA). Culture flasks (75 cm2 growth area), polyester Transwell inserts (pore size 0.22 μmol/L and 12 mm diameter) and 96-well plates (0.32 cm2 growth area per well) were obtained from Corning Costar Corp (Cambridge, MA, USA). The CellTiter-Glo Luminescent Cell Viability Assay and Pgp-GloTM Assay systems were purchased from Promega (Madison, WI, USA). Fetal bovine serum (FBS), Hank's balanced salt solution (HBSS), and trypsin–EDTA were purchased from Gibco Invitrogen Corp (Grand Island, NY, USA). Dulbecco's modified Eagle's medium (DMEM), non-essential amino acids (NEAA), phosphate-buffered saline (PBS), and penicillin–streptomycin solution were obtained from HyClone (Logan, UT, USA). All other chemicals and reagents were of the highest grade available.
Caco-2, MDCKII-WT, and MDCKII-MDR1 cell culture
Cell lines and maintenance
The Caco-2 cell line was obtained from American Type Culture Collection (Rockville, MD, USA). The MDCKII-WT and MDCKII-MDR1 cell lines were generously provided by the Netherland Cancer Institute (Amsterdam, NL, USA). Caco-2 and MDCKII cells were cultured at 37 °C, 95% relative humidity and 5% CO2 atmosphere and maintained in DMEM supplemented with 10% FBS, 1% penicillin and streptomycin and 0.1 mmol/L NEAA27. The culture medium was changed every 2–3 d. After reaching a confluence level of 80%–90%, the cells were detached from the culture flask by introducing a 0.25% trypsin–0.02% EDTA solution.
Growth of cell monolayers
For the transport studies, cells were seeded onto Transwell inserts pre-coated with collagen at a density of 2.5×104 cells/well and maintained by providing 0.5 mL of culture medium to the apical (A) chamber and 1.5 mL to the basolateral (B) chamber. Cells grown on the Transwell membranes were maintained until use on d 21–23 (Caco-2, passages 25–30) and d 6–7 (MDCKII-MDR1 and MDCKII WT) to obtain differentiated monolayers and an expected higher expression of transport proteins.
Cell monolayer integrity
The formation and perpetuation of confluent cell monolayers in the Transwell inserts were ensured before and after the transport studies by measuring the transepithelial electrical resistance (TEER) using a World Precision Instrument, EVOM (Sarasota, FL, USA). The resistance of the cell monolayer was determined by subtracting the total resistance from the membrane support resistance. The approximate MDCKII-WT TEER values were 220–250 Ω·cm2. MDCKII–MDR1 TEER values ranged from 300 to 380 Ω·cm2, and Caco-2 TEER values ranged from 250 to 340 Ω·cm2. These data indicated that the cell monolayers were not comprised during the experiment.
Cellular viability study
The MTT assay was used to determine the cytotoxic effects of DIG, VER, PMA, and 4α-PMA on the cells. Caco-2, MDCKII-MDR1 and MDCKII-WT cells were seeded in 96-well plates at a density of 1×104 cells/well and subsequently treated with different concentrations of the studied compounds for 120 min, with HBSS (pH 7.4) as the control. Cell viability was assessed by adding 20 μL of MTT reagent (5 mg/mL) for a 4-h incubation. Absorbance was read at 570 nm on a multifunctional microplate reader (Thermo Fisher Scientific, USA). Cell viability was expressed as a percentage of HBSS control28.
Transport studies
Bidirectional transport studies with digoxin
All transport experiments were conducted in transport medium consisting of HBSS buffered with 10 mmol/L 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (pH 7.4) at 37 °C. Prior to the transport studies, the cell monolayers were washed twice with pre-warmed transport buffer and pre-incubated with the test compounds (PMA, 4α-PMA, or verapamil) for 20 min in HBSS. The test compounds were added to both the apical and basolateral sides of the monolayers at equal concentrations to maintain osmotic pressure for the duration of study. VER (100 μmol/L) was used as a positive control. The apical side had a final volume of 0.5 mL, while the basolateral sides contained 1.5 mL.
In the absorptive (A-B) transport studies, 0.5 mL of transport medium containing 1 μmol/L digoxin was added in the donor (A) chamber and 1.5 mL of transport medium in receiver (B) chamber. For the secretory (B-A) transport studies, 1.5 mL of transport medium containing 1 μmol/L digoxin was added in the donor (B) chamber and 0.5 mL of transport medium was added in the receiver (A) chamber. One hundred microliter samples were collected from the receiver chamber at time intervals of 15, 30, 60, 90, and 120 min, and the removed volume was appropriately replenished with a corresponding volume of pre-warmed transport medium at 37 °C29.
LC/MS/MS measurement of digoxin
To determine the amount of digoxin transported, a 100-μL sample of transport medium from the recipient compartment was mixed with 50 μL NH3·H2O by vortexing for 2 min. A volume of 10 μL of digitoxin was added to the mixture as the internal standard and vortexed for 1 min. Subsequently, the extraction of digoxin was carried out with 1 mL of tert-butyl methylether/dichloromethane (75:25, v/v). After centrifugation at 23 755×g for 5 min at 4 °C, the organic phase was transferred to a new test tube and evaporated to dryness. The residues were dissolved in 200 μL of mobile phase, and the concentration of the digoxin was determined as per an established LC/MS/MS method30.
Data analysis
The apparent permeability coefficient (Papp) of digoxin was calculated according to the following equation31:
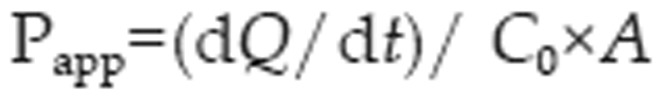 |
where dQ/dt is the cumulative rate of digoxin in the receiver chamber, C0 is the initial drug concentration of in the donor compartment, and A represents the membrane surface area (1.12 cm2) of the Caco-2 and MDCKII–MDR1 cell monolayers.
The P-gp potential to inhibit compounds or drugs is indicated by a decreased secretory flux and/or an enhanced absorptive flux compared to the untreated control wells. This can be deduced from an efflux ratio derived from the bidirectional Papp of digoxin in the presence and absence of PMA and VER.
The efflux ratio (ER) was obtained as
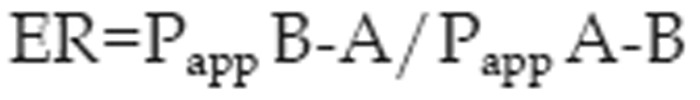 |
where Papp B-A and Papp A-B are the mean permeability coefficients obtained for transport in the basolateral to apical (B-A) direction and the apical to basolateral (A-B) direction, respectively.
ATP assay
For the ATP assay, MDCKII-WT, MDCKII-MDR1 and Caco-2 cells seeded in 96-well plates (approximately 10 000 cells/well) were treated with various concentrations of PMA for 48 h. Following treatment, cells were then washed twice with ice-cold PBS (200 μL per well). The intracellular level of ATP was determined using a firefly luciferase assay following the CellTiter-Glo luminescent cell viability assay standard procedure in opaque-walled plates32. For this purpose, 100 μL of ATP detection reagent was added to each well. The plate was mixed on an orbital shaker for 2 min and then further incubated for 10 min at room temperature. The luminescence was read in a multimode microplate reader, and the intracellular level of ATP in cells was expressed as the intensity of the emitted relative luminescence units (RLU). Samples were measured in triplicate, and values are given as the mean±SD.
P-gp ATPase activity measurement
P-gp ATPase activity was determined by measuring the release of inorganic phosphate (Pi) from the consumption of ATP using the light-generating reaction of firefly luciferase33. According to the manufacturer's protocol for the Pgp-GloTM assay system with recombinant human P-gp-containing membrane, the P-gp-containing membrane (25 μg/well) was pre-incubated in a 96-well with 100 nmol/L 4α-PMA and various concentrations of PMA for 5 min at 37 °C. The reaction was initiated by adding 10 μL of 25 mmol/L MgATP to all wells except the ATP standards. At this point, each P-gp reaction contained 5 mmol/L ATP. The reaction system was mixed briefly on a plate shaker and then incubated for 40 min at 37 °C. The plate was removed from the 37 °C incubator, and luminescence was initiated by adding 50 μL of the ATP detection reagent. After incubating the plate at room temperature for 20 min to develop the luminescent signal, the luminescence was determined in a multimode microplate reader.
Drug-stimulated ATPase activity (nmol Pi/min/mg protein) was calculated as the difference between the amounts of Pi released from ATP in the absence and presence of Na3VO4. MgATP standards were prepared for each plate, and VER served as a positive control for the drug stimulation of P-gp ATPase activity. The effect of a compound was considered to be stimulatory if the attained ATPase activity was higher than the basal P-gp ATPase activity incubated with the pure assay buffer. Samples were measured in triplicate, and values are given as the mean±SD.
Statistical analysis
Data are described as the mean±SD and were analyzed by Student's t-test with SPSS (Statistical Package for the Social Sciences) version 13.0 software (SPSS Inc, Chicago, IL, USA). Probability values of P<0.05 were considered to be statistically significant.
Results
Cell viability of Caco-2 and MDCKII cells
Prior to the transport studies, the in vitro cytotoxicity of the studied compounds in the transport buffer was tested in all three cell lines. The MTT assay was performed to investigate the concentration-dependent cytotoxic effects of PMA, 4α-PMA, VER, and DIG. As shown in Figure 1, a statistically significant increase in Caco-2 cell viability for 4α-PMA (5 and 10 μmol/L) was observed when compared with the HBSS control. There was no significant cytotoxic reduction in cell viability for all three cell lines. VER and DIG did not significantly alter cell viability (data not shown). Based on these data, none of the compounds was found to affect the growth of Caco-2, MDCKII-MDR1, MDCKII-WT cells at the examined concentrations.
Figure 1.

Effect of PMA (A) and 4α-PMA (B) on the growth of Caco-2, MDCKII-MDR1 and MDCKII-WT cells. Cells were treated with various concentrations of PMA and 4α-PMA for 120 min. Viable cells were evaluated by the MTT assay and are denoted as a percentage of untreated controls at the same concentration. Data are presented as the mean±SD (n=3).
Transepithelial transport of digoxin across Caco-2 and MDCKII-MDR1 cell monolayers
The amount of the transported P-gp substrate digoxin reflected the extent of interference in P-gp activity caused by the test compounds. This effect was displayed either by enhancing absorption and/or impairing secretion across the cell monolayers.
Effect of PMA on the bidirectional transport of digoxin in Caco-2 cell model
The Caco-2 cell monolayer model was validated by examining the bidirectional transport of digoxin. Figure 2 illustrates the basic transport characteristics of digoxin across the Caco-2 cell monolayer. The transport rate of digoxin in the B-A direction was much greater than that in the A-B direction. These data confirmed the high expression level of P-gp in the Caco-2 cell monolayer as well as the predominance of P-gp-mediated transport. P-gp inhibition was investigated by assessing bidirectional transport in the absence or presence of VER (100 μmol/L), a typical P-gp inhibitor. The introduction of VER significantly decreased the mean B-A transport of digoxin (P<0.05) and slightly increased the mean A-B transport of digoxin, further confirming the inhibitory effect of VER on P-gp.
Figure 2.

The time profiles of digoxin transport across Caco-2 cell monolayers. The bidirectional transport of digoxin with or without verapamil (VER, 100 μmol/L) across Caco-2 cell monolayers was determined as a function of time. Each data point represents the mean±SD (n=4–6).
The effect of PMA and 4α-PMA (100 nmol/L) on P-gp-mediated digoxin transport was then investigated. Transepithelial bidirectional transport of digoxin was time dependent and initially carried out for PMA at a concentration range of 1–100 nmol/L. The Papp values for the permeation of digoxin across Caco-2 cell monolayers in the absorptive and secretory directions in the presence or absence of various concentrations of PMA are shown in Figure 3. In the B-A direction, a significant 55% decrease in the transport rate of digoxin was observed at a concentration of 100 nmol/L PMA, suggesting significant P-gp inhibition (P<0.05). Subsequently, the effect of PMA on the A-B transport of digoxin was further investigated. However, transport was only slightly affected by PMA in the A-B direction. Here, the negative control of PMA, 4α-PMA, had no significant impact on either the A-B or B-A transport of digoxin. An increase in the A-B transport of digoxin was also observed with co-administration of VER (100 μmol/L) in Caco-2 cells, suggesting that P-gp mediated the efflux transport of digoxin in the apical membranes of Caco-2 cells. PMA and VER strongly suppressed the net secretory transport of digoxin, as evidenced by the decrease in ER when compared to the control (DIG alone).
Figure 3.

Effect of PMA on the bidirectional transport of digoxin across Caco-2 cell monolayers. The apparent permeability coefficient (Papp; mean±SD, n=3) and efflux ratio (ER) of digoxin in combination with PMA, 4α-PMA, and verapamil (VER) at the indicated concentrations in the apical-to-basolateral (AP-BL) and basolateral-to-apical (BL-AP) direction across a Caco-2 cell monolayer. bP<0.05, cP<0.01 vs untreated control.
Effect of PMA on the bidirectional transport of digoxin in MDCKII cells
The MDR1-transfected, P-gp-overexpressing cell line MDCKII-MDR1 was used to confirm the results observed in Caco-2 cells. The MDCKII-MDR1 cell monolayer model was also validated by examining the bidirectional transport of digoxin. The basic transport characteristics of digoxin across the MDCKII-MDR1 cell monolayer are shown in Figure 4A. The MDCKII-MDR1 cells displayed digoxin transport that was predominantly secretory (ER=28.3) and significantly reduced by 100 μmol/L VER, proving the suitability of MDCKII-MDR1 cell monolayer for use in investigating the effect of P-gp in vitro. Consistent with the results observed in the Caco-2 cell monolayer, the secretory transport of digoxin was strongly diminished in the presence of PMA, while absorptive transport remained largely unaffected. In addition, the efflux ratio for the permeation of digoxin across the MDCKII-MDR1 cell monolayer was markedly decreased in a concentration-dependent manner in the presence of PMA compared to the control, although the efflux was not totally abolished (Figure 5A).
Figure 4.

The time profiles of digoxin transport across MDCKII-MDR1 (A) and MDCKII-WT (B) cell monolayers. The bidirectional transport of digoxin with or without verapamil (VER, 100 μmol/L) across MDCKII-MDR1 and MDCKII-WT cell monolayers was determined as a function of time. Each data point represents the mean±SD (n=4–6).
Figure 5.

Effect of PMA on the bidirectional transport of digoxin across MDCKII-MDR1 (A) and MDCKII-WT (B) cell monolayers. The apparent permeability coefficient (Papp; mean±SD, n=3) and efflux ratio (ER) of digoxin in combination with PMA, 4α-PMA, and verapamil (VER) at the indicated concentrations in the apical-to-basolateral (AP-BL) and basolateral-to-apical (BL-AP) direction across MDCKII-MDR1 and MDCKII-WT cell monolayers. bP<0.05, cP<0.01 vs untreated control.
We further examined the effect of PMA on the bidirectional transport of digoxin in the corresponding MDCKII-WT cells. As shown in Figure 5B, the efflux ratio of digoxin in untreated MDCKII-WT cells (ER=8.2) was much lower than that in MDCKII- MDR1 cells. Furthermore, a non-significant inhibition of the transport of digoxin was found after treatment with verapamil, indicating the lower expression of P-gp in MDCKII-WT cell line. In contrast to the results observed in Caco-2 and MDCKII-MDR cell monolayer, a significant reduction in the efflux ratio of digoxin did not occur in the non-transfected cells in the presence of PMA.
Effect of PMA or 4α-PMA on intracellular ATP levels
Intracellular ATP levels of Caco-2, MDCKII-MDR1, and MDCKII-WT cells were examined using the CellTiter-Glo luminescent cell viability assay after treatment with PMA (1–100 nmol/L) for 2 h. As indicated in Figure 6, a significant and dose-dependent decrease of intracellular ATP levels in Caco-2 cells was observed in the presence of PMA. Compared to the control, PMA at concentrations of 10 and 100 nmol/L significantly reduced the relative luminescence units (RLU) of ATP in Caco-2 cells by 19.9% and 38.1%, respectively. In addition, we found that PMA at concentrations of 1, 10, and 100 nmol/L significantly decreased the intracellular ATP concentrations in MDCKII-MDR1 cells. However, no apparent decrease of the RLU of ATP in MDCKII-WT cells was observed at low concentrations (1 and 10 nmol/L), but there was a significant effect (P<0.05) at high PMA concentrations up to 100 nmol/L. Completely different cell behavior and dose response (1 to 100 nmol/L) were observed in the PMA treatment of MDCKII-MDR1 and MDCKII-WT cells. No effect was observed with 100 nmol/L 4α-PMA (the negative control of PMA) on the intracellular ATP concentrations of all three cell lines.
Figure 6.

Intracellular ATP levels in Caco-2, MDCKII-MDR1, and MDCKII-WT cells. Cells were incubated with PMA (1–100 nmol/L) and 4α-PMA (100 nmol/L) for 2 h. The relative luminescence units (RLU) were measured according to that manufactures' instructions of the CellTiter-Glo luminescent cell viability assay. Intracellular levels of ATP in cells are expressed as the intensity of the emitted RLU. Data are presented as the mean±SD (n=3). bP<0.05, cP<0.01 vs untreated control.
Effect of PMA and 4α-PMA on P-gp ATPase activity
To confirm the transporter inhibition in the cellular studies and elucidate whether PMA exerted its effect via direct P-gp inhibition, the effect of PMA on P-gp ATPase activity was investigated by determining the release of inorganic phosphate (Pi) from the consumption of ATP using the Pgp-GloTM assay system with recombinant human Pgp membrane. P-gp ATPase activity was apparently stimulated after treatment with PMA. As shown in Figure 7A, PMA at concentrations of 10 and 100 nmol/L significantly increased the Pi levels by 1.3 fold and 2.5 fold, respectively, compared to the basal group. However, all results varied around basal values, and no significant effects were observed even at 4α-PMA concentrations up to 500 nmol/L (Figure 7B).
Figure 7.

P-gp ATPase activity in P-gp-containing membranes. Drug-stimulated P-gp ATPase activity was reported as fold-stimulation relative to the basal P-gp ATPase activity in the absence of drug (A). The drug-stimulated ATPase activity (nmol Pi/min/mg protein) after incubation with PMA (1–500 nmol/L) and 4α-PMA (1–500 nmol/L) was calculated as the difference between the amounts of Pi released from ATP in the absence and the presence of Na3VO4 (B). Data are presented as the mean±SD (n=3). bP<0.05, cP<0.01 vs untreated control.
Discussion
Membrane transporters play a crucial role in the modulation of absorption, distribution, metabolism, and excretion of drugs34. P-gp, a member of the ATP-binding cassette (ABC) superfamily encoded by the ABCB1 gene, is well known among these transporters. P-gp functions as a barrier protein for the efflux of drugs and is a determinant of the pharmacokinetics, efficacy, and toxicity of xenobiotics35. In the intestine, P-gp is predominantly located in the apical membrane of mucosal cells and can extrude orally administered drugs back into the lumen, resulting in a reduction of the bioavailability of pharmaceutical agents36. Thus, the effect of PKC activation on the P-gp-mediated efflux of digoxin in Caco-2, MDCKII-MDR1, and MDCKII-WT cell transport models was investigated in the present study.
Prior to the investigation of PMA on the bidirectional transport of digoxin in Caco-2 and MDCKII cell monolayers, the Caco-2 cell monolayer model and MDCKII-MDR1 cell monolayer model were validated by examining the basic transport characteristics of digoxin. The results showed the suitability of these two cell monolayer models to investigate P-gp function in vitro and exhibited similar basic transport characteristics of digoxin compared to data from several reports37,38.
Although there are well-documented reports on the use of PMA to induce the PKC signaling pathway and its potent effect on drug resistance in cancer cells, this is the first report to investigate the effect of PKC activation by PMA on the P-gp-mediated efflux of digoxin using the Caco-2 and MDCKII-MDR1 cell transport models. In the current study, PMA showed an inhibitory effect on the P-gp-mediated efflux transport of digoxin in a concentration-dependent manner in a Caco-2 cell monolayer. In addition, the marked inhibition of PMA and VER on the net secretory transport of digoxin was further exemplified by the decrease in ER compared to the control. Previous studies have reported that stimulation of PKC induced the efflux function of P-gp in multidrug-resistant cells through P-g phosphorylation in human glioma cells and human breast cancer cells23,24. However, the results of this study suggested that PKC activation by PMA could inhibit the efflux function of P-gp through competitive inhibition in Caco-2 and MDCKII-MDR1 cells. This distinct difference in the effect of PKC on P-gp mediated efflux function may result from difference in cell lines and modulation mechanisms.
The P-gp-overexpressing cell line MDCKII-MDR1 was used to carry out the transport assay to confirm the results obtained in the Caco-2 cell monolayer. The results showed that PMA significantly inhibited the P-gp-mediated efflux transport of digoxin in the MDCKII-MDR1 cell model. To verify that these results were truly caused by P-gp, transport studies of digoxin in the presence of PMA were performed in the corresponding MDCKII-WT cells. In the non-transfected MDCKII-WT cells, PMA did not show a similar reduction of the ER ratio as in MDCKII-MDR1 cells. We speculated that the distinctly different level of P-gp expression leads to the completely different behavior in the two MDCKII cell lines. Additionally, the negative control for PMA, 4α-PMA, had no significant impact on A-B or B-A transport of digoxin in all three cell lines, confirming the key role of the PKC signaling pathway in the inhibition of P-gp-mediated transport of digoxin.
Based on the observed inhibition of the P-gp-mediated transport of digoxin, the effect of various concentrations of PMA on the intracellular ATP level was evaluated. A significant decrease of intracellular ATP levels was observed in the Caco-2 cells and MDCKII-MDR1 cells but not MDCKII-WT cells after PMA treatment. This type of phenomenon in different cell types (MDCKII-MDR1 cells and MDCKII-WT cells) may result from different expression levels of P-gp.
To further investigate the interaction between PMA and P-gp, the effect of PMA on the P-gp ATPase activity was examined using a P-gp ATPase activity kit in vitro. PMA significantly stimulated ATPase activity compared to the basal group, suggesting that PMA modulated P-gp-mediated efflux in cells via the inhibition of ATP-driven P-gp activity. It is well documented that P-gp-mediated drug efflux transport depends on the energy from ATP hydrolysis39,40. The hydrolysis and energy release of ATP derived from the synergistic action of drug binding sites and nucleotide-binding domains (NBDs) help P-gp to transport a wide variety of compounds across the cell membrane and is also assumed to influence each other by alterations of conformation41,42. In the present study, PMA was found to stimulate P-gp ATPase activity in a P-gp-enriched cell membrane fraction. It is well known that stimulators of ATPase activity are likely substrates of P-gp-mediated efflux that could competitively inhibit the transport of other P-gp substrates. Therefore, these results indicated that PMA is likely a substrate for P-gp-mediated efflux that could lead to a decrease in P-gp-dependent digoxin efflux through competitive inhibition. This finding is similar to previous reports that ATPase activity can be stimulated by verapamil in Caco-2 cells. Drugs and modulators may affect ATPase activity in cells in a complex manner, and their exact mechanisms of action have yet to be completely elucidated43.
In summary, the present study demonstrated that PMA has an inhibitory effect on the P-gp-mediated efflux of digoxin in Caco-2 and MDCKII-MDR1 cells. The present study provides useful information for predicting and understanding potential DDIs when P-gp substrates, such as digoxin, are co-administered with clinical drugs and other xenobiotics that could activate PKC. Further studies are required to verify the inhibition of P-gp-mediated efflux transport in vivo and clarify whether the P-gp ATPase inhibition is due to direct interaction with P-gp NBDs, heterotropic allosteric modulation, steric blocking of substrate binding sites, or a combination thereof.
Author contribution
Min HUANG designed the research study; Yu-hua LI and Xu-nian ZHOU performed the research; Yu-hua LI wrote the paper; Hui-chang BI, Jing JIN, and Guo-ping ZHONG helped analyze the data; Hui-chang BI and Ling HUANG revised the paper.
Acknowledgments
This project was supported by the Natural Major Projects for Science and Technology Developments from the Science and Technology Ministry of China (Grant 2012ZX09506001–004) and National Natural Science Foundation of China (Grant 81001685).
References
- 1Kumar YS, Adukondalu D, Sathish D, Vishnu YV, Ramesh G, Latha AB, et al. P-Glycoprotein-and cytochrome P-450-mediated herbal drug interactions. Drug Metabol Drug Interact 2010; 25: 3–16. [DOI] [PubMed] [Google Scholar]
- 2Chen KC, Li C, Uss AS. Prediction of oral drug absorption in humans — From cultured cell lines and experimental animals. Expert Opin Drug Metab Toxicol 2008; 4: 581–90. [DOI] [PubMed] [Google Scholar]
- 3Benet LZ, Cummins CL, Wu CY. Unmasking the dynamic interplay between efflux transporters and metabolic enzymes. Int J Pharm 2006; 277: 3–9. [DOI] [PubMed] [Google Scholar]
- 4Zhang LL, Lu L, Jin S, Jing XY, Yao D, Hu N, et al. Tissue-specific alterations in expression and function of P-glycoprotein in streptozotocin-induced diabetic rats. Acta Pharmacol Sin 2011; 32: 956–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5Kim WY, Benet LZ. P-glycoprotein (P-gp/MDR1)-mediated efflux of sex-steroid hormones and modulation of P-gp expression in vitro. Pharm Res 2004; 21: 1284–93. [DOI] [PubMed] [Google Scholar]
- 6Higgins CF. ABC transporters: from microorganisms to man. Annu Rev Cell Biol 1992; 8: 67–113. [DOI] [PubMed] [Google Scholar]
- 7Mizuno N, Niwa T, Yotsumoto Y, Suqiyama Y. Impact of drug transporter studies on drug discovery and development. Pharmacol Rev 2003; 55: 425–61. [DOI] [PubMed] [Google Scholar]
- 8Pang KS, Rodrigues AD, Peter R, eds. Enzyme- and Transporter-based drug-drug interactions, Progress and Future Challenges. New York: Springer; 2010. p 299–315.
- 9Lin JH. Transporter-mediated drug interactions: clinical implication and in vitro assessment. Expert Opin Drug Metab Toxicol 2007; 3: 81–92. [DOI] [PubMed] [Google Scholar]
- 10Horn JR, Hansten PD. Drug interactions with digoxin: the role of P-glycoprotein. Pharm Times 2004. p 45–6.
- 11Artursson P, Palm K, Luthman K. Caco-2 monolayers in experimental and theoretical predictions of drug transport. Adv Drug Deliv Rev 2001; 46: 27–43. [DOI] [PubMed] [Google Scholar]
- 12Cogburn JN, Donovan MG, Schasteen CS. A model of human small intestinal absorptive cells. 1. Transport barrier. Pharm Res 1991; 8: 210–6. [DOI] [PubMed] [Google Scholar]
- 13Kataoka M, Masaoka Y, Yamazaki Y, Sakane T, Sezaki H, Yamashita S. In vitro system to evaluate oral absorption of poorly water-soluble drugs: simultaneous analysis on dissolution and permeation of drugs. Pharm Res 2002; 20: 1674–80. [DOI] [PubMed] [Google Scholar]
- 14Zhou W, Di LQ, Wang J, Shan JJ, Liu SJ, Ju WZ, et al. Intestinal absorption of forsythoside A in in situ single-pass intestinal perfusion and in vitro Caco-2 cell models. Acta Pharmacol Sin 2012; 33: 1069–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15Ingels FM, Augustijns PF. Biological, pharmaceutical, and analytical considerations with respect to the transport media used in the absorption screening system, Caco-2. J Pharm Sci 2003; 92: 1545–58. [DOI] [PubMed] [Google Scholar]
- 16Tang F, Horie K, Borchardt RT. Are MDCK cells transfected with the human MDR1 gene a good model of the human intestinal mucosa? Pharm Res 2002; 19: 765–72. [DOI] [PubMed] [Google Scholar]
- 17de Souza J, Benet LZ, Huang Y, Storpirtis S. Comparison of bidirectional lamivudine and zidovudine transport using MDCK, MDCK-MDR1, and Caco-2 cell monolayers. J Pharm Sci 2009; 98: 4413–9. [DOI] [PubMed] [Google Scholar]
- 18Blumberg PM. Protein kinase C as the receptor for the phorbol ester tumor promoters: sixth Rhoads memorial award lecture. Cancer Res 1988; 48: 1–8. [PubMed] [Google Scholar]
- 19Petiti JP, De Paul AL, Gutierrez S, Palmeri CM, Mukdsi JH, Torres AI. Activation of PKC epsilon induces lactotroph proliferation through ERK1/2 in response to phorbol ester. Mol Cell Endocrinol 2008; 289: 77–84. [DOI] [PubMed] [Google Scholar]
- 20Mason SA, Cozzi SJ, Pierce CJ, Pavey SJ, Parsons PG, Boyle GM. The induction of senescence-like growth arrest by protein kinase C-activating diterpene esters in solid tumor cells. Invest New Drugs 2010; 28: 575–86. [DOI] [PubMed] [Google Scholar]
- 21O'Neill AK, Gallegos LL, Justilien V, Garcia EL, Leitges M, Fields AP, et al. Protein kinase Calpha promotes cell migration through a PDZ-dependent interaction with its novel substrate discs large homolog 1 (DLG1). J Biol Chem 2011; 286: 43559–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22Eĭdus LKh, Emel'ianov MO, Korystova AF, Kublik LN, Shaposhnikova VV, Korystov IuN. Multidrug resistance increase of tumor cells at the effect of radiation and phorbol ether depends on protein kinase C and reactive oxygen species. Radiats Biol Radioecol 2010; 50: 37–41. [PubMed] [Google Scholar]
- 23Fine RL, Patel J, Chabner BA. Phorbol esters induce multidrug resistance in human breast cancer cells. Proc Natl Acad Sci U S A 1988; 85: 582–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24Matsumoto T, Tani E, Yamaura I, Miyaji K, Kaba K. Effects of protein kinase C modulators on multidrug resistance in human glioma cells. Neurosurgery 1995; 36: 565–71. [DOI] [PubMed] [Google Scholar]
- 25Pondugula SR, Dong H, Chen T. Phosphorylation and protein-protein interactions in PXR-mediated CYP3A repression. Expert Opin Drug Metab Toxicol 2009; 5: 861–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26Ding X, Staudinger JL. Repression of PXR-mediated induction of hepatic CYP3A gene expression by protein kinase C. Biochem Pharmacol 2005; 69: 867–73. [DOI] [PubMed] [Google Scholar]
- 27Evers R, Zaman GJ, van Deemter L, Jansen H, Calafat J, Oomen LC, et al. Basolateral localization and export activity of the human multidrug resistance-associated protein in polarized pig kidney cells. J Clin Invest 1996; 97: 1211–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28Laitinen LA, Tammela PS, Galkin A, Vuorela HJ, Marvola ML, Vuorela PM. Effect of extracts of commonly consumed food supplements and food fraction on the permeability of drugs across Caco-2 cell monolayers. Pharm Res 2004; 21: 1904–16. [DOI] [PubMed] [Google Scholar]
- 29Simon S, Schubert R. Inhibitory effect of phospholipids on P-glycoprotein: cellular studies in Caco-2, MDCKII mdr1 and MDCKII wildtype cells and P-gp ATPase activity measurements. Biochim Biophys Acta 2012; 1821: 1211–23. [DOI] [PubMed] [Google Scholar]
- 30Xue XP, Huang M, Xiao HY, Qin XL, Huang L, Zhong GP, et al. Rapid and simultaneous measurement of midazolam, 1′-hydroxymidazolam and digoxin by liquid chromatography/tandem mass spectrometry: application to an in vivo study to simultaneously measure P-glycoprotein and cytochrome P450 3A activity. J Pharm Biomed Anal 2011; 55: 187–93. [DOI] [PubMed] [Google Scholar]
- 31Taub ME, Podila L, Ely D, Almeida L. Functional assessment of multiple P-glycoprotein (P-gp) probe substrates: Influence of cell line and modulator concentration on P-gp activity. Drug Metab Dispos 2005; 33: 1679–87. [DOI] [PubMed] [Google Scholar]
- 32Sprague RS, Hanson MS, Achilleus D, Bowles EA, Stephenson AH, Sridharan M, et al. Rabbit erythrocytes release ATP and dilate skeletal muscle arterioles in the presence of reduced oxygen tension. Pharmacol Rep 2009; 61: 183–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33Matsunaga T, Kose E, Yasuda S, Ise H, Ikeda U, Ohmori S. Determination of P-glycoprotein ATPase activity using luciferase. Biol Pharm Bull 2006; 29: 560–4. [DOI] [PubMed] [Google Scholar]
- 34Giacomini KM, Huang SM, Tweedie DJ, Benet LZ, Brouwer KL, Chu X, et al. Membrane transporters in drug development. Nat Rev Drug Discov 2010; 9: 215–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35Taniqawara Y. Role of P-glycoprotein in drug disposition. The Drug Monit 2000. P 137–40. [DOI] [PubMed]
- 36Sharom FJ. Shedding light on drug transport: structure and function of the P-glycoprotein multidrug transporter (ABCB1). Biochem Cell Biol 2006; 84: 979–92. [DOI] [PubMed] [Google Scholar]
- 37Oga EF, Sekine S, Shitara Y, Horie T. P-glycoprotein mediated efflux in Caco-2 cell monolayers: the influence of herbals on digoxin transport. J Ethnopharmacol 2012; 144: 612–7. [DOI] [PubMed] [Google Scholar]
- 38Oga EF, Sekine S, Shitara Y, Horie T. Potential P-glycoprotein-mediated drug-drug interactions of antimalarial agents in Caco-2 cells. Am J Trop Med Hyg 2012; 87: 64–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39Loo TW, Bartlett M, Clarke DM. Nucleotide binding, ATP hydrolysis, and mutation of the catalytic carboxylates of human P-glycoprotein cause distinct conformational changes in the transmembrane segments. Biochemistry 2007; 46: 9328–36. [DOI] [PubMed] [Google Scholar]
- 40Jones PM, George AM. Opening of the ADP-bound active site in the ABC transporter ATPase dimer: evidence for a constant contact, alternating sites model for the catalytic cycle. Proteins 2009; 75: 387–96. [DOI] [PubMed] [Google Scholar]
- 41Martin C, Berridge G, Higgins CF, Mistry P, Charlton P, Callaghan R. Communication between multiple drug binding sites on P-glycoprotein. Mol Pharmacol 2000; 58: 624–32. [DOI] [PubMed] [Google Scholar]
- 42Dubielecka PM, Jaźwiec B, Potoczek S, Wróbel T, Miłoszewska J, Haus O, et al. Changes in spectrin organisation in leukaemic and lymphoid cells upon chemotherapy. Biochem Pharmacol 2005; 69: 73–85. [DOI] [PubMed] [Google Scholar]
- 43Barakat S, Gayet L, Dayan G, Labialle S, Lazar A, Oleinikov V, et al. Multidrug-resistant cancer cells contain two populations of P-glycoprotein with differently stimulated P-gp ATPase activities: evidence from atomic force microscopy and biochemical analysis. Biochem J 2005; 388: 563–71. [DOI] [PMC free article] [PubMed] [Google Scholar]