

Abstract
Dying cells initiate adaptive immunity by providing both antigens and inflammatory stimuli for dendritic cells (DCs), which in turn activate CD8+ T cells through a process called antigen cross-priming. To define how different forms of programmed cell death influence immunity, we established models of necroptosis and apoptosis, where dying cells are generated by RIPK3 and CASP8 dimerization, respectively. We found that release of inflammatory mediators such as damage-associated molecular patterns (DAMPs) by dying cells was not sufficient for CD8+ T cell cross-priming. Instead, robust cross-priming required RIPK1 signaling and NF-κB-induced transcription within dying cells. Decoupling NF-κB signaling from necroptosis or inflammatory apoptosis reduced priming efficiency and tumor immunity. Our results reveal that coordinated inflammatory and cell death signaling pathways within dying cells orchestrate adaptive immunity.
Phagocytosis of dying cells by dendritic cells (DCs) results in cross-presentation of cell-associated antigen, and the priming of CD8+ T cells (1). This pathway mediates the processing and presentation of tumor antigens (2) as well as viral- and self-proteins in instances where expression is restricted to non-hematopoietic cells (3, 4). However, the manner by which different forms of programmed cell death (PCD) influence the ability of DCs to cross-present and initiate CD8+ T cell responses is still poorly understood.
Until recently apoptosis was thought to be immunologically quiescent, in contrast to necrosis, which is characterized by rapid membrane permeabilization and release of inflammatory mediators termed damage-associated molecular patterns (DAMPs). Paradoxically, the inflammatory nature of necrotic cells (defined by their ability to activate innate immune cells) (5–8) does not correlate with their ability to serve as a source of antigen for the initiation of CD8+ T cell immunity (defined as immunogenicity) (1, 9–12). Moreover, immunogenic cell death has often been associated with apoptotic pathways (1, 10, 13–15). Several recent studies highlighted the interconnections between cell death and inflammatory signal transduction. For example, proteins such as receptor-interacting protein kinase 3 (RIPK3) and caspase-8, which initiate necroptosis or apoptosis respectively, are incorporated into dynamic innate immune signaling modules (e.g., ripoptosome) (16–19). These cytosolic scaffolds establish the crosstalk between innate immune and cell death programs, and in some instances both pathways may be simultaneously engaged (fig. S1A). This integration of pathways, combined with the recent discovery of necroptosis, a regulated form of necrosis, prompted us to re-evaluate how different PCD pathways impact cross-priming of CD8+ T cells.
To selectively induce apoptosis or necroptosis, we constructed “pure” cell death systems in which death effector proteins, caspase-8 or RIPK3, were fused to a modified FK506 binding protein (FKBP) domain (Fv-ΔN-Caspase-8 and RIPK3-2xFv, respectively) (20–22) (fig. S1B). RIPK3 oligomerization results in the recruitment of RIPK1 via RIPK3 RHIM (RHIMRIPK3) domain interactions, leading to the formation of a cytosolic ripoptosome-like complex (21, 23). Therefore, we also generated a C-terminally truncated construct (RIPK3ΔC-2xFv) (fig. S1B), which lacks the RHIMRIPK3 domain and does not recruit RIPK1 (21). NIH-3T3 cells were stably transduced with these activatable constructs (referred to herein as acC8, acR3, and acR3ΔC). Dimerization of caspase-8 resulted in the induction of apoptosis; whereas oligomerization of full length RIPK3 and RHIM-less RIPK3 induced rapid cell swelling and membrane rupture (<3 hours) in the absence of caspase activation (Fig. 1, A and B; fig. S2, A to C; and movies S1 to S3). The ability to induce necroptosis in the absence of the RHIMRIPK3 domain enabled us to decouple RIPK1-dependent ripoptosome complex formation from cell death (21), hence eliminating the activation of other pathways emanating from the ripotosome.
Fig. 1. Necroptotic cells release DAMPs and induce dendritic cell maturation.
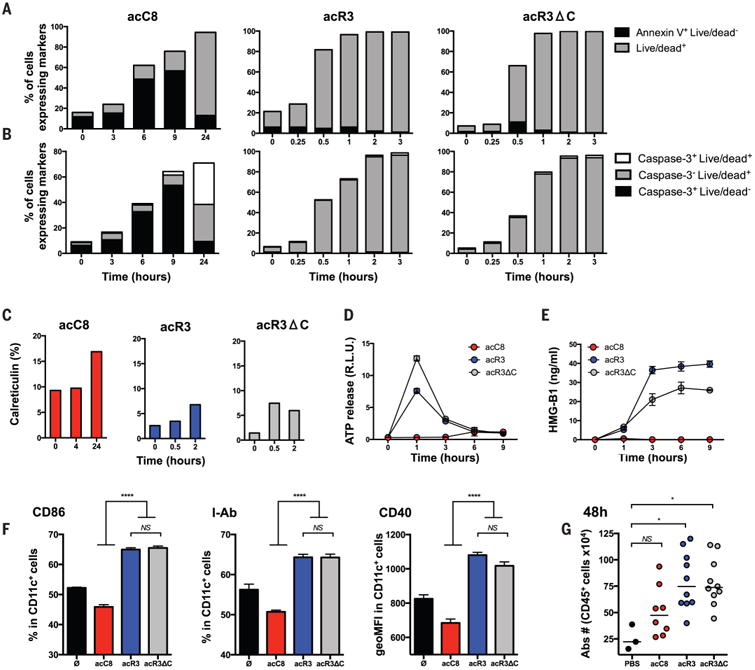
(A to C) NIH-3T3 cells expressing the death constructs were stimulated with dimerizer and cells harvested at the indicated time points and stained with Annexin-V and Live/Dead reagent (A); cleaved caspase-3 antibody and Live/Dead reagent (B); or calreticulin antibody (C). Cells that are Annexin V+ Live/Dead (indicating phosphatidylserine exposure prior to membrane permeabilization) or cleaved-caspase-3+ (indicating the activation of executionner caspases) are undergoing apopotsis. At later time points (24hrs), staining with Live/Dead reagent indicates loss of plasma membrane integrity and characterizes secondary necrotic cells. Rapid membrane permeabilzation without activation of executionner caspases (Live-Dead+ Caspase-3-) is a feature of necroptosis. N=2, results represent one representative experiment (D and E) ATP and HMGB1 were quantified from dying cell culture supernatants. N=3, results are reported as mean +/− SEM of triplicates of one representative experiment. (F) BMDCs were co-cultured with dimerizer treated acC8-, acR3- and acR3ΔC-expressing cells for 24h and DC maturation phenotype was assessed by flow cytometry. N=4, results are reported as mean +/− SEM of triplicates of one representative experiment. (G) 2×106 dimerizer treated cells were injected into the peritoneal cavity of WT C57BL/6 mice. 48h later, peritoneal cells were collected and immune cells were enumerated by cytometry. N=2, bars indicate mean of two pooled independent experiment with 4-5 mice per group (except PBS group). Each circle represent one mouse. p values were determined using one-way ANOVA test for (F) and Kruskal-Wallis test (KW - multigroup comparision) followed by a Dunn's post-test, comparing each group to PBS group for (G). *P < 0.05; ****P < 0.0001. acC8 = Casp8 apoptosis; acR3 = RIPK3 necroptosis; acR3ΔC = RIPK3RHIMless necroptosis; ATP= Adenosine triphosphate; HMGB1= High mobility group box 1; PBS= Phospahte-buffered saline.
Cell death-associated molecules such as calreticulin (CRT), ATP and HMGB1, have been shown to trigger inflammation and to regulate immunogenic cell death (8, 15, 24-26). We thus quantified CRT surface exposure and the release of both ATP and HMGB1 by apoptotic or necroptotic cells. Only low levels of CRT exposure were observed during the three forms of cell death, whereas only the acR3 and acR3ΔC-expressing NIH-3T3 cells rapidly released high concentrations of ATP and HMGB1 upon treatment (Fig. 1, C to E). In all cases, there were no detectable levels of interleukin (IL)-1α, IL-1β or uric acid released. We next evaluated phagocytosis by DCs (i.e., acquisition of antigen) and subsequent DC maturation - two steps required for achieving CD8+ T cell cross-priming (27, 28). We found that bone marrow derived dendritic cells (BMDCs) and a CD8α+ DC derived cell line (MuTuDC) acquired similar amounts of dimerizer-treated acC8-, acR3- and acR3ΔC-expressing NIH-3T3 cells, but did not efficiently phagocytose live cells (fig. S4, A and B). Moreover, both acR3 and acR3ΔC induced the up-regulation of DC activation markers; whereas acC8 NIH-3T3 cells did not (Fig. 1F and fig. S4C). Similarly, intraperitoneal injection of dimerizer-treated acR3- or acR3ΔC-expressing cells induced higher recruitment of immune cells, as compared to acC8 cells (Fig. 1G). Together, these data suggested that necroptotic cells released DAMPs, induced DCs maturation in vitro and inflammation in vivo.
To assess the respective immunogenicity of apoptotic and necroptotic cells, we immunized C57BL/6 mice by intradermally injecting 106 dimerizer-treated NIH-3T3 cells that stably expressed a non-secretable form of ovalbumin (OVA) (29) (fig. S5A). Cells were exposed to dimerizer immediately prior to injection, thereby permitting them to undergo cell death in situ. We observed significantly higher CD8+ T cell cross-priming when mice were immunized with cells undergoing RIPK3-mediated necroptosis compared to caspase-8-mediated apoptosis (Fig. 2A and fig. S5B; P < 0.0001). Immunization with acR3ΔC-OVA NIH-3T3 cells did not result in robust CD8+ T cell priming (Fig. 2A and fig. S5B; P < 0.01 as compared to acR3-OVA) indicating that RHIM-dependent interactions are required for immunogenicity of necroptotic cells.
Fig. 2.
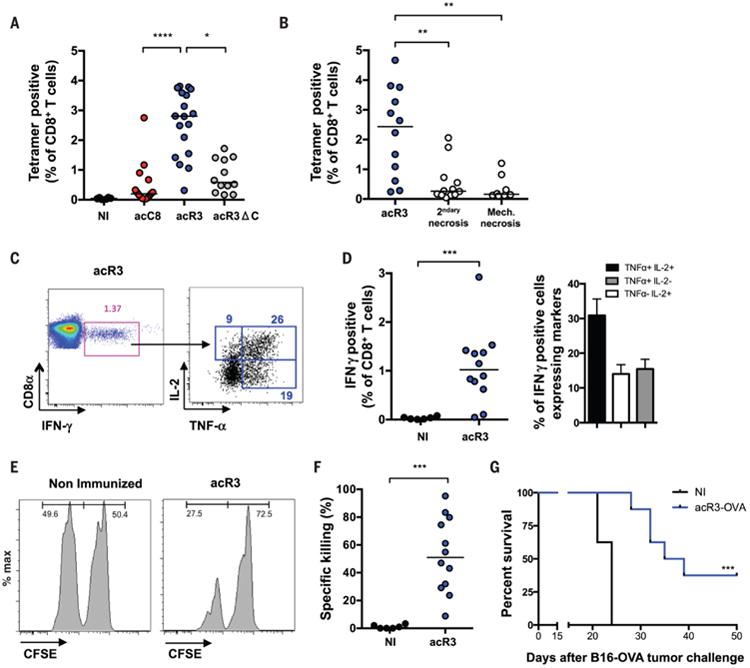
Necroptotic cells are immunogenic and require RHIM-dependent ripoptosome formation for efficient cross-priming of CD8+ T cells. (A to G) To elicit cross-priming we intradermally injected (i.d.) OVA-expressing dying cells (H-2q) into mice (H-2b), and analyzed on day 9 post-immunization (p.i.). (A and B) Using Kb-SIINFEKL-tetramers, OVA-specific CD8+ T cells were quantified and plotted as a percentage of total CD8+ T cells. N=4 (A), N=3 (B) Bars indicate median and results are pooled from three independent experiments with 3-6 mice per group (each circle represent one mouse) (C and D) IFN-γ, TNF-α and IL-2 production in response to ex-vivo SIINFEKL peptide re-stimulation was determined. Representative FACS plots are shown and numbers indicate the percentage of gated cells (C); and frequency of IFN-γ expressing and polyfunctional cells are plotted (D) N=3, results are pooled from three independent experiments with 3-6 mice per group and reported as individual mice (each circle represent one mouse) and bars indicate median (IFN-γ) or as histogram and mean+/− SEM (TNFα and IL-2). (E and F) In vivo cytotoxicity assay was performed in acR3-OVA immunized mice. At day 8 p.i., mice were adoptively transfered with CFSE-labeled splenocytes and the frequency of CSFEhi (irrelevant peptide control) and CFSElow (SIINFEKL loaded) splenocytes (injected at a 1:1 ratio) was determined at day 9. Representative FACS plots are shown (E) and the percent of specific killing plotted (F). N=3, Bars indicate median and results are pooled from three independent experiments with 4 mice per group (each circle represent one mouse). (G) Tumor challenge experiments were performed, injecting 5×105 B16F10-OVA cells on day 12 p.i., N=2, and results are reported as a survival curve from one representative experiment with 8-11 mice per group ; OVA = ovalbumin. p values were determined using KW test followed by Dunn's post-test for (A and B), Mann-Witney (MW) test for (D and F) and mice survival (G) was compared by log-rank test. *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001.
We next compared necroptosis to cells undergoing unregulated necrosis such as cells killed by “mechanical” necrosis (achieved by repeated freeze/thaw); or cells undergoing “secondary” necrosis (achieved by incubating apoptotic cells for 24h prior to immunization). We found that primary and secondary necrotic cells induced only weak CD8+ T cell responses (Fig. 2B; P < 0.01). Although the latter results could be partially explained by the loss of antigen after necrotic membrane permeabilisation (fig. S6), the findings suggested that in vivo necroptosis is a more efficient inducer of cross-priming, as compared to apoptotic or necrotic cells.
The efficiency and outcome of antigen cross-presentation has been shown to depend on a subset of CD8α+ / CD103+ DCs, whose differentiation is driven by the Batf3 transcription factor (30). We found that immunization of batf3−/− mice with acR3-OVA cells failed to elicit a CD8+ T cell response (fig. S7, A and B), confirming that cross-presentation of necroptotic cells-associated antigen is mediated by this DC lineage. We next characterized the CD8+ T cells induced by acR3-OVA immunization. CD8+ T cells primed by immunization with necroptotic cells produced multiple effector cytokines (Fig. 2, C and D), possessed in vivo cytolytic activity (Fig. 2, E and F), and protected mice from tumor challenge (Fig. 2G). Together, these data indicated that necroptotic cells are able to provide both antigen and immune stimulation, in turn supporting DC-mediated cross-priming of CD8+ T cells. The requirement of RHIMRIPK3 for the immunogenicity of necroptotic cells suggested that classical DAMPs (e.g., HMGB1) are insufficient to achieve robust cross-priming and supported a critical role for RIPK1 independent of cell death.
To understand the requirement for RHIM for immunogenic necroptosis, we studied the signaling pathways engaged during the different forms of cell death and assessed MAPK and NF-κB activation following dimerizer treatment. Oligomerization of RIPK3 in acR3-expressing NIH-3T3 cells resulted in the rapid phosphorylation of p38 and ERK1/2, and degradation of IκB (Fig. 3A). Activation of these inflammatory pathways was not observed after dimerization of Caspase-8 and was attenuated in acR3ΔC-expressing NIH-3T3 cells, with the greatest difference mapping to the NF-κB pathway (Fig. 3A). To determine the impact on transcriptional profile, we quantified the mRNA expression of 179 immune-related genes at different stages of cell death (Fig. 3B). Despite the rapid cell death kinetics, full-length RIPK3 activation triggered a significant upregulation of 72 inflammatory genes, many of which are regulated by NF-κB and MAPK activation (q < 0.05; fig. S8A and table S1). By contrast, we observed only modest changes in dimerizer-treated acC8-expressing cells (3 genes differentially expressed) and acR3ΔC-expressing cells (17 genes) (fig. S8A and table S1). We next measured inflammatory cytokines in the supernatant from dimerizer treated cell cultures and found that acR3 cells released high amounts of IL-6 (Fig. 3C) and CXCL1 (Fig. 3D), validating our transcriptional analysis. IL-6 production was inhibited in a time-dependent manner by treating the cells with actinomycin-D (ActD) or cycloheximide (CHX) (Fig. 3E). These data suggest that necroptotic cells actively transcribe and translate inflammatory cytokines during cell death (fig. S8B). Moreover, chemical inhibition of IκK kinase activity diminished IL-6 release (Fig. 3F) and stable expression of an IkB dominant negative protein (NFκB (S32A, S36A) super repressor, SR) also inhibited cytokine secretion (Fig. 3G). To formally test the contribution of RIPK1, we deleted RIPK1 from NIH-3T3 cells and stably expressed RIPK3-2xFv construct under a tetracycline inducible promoter (Tet-acR3) (fig. S9, A and B). Addition of dimerizer triggered necroptosis in both cell lines (fig. S9C), however NF-κB activation (fig. S9D) and IL-6 production (Fig. 3H) were reduced in the cells lacking RIPK1. These results revealed an NF-κB transcriptional and translational activity that is engaged during RIPK3 necroptosis.
Fig. 3. RIPK3 oligomerization results in RIPK1-dependent activation of NF-κB mediated gene expression.
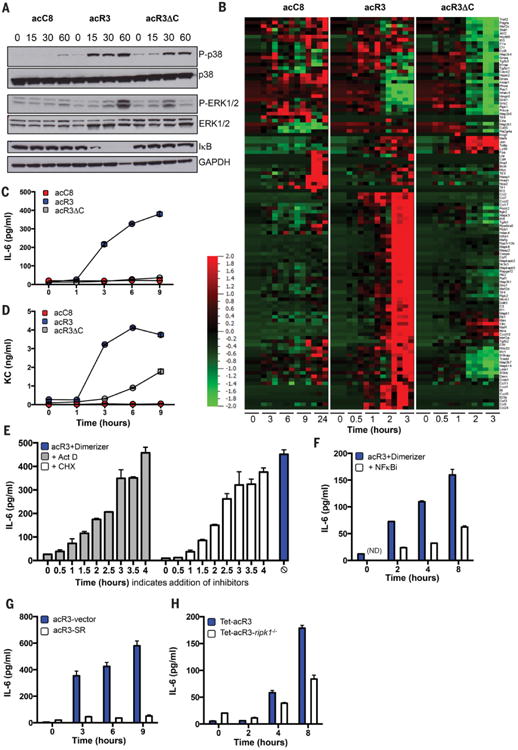
(A to D), acC8-, acR3- and acR3ΔC-expressing NIH-3T3 cells were treated with dimerizer, and at indicated time points: protein extracts were analyzed by Western blot, N=2 (A); RNA was extracted for transcriptional profiling, N=3 (B); or culture supernatants were collected for luminex analysis (C and D). (E to H) IL-6 release upon addition of dimerizer was determined, after treating the cells with (E), or (F). In (E), 2μg/ml actinomycin D (Act D) or 2.5μg/ml cycloheximide (CHX) were added at the indicated time points after addition of dimerizer (t=0), and IL-6 was measured at 6h. In (F), cells were pre-teated with 10μM Wedelolactone (NFKBi) and IL-6 measured at the indicated time points. In (G), acR3 cells stably expressing control vector (acR3-vector) or mutant super-repressor IκB (acR3-SR) were used. In (H), control NIH-3T3 cells (Tet-acR3), and cells lacking RIPK1 and expressing RIPK3 2xFv under a tetracycline promoter (Tet-acR3 ripk1−/−), were treated overnight with 500ng/ml of doxycycline (Dox) before addition of dimerizer. In (C to H), N≥2, and data are presented as mean +/− SEM of triplicates from one representative experiment. Heat map indicates the relative expression of the indicated transcript (red indicating high levels and green indicating low levels of expression).
We next tested the hypothesis that RIPK1 signaling and NFκB-dependent gene expression within the dying cell are critical for cross-priming. We immunized mice using necroptotic cells that lacked RIPK1 (Tet-acR3-ripk1−/−) (Fig. 4A), cells that lacked NF-κB signaling (pre-treated with NFκBi (Fig. 4B), or overexpressing the NF-κB SR (Fig. 4C)), or cells in which transcription was inhibited (pre-treated with ActD) (Fig. 4D). Cross-priming was significantly reduced in all instances, thus establishing that active RIPK1-NF-κB signaling is essential for the immunogenicity of necroptotic cells.
Fig. 4. RIPK1 expression and NF-κB activation during cell death are required for efficient cross-priming and anti-tumor immunity.
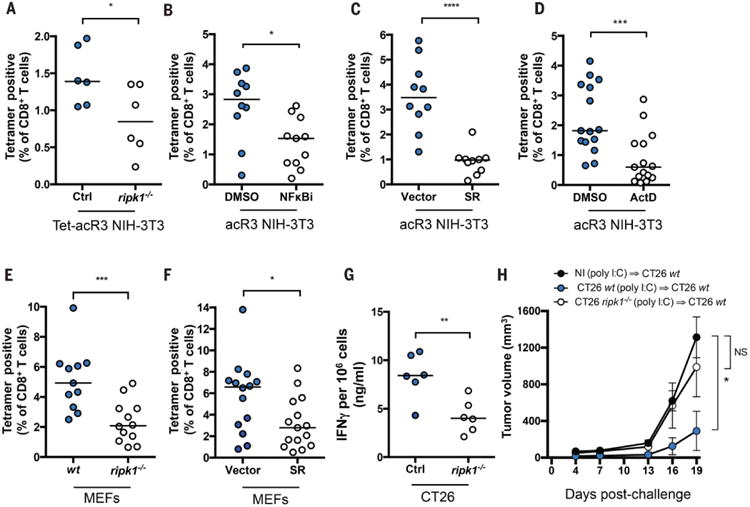
In (A) mice were immunized with Tet-acR3-OVA and Tet-acR3-OVA ripk1−/− NIH-3T3 cells. Data represent one experiment with six mice per group, bars indicate median. In (B) acR3-OVA NIH-3T3 cells were pre-treated with DMSO or BAY 11-7085 (NFκBi-10μM) for 10 min prior to addition of dimerizer and immunization; In (C) mice were immunized with acR3-OVA cells expressing NF-κB-SR or control vector. In (D) acR3-OVA were pre-treated with DMSO or ActD for 45 min prior to immunization. (E and F) OVA-expressing MEFs were transfected with 10μg/ml poly I:C and after 6h used for immunization. WT or cells lacking ripk1−/− were used in (E); or cells expressing control vector or NFκB-SR were used in (F). Cross-priming was assessed on day 9 p.i. In (B to F), N=3 and results shown are pooled from three independent experiments with 3 to 6 mice per group. (G and H) CT26 Ctrl or a CRISPR/cas9-modified line that lacks RIPK1 expression (CT26 ripk1−/−) were pIC-transfected and injected into Balb/cByJ mice. 7 days later, spleen and lymph nodes were harvested and IFNγ production was quantified (G) or mice were challenged with 5×105 WT CT26 injected in the opposite flank and tumor growth was monitored every 3 days (H). N=3 and results are from one representative experiment with 6 mice per group. p values were determined using MW test (A to G) or 2-way ANOVA test (multipe group comaprison) comparing each group to nonimmunized group (NI) (J). *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001.
To extend our findings in a second model that leads to simultaneous RIPK1-dependent NF-κB activation and cell death, we utilized transfection of polyinosinic-polycytidylic acid (poly I:C) (fig. S10A), which engages the cytosolic RNA sensors RIG-I and MDA5, in turn recruiting the adaptor proteins IPS-1, RIPK1, TRADD and FADD (31). We confirmed that in this model, RIPK1 was essential for NF-κB activation (fig. S10, B and C), and cytokine secretion (fig. S10D) (31). Moreover, poly I:C transfection results in intrinsic apoptosis rather than necroptosis (32). We found that in both WT and ripk1−/− cells, poly I:C is capable of inducing similar levels of caspase-3 activation and cell death (fig. S10, E and F). Thus, we were able to decouple NF-κB activation from apoptosis induction downstream of double stranded (ds)RNA sensors. We next tested the hypothesis that immunogenic apopotosis induced by poly I:C(33) was regulated by the RIPK1-NF-κB axis and immunized mice with poly I:C-transfected OVA-expressing mouse embryonic fibroblasts (MEFs) (Fig. 4, E and F). Compared to WT cells, immunization with ripk1−/− (Fig. 4E) or NF-κB SR-expressing cells (Fig. 4F) showed a significant reduction in CD8+ T cell priming. These data reinforce the crucial role of RIPK1-mediated NF-κB activation within dying cells during the initiation of CD8+ T cell immune responses, despite the presence of a strong inflammatory PAMP such a poly I:C.
Finally, we tested the relevance of our findings in the context of tumor immunity. Deletion of RIPK1 from poly I:C-transfected CT26 colon carcinoma cells (fig. S10G) rendered them poorly immunogenic in comparison to WT cells, as measured by interferon (IFN)γ production (Fig. 4G) and protection from tumor challenge (Fig. 4H). Overall, our results reveal RIPK1-induced NF-κB as the critical determinant of CD8+ T cell immunity to cell-associated antigens.
The danger model predicts that cell death resulting from tissue damage and stress induces the passive release of preformed danger molecules that mediate subsequent immune responses (34). Breaking from this model, the present study reveals an unexpected role for RIPK1- and NF-κB-driven gene expression during cell death as a key determinant for cross-priming of CD8+ T cells. Thus, while the release of DAMPs can trigger inflammatory responses, we show that RIPK1-mediated induction of NF-κB and its' downstream target genes are necessary for initiating CD8+ T cell adaptive immunity. To date, PCD pathways are defined by morphological and biochemical methods; our results highlight the need for a transcriptional definition of cell death as a means for understanding the relationship between dying cells and immunity. Whether these findings apply to other aspects of adaptive immunity (e.g., B cell or CD4 T cell priming) remains to be determined (7, 35).
NF-κB is a critical regulator of innate immune responses and is a prime target of pathogen interference, our results suggest an additional benefit for microbes that interfere with both NF-κB signaling (36) and cell death pathways (37, 38). For example, viral inhibitors of RHIM-dependent interactions (e.g., MCMV M45) may have evolved to subvert CD8+ T cell cross-priming. In turn, scaffold proteins such as RIPK1, which are an assemblage of multiple domains (RHIM domain, death domain and kinase domain), may have evolved to coordinate cell death and innate signaling modules (39), together orchestrating adaptive immunity. Thus, investigation and targeting of scaffold proteins at the crossroad of cell death and host defense pathways may provide new therapeutic opportunities in the field of immunotherapy.
Supplementary Material
Acknowledgments
The authors would like to thank S. Zelenay for the LA-ΔOVA-mCherry and ΔOVA-mCherry constructs, R. Weil for the dominant negative NFκB plasmid, P.O. Vidalain for the NFκB-Luciferase reporter plasmid and P. Fitzgerald for cell lines and M.A. Ingersoll, D. Duffy and O. Schwartz, J. Boussier and A. Yatim for their critical reading of the manuscript and advice. We also thank MAI for mouse protocols, M. Fontes for assistance with Qlucore Omics software and the Centre for Human Immunology (CIH) for providing technical support and project management. The data presented in this manuscript are tabulated in the main paper and in the supplementary materials. Dimerizable RIPK3 and caspase-8 constructs are available from Dr. Oberst under an MTA with the University of Washington. Patent PCT/US2014/036196 has been filed by St. Jude Children's Research Hospital covering the induction of necroptosis using RIPK3 oligomerization. Funding for the work was provided by the Agence nationale de recherches sur le sida et les hepatitis (ANRS), the Agence Nationale de la Recherche - projet ANUBIS (ANR-12-BSV3-0011-01) and LabEx Immuno-Onco grant (ANR-11-IDEX-0005-02) (MLA, NY, HS and RbdS). by INCa (Plan Cancer 2014-2019) and Ecole de l'Inserm Liliane Bettencourt (NY), NIH grants R21CA185681 and R01AI108685 (AO), NIH grant AI44848 (DRG), NIH award 5R01AI108685-02 (SO). CRS and OS were supported by grants from Cancer Research UK, The Francis Crick Institute and European Research Council. The authors thanks the Genomic platform from the translational research department of Institut Curie, for experiments conducted using Nanostring tools (Grant INCa-DGOS- 4654, SIRIC11-002, ANR-10-IDEX-0001-02 PSL and ANR-11-LBX-0044).
Footnotes
References and Notes
- 1.Albert ML, Sauter B, Bhardwaj N. Dendritic cells acquire antigen from apoptotic cells and induce class I-restricted CTLs. Nature. 1998;392:86–89. doi: 10.1038/32183. Medline. [DOI] [PubMed] [Google Scholar]
- 2.Albert ML, Darnell JC, Bender A, Francisco LM, Bhardwaj N, Darnell RB. Tumor-specific killer cells in paraneoplastic cerebellar degeneration. Nat Med. 1998;4:1321–1324. doi: 10.1038/3315. Medline. [DOI] [PubMed] [Google Scholar]
- 3.Bevan MJ. Cross-priming for a secondary cytotoxic response to minor H antigens with H-2 congenic cells which do not cross-react in the cytotoxic assay. J Exp Med. 1976;143:1283–1288. doi: 10.1084/jem.143.5.1283. Medline. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Turley S, Poirot L, Hattori M, Benoist C, Mathis D. Physiological beta cell death triggers priming of self-reactive T cells by dendritic cells in a type-1 diabetes model. J Exp Med. 2003;198:1527–1537. doi: 10.1084/jem.20030966. Medline. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Iyer SS, Pulskens WP, Sadler JJ, Butter LM, Teske GJ, Ulland TK, Eisenbarth SC, Florquin S, Flavell RA, Leemans JC, Sutterwala FS. Necrotic cells trigger a sterile inflammatory response through the Nlrp3 inflammasome. Proc Natl Acad Sci USA. 2009;106:20388–20393. doi: 10.1073/pnas.0908698106. Medline. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shi Y, Evans JE, Rock KL. Molecular identification of a danger signal that alerts the immune system to dying cells. Nature. 2003;425:516–521. doi: 10.1038/nature01991. Medline. [DOI] [PubMed] [Google Scholar]
- 7.Kazama H, Ricci JE, Herndon JM, Hoppe G, Green DR, Ferguson TA. Induction of immunological tolerance by apoptotic cells requires caspase-dependent oxidation of high-mobility group box-1 protein. Immunity. 2008;29:21–32. doi: 10.1016/j.immuni.2008.05.013. Medline. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Scaffidi P, Misteli T, Bianchi ME. Release of chromatin protein HMGB1 by necrotic cells triggers inflammation. Nature. 2002;418:191–195. doi: 10.1038/nature00858. Medline. [DOI] [PubMed] [Google Scholar]
- 9.Sauter B, Albert ML, Francisco L, Larsson M, Somersan S, Bhardwaj N. Consequences of cell death: Exposure to necrotic tumor cells, but not primary tissue cells or apoptotic cells, induces the maturation of immunostimulatory dendritic cells. J Exp Med. 2000;191:423–434. doi: 10.1084/jem.191.3.423. Medline. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Scheffer SR, Nave H, Korangy F, Schlote K, Pabst R, Jaffee EM, Manns MP, Greten TF. Apoptotic, but not necrotic, tumor cell vaccines induce a potent immune response in vivo. Int J Cancer. 2003;103:205–211. doi: 10.1002/ijc.10777. Medline. [DOI] [PubMed] [Google Scholar]
- 11.Ochsenbein AF, Klenerman P, Karrer U, Ludewig B, Pericin M, Hengartner H, Zinkernagel RM. Immune surveillance against a solid tumor fails because of immunological ignorance. Proc Natl Acad Sci USA. 1999;96:2233–2238. doi: 10.1073/pnas.96.5.2233. Medline. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gamrekelashvili J, Kapanadze T, Han M, Wissing J, Ma C, Jaensch L, Manns MP, Armstrong T, Jaffee E, White AO, Citrin DE, Korangy F, Greten TF. Peptidases released by necrotic cells control CD8+ T cell cross-priming. J Clin Invest. 2013;123:4755–4768. doi: 10.1172/JCI65698. Medline. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Albert ML. Death-defying immunity: Do apoptotic cells influence antigen processing and presentation? Nat Rev Immunol. 2004;4:223–231. doi: 10.1038/nri11308. Medline. [DOI] [PubMed] [Google Scholar]
- 14.Casares N, Pequignot MO, Tesniere A, Ghiringhelli F, Roux S, Chaput N, Schmitt E, Hamai A, Hervas-Stubbs S, Obeid M, Coutant F, Métivier D, Pichard E, Aucouturier P, Pierron G, Garrido C, Zitvogel L, Kroemer G. Caspase-dependent immunogenicity of doxorubicin-induced tumor cell death. J Exp Med. 2005;202:1691–1701. doi: 10.1084/jem.20050915. Medline. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Obeid M, Tesniere A, Ghiringhelli F, Fimia GM, Apetoh L, Perfettini JL, Castedo M, Mignot G, Panaretakis T, Casares N, Métivier D, Larochette N, van Endert P, Ciccosanti F, Piacentini M, Zitvogel L, Kroemer G. Calreticulin exposure dictates the immunogenicity of cancer cell death. Nat Med. 2007;13:54–61. doi: 10.1038/nm1523. Medline. [DOI] [PubMed] [Google Scholar]
- 16.Feoktistova M, Geserick P, Kellert B, Dimitrova DP, Langlais C, Hupe M, Cain K, MacFarlane M, Häcker G, Leverkus M. cIAPs block Ripoptosome formation, a RIP1/caspase-8 containing intracellular cell death complex differentially regulated by cFLIP isoforms. Mol Cell. 2011;43:449–463. doi: 10.1016/j.molcel.2011.06.011. Medline. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tenev T, Bianchi K, Darding M, Broemer M, Langlais C, Wallberg F, Zachariou A, Lopez J, MacFarlane M, Cain K, Meier P. The Ripoptosome, a signaling platform that assembles in response to genotoxic stress and loss of IAPs. Mol Cell. 2011;43:432–448. doi: 10.1016/j.molcel.2011.06.006. Medline. [DOI] [PubMed] [Google Scholar]
- 18.Blander JM. A long-awaited merger of the pathways mediating host defence and programmed cell death. Nat Rev Immunol. 2014;14:601–618. doi: 10.1038/nri3720. Medline. [DOI] [PubMed] [Google Scholar]
- 19.Martin SJ, Henry CM, Cullen SP. A perspective on mammalian caspases as positive and negative regulators of inflammation. Mol Cell. 2012;46:387–397. doi: 10.1016/j.molcel.2012.04.026. Medline. [DOI] [PubMed] [Google Scholar]
- 20.Oberst A, Pop C, Tremblay AG, Blais V, Denault JB, Salvesen GS, Green DR. Inducible dimerization and inducible cleavage reveal a requirement for both processes in caspase-8 activation. J Biol Chem. 2010;285:16632–16642. doi: 10.1074/jbc.M109.095083. Medline. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Orozco S, Yatim N, Werner MR, Tran H, Gunja SY, Tait SW, Albert ML, Green DR, Oberst A. RIPK1 both positively and negatively regulates RIPK3 oligomerization and necroptosis. Cell Death Differ. 2014;21:1511–1521. doi: 10.1038/cdd.2014.76. Medline. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tait SWG, Oberst A, Quarato G, Milasta S, Haller M, Wang R, Karvela M, Ichim G, Yatim N, Albert ML, Kidd G, Wakefield R, Frase S, Krautwald S, Linkermann A, Green DR. Widespread mitochondrial depletion via mitophagy does not compromise necroptosis. Cell Reports. 2013;5:878–885. doi: 10.1016/j.celrep.2013.10.034. Medline. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dillon CP, Weinlich R, Rodriguez DA, Cripps JG, Quarato G, Gurung P, Verbist KC, Brewer TL, Llambi F, Gong YN, Janke LJ, Kelliher MA, Kanneganti TD, Green DR. RIPK1 blocks early postnatal lethality mediated by caspase-8 and RIPK3. Cell. 2014;157:1189–1202. doi: 10.1016/j.cell.2014.04.018. Medline. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ghiringhelli F, Apetoh L, Tesniere A, Aymeric L, Ma Y, Ortiz C, Vermaelen K, Panaretakis T, Mignot G, Ullrich E, Perfettini JL, Schlemmer F, Tasdemir E, Uhl M, Génin P, Civas A, Ryffel B, Kanellopoulos J, Tschopp J, André F, Lidereau R, McLaughlin NM, Haynes NM, Smyth MJ, Kroemer G, Zitvogel L. Activation of the NLRP3 inflammasome in dendritic cells induces IL-1beta-dependent adaptive immunity against tumors. Nat Med. 2009;15:1170–1178. doi: 10.1038/nm.2028. Medline. [DOI] [PubMed] [Google Scholar]
- 25.Tesniere A, Schlemmer F, Boige V, Kepp O, Martins I, Ghiringhelli F, Aymeric L, Michaud M, Apetoh L, Barault L, Mendiboure J, Pignon JP, Jooste V, van Endert P, Ducreux M, Zitvogel L, Piard F, Kroemer G. Immunogenic death of colon cancer cells treated with oxaliplatin. Oncogene. 2010;29:482–491. doi: 10.1038/onc.2009.356. Medline. [DOI] [PubMed] [Google Scholar]
- 26.Michaud M, Martins I, Sukkurwala AQ, Adjemian S, Ma Y, Pellegatti P, Shen S, Kepp O, Scoazec M, Mignot G, Rello-Varona S, Tailler M, Menger L, Vacchelli E, Galluzzi L, Ghiringhelli F, di Virgilio F, Zitvogel L, Kroemer G. Autophagy-dependent anticancer immune responses induced by chemotherapeutic agents in mice. Science. 2011;334:1573–1577. doi: 10.1126/science.1208347. Medline. [DOI] [PubMed] [Google Scholar]
- 27.Schulz O, Reis e Sousa C. Cross-presentation of cell-associated antigens by CD8α+ dendritic cells is attributable to their ability to internalize dead cells. Immunology. 2002;107:183–189. doi: 10.1046/j.13652567.2002.01513.x. Medline. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kurts C, Robinson BWS, Knolle PA. Cross-priming in health and disease. Nat Rev Immunol. 2010;10:403–414. doi: 10.1038/nri2780. Medline. [DOI] [PubMed] [Google Scholar]
- 29.Sancho D, Joffre OP, Keller AM, Rogers NC, Martínez D, Hernanz-Falcón P, Rosewell I, Reis e Sousa C. Identification of a dendritic cell receptor that couples sensing of necrosis to immunity. Nature. 2009;458:899–903. doi: 10.1038/nature07750. Medline. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hildner K, Edelson BT, Purtha WE, Diamond M, Matsushita H, Kohyama M, Calderon B, Schraml BU, Unanue ER, Diamond MS, Schreiber RD, Murphy TL, Murphy KM. Batf3 deficiency reveals a critical role for CD8α+ dendritic cells in cytotoxic T cell immunity. Science. 2008;322:1097–1100. doi: 10.1126/science.1164206. Medline. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Michallet MC, Meylan E, Ermolaeva MA, Vazquez J, Rebsamen M, Curran J, Poeck H, Bscheider M, Hartmann G, König M, Kalinke U, Pasparakis M, Tschopp J. TRADD protein is an essential component of the RIG-like helicase antiviral pathway. Immunity. 2008;28:651–661. doi: 10.1016/j.immuni.2008.03.013. Medline. [DOI] [PubMed] [Google Scholar]
- 32.Besch R, Poeck H, Hohenauer T, Senft D, Häcker G, Berking C, Hornung V, Endres S, Ruzicka T, Rothenfusser S, Hartmann G. Proapoptotic signaling induced by RIG-I and MDA-5 results in type I interferon-independent apoptosis in human melanoma cells. J Clin Invest. 2009;119:2399–2411. doi: 10.1172/JCI37155. Medline. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Schulz O, Diebold SS, Chen M, Näslund TI, Nolte MA, Alexopoulou L, Azuma YT, Flavell RA, Liljeström P, Reis e Sousa C. Toll-like receptor 3 promotes cross-priming to virus-infected cells. Nature. 2005;433:887–892. doi: 10.1038/nature03326. Medline. [DOI] [PubMed] [Google Scholar]
- 34.Matzinger P. Tolerance, danger, and the extended family. Annu Rev Immunol. 1994;12:991–1045. doi: 10.1146/annurev.iy.12.040194.005015. Medline. [DOI] [PubMed] [Google Scholar]
- 35.Gallucci S, Lolkema M, Matzinger P. Natural adjuvants: Endogenous activators of dendritic cells. Nat Med. 1999;5:1249–1255. doi: 10.1038/15200. Medline. [DOI] [PubMed] [Google Scholar]
- 36.Rahman MM, McFadden G. Modulation of NF-κB signalling by microbial pathogens. Nat Rev Microbiol. 2011;9:291–306. doi: 10.1038/nrmicro2539. Medline. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lamkanfi M, Dixit VM. Manipulation of host cell death pathways during microbial infections. Cell Host Microbe. 2010;8:44–54. doi: 10.1016/j.chom.2010.06.007. Medline. [DOI] [PubMed] [Google Scholar]
- 38.Yatim N, Albert ML. Dying to replicate: The orchestration of the viral life cycle, cell death pathways, and immunity. Immunity. 2011;35:478–490. doi: 10.1016/j.immuni.2011.10.010. Medline. [DOI] [PubMed] [Google Scholar]
- 39.Weinlich R, Green DR. The two faces of receptor interacting protein kinase-1. Mol Cell. 2014;56:469–480. doi: 10.1016/j.molcel.2014.11.001. Medline. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fuertes Marraco SA, Grosjean F, Duval A, Rosa M, Lavanchy C, Ashok D, Haller S, Otten LA, Steiner QG, Descombes P, Luber CA, Meissner F, Mann M, Szeles L, Reith W, Acha-Orbea H. Novel murine dendritic cell lines: A powerful auxiliary tool for dendritic cell research. Front Immunol. 2012;3:331. doi: 10.3389/fimmu.2012.00331. Medline. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cullen SP, Henry CM, Kearney CJ, Logue SE, Feoktistova M, Tynan GA, Lavelle EC, Leverkus M, Martin SJ. Fas/CD95-induced chemokines can serve as “find-me” signals for apoptotic cells. Mol Cell. 2013;49:1034–1048. doi: 10.1016/j.molcel.2013.01.025. Medline. [DOI] [PubMed] [Google Scholar]
- 42.Kearney CJ, Cullen SP, Tynan GA, Henry CM, Clancy D, Lavelle EC, Martin SJ. Necroptosis suppresses inflammation via termination of TNF- or LPS-induced cytokine and chemokine production. Cell Death Differ. 2015;22:1313–1327. doi: 10.1038/cdd.2014.222. Medline. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kang TB, Yang SH, Toth B, Kovalenko A, Wallach D. Caspase-8 blocks kinase RIPK3-mediated activation of the NLRP3 inflammasome. Immunity. 2013;38:27–40. doi: 10.1016/j.immuni.2012.09.015. Medline. [DOI] [PubMed] [Google Scholar]
- 44.Ea CK, Deng L, Xia ZP, Pineda G, Chen ZJ. Activation of IKK by TNFalpha requires site-specific ubiquitination of RIP1 and polyubiquitin binding by NEMO. Mol Cell. 2006;22:245–257. doi: 10.1016/j.molcel.2006.03.026. Medline. [DOI] [PubMed] [Google Scholar]
- 45.Kelliher MA, Grimm S, Ishida Y, Kuo F, Stanger BZ, Leder P. The death domain kinase RIP mediates the TNF-induced NF-κB signal. Immunity. 1998;8:297–303. doi: 10.1016/S1074-76130080535-X. Medline. [DOI] [PubMed] [Google Scholar]
- 46.Wong WWL, Gentle IE, Nachbur U, Anderton H, Vaux DL, Silke J. RIPK1 is not essential for TNFR1-induced activation of NF-κB. Cell Death Differ. 2010;17:482–487. doi: 10.1038/cdd.2009.178. Medline. [DOI] [PubMed] [Google Scholar]
- 47.Meylan E, Burns K, Hofmann K, Blancheteau V, Martinon F, Kelliher M, Tschopp J. RIP1 is an essential mediator of Toll-like receptor 3-induced NF-κB activation. Nat Immunol. 2004;5:503–507. doi: 10.1038/ni1061. Medline. [DOI] [PubMed] [Google Scholar]
- 48.Rebsamen M, Heinz LX, Meylan E, Michallet MC, Schroder K, Hofmann K, Vazquez J, Benedict CA, Tschopp J. DAI/ZBP1 recruits RIP1 and RIP3 through RIP homotypic interaction motifs to activate NF-κB. EMBO Rep. 2009;10:916–922. doi: 10.1038/embor.2009.109. Medline. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.