

Abstract
In portions of South Asia, vectors and patients co-infected with dengue (DENV) and chikungunya (CHIKV) are on the rise, with the potential for this occurrence in other regions of the world, for example the United States. Therefore, we engineered an antiviral approach that suppresses the replication of both arboviruses in mosquito cells using a single antiviral group I intron. We devised unique configurations of internal, external, and guide sequences that permit homologous recognition and splicing with conserved target sequences in the genomes of both viruses using a single trans-splicing Group I intron, and examined their effectiveness to suppress infections of DENV and CHIKV in mosquito cells when coupled with a proapoptotic 3' exon, ΔN Bax. RT-PCR demonstrated the utility of these introns in trans-splicing the ΔN Bax sequence downstream of either the DENV or CHIKV target site in transformed Aedes albopictus C6/36 cells, independent of the order in which the virus specific targeting sequences were inserted into the construct. This trans-splicing reaction forms DENV or CHIKV ΔN Bax RNA fusions that led to apoptotic cell death as evidenced by annexin V staining, caspase, and DNA fragmentation assays. TCID50-IFA analyses demonstrate effective suppression of DENV and CHIKV infections by our anti-arbovirus group I intron approach. This represents the first report of a dual-acting Group I intron, and demonstrates that we can target DENV and CHIKV RNAs in a sequence specific manner with a single, uniquely configured CHIKV/DENV dual targeting group I intron, leading to replication suppression of both arboviruses, and thus providing a promising single antiviral for the transgenic suppression of multiple arboviruses.
Introduction
The WHO estimates hundreds of millions of infections and tens of thousands of deaths each year are attributed to mosquito-borne virus related diseases, with well over half the world’s population remaining at risk for infection [1–7]. Individual outbreaks along with instances of co-infections of dengue (DENV) and chikungunya viruses (CHIKV), are on the rise due to the presence of both pathogens in a shared mosquito vector, Aedes albopictus [2,8–12].
Infection with one of four orthologous, but antigenically distinct DENV serotypes (designated DENV 1 through 4) can result in dengue fever (DF) or dengue hemorrhagic fever (DHF) [1]. DF and DHF are endemic to tropical and subtropical regions of the world, but global changes in climate, rapid dispersal of virus due to world travel, and migration of humans to non-tropical regions has resulted in epidemic DENV outbreaks in areas that are non-endemic for these viruses [13,14]. There are currently no consistently effective preventive control measures or approved tetravalent vaccines to combat DENV.
CHIKV is an emerging pathogen that infects humans with the principle mosquito vectors being Ae. aegypti, Ae. albopictus, and Ae. vigilax [15], the same vectors responsible for dengue virus spread [16–18]. Following a 2–12 day incubation period, clinical symptoms develop that are similar to dengue fever including high fever, a prominent rash on the thorax and face, headache, back pain, and myalgia. An intense arthralgia distinguishes CHIK fever from DF. Hemorrhagic fever resulting from CHIKV infection, has been reported during outbreaks in Thailand [2].
CHIKV has been transmitted throughout Asia and Africa since the initial discovery of this virus in Tanzania in 1952 [2,19–25]. Importation of this virus into Europe and the USA resulted from infected travelers returning from endemic areas with high incidences of CHIKV infection and Ae. albopictus transmission [26], underscoring the potential for a worldwide CHIKV epidemic and the need for novel therapies to effectively combat the spread of this virus. Most recently CHIKV transmission has occurred in the French Riviera [27], the Caribbean islands, and the United States [28–30].
Our lab has been surveying ribozymes as suppressive agents against arbovirus infection for potential use in generating refractory transgenic mosquitoes. We previously examined the effectiveness of hammerhead ribozymes in suppressing DENV infections in retrovirus transduced mosquito cells [31]. This led to the identification of several hammerhead ribozymes effective in significantly reducing DENV serotype 2 New Guinea strain (DENV2-NGC) infection of Ae. albopictus C6/36 cells. However, due to the relatively strict triplet nucleotide sequence requirements for catalysis, engineering a single hammerhead ribozyme possessing the ability to target all DENV serotypes as well as CHIKV is not practical. This necessitated exploration of ribozymes that have an increased potential for broader specificity and utility.
Trans-splicing group I introns provide a versatile tool for repairing defective RNA [32–44]. Trans-splicing group I introns have also demonstrated utility in targeting the RNA genomes of a number of viruses such as HIV-1 tat [45], cucumber mosaic virus coat protein mRNAs [32], and the hepatitis C virus internal ribosome entry site (HCV-IRES; [46,47]). In recent years, our lab has demonstrated the effectiveness of a group I intron trans-splicing strategy to target conserved sequences among all DENV genomes [48].
In a previous report, we noted that the 5' end of the transcript encoding our anti-DENV group I intron was composed of a 56 nt untranslated RNA leader sequence expressed from the Actin 5c promoter [49]. This observation led us to consider the possibility that additional sequences might be appended to the 5' terminus of our anti-DENV group I intron in a way that allows the recognition and catalysis of another virus genome.
This report describes the development of dual targeting group I introns for the replication suppression of all DENV serotypes and CHIKV through a “death by infection” strategy that involves the inclusion of a proapoptotic effector gene that is active only when fused to CHIKV or DENV RNA. These introns are based upon the previously described anti-DENV group I intron [48] that is modified by the addition of CHIKV targeting external and internal guide sequences, along with a corresponding interacting P10 sequence, that allows targeting of either CHIKV or DENV genomic RNAs. When coupled with an apoptosis-inducing ΔN Bax 3’ exon, these constructs trans-splice conserved sequences of either CHIKV or DENV to the ΔN Bax 3’ exon, leading to the induction of apoptotic cell death upon virus infection. We demonstrate antiviral and proapoptotic activity for these uniquely constructed group I introns in transformed mosquito cell cultures challenged with infectious DENV serotypes 1 through 4 and CHIKV.
The trans-splicing reaction of these dual targeting intron constructs was designed to attack the conserved circularization sequence (CS) of the DENV genome as well as a highly conserved region of the CHIKV NS1 gene. Splicing appends a 3’ exon RNA sequence encoding the ΔN Bax sequence to the capsid (DENV) or the NS1 (CHIKV) protein coding regions of the respective genomic RNAs resulting in translation of a chimeric protein that induces premature cell death upon infection by either DENV or CHIKV. TCID50-IFA analyses demonstrate suppression of each of the four DENV serotypes, and CHIKV 181/25 vaccine test strain. Results obtained from annexin V staining, effector caspase assays, and DNA ladder assays confirm that the resulting DENV CA-ΔN Bax (DCA- ΔN Bax) or CHIKV NS1- ΔN Bax (CNS1-ΔN Bax) fusion proteins induce apoptotic cell death. Our cumulative results confirm the effectiveness of our dual targeting (DENV and CHIKV) group I intron as a sequence specific antiviral that should be useful for suppression of DENV and CHIKV replication in transgenic mosquitoes.
Results
Basic trans-splicing mechanism of the group I intron
The Tetrahymena thermophila group I intron trans-splicing mechanism is characterized by two independent transesterification reactions ([50]; Fig 1). The ribozyme-mediated targeting of substrate RNAs forms the P1 and extended antisense helices, which promotes guanosine-mediated transesterification resulting in cleavage of the target virus RNA (Step 1). An internal guide sequence (IGS) of 9 bases complimentary base pairs with the target RNA, except a reactive guanosine that forms a wobble base pair with the targeted uracil to initiate the cleavage reaction. The external guide sequence (EGS) Watson-Crick base pairs with the substrate RNA nucleotides, downstream from the reactive uracil. This interaction provides additional stability for the trans-splicing reaction. Sequences from the 3’ exon (ΔN Bax) displace the distal portion of the P1 helix to form the P10 helix (Step 2). This allows the second transesterification to proceed, resulting in ligation of the DENV or CHIKV capsid and ΔN Bax. The end result is a new RNA fusion molecule that, if appropriately configured, can be translated into a new proapoptotic protein (Step 3).
Fig 1. Trans-splicing reaction catalyzed by the group I intron.
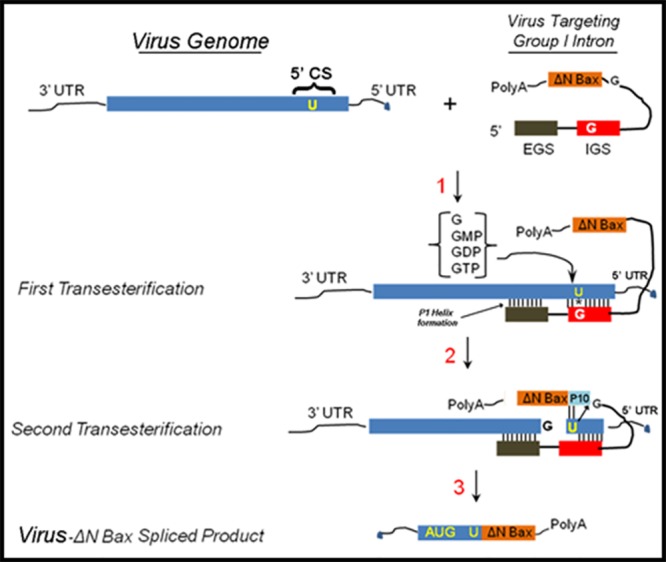
First step → Intron finds its target RNA sequence through complimentary base pairing with the internal and external guide sequences (IGS and EGS, respectively). The intron then sequesters the 3’-OH of a free-floating guanosine (GTP, GDP, GMP, or guanosine alone). The 3’ GNP OH attacks the phosphodiester backbone directly downstream of the reactive uracil on the 5’ exon. Second step → The 3’ exon is brought into proximity with the newly freed 3’-OH on the cleavage uracil, guided by the P10 helix. The 3’-OH attacks the phosphodiester backbone just upstream of the 3’ exon in another transesterification reaction, resulting in the 5’ exon and the 3’ exon being joined covalently. Third step → The end result is a new mRNA molecule, functional and ready for translation, formed of two separate RNA molecules. Adopted from Figure 1 in reference [49].
Our lab has demonstrated the effectiveness of group I introns targeting sequences in the DENV 5’ CS [48]. While these group I introns efficiently cleave this conserved region, the use of these molecules as simple catalytic cleavage agents requires levels of expression that match or exceed those for the generation of virus in infected cells. Escape events may be possible in the event virus replication exceeds group I intron catalytic suppression. As a result, we presumed that expression of anti-viral group I introns alone in cells may not be the ideal method for suppressing DENV expression and decreasing the probability of generating escape mutants. We reasoned that coupling the splicing activity of the group I intron to a death-upon-infection strategy would provide an added level of insurance against the development of escape mutants. We therefore designed anti-DENV group I introns coupled with the apoptosis-inducing genes as the 3’ exon to induce cell death upon infection with DENV and validated this approach as an effective antiviral in C6/36 cells [47].
Construction of CHIKV targeting Group I intron
ClustalX alignments of available CHIKV genome sequences revealed the presence of a conserved region located within the NS1 gene of CHIKV (S5 Fig). This conserved sequence occurs in genomic forms of CHIKV RNA that are present during the early and late stages of alphavirus infection [51], and represents an ideal target for our antiviral group I strategy (Fig 2A). The anti-CHIKV group I intron was produced through PCR amplification of the anti-DENV group I intron [48,49], with a forward primer that possessed EGS and IGS sequences complimentary to nucleotides C201 to C209 and G188 to U196, respectively, of the CHIKV RNA genome. The CHIKV P10 helix forming sequences (S1 Table) and a UAA stop codon were inserted into our anti-CHIKV group I intron as previously described [48,49]. The UAA triplet was inserted immediately upstream of the UCG splice site to prevent inadvertent expression of the 3’ exon, ΔN-Bax [48,49]. Though the conserved region of the CHIKV target RNA does not possess an invariant base, a single variable base at nucleotide 152 of the DENV RNA was positioned within a non-homologous bulge loop (BL) structure that separates the IGS and EGS, and therefore did not influence the targeting of the intron (42). This BL structure promotes the formation of the P10 helix, which increases the catalytic efficiency of the intron (45).
Fig 2. Schematic Representation of the A. CHIKV and B. DENV genomes, respectively, with regions targeted by the Dual Targeting Introns.
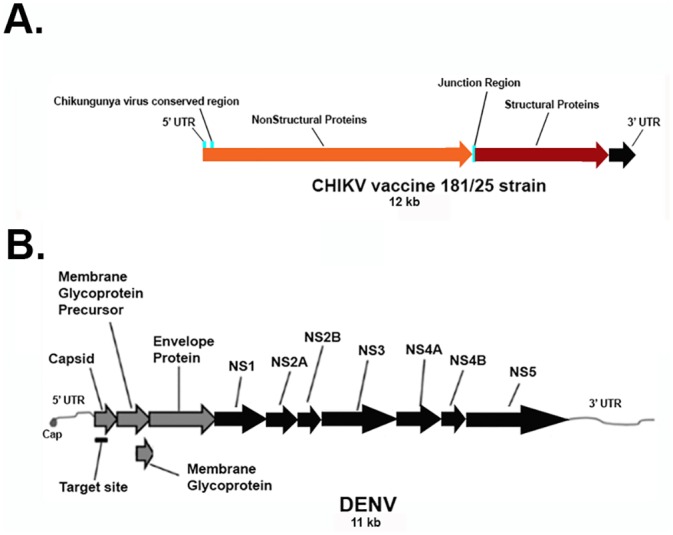
A. CHIKV genomic RNA diagram with the indicated conserved region targeted for viral suppression. The CHIKV conserved domain, labeled as the CHIKV Conserved Region, was determined through a Clustal X alignment of GenBank obtained CHIKV sequences (see Materials and Methods and S5 Fig). B. The most invariant segments of the DENV RNA are known as the 5’ and 3’ cyclization sequences (labeled as the Group I Intron target site). These sequences are so named because they are complementary to each other and are thought to be involved in the formation of a panhandle structure during genome replication.
Construction of anti-DENV and anti-DENV/CHIKV dual targeting group I intron-ΔN Bax vectors
In previous reports, we described the construction and trans-splicing activity of an anti-DENV group I intron that was designed to effectively trans-splice all known DENV sequences by targeting the uracil at position 143 within the DENV 5’CS [48,49] (Fig 2B; see Materials and Methods). This trans-splicing anti-DENV group I intron included a 9 nucleotide EGS that Watson-Crick base pairs to sequences that are fully conserved among all known DENV genomes to improve catalytic efficiency and minimize potential off-target splicing interactions. A 9 base IGS was also present in this antiviral intron, possessing a reactive guanosine that forms a wobble base pair with the targeted uracil on the substrate DENV RNA (Fig 1). The anti-DENV group I intron was used in this study as a positive control for DENV trans-splicing and a negative control for CHIKV trans-splicing. It also served as a PCR template for the production of anti-CHIKV and DENV/CHIKV dual targeting group I introns.
The antiviral group I intron constructs that we refer to as “dual targeting introns” were constructed by engineering the DENV-targeting antiviral group I intron [48,49], to include EGS, IGS, and P10 helix forming sequences specific for CHIKV (Fig 3). This was accomplished by incorporating IGS, EGS, and P10 sequences specific to the highly conserved NS1 region of the CHIKV RNA into the previously successful anti-DENV 9v1 construct [48,49], allowing targeting of genomic CHIKV RNA as well as DENV genomic RNAs by a single antivirus group I intron (Fig 3). Excluding the wobble base with the uracil at nucleotide position 143 (for DENV RNA targeting) or nucleotide position 193 (for CHIKV RNA targeting), which are required for proper cleavage [52–55], 17 bases of each set of targeting sequences from each dual targeting intron interacted directly with the intended target sequence.
Fig 3. anti-CHIKV/DENV Dual Targeting Introns in a Bicistronic Configuration.

Each of the trans-splicing dual targeting introns was tagged downstream of the ΔN Bax 3’ exon (active, further truncated version of tBax) with the mCherry fluorescent marker gene expressed from an IRES sequence of the Drosophila C Virus (DCV). Expression of these constructs was driven by the Drosophila melanogaster Actin 5c promoter. Each of these Dicistrovirus IRES sequences was previously determined to yield the highest levels of expression in Ae. aegypti mosquito cells [48]. The bicistronic configuration allowed monitoring for the presence and expression of these constructs within cell cultures through dual expression of antiviral intron-ΔN Bax and mCherry cistronic portions of the plasmid. Version 1 (v1) and Version 2 (v2) refer to the order of the CHIKV and DENV specific P10 helix sequences. CHIKV = Chikungunya virus targeting sequences; DENV = Dengue virus targeting sequences; IRES = internal ribosome entry site; EGS = external guide sequence; IGS = internal guide sequence; BL = bulge loop; TSD = trans splicing domain; P10 = P10 helix.
The CHIKV/DENV dual targeting version 1 (v1) and version 2 (v2) group I intron constructs differ by the 5’ to 3’ arrangement of the p10 helix forming sequences that are specific for CHIKV and DENV (Fig 3). For example, the dual targeting introns with the v1 designation possess the CHIKV p10 immediately upstream (5’) of the DENV p10. The vice versa is true for the v2 dual targeting intron configurations.
The ΔN Bax coding sequence, corresponding to amino acids 112–192 of the Bax protein, was included as the 3' exon (Fig 3). The ΔN Bax protein has been shown to induce cell death in A549 and NCI-H1299 cell lines more efficiently than tBax [56]. The increased induction of apoptosis is due to deletion of the Bax BH-3 domain that facilitates protein-protein interactions between Bax and Bcl-2 or other anti-apoptotic regulators. To insure that this potent apoptosis inducer was not expressed from the intron construct, we inserted a UAA stop codon into the P9.0 helix of the group I intron immediately upstream of the UCG splice donor [48,49]. Linking the 3’exon, ΔN-Bax, to these dual targeting, antivirus group I introns was designed to insure replication suppression of two different viral RNA genomes, in this case DENV and CHIKV, with a single group I intron.
We previously constructed negative controls for trans-splicing activity by removing the entire catalytic core, domains P4 through P6, of the trans-splicing intron [48]. Using this negative control group I intron as a PCR template, we engineered two negative control group I introns, ΔCHIKV/DENVv1 and ΔDENV/CHIKVv1, as described in Materials and Methods.
The DENV/CHIKV dual targeting intron-ΔN Bax constructs were further modified by attaching a Drosophila C Virus (DCV) IRES-driven mCherry fluorescent marker gene immediately downstream of the ΔN Bax 3’ exon (Fig 3). This bi-cistronic configuration allowed monitoring for the presence and expression of the dual targeting CHIKV/DENVgroup I intron constructs within mosquito cell cultures. Expression of the entire construct was driven in mosquito cells by the Drosophila melanogaster Actin 5c promoter (A5c).
Dual targeting CHIKV/DENV-ΔN Bax intron constructs effectively target CHIKV and all DENV serotypes analyzed.
Our previous studies demonstrated that anti-DENV group I intron-firefly luciferase (FL) and anti-DENV group I intron-ΔN Bax constructs were capable of effectively targeting the 5’ CS region located within the RNA of all DENV serotypes [48,49]. We followed a similar protocol to examine the ability of our dual targeting constructs to effectively target, splice, and suppress both DENV and CHIKV in transformed cells (Fig 4).
Fig 4. CHIKV/DENV dual targeting intron–ΔN Bax constructs effectively target CHIKV and all four DENV serotypes tested.

Clonal Ae. albopictus C6/36 cells transformed with constructs bearing anti-CHIKV, anti-DENV, or the catalytically active and inactive forms of the anti-CHIKV/DENV dual targeting intron constructs were infected with one of the arboviruses indicated on each figure panel and analyzed for the presence of splice product by RT-PCR with heterologous primers as stated in Materials and Methods. The title of each figure panel indicates the virus the CHIKV/DENV dual targeting intron–ΔN Bax constructs were tested against. CHIKV/DENV-ΔN Bax or DENV/CHIKV-ΔN Bax refers to the CHIKV/DENV dual targeting trans-splicing group I introns that were designed for dual targeting of CHIKV and DENV. These introns are coupled to the proapoptotic ΔN Bax as the 3’ exon. ‘v1’ and ‘v2’ refer to the order of the CHIKV and DENV specific P10 helix-forming sequences, as explained in Materials and Methods ΔCHIKV/DENVv1-ΔN Bax (ΔCD) or ΔDENV/CHIKVv1-ΔN Bax (ΔCD) refer to the anti- CHIKV/DENV dual targeting introns possessing the inactive deletion mutation of the trans-splicing domain that is linked to the ΔN Bax 3’ exon. The deletion mutation of the trans-splicing domain is designed to knock out trans-splicing function, providing a negative control [76]. Control RT-PCR experiments were performed with primers for actin to confirm similar RNA loading. Heterolgous primers to the intron-ΔN Bax segment of the dual targeting intron construct were used to confirm the presence of our anti-CHIKV/DENV introns. All constructs are linked to the DCV IRES-mCherry configuration as shown in Fig 3. The identity of spliced products was confirmed by sequencing (Data not shown).
Intron-expressing stable lines were generated by co-transfection of C6/36 cells with a hygromycin selectable marker plasmid along with one of the A5c promoter plasmids expressing each of the dual targeting CHIKV/DENV group I introns, or with a negative control intron coupled to the 3’ ΔN Bax exon, as described in Materials and Methods and previous reports [48,49]. Post-spliced products of approximately 300 or 310 bp were recovered as a result of targeting either DENV or CHIKV by the respective singlet antiviral introns (lanes 3 and 4), or dual antivirus intron (lanes 5 through 8) targeting of DENV or CHIKV (Fig 4). The identity of these presumptive spliced product bands was confirmed by sequencing (Data not shown).
As expected we did not detect spliced products by RT-PCR of RNAs extracted from cells transfected with the negative control dual ΔCHIKV/DENV-ΔN Bax vector constructs (ΔC/D and ΔD/C, Lanes 1 and 2, respectively; Fig 4). Moreover, DENV CA-ΔN Bax spliced product was not detected when cells transformed with the antiviral intron designed to target only CHIKV were challenged with DENV (lane 3), nor was the CHIKV NS1 ΔN Bax spliced product detected when anti-DENV-ΔN Bax transformed C6/36 cells were challenged with CHIKV (lane 4).
CHIKV/DENV dual targeting intron-ΔN Bax constructs initiate apoptosis upon arbovirus infection
Annexin V apoptosis assays were performed to establish the ability of expressed DENV CA-ΔN Bax or CHIKV NS1-ΔN Bax fusion proteins to initiate apoptosis [57–59]. C6/36 clonals stably expressing CHIKV-ΔN Bax, DENV-ΔN Bax, or the dual targeting intron-ΔN Bax constructs were challenged with each DENV serotype indicated, or CHIKV strain and analyzed by annexin V staining as described in Materials and Methods (Fig 5). As a control, C6/36 cell lines stably expressing CHIKV-ΔN Bax, DENV-ΔN Bax, and CHIKV/DENV dual targeting intron-ΔN Bax constructs were tested in the absence of virus for annexin V activity to ensure that stable expression of the intron itself does not trigger apoptosis.
Fig 5. Promotion of the initial stages of apoptosis by the activation of our Dual Targeting Intron Constructs.

Clonal Ae. albopictus C6/36 cells transformed with the single or dual targeting antiviral intron constructs indicated were challenged with eeach of the arboviruses shown on the figure. Cells were stained with FITC conjugated annexin V and analyzed the manufacturer’s instructions (Cayman Chemical Company; see Materials and Methods). ΔC/D- or ΔD/C refer to the anti- CHIKV/DENV dual targeting introns possessing the inactive deletion mutation of the trans-splicing domain that is linked to the ΔN Bax 3’ exon. Wt (wild type) refers to a negative control C6/36 cell line that does not express antiviral intron constructs. Six individual clonal cell populations per single or dual virus targeting intron construct are shown. Each number corresponds to the individual clonal cell population tested. Three wells were infected per clonal cell population and analyzed for annexin V-FITC staining. The experiment was performed at three independent times resulting in a total of nine replicates for each dual-targeting intron-ΔN Bax tested.
As expected, non-transfected (designated as "wild type") C6/36 cells, as well as clonal cells expressing the inactive anti-CHIKV/DENVv1 group I intron-ΔN Bax constructs (ΔCHIKV/DENVv1-ΔN Bax or ΔDENV/CHIKV v1-ΔN Bax), displayed background annexin V-FITC staining following DENV or CHIKV infection (Fig 5). Expression of the active dual targeting intron constructs with ΔN Bax as the 3’ exon, irrespective of the DENV serotype or CHIKV challenge, led to the activation of cellular apoptotic pathways as indicated by the binding of FITC-conjugated annexin V to the PS externalized on the cell surface [59].
All CHIKV/DENV dual targeting-ΔN Bax clonal cell lines displayed 2 fold greater initiation of apoptosis than negative control cells
Although C6/36 cell lines stably expressing the anti-CHIKV -ΔN Bax construct did not exhibit annexin V staining above background in the presence of DENV infection, CHIKV challenge resulted in significant annexin V staining (Fig 5). Similarly, C6/36 cell lines stably expressing the anti-DENV-ΔN Bax construct did not exhibit annexin V staining above background in the presence of CHIKV infection, while DENV challenge resulted in positive staining (Fig 5). These results further demonstrate the specificity of our anti-CHIKV/DENV dual targeting intron approach.
Activity of CHIKV/DENV dual targeting intron-ΔN Bax constructs against DENV and CHIKV results in further progression of apoptosis
Caspases are associated with the initiation of the “death cascade” and are important markers of the cell’s entry point into apoptosis [60,61]. Caspase-3 is a mammalian-specific effector caspase. However, mosquitoes possess several “effector caspases”, and it is not clear which caspase(s) is/are involved in apoptosis in Aedes mosquito cells [62]. Therefore, for the purposes of this paper, we will simply refer to these mosquito caspases as ‘effector caspases’.
Clonal C6/36 cell populations expressing anti-CHIKV/DENV-ΔN Bax constructs, linked to a DCV IRES-mCherry configuration, were challenged with each of the four DENV serotypes or the CHIKV strain 181/25, and analyzed (Fig 6), as described in Materials and Methods. As a control, C6/36 cell lines stably expressing antiviral intron constructs were tested in the absence of virus for effector caspase activity to ensure that stable expression of the intron itself does not trigger apoptosis.
Fig 6. Effector caspase activation confirms the induction of apoptosis by the activation of Dual Targeting Intron Constructs.

Clonal Ae. albopictus C6/36 cells transformed with the single or anti-CHIKV/DENV dual targeting antiviral intron constructs indicated were challenged with either of the four DENV serotypes or CHIKV vaccine strain 181/25 (MOI 0.01). 1x106 cells were assessed for effector caspase activity, per the manufacturer’s instructions (see Materials and Methods). ΔC/D-ΔN Bax or Δ/C-ΔN Bax refer to the anti- CHIKV/DENV dual targeting introns possessing the inactive deletion mutation of the trans-splicing domain that is linked to the ΔN Bax 3’ exon. Wt (wild type) refers to a negative control C6/36 cell line that does not express antiviral intron constructs. Numbered lanes indicate clonal cell populations expressing single or dual virus targeting intron constructs. Three wells were infected per clonal cell population and analyzed for “effector caspase” activity. The experiment was performed at three independent times resulting in a total of nine replicates for each dual-targeting intron-ΔN Bax tested.
In the presence of either CHIKV or DENV challenge, all CHIKV/DENVv1-ΔN Bax clonal cell lines displayed 10 fold greater initiation of apoptosis than negative control cells. Five out of 6 DENV/CHIKVv1 clonal cell lines initiated apoptosis at levels similar to CHIKV/DENVv1 clonals, as was the case for 4 out of 6 for CHIKV/DENVv2 and 5 out of 6 DENV/CHIKVv2 clonal cell populations tested. As expected, wild type C6/36 or ΔCHIKV/DENVv1-ΔN Bax or ΔDENV/CHIKV v1-ΔN Bax constructs displayed only background levels of effector caspase activity, following DENV or CHIKV infection.
Uninfected wild type C6/36 displayed minimal effector caspase activity, comparable to background levels following CHIKV or DENV infection. Similar results were also observed for cell clones transformed with the active anti-DENV group I intron following CHIKV challenge, and the active anti-CHIKV group I intron following DENV challenge, demonstrating virus specificity of our anti-arbovirus approach. The absence of significant effector caspase activity observed with the inactive dual targeting introns ΔCHIKV/DENVv1-ΔN Bax or ΔDENV/CHIKV v1 following CHIKV or DENV challenge demonstrated effective suppression of read through of the ΔN Bax 3’ exon linked to these intron constructs.
A hallmark of apoptosis is the degradation of nuclear DNA into nucleosomal units of approximately 180 bp in length [63], yielding a DNA ladder appearance that can be visualized on an agarose gel. DNA fragmentation analysis was performed on infected anti-CHIKV/DENV dual targeting intron-ΔN Bax expressing clonal cell lines, as described previously [64,65] and in Materials and Methods, to further demonstrate the apoptosis inducing capabilities of our antiviral constructs (Fig 7 and S1 Fig through S5 Fig).
Fig 7. DNA Fragmentation Assay.

Clonal Aedes albopictus C6/36 cells transformed with the anti-CHIKV, anti-DENV, anti-CHIKV/DENV or anti-DENV/CHIKV dual targeting antiviral intron constructs indicated were each challenged with each arbovirus indicated. Following virus challenge cells were pelleted, lysed, and analyzed as described in Materials and Methods. DNA fragmentation results obtained following clonal C6/36 cells challenged with A. CHIKV or B. DENV-2 are shown. See S1 Fig through S5 Fig for DNA fragmentation assays performed following challenge with DENV-1, DENV-3, DENV-4, or DNA fragmentation assays performed on uninfected clonal cells expressing anti-CHIKV, anti-DENV or anti-CHIKV/DENV dual targeting antiviral intron constructs. Numbered lanes indicate clonal cell populations expressing single or dual virus targeting intron constructs. Commercial DNA ladder served as size standards for each gel. Size of DNA ladder is indicated on the right of each gel. I = Wt C6/36 mosquito cells infected with virus indicated; U = uninfected; Wt C6/36 mosquito cells; ΔC/Dv1 = ΔCHIKV/DENVv1; ΔD/Cv1 = ΔDENV/CHIKVv1; ΔC/Dv2 = ΔCHIKV/DENVv2; ΔD/Cv2 = ΔDENV/CHIKVv2.
As expected, infected or uninfected wild type C6/36 cells did not display fragmentation of nuclear DNA. Clonal Ae. albopictus C6/36 cells stably expressing the trans-splicing deficient ΔCHIKV/DENVv1-ΔN Bax, ΔDENV/CHIKVv1-ΔN Bax, ΔCHIKV/DENVv1-ΔN Bax, or ΔDENV/CHIKVv1-ΔN Bax constructs did not display observable DNA fragmentation upon infection with CHIKV (Fig 7A) or DENV (Fig 7B and S1 Fig through S4 Fig), further demonstrating that the insertion of a UAA codon in the P9.0 helix of the group I intron prevents premature expression of the ΔN Bax effector gene. DNA fragmentation was evident in all CHIKV/DENVv1 dual targeting intron-ΔN Bax clonal cell populations infected with CHIKV or DENV. 5 out of 6 DENV/CHIKVv1 clonal cell lines displayed DNA fragmentation similar to CHIKV/DENVv1 clonals, as was the case for 4 out of 6 for CHIKV/DENVv2 and 5 out of 6 DENV/CHIKVv2 clonal cell populations tested.
Wild type C6/36 or the inactive controls (ΔCHIKV/DENVv1-ΔN Bax, ΔDENV/CHIKV v1-ΔN Bax, ΔCHIKV/DENVv1-ΔN Bax, ΔDENV/CHIKV v1-ΔN Bax) displayed no detectable DNA fragmentation following DENV or CHIKV infection. Uninfected wild type C6/36 also displayed no DNA fragmentation. Similar results were also observed for cell clones transformed with the active anti-DENV group I intron following CHIKV challenge, and the active anti-CHIKV group I intron following DENV challenge, demonstrating the specificity of our anti arbovirus approach. The absence of observable DNA fragmentation with the dual targeting CHIKV/DENV introns, without CHIKV or DENV challenge, demonstrated effective suppression of read through of the ΔN Bax 3’ exon linked to these intron constructs.
All DNA fragmentation assay results directly correlated with the clonal cell populations displaying high levels of effector caspase activity, and helped confirm that optimal antiviral group I intron mediated DENV CA-ΔN Bax or CHIKV NS1-ΔN Bax splice formation and subsequent protein expression leads to full induction of apoptosis in the presence of arbovirus infection.
Anti-CHIKV/DENV dual targeting intron-ΔN Bax activation leads to full suppression of DENV replication
Effective targeting and suppression of DENV replication was previously demonstrated through the expression of anti-DENV group I intron-FL and anti-DENV group I intron-ΔN Bax constructs [48,49]. We expected that clonal cell expression of a ΔN Bax product following DENV or CHIKV targeting by our dual-targeting intron constructs would demonstrate an enhanced suppressive effect. C6/36 cell clones stably expressing dual targeting intron constructs bearing ΔN Bax as a 3’ exon were challenged with each DENV serotype indicated or CHIKV 181/25, and virus production was quantified by TCID50-immunofluorescence antibody assays as described in Materials and Methods (Fig 8; [31]).
Fig 8. Suppression of CHIKV and DENV replication is evident in clonal cell populations expressing antiviral Dual Virus Targeting Group I Intron Constructs.

Clonal Ae. albopictus C6/36 cells, transformed with the single or dual targeting antiviral intron constructs indicated, were challenged with each arbovirus indicated. Infected cell supernatants were collected, and viral titers were determined by TCID50-IFA. I = wild type C6/36 mosquito cells infected with virus indicated; U = uninfected wild type C6/36 mosquito cells; ΔC/D = ΔCHIKV/DENVv1; ΔD/C = ΔDENV/CHIKVv1. Supernatants originating from each clonal cell population were used as inoculum for three wells of fresh C6/36 cells and analyzed for the presence of each DENV serotype of CHIKV vaccine strain 181/25 by TCID50-IFA. The experiment was performed at three independent times resulting in a total of nine replicates for each dual-targeting intron-ΔN Bax tested.
In the presence of either CHIKV or DENV challenge, all CHIKV/DENVv1-ΔN Bax clonal lines demonstrated complete suppression of DENV and CHIKV replication. More specifically, in all 6 clonal lines that stably expressed CHIKV/DENVv1, none displayed detectable levels of DENV or CHIKV, exhibiting up to 7 logs reduction in viral titer when compared to the wild type C6/36 infection control (I). 5 out of 6 DENV/CHIKVv1 clonal lines fully suppressed these arboviruses, as was the case for 4 out of 6 for CHIKV/DENVv2 and 5 out of 6 DENV/CHIKVv2 clonal cell populations tested. As expected, virus infected wild type C6/36 (I) or ΔCHIKV/DENVv1-ΔN Bax or ΔDENV/CHIKV v1-ΔN Bax constructs did not exhibit suppression of DENV or CHIKV replication. This result was also observed for clonal cells transformed with either the active anti-DENV group I intron following CHIKV challenge and active anti-CHIKV group I intron following DENV challenge.
Discussion
Our results demonstrate, for the first time, that a single group I intron can be configured to target and trans splice two different RNA sequences. In this instance we establish the effectiveness of a constitutively expressed group I intron that targets and trans-splices conserved sequences within CHIKV and DENV, viruses representing two distantly related families of arboviruses. These trans-splicing, dual targeting group I intron configurations employ a 3' exon, ΔN Bax, that, when appropriately spliced with coding domains from each virus, induces apoptotic cell death upon infection in transformed mosquito cells.
Group I trans-splicing introns have an established potential for targeting RNA virus genomes in infected cells [45,46,48]. In previous reports we determined an optimal group I intron target sequence following an alignment of 98 instances of DENV that identified one highly conserved region positioned within the capsid coding sequence at nucleotides C131 to G151. These nucleotides are a part of the 5’-3’ CS domain of the DENV genome [66,67] that is essential for DENV replication [67] and include the amino terminal portion of the CA protein coding region. In this report we also identified a highly conserved region within the CHIKV NS1 gene at nucleotides G188 to C216 (S5 Fig) that we demonstrate serves as an effective target for trans splicing.
The unique configuration of our anti-CHIKV/DENV dual targeting group I introns catalyzes trans-splicing of the 5’ conserved target sequences of the DENV and CHIKV genomes to a 3’ ΔN Bax exon to effectively induce apoptotic death of cells following infection, thus preventing viral spread. A UAA stop codon was inserted in the trans-splicing domains of these introns to prevent premature expression of ΔN Bax that may result in cell death prior to infection. Following CHIKV/DENV dual targeting intron catalysis of DENV genomes at uracil 143, or CHIKV RNA genomes at U193, a chimeric mRNA is formed that consists of the 5’ cap, 5' UTR, and either 144 nucleotides of the DENV Capsid (CA) or 117 nucleotides of the CHIKV NS1 coding sequences appended to the 3’ ΔN Bax exon. These chimeric RNAs express a DENV CA-ΔN Bax or CHIKV NS1-ΔN Bax fusion protein that is demonstrated capable of inducing apoptotic cell death instead of allowing a productive virus infection.
Effective suppression of arbovirus replication with induction of apoptosis has been previously demonstrated in mosquitoes, most notably with Sindbis viruses [68]. Therefore, providing an apoptotic response in addition to genome targeting should enhance suppression of DENV or CHIKV replication as well. The ability to induce apoptosis following targeted splicing of viral genomes is an important advantage of this antiviral approach. While our group I introns demonstrated the capacity to cleave the highly conserved DENV 5’ CS region and a highly conserved region found within the CHIKV NS1 gene, the utility of these molecules as simple catalytic cleavage agents requires levels of expression that are equal to or greater than those generated by virus in infected cells.
Since escape events may arise if the rate of virus replication exceeds the rate of group I intron catalytic suppression, coupling the splicing activity of group I introns to a death-upon-infection strategy provides an added level of insurance against the development of escape mutants by insuring virus replication rates do not exceed group I intron expression and catalytic rates.
The potential emergence of escape mutants resulting from changes in the viral RNA, preventing targeting by antiviral ribozymes, is a possibility. The potential inability of the CHIKV/DENV dual targeting group I introns to suppress either CHIKV or DENV escape mutants can occur if the ribonucleic acid residues that constitute the IGS are not able to successfully target CHIKV or DENV RNA. This can be due to a mutation of the targeted uracil or the viral RNA residues targeted by the IGS. Mutation of viral RNA residues targeted by the EGS may not have a detrimental effect on group I intron catalysis of the viral RNA genome. It has been demonstrated that mutation of any residue in the sequence targeted by the EGS domain of group I introns does not eliminate catalysis or trans-splicing of the viral RNA sequences to the 3’ exon RNA.
We constructed a set of anti-arbovirus group I trans-splicing intron constructs (S1 Table, Fig 3) that are coupled to the proapoptotic ΔN Bax 3’ exon to assess whether the relative order of virus specific targeting sequences and P10 helices in these constructs affected activity against each virus. The results confirm that these constructs effectively trans-splice the RNA of an infecting DENV or CHIKV to the ΔN Bax 3’ exon when constitutively expressed as RNA in transformed C6/36 cells, irrespective of the relative position of the targeting sequences. These intron constructs effectively suppress DENV or CHIKV infection of transformed mosquito cells. Clonal C6/36 cell populations expressing the anti-CHIKV/DENV group I intron-ΔN Bax constructs, linked with the IRES driven mCherry as a bicistronic transcript (Fig 3), resisted infection with either CHIKV or DENV. Furthermore, addition of these IRES/mCherry configurations immediately downstream of the 3’ ΔN Bax exon did not appear to alter the trans-splicing capabilities of these trans-splicing introns (Fig 4), or affect the ability of ΔN Bax to initiate apoptosis in DENV or CHIKV infected cells (Figs 5, 6 and 7).
The expression and pro-apoptotic function of ΔN Bax is not inhibited by the 15 amino acids of CHIKV NS1 or 19 amino acids of dengue CA proteins fused to its N-terminus. Expression and activity of ΔN Bax, CNS1- ΔN Bax or DCA-ΔN Bax expressed in cells are not significantly different, and trans-splicing of the CHIKV and DENV RNA genomes by CHIKV-ΔN Bax (against CHIKV RNA), DENV-ΔN Bax (against DENV RNA), or CHIKV/DENV-ΔN Bax leads to the activation of cellular apoptosis as indicated by annexin V-FITC (Fig 5), effector caspase assays (Fig 6), and DNA ladder analysis (Fig 7 and S1 Fig through S5 Fig). None of these assays indicate apoptotic cell death when the trans-splicing negative ΔCHIKV/DENV or ΔDENV/CHIKV intron-bearing constructs are expressed or when cultured wild type C6/36 mosquito cells are infected with either CHIKV or DENV, confirming our results are a consequence of the presence of a trans-spliced RNA encoding the CNS1-ΔN Bax or DCA-ΔN Bax. Results obtained also demonstrate that fusion of the N-terminal amino acids of the DENV CA or CHIKV NS1 proteins to the N-terminus of the ΔN Bax protein does not eliminate the apoptotic inducing capacity of ΔN Bax, as may have been expected. The C-terminal residues of Bax possess the pore forming function of this pro-apoptotic protein [69], and may be the reason ΔN Bax retains proapoptotic activity in the presence of an N-terminal addition. Other researchers have analyzed N-terminal epitope-tagged variants of tBax with little alteration in activity [56,70,71].
Lower effector caspase and inhibition of viral replication may be due to the hindrance of dual targeting intron-ΔN Bax mRNA translocation out of the nucleus, leading to a decrease in the number of trans-spliced mRNA molecules that are produced following dual targeting intron-ΔN Bax targeting of CHIKV or DENV RNA in the cytoplasm of clonal cells.
Several labs have demonstrated that differences observed between annexin V and effector caspase/DNA fragmentation assays, may reflect the fact that the early stages of apoptosis can be reversed [72–76], preventing the progression of several clonal cell populations from the early stages of apoptosis (positive annexin V staining) to the latter stages of apoptosis (negative effector caspase activity and DNA fragmentation). Additionally, variability seen among the individual clones can be attributed to differences in expression of the group I introns due to position effects resulting from random integration of the transgene.
Additional support for the utility of these anti-CHIKV/DENV dual arbovirus targeting constructs as potent antiviral effectors is evidenced by the TCID50-IFA results that demonstrate suppression of infectious virus production from transformed clonal cell lines upon challenge with either CHIKV or each of the four serotypes tested (Fig 8). These results validate the utility of this single antiviral effector gene as a potential means for producing transgenic mosquitoes that will suppress DENV and CHIKV transmission.
We now have a set of anti-CHIKV/DENV group I introns that each have the ability to target four DENV serotypes and CHIKV. Our results demonstrate for the first time that group I introns may be configured to mediate trans-splicing against two different RNA targets. In this case, we successfully used this approach to create antiviral group I intron constructs that could arm mosquito cells to suppress two mosquito-borne viruses (CHIKV and DENV). Targeting these co-endemic viruses with a single catalytic ribozyme eliminates the necessity to construct and test separate catalytic RNAs or siRNA molecules, and could greatly facilitate the establishment of effective transgenic mosquito lines for potential release. The induction of cellular apoptosis by our anti-CHIKV/DENV constructs following DENV or CHIKV trans-splicing reduces the opportunity for escape mutants that may evolve in the infected cell and prevents the virus replication from overriding catalytic intron activity if the replication of DENV or CHIKV supersedes the catalytic activity of the anti-CHIKV/DENV group I intron.
Materials and Methods
Cells, Virus and Antibody
The Ae. albopictus C6/36 cells used in this study were obtained from ATCC, and maintained in Leibovitz’s L-15 media (Atlanta Biologicals) supplemented with 10% FBS (Atlanta Biologicals), 10% TPB (triptose phosphate broth; Invitrogen/Gibco), penicillin G (100U/ml; Invitrogen/Gibco) and streptomycin (100μU/ml; Invitrogen/Gibco), and grown in a 28°C incubator and passaged every 4 days. Assays involving DENV and CHIKV infections required L-15 media supplemented with 2% FBS and 10% TPB. Viral stocks were prepared as previously described [31,48,77].
CHIKV and DENV sequence data were provided by NCBI. Genbank GenInfo identifiers for the four DENV serotypes and CHIKV used in this study comprise the following: DENV type 1 Hawaii: DQ672564.1, DENV type 2 strain New Guinea C (NGC): AF038403.1, DENV type 3 strain ThD3 0010 87(strain H87): AY676352.1, DENV 4 strain DENV-4/SG/06K2270DK1/2005 (strain H241): GQ398256.1, and CHIKV vaccine strain 181/25: L37661.
TCID50-IFA analyses involving DENV infection were performed using monoclonal antibody (MAb) 4G2 (kindly provided by Dr. Stephen Higgs, Kansas State University), while analyses involving CHIKV infection were performed using a Chikungunya 181/25 vaccine strain specific antibody (IBT Bioservices, Gaithersburg, Maryland, USA).
Plasmid construction
Assurance of identity and integrity of all plasmids used was established through sequencing and restriction analysis. All restriction enzymes were obtained from New England Bio Labs (NEB).
The Drosophila melanogaster actin 5c (A5c) promoted anti- DENV 9v1 trans-splicing intron construct employed in this study was used previously to trans-splice DENV type 2-NGC targets with either the firefly luciferase (FL) [48] or ΔN Bax [49] 3’exons, and was used as a template for the production of anti-CHIKV/DENV dual targeting trans-splicing introns by PCR amplification. Our negative controls for trans-splicing activity, ΔCHIKV/DENVv1 and ΔDENV/CHIKVv1, were produced by a two-step PCR amplification process. First, the entire catalytic core [52], domains P4 through P6 [78], of the anti-DENV group I intron were removed by PCR amplification with Platinum Taq polymerase (Invitrogen) using the forward and reverse primers listed in S2 Table. The PCR product was used to replace the catalytic core of the anti-DENV group I intron using the enzymes MluI and NheI. Then, the resulting inactive intron was used as a template to produce ΔCHIKV/DENVv1 and ΔDENV/CHIKVv1 negative control introns using forward and reverse primers that possess the viral targeting sequences and P10 helix forming sequences (S2 Table). This resulted in control introns that lacked trans-splicing activity.
Group I introns that possess efficacy in targeting of CHIKV and DENV were produced by PCR amplification of the anti-DENV group I intron previously described [48]. See S2 Table for sequences of primers used to produce these dual arbovirus targeting introns. Amplification of the CHIKV/DENV introns was achieved with forward primers possessing the IGS and EGS complementary to the RNAs of CHIKV and DENV, respectively, were tailed with the MluI restriction site sequence. The dual arbovirus targeting introns referred to as DENV/CHIKV were produced by PCR amplification with forward primers possessing IGS and EGS complementary to the RNAs of DENV and CHIKV, respectively, were tailed with the MluI restriction site sequence.
Any active dual targeting CHIKV/DENV dual targeting intron listed with a “version 1” (i.e. v1) or “version 2” (i.e. v2) designation were each produced by a 2 step PCR process. Version 1 (v1) introns possess 6 nucleotides responsible for the formation of the CHIKV specific P10 helix immediately followed by 6 nucleotides responsible for the formation of the DENV specific P10 helix (Fig 3). Production of these “v1” introns was achieved by PCR with each respective forward primer possessing either CHIKV/DENV or DENV/CHIKV targeting sequences and a reverse primer possessing the CHIKV P10 helix forming sequences (also functions as a PstI site), and tailed with a XhoI site that is immediately upstream of the PstI restriction site/CHIKV P10 helix (S2 Table). The XhoI site was removed, and DENV P10 helix forming sequences inserted, by PCR amplification of the ΔN Bax 3’ exon with forward and reverse primers tailed with PstI and XbaI, respectively. This final PCR step resulted in insertion of DENV P10 helix forming sequences immediately upstream of the ΔN Bax 3’ exon and immediately downstream of the CHIKV P10 helix forming sequences.
Version 2 (v2) introns possess 6 nucleotides responsible for the formation of the DENV specific P10 helix immediately followed by 6 nucleotides responsible for the formation of the CHIKV specific P10 helix (Fig 3). Production of these v2 introns was achieved by PCR with each respective forward primer possessing either CHIKV/DENV or DENV/CHIKV targeting sequences and a reverse primer possessing the DENV P10 helix forming sequences immediately followed by CHIKV P10 helix forming sequences (also a PstI site), and tailed with a XhoI site that is immediately upstream of the PstI restriction site/CHIKV P10 helix (S2 Table). The XhoI site was removed by PCR amplification of the ΔN Bax 3’ exon with forward and reverse primers tailed with PstI and XbaI, respectively.
The anti-CHIKV group I intron was produced through PCR amplification of the anti-DENV group I intron [48] with a forward primer that possessed the EGS and IGS sequences corresponding nucleotides that are complimentary to nucleotides C201 to C209 and G188 to U196, respectively, of the CHIKV RNA genome and tailed with MluI. The CHIKV P10 helix forming sequences were inserted immediately downstream of the P9.0 helix. Production of these anti-CHIKV introns was achieved by PCR with a forward primer possessing CHIKV targeting sequences and a reverse primer possessing the CHIKV P10 helix forming sequences (also a PstI site), and tailed with a XhoI site that is immediately upstream of the PstI restriction site/CHIKV P10 helix (S2 Table). The XhoI site was removed by PCR amplification of the ΔN Bax 3’ exon with forward and reverse primers tailed with PstI and XbaI, respectively.
The anti-DENV intron construct used in this report as a negative control for CHIKV and a positive control for DENV targeting and trans-splicing is the same antiviral intron as the previously tested 9v1 intron [48], but was renamed “U143” for clarity [49].
Production of anti-CHIKV, anti- DENV, and anti-CHIKV/DENV dual targeting group I intron constructs possessing the DCV intergenic IRES site driving an mCherry fluorescent marker was achieved by subcloning DCV-mCherry from the previously described 9v1 construct into the antiviral group I intron constructs described above using NotI and SacI restriction sites ([48,49]; Fig 3).
Establishment of clonal cell populations
Clonal cell populations were produced as previously described [79]. Briefly, C6/36 cells stably expressing anti-CHIKV, anti-DENV, CHIKV/DENV dual targeting intron constructs, or the inactive intron versions ΔCHIKV/DENVv1 or ΔDENV/CHIKVv1 were grown to 1x104 cells/mL and then diluted to 5 cells/mL. 100μl of this cell suspension was placed in each of a 96 well plate and grown to confluency. Twelve wells of each plate were scraped and transferred to individual wells of a 24 well plate. Once confluent, cells were then transferred to a 12 well plate, then a 6 well plate, and lastly T-25 flasks. Following each transfer step, cells were maintained with 1mL L-15 complete media supplemented with 100 μg/mL hygromycin. In order to guarantee clonability, 3 cloning cycles were carried out.
Reverse Transcription-PCR of DENV-ΔN Bax Splice Products Derived from Cell Culture
Intron-expressing stable lines were generated by co-transfection of C6/36 cells with a hygromycin selectable marker plasmid and one of either A5c promoter plasmids expressing dual targeting CHIKV/DENV group I introns or negative control introns coupled to the 3’ ΔN Bax exon, as previously described [48,49]. Following hygromycin selection, the transformed C6/36 cells were seeded at a density of 5x106 cells/ml in T25 flasks and were challenged with either DENV (MOI = 0.1) or CHIKV (MOI = 0.01). Total cellular RNAs were isolated at 96 hours post-infection and assessed for CHIKV/DENV group I intron activity by RT-PCR detection of trans-spliced products. Post-spliced products of approximately 300 or 310 bp were recovered as a result of DENV or CHIKV targeting by the respective singlet antiviral intron (lanes 3 and 4) and dual antivirus intron (lanes 5 through 8) targeting of DENV or CHIKV (Fig 4). The identity of these presumptive spliced product bands was confirmed by sequencing (Data not shown).
Extraction of RNA from CHIKV or DENV infected and uninfected cells was performed using the Qiashredder and RNeasy Mini kits (QIAGEN Inc., Valencia, CA, USA). Extracted RNAs (5 μg) were treated with Turbo DNA-free DNAse (Applied Biosystems/Ambion, Inc. Austin, TX USA). RT-PCR was performed using the SuperScript III One-Step RT-PCR kit (Invitrogen) as directed. cDNA synthesis and PCR amplification were also performed as previously indicated [48,49].
A forward primer with the sequence 5’-GTGGACATAGACGCTGACA-3’ and a reverse primer to ΔN Bax with the sequence 5’-CACTCCCGCCACAAAGATG-3’ were used for the RT-PCR of CHIKV NS1-ΔN Bax (CNS1-ΔN Bax) splice products. For the RT-PCR of DENV CA-ΔN Bax (DCA-ΔN Bax) splice products, previously described [49] forward primers specific to each DENV serotype were used with a reverse primer possessing the sequence 5’-CACTCCCGCCACAAAGATG-3’.
Annexin V Assay
Binding of annexin V to translocated phospholipid phosphotidylserine (PS) allows for the detection and analysis of apoptotic cells [57–59]. These assays were performed using the Enhanced Apoptosis Kit as indicated by the manufacturer (Cayman Chemical Co.) with a few modifications. Briefly, C6/36 clonal cell lines stably expressing the CHIKV-, DENV-, CHIKV/DENV, and DENV/CHIKV dual targeting intron-ΔN Bax bearing constructs were infected with DENV serotypes 1 through 4 (MOI = 0.1) or CHIKV strain 181/25 (MOI = 0.01) and assayed for annexin V binding at 24 hpi (with CHIKV) or 48 hpi (with DENV). 1x106 clonal cells were scraped and placed in a well of a 96 well black opaque microtiter plate in triplicate for each clonal cell type assayed. FITC-annexin V stained microtiter plates were assayed for FITC-annexin V binding at 485 nm with the Spectra max M2 luminometer (Molecular Devices) and analyzed with Softmax Pro 5.4.5.
Three wells per clonal cell population indicated were infected with the arbovirus shown and analyzed for annexin V-FITC staining. This experiment was performed three times to give a total of nine replicates for each dual -targeting intron-ΔN Bax tested. Assay were performed in triplicate. Error bars indicate standard deviation of three independent experiments.
Effector Caspase Assay
Further validation of apoptosis induction was performed by assaying for increases in effector caspase and other DEVD-specific protease activities using the EnzChekCaspase-3 Assay Kit #2 Kit (Life Technologies) as directed by the manufacturer. Briefly, C6/36 clonal cell lines stably expressing the CHIKV-, DENV-, CHIKV/DENV, and DENV/CHIKV dual targeting intron-ΔN Bax bearing constructs were infected with DENV serotypes 1–4 (MOI = 0.1) or CHIKV strain 181/25 (MOI = 0.01) and assayed for effector caspase activity at 4d.pi (for DENV) or 3dpi (for CHIKV). 1x106 clonal cells were lysed, cell debris was pelleted, and lysates were placed in a well of a 96 well black opaque microtiter plate in triplicate for each clonal cell type assayed. Following addition of the Z-DEVD–R110 substrate, microtiter plates were assayed for effector caspase activity at 496 nm with the Spectra max M2 luminometer (Molecular Devices) and analyzed with Softmax Pro 5.4.5.
Three wells per clonal cell population indicated were infected with the arbovirus shown and analysed for “effector caspase” activity. This experiment was performed three times to give a total of nine replicates for each dual-targeting intron-ΔN Bax tested.
DNA Fragmentation Assay
Clonal C6/36 cells (1x107) stably expressing the CHIKV-, DENV-, CHIKV/DENV or DENV/CHIKV dual targeting intron-ΔN Bax constructs were infected with one of four known DENV serotypes (MOI = 0.1) or CHIKV strain 181/25 (MOI = 0.01) and assayed for DNA fragmentation as previously described [49]. Briefly, at 4 dpi (for DENV) or 3dpi (for CHIKV) infected and uninfected cells were scraped, pelleted and lysed overnight at 50°C in lysis buffer [1.67mg/ml Proteinase K, 10mM Tris (pH8.0), 100mM NaCl, 0.5% SDS 25mM EDTA]. Genomic DNA was extracted with 200 μl Phenol:Chloroform:IAA (25:24:1) and sodium acetate/ethanol precipitated. DNA pellets were resuspended in 20 μl TE buffer, RNase A treated (6.0 mg/ml) at 37°C for 3 hours, and analyzed by 2% agarose gel electrophoresis at 5v/cm and visualized under UV light. DNA fragmentation is demonstrated by the appearance of a DNA ladder-like pattern.
TCID50-IFA analysis of dengue viruses
Immunofluorescence detection of cell surface expressed CHIKV or DENV envelope (E) proteins in C6/36 cultures infected with serial 10 fold dilutions were used to assess the titer of CHIKV (MOI 0.01) or each DENV serotype (MOI 0.1) tested as previously described [31,48]. 10 fold serial dilutions of infected C6/36 cell culture supernatants were harvested at 24 hpi (CHIKV) or 48 hpi (DENV) and used as inoculum for 96 well plate cultures of C6/36 cells. Plates were incubated for 4 days at 28°C without CO2, washed, fixed with acetone:DPBS (3:1), and stained with a primary DENV E antibody (1:200) [80], followed by a biotinylated-streptavidin detection system conjugated with FITC (Amersham Biosciences, Piscataway, NJ). Wells displaying cellular fluorescence were scored as positive for DENV infection. The number of positive wells were counted and the virus titers calculated according to Karber’s method [81].
Supernatants originating from each clonal cell population indicated were used as inoculum for three wells of naïve C6/36 cells and analyzed for the presence of each DENV serotype or CHIKV vaccine strain 181/25 by TCID50-IFA. This experiment was performed three times to give a total of nine replicates for each dual-targeting intron-ΔN Bax tested.
Supporting Information
An alignment was performed of twenty five (25) CHIKV genomic sequences to determine the most optimal regions for the design of the chikungunya virus specific and DENV/CHIKV dual targeting antiviral group I introns by determining the region with the greatest conservation within the CHIKV RNA genomes. Nucleotide sequences in yellow indicate complete conservation. Nucleotide sequences in blue indicate partial conservation. Nucleotide sequence position is indicated at the top of the figure. GenBank Accession Numbers at the left of the figure indicate the CHIKV sequences aligned.
(TIF)
Clonal Ae. albopictus C6/36 cells transformed with the anti-CHIKV, anti-DENV or anti-CHIKV/DENV dual targeting antiviral intron constructs indicated were each challenged with dengue virus serotype 1, processed, and analyzed as described for Fig 7 and in Materials and Methods. I = Wt C6/36 mosquito cells infected with virus indicated; U = uninfected Wt C6/36 mosquito cells; ΔC/Dv1 = ΔCHIKV/DENVv1; ΔD/Cv1 = ΔDENV/CHIKVv1; ΔC/Dv2 = ΔCHIKV/DENVv2; ΔD/Cv2 = ΔDENV/CHIKVv2.
(TIF)
Clonal Ae. albopictus C6/36 cells transformed with the anti-CHIKV, anti-DENV or anti-CHIKV/DENV dual targeting antiviral intron constructs indicated were each challenged with dengue virus serotype 3, processed, and analyzed as described for Fig 7 and in Materials and Methods. I = Wt C6/36 mosquito cells infected with virus indicated; U = uninfected Wt C6/36 mosquito cells; ΔC/Dv1 = ΔCHIKV/DENVv1; ΔD/Cv1 = ΔDENV/CHIKVv1; ΔC/Dv2 = ΔCHIKV/DENVv2; ΔD/Cv2 = ΔDENV/CHIKVv2.
(TIF)
Clonal Ae. albopictus C6/36 cells transformed with the anti-CHIKV, anti-DENV or anti-CHIKV/DENV dual targeting antiviral intron constructs indicated were each challenged with dengue virus serotype 4, processed, and analyzed as described for Fig 7 and in Materials and Methods. I = Wt C6/36 mosquito cells infected with virus indicated; U = uninfected Wt C6/36 mosquito cells; ΔC/Dv1 = ΔCHIKV/DENVv1; ΔD/Cv1 = ΔDENV/CHIKVv1; ΔC/Dv2 = ΔCHIKV/DENVv2; ΔD/Cv2 = ΔDENV/CHIKVv2.
(TIF)
Clonal Ae. albopictus C6/36 cells transformed with the anti-CHIKV, anti-DENV or anti-CHIKV/DENV dual targeting antiviral intron constructs indicated were each mock infected, processed, and analyzed as described for Fig 7 and in Materials and Methods. I = Wt C6/36 mosquito cells infected with virus indicated; U = uninfected Wt C6/36 mosquito cells; ΔC/Dv1 = ΔCHIKV/DENVv1; ΔD/Cv1 = ΔDENV/CHIKVv1; ΔC/Dv2 = ΔCHIKV/DENVv2; ΔD/Cv2 = ΔDENV/CHIKVv2.
(TIF)
The ribonucleotide sequences of each antiviral group I intron are shown. The left column lists the viruses targeted by the dual targeting introns containing respective targeting sequences shown. Targeting sequences specific for each virus are indicated. See methods for description of assembly. CHIKV = Chikungunya virus targeting sequences; DENV = Dengue virus targeting sequences; EGS = external guide sequence; IGS = internal guide sequence; BL = bulge loop; TSD = trans splicing domain; P10 = P10 helix.
(TIF)
Listed are the forward and reverse primer sets used to produce the PCR fragments of anti-CHIKV/DENV introns, negative controls, and the ΔN Bax 3’ exon for plasmid insertion. Restriction sites used are indicated by lowercase nucleic acids. See Materials and Methods for description of vector construct assembly.
(TIF)
Acknowledgments
Our thanks to Dr. Thomas Cech (University of Colorado, Boulder) for the pTT1A3-T7 plasmid. Chikungunya virus strain 181/25 used in this study was a kind gift from Dr. Scott Weaver (UTMB). This research was supported by research grant NIH/NIAID AI097554 to MJF.
Data Availability
All relevant data are within the paper and its Supporting Information files.
Funding Statement
This work was supported by the US National Institutes of Health (NIH) grant AI097554-04 to MJF.
References
- 1. Clyde K, Kyle JL, Harris E (2006) Recent advances in deciphering viral and host determinants of dengue virus replication and pathogenesis. J Virol 80: 11418–11431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Pialoux G, Gauzere BA, Jaureguiberry S, Strobel M (2007) Chikungunya, an epidemic arbovirosis. Lancet Infect Dis 7: 319–327. [DOI] [PubMed] [Google Scholar]
- 3. Randolph SE, Rogers DJ (2010) The arrival, establishment and spread of exotic diseases: patterns and predictions. Nat Rev Microbiol. [DOI] [PubMed] [Google Scholar]
- 4. Rigau-Perez JG, Clark GG, Gubler DJ, Reiter P, Sanders EJ, Vorndam AV(1998) Dengue and dengue haemorrhagic fever. Lancet 352: 971–977. [DOI] [PubMed] [Google Scholar]
- 5. Weaver SC, Reisen WK (2010) Present and future arboviral threats. Antiviral Res 85: 328–345. 10.1016/j.antiviral.2009.10.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bhatt S, Gething PW, Brady OJ, Messina JP, Farlow AW, Moyes AL, et al. The global distribution and burden of dengue. Nature 496: 504–507. 10.1038/nature12060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Singh P, Mittal V, Rizvi MM, Chhabra M, Sharma P, Rawat DS, et al. The first dominant co-circulation of both dengue and chikungunya viruses during the post-monsoon period of 2010 in Delhi, India. Epidemiol Infect 140: 1337–1342. 10.1017/S0950268811001671 [DOI] [PubMed] [Google Scholar]
- 8. Maddox N (2011) Time to prepare for dengue testing? Florida Bureau of Labs is already on the job. MLO Med Lab Obs 43: 44–45. [PubMed] [Google Scholar]
- 9. Graham AS, Pruszynski CA, Hribar LJ, DeMay DJ, Tambasco AN, Hartley AE, et al. (2011) Mosquito-associated dengue virus, Key West, Florida, USA, 2010. Emerg Infect Dis 17: 2074–2075. 10.3201/eid1711.110419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Radke EG, Gregory CJ, Kintziger KW, Sauber-Schatz EK, Hunsperger EA, Gallagher GR, et al. (2012) Dengue outbreak in Key West, Florida, USA, 2009. Emerg Infect Dis 18: 135–137. 10.3201/eid1801.110130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Myers RM, Carey DE (1967) Concurrent isolation from patient of two arboviruses, Chikungunya and dengue type 2. Science 157: 1307–1308. [DOI] [PubMed] [Google Scholar]
- 12. Leroy EM, Nkoghe D, Ollomo B, Nze-Nkogue C, Becquart P, Grard G, et al. (2009) Concurrent chikungunya and dengue virus infections during simultaneous outbreaks, Gabon, 2007. Emerg Infect Dis 15: 591–593. 10.3201/eid1504.080664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. James AA (2005) Gene drive systems in mosquitoes: rules of the road. Trends Parasitol 21: 64–67. [DOI] [PubMed] [Google Scholar]
- 14. Sinkins SP, Gould F (2006) Gene drive systems for insect disease vectors. Nat Rev Genet 7: 427–435. [DOI] [PubMed] [Google Scholar]
- 15. van den Hurk AF, Hall-Mendelin S, Pyke AT, Smith GA, Mackenzie JS (2009) Vector Competence of Australian Mosquitoes for Chikungunya Virus. Vector Borne Zoonotic Dis. [DOI] [PubMed] [Google Scholar]
- 16. Fauran P (1996) [New epidemiological aspects of dengue]. Bull Soc Pathol Exot 89: 163–164; discussion 165. [PubMed] [Google Scholar]
- 17. McMeniman CJ, Lane RV, Cass BN, Fong AW, Sidhu M, Wang YF, et al. (2009) Stable introduction of a life-shortening Wolbachia infection into the mosquito Aedes aegypti. Science 323: 141–144. 10.1126/science.1165326 [DOI] [PubMed] [Google Scholar]
- 18. Rudnick A, Chan YC (1965) Dengue Type 2 Virus in Naturally Infected Aedes albopictus Mosquitoes in Singapore. Science 149: 638–639. [DOI] [PubMed] [Google Scholar]
- 19. Chusri S, Siripaitoon P, Silpapojakul K, Hortiwakul T, Charernmak B, Chinnawirotpisan P, et al. (2014) Kinetics of chikungunya infections during an outbreak in Southern Thailand, 2008–2009. Am J Trop Med Hyg 90: 410–417. 10.4269/ajtmh.12-0681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Thavara U, Tawatsin A, Pengsakul T, Bhakdeenuan P, Chanama S, Anantapreecha S, et al. (2009) Outbreak of chikungunya fever in Thailand and virus detection in field population of vector mosquitoes, Aedes aegypti (L.) and Aedes albopictus Skuse (Diptera: Culicidae). Southeast Asian J Trop Med Public Health 40: 951–962. [PubMed] [Google Scholar]
- 21. Pastorino B, Muyembe-Tamfum JJ, Bessaud M, Tock F, Tolou H, Durand JP, et al. (2004) Epidemic resurgence of Chikungunya virus in democratic Republic of the Congo: identification of a new central African strain. J Med Virol 74: 277–282. [DOI] [PubMed] [Google Scholar]
- 22. Powers AM, Brault AC, Tesh RB, Weaver SC (2000) Re-emergence of Chikungunya and O'nyong-nyong viruses: evidence for distinct geographical lineages and distant evolutionary relationships. J Gen Virol 81: 471–479. [DOI] [PubMed] [Google Scholar]
- 23. Higgs S, Ziegler SA (2010) A nonhuman primate model of chikungunya disease. J Clin Invest 120: 657–660. 10.1172/JCI42392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Reiter P, Fontenille D, Paupy C (2006) Aedes albopictus as an epidemic vector of chikungunya virus: another emerging problem? Lancet Infect Dis 6: 463–464. [DOI] [PubMed] [Google Scholar]
- 25. Schuffenecker I, Iteman I, Michault A, Murri S, Frangeul L, Vaney MC, et al. (2006) Genome microevolution of chikungunya viruses causing the Indian Ocean outbreak. PLoS Med 3: e263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. de Lamballerie X, Leroy E, Charrel RN, Ttsetsarkin K, Higgs S, Gould EA. (2008) Chikungunya virus adapts to tiger mosquito via evolutionary convergence: a sign of things to come? Virol J 5: 33 10.1186/1743-422X-5-33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Press AF (2010) French Riviera sees new case of chikungunya virus. American Free Press. [Google Scholar]
- 28. Morens DM, Fauci AS (2014) Chikungunya at the Door—Deja Vu All Over Again? N Engl J Med. [DOI] [PubMed] [Google Scholar]
- 29. Johansson MA, Powers AM, Pesik N, Cohen NJ, Staples JE (2014) Nowcasting the Spread of Chikungunya Virus in the Americas. PLoS One 9: e104915 10.1371/journal.pone.0104915 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Van Bortel W, Dorleans F, Rosine J, Blateau A, Rousset D, Matheus S, et al. (2014) Chikungunya outbreak in the Caribbean region, December 2013 to March 2014, and the significance for Europe. Euro Surveill 19. [DOI] [PubMed] [Google Scholar]
- 31. Nawtaisong P, Keith J, Fraser T, Balaraman V, Kolokoltsov A, Davey RA, et al. (2009) Effective suppression of Dengue fever virus in mosquito cell cultures using retroviral transduction of hammerhead ribozymes targeting the viral genome. Virol J 6: 73 10.1186/1743-422X-6-73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ayre BG, Kohler U, Goodman HM, Haseloff J (1999) Design of highly specific cytotoxins by using trans-splicing ribozymes. Proc Natl Acad Sci U S A 96: 3507–3512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Byun J, Lan N, Long M, Sullenger BA (2003) Efficient and specific repair of sickle beta-globin RNA by trans-splicing ribozymes. Rna 9: 1254–1263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Jung HS, Kwon BS, Lee SW (2005) Tumor-specific gene delivery using RNA-targeting Tetrahymena group I intron. Biotechnol Lett 27: 567–574. [DOI] [PubMed] [Google Scholar]
- 35. Kastanos E, Hjiantoniou E, Phylactou LA (2004) Restoration of protein synthesis in pancreatic cancer cells by trans-splicing ribozymes. Biochem Biophys Res Commun 322: 930–934. [DOI] [PubMed] [Google Scholar]
- 36. Mouse Genome Sequencing Consortium, Waterston RH, Lindblad-Toh K, Birney E, Rogers J, Abril JF, et al. (2002) Initial sequencing and comparative analysis of the mouse genome. Nature 420: 520–562. [DOI] [PubMed] [Google Scholar]
- 37. Ryu KJ, Lee SW (2003) Identification of the most accessible sites to ribozymes on the hepatitis C virus internal ribosome entry site. J Biochem Mol Biol 36: 538–544. [DOI] [PubMed] [Google Scholar]
- 38. Ryu KJ, Lee SW (2004) Comparative analysis of intracellular trans-splicing ribozyme activity against hepatitis C virus internal ribosome entry site. J Microbiol 42: 361–364. [PubMed] [Google Scholar]
- 39. Sullenger BA, Cech TR (1994) Ribozyme-mediated repair of defective mRNA by targeted, trans-splicing. Nature 371: 619–622. [DOI] [PubMed] [Google Scholar]
- 40. Lander ES, Linton LM, Birren B, Nusbaum C, Zody MC, Baldwin J, et al. (2001) Initial sequencing and analysis of the human genome. Nature 409: 860–921. [DOI] [PubMed] [Google Scholar]
- 41. Jeong JS, Lee SW, Hong SH, Lee YJ, Jung HI, Cho KS, et al. (2008) Antitumor effects of systemically delivered adenovirus harboring trans-splicing ribozyme in intrahepatic colon cancer mouse model. Clin Cancer Res 14: 281–290. 10.1158/1078-0432.CCR-07-1524 [DOI] [PubMed] [Google Scholar]
- 42. Kwon BS, Jung HS, Song MS, Cho KS, Kim SC, Kimm K, et al. (2005) Specific regression of human cancer cells by ribozyme-mediated targeted replacement of tumor-specific transcript. Mol Ther 12: 824–834. [DOI] [PubMed] [Google Scholar]
- 43. Watanabe T, Sullenger BA (2000) Induction of wild-type p53 activity in human cancer cells by ribozymes that repair mutant p53 transcripts. Proc Natl Acad Sci U S A 97: 8490–8494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Rogers CS, Vanoye CG, Sullenger BA, George AL Jr. (2002) Functional repair of a mutant chloride channel using a trans-splicing ribozyme. J Clin Invest 110: 1783–1789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Kohler U, Ayre BG, Goodman HM, Haseloff J (1999) Trans-splicing ribozymes for targeted gene delivery. J Mol Biol 285: 1935–1950. [DOI] [PubMed] [Google Scholar]
- 46. Ryu KJ, Kim JH, Lee SW (2003) Ribozyme-mediated selective induction of new gene activity in hepatitis C virus internal ribosome entry site-expressing cells by targeted trans-splicing. Mol Ther 7: 386–395. [DOI] [PubMed] [Google Scholar]
- 47. Nawtaisong P, Fraser ME, Carter JR, Fraser MJ Jr. (2015) Trans-splicing group I intron targeting hepatitis C virus IRES mediates cell death upon viral infection in Huh7.5 cells. Virology 481: 223–234. 10.1016/j.virol.2015.02.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Carter JR, Keith JH, Barde PV, Fraser TS, Fraser MJ Jr. (2010) Targeting of highly conserved Dengue virus sequences with anti-Dengue virus trans-splicing group I introns. BMC Mol Biol 11: 84 10.1186/1471-2199-11-84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Carter JR, Keith JH, Fraser TS, Dawson JL, Kucharski CA, Higgs S, et al. (2014) Effective suppression of Dengue virus using a novel group-I intron that induces apoptotic cell death upon infection through conditional expression of the Bax C-terminal domain. Virol J 11: 111 10.1186/1743-422X-11-111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Cech TR (1991) RNA editing: world's smallest introns? Cell 64: 667–669. [DOI] [PubMed] [Google Scholar]
- 51. Strauss JH, Strauss EG (1994) The alphaviruses: gene expression, replication, and evolution. Microbiol Rev 58: 491–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Cech TR (1990) Self-splicing of group I introns. Annu Rev Biochem 59: 543–568. [DOI] [PubMed] [Google Scholar]
- 53. Strobel SA, Cech TR (1993) Tertiary interactions with the internal guide sequence mediate docking of the P1 helix into the catalytic core of the Tetrahymena ribozyme. Biochemistry 32: 13593–13604. [DOI] [PubMed] [Google Scholar]
- 54. Campbell TB, Cech TR (1996) Mutations in the Tetrahymena ribozyme internal guide sequence: effects on docking of the P1 helix into the catalytic core and correlation with catalytic activity. Biochemistry 35: 11493–11502. [DOI] [PubMed] [Google Scholar]
- 55. Guo F, Cech TR (2002) In vivo selection of better self-splicing introns in Escherichia coli: the role of the P1 extension helix of the Tetrahymena intron. RNA 8: 647–658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Usui K, Saijo Y, Narumi K, Koyama S, Maemondo M, Kikuchi T, et al. (2003) N-terminal deletion augments the cell-death-inducing activity of BAX in adenoviral gene delivery to nonsmall cell lung cancers. Oncogene 22: 2655–2663. [DOI] [PubMed] [Google Scholar]
- 57. Muhlenfeld K, Langner A (1998) Biotransformation and toxicity of the lipoxygenase inhibitor 2-hydroxy-5-methyllaurophenone oxime (FLM 5011) on Hep G2 cells. Arch Pharm (Weinheim) 331: 259–261. [DOI] [PubMed] [Google Scholar]
- 58. Koopman G, Reutelingsperger CP, Kuijten GA, Keehnen RM, Pals ST, van Oers MH (1994) Annexin V for flow cytometric detection of phosphatidylserine expression on B cells undergoing apoptosis. Blood 84: 1415–1420. [PubMed] [Google Scholar]
- 59. Martin SJ, Reutelingsperger CP, McGahon AJ, Rader JA, van Schie RC, LaFace DM, et al. (1995) Early redistribution of plasma membrane phosphatidylserine is a general feature of apoptosis regardless of the initiating stimulus: inhibition by overexpression of Bcl-2 and Abl. J Exp Med 182: 1545–1556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Vermeulen K, Berneman ZN, Van Bockstaele DR (2003) Cell cycle and apoptosis. Cell Prolif 36: 165–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Vermeulen K, Van Bockstaele DR, Berneman ZN (2003) The cell cycle: a review of regulation, deregulation and therapeutic targets in cancer. Cell Prolif 36: 131–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Liu Q, Clem RJ (2011) Defining the core apoptosis pathway in the mosquito disease vector Aedes aegypti: the roles of iap1, ark, dronc, and effector caspases. Apoptosis 16: 105–113. 10.1007/s10495-010-0558-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Coupe SA, Watson LM, Ryan DJ, Pinkney TT, Eason JR (2004) Molecular analysis of programmed cell death during senescence in Arabidopsis thaliana and Brassica oleracea: cloning broccoli LSD1, Bax inhibitor and serine palmitoyltransferase homologues. J Exp Bot 55: 59–68. [DOI] [PubMed] [Google Scholar]
- 64. Nava VE, Rosen A, Veliuona MA, Clem RJ, Levine B, Hardwick JM (1998) Sindbis virus induces apoptosis through a caspase-dependent, CrmA-sensitive pathway. J Virol 72: 452–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Wang H, Blair CD, Olson KE, Clem RJ (2008) Effects of inducing or inhibiting apoptosis on Sindbis virus replication in mosquito cells. J Gen Virol 89: 2651–2661. 10.1099/vir.0.2008/005314-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Alvarez DE, Lodeiro MF, Luduena SJ, Pietrasanta LI, Gamarnik AV (2005) Long-range RNA-RNA interactions circularize the dengue virus genome. J Virol 79: 6631–6643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Alvarez DE, Filomatori CV, Gamarnik AV (2008) Functional analysis of dengue virus cyclization sequences located at the 5' and 3'UTRs. Virology 375: 223–235. 10.1016/j.virol.2008.01.014 [DOI] [PubMed] [Google Scholar]
- 68. O'Neill K, Olson BJ, Huang N, Unis D, Clem RJ (2015) Rapid selection against arbovirus-induced apoptosis during infection of a mosquito vector. Proc Natl Acad Sci U S A 112: E1152–1161. 10.1073/pnas.1424469112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Nechushtan A, Smith CL, Hsu YT, Youle RJ (1999) Conformation of the Bax C-terminus regulates subcellular location and cell death. EMBO J 18: 2330–2341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Toyota H, Kondo S, Kyo S, Mizuguchi J (2006) Enforced expression of a truncated form of Bax-alpha (tBax) driven by human telomerase reverse transcriptase (hTERT) promoter sensitizes tumor cells to chemotherapeutic agents or tumor necrosis factor-related apoptosis-inducing ligand (TRAIL). Anticancer Res 26: 99–105. [PubMed] [Google Scholar]
- 71. Toyota H, Yanase N, Yoshimoto T, Moriyama M, Sudo T, Mizuguchi J. (2003) Calpain-induced Bax-cleavage product is a more potent inducer of apoptotic cell death than wild-type Bax. Cancer Lett 189: 221–230. [DOI] [PubMed] [Google Scholar]
- 72. Geske FJ, Lieberman R, Strange R, Gerschenson LE (2001) Early stages of p53-induced apoptosis are reversible. Cell Death Differ 8: 182–191. [DOI] [PubMed] [Google Scholar]
- 73. Tang HL, Tang HM, Mak KH, Hu S, Wang SS, Wong KM, et al. (2012) Cell survival, DNA damage, and oncogenic transformation after a transient and reversible apoptotic response. Mol Biol Cell 23: 2240–2252. 10.1091/mbc.E11-11-0926 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Balasubramanian K, Mirnikjoo B, Schroit AJ (2007) Regulated externalization of phosphatidylserine at the cell surface: implications for apoptosis. J Biol Chem 282: 18357–18364. [DOI] [PubMed] [Google Scholar]
- 75. O'Brien IE, Murray BG, Baguley BC, Morris BA, Ferguson IB (1998) Major changes in chromatin condensation suggest the presence of an apoptotic pathway in plant cells. Exp Cell Res 241: 46–54. [DOI] [PubMed] [Google Scholar]
- 76. Blankenberg FG (2008) In vivo imaging of apoptosis. Cancer Biol Ther 7: 1525–1532. [DOI] [PubMed] [Google Scholar]
- 77. Nuckols JT, Ziegler SA, Huang YJ, McAuley AJ, Vanlandingham DL, Klowden MJ et al. (2013) Infection of Aedes albopictus with chikungunya virus rectally administered by enema. Vector Borne Zoonotic Dis 13: 103–110. 10.1089/vbz.2012.1013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Johnson TH, Tijerina P, Chadee AB, Herschlag D, Russell R (2005) Structural specificity conferred by a group I RNA peripheral element. Proc Natl Acad Sci U S A 102: 10176–10181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Adelman ZN, Sanchez-Vargas I, Travanty EA, Carlson JO, Beaty BJ, Blair CD, et al. (2002) RNA silencing of dengue virus type 2 replication in transformed C6/36 mosquito cells transcribing an inverted-repeat RNA derived from the virus genome. J Virol 76: 12925–12933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Henchal EA, McCown JM, Burke DS, Seguin MC, Brandt WE (1985) Epitopic analysis of antigenic determinants on the surface of dengue-2 virions using monoclonal antibodies. Am J Trop Med Hyg 34: 162–169. [DOI] [PubMed] [Google Scholar]
- 81. Karber G (1931) 50% end-point calculation. Arch Exp Pathol Pharmk 162: 480–483. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
An alignment was performed of twenty five (25) CHIKV genomic sequences to determine the most optimal regions for the design of the chikungunya virus specific and DENV/CHIKV dual targeting antiviral group I introns by determining the region with the greatest conservation within the CHIKV RNA genomes. Nucleotide sequences in yellow indicate complete conservation. Nucleotide sequences in blue indicate partial conservation. Nucleotide sequence position is indicated at the top of the figure. GenBank Accession Numbers at the left of the figure indicate the CHIKV sequences aligned.
(TIF)
Clonal Ae. albopictus C6/36 cells transformed with the anti-CHIKV, anti-DENV or anti-CHIKV/DENV dual targeting antiviral intron constructs indicated were each challenged with dengue virus serotype 1, processed, and analyzed as described for Fig 7 and in Materials and Methods. I = Wt C6/36 mosquito cells infected with virus indicated; U = uninfected Wt C6/36 mosquito cells; ΔC/Dv1 = ΔCHIKV/DENVv1; ΔD/Cv1 = ΔDENV/CHIKVv1; ΔC/Dv2 = ΔCHIKV/DENVv2; ΔD/Cv2 = ΔDENV/CHIKVv2.
(TIF)
Clonal Ae. albopictus C6/36 cells transformed with the anti-CHIKV, anti-DENV or anti-CHIKV/DENV dual targeting antiviral intron constructs indicated were each challenged with dengue virus serotype 3, processed, and analyzed as described for Fig 7 and in Materials and Methods. I = Wt C6/36 mosquito cells infected with virus indicated; U = uninfected Wt C6/36 mosquito cells; ΔC/Dv1 = ΔCHIKV/DENVv1; ΔD/Cv1 = ΔDENV/CHIKVv1; ΔC/Dv2 = ΔCHIKV/DENVv2; ΔD/Cv2 = ΔDENV/CHIKVv2.
(TIF)
Clonal Ae. albopictus C6/36 cells transformed with the anti-CHIKV, anti-DENV or anti-CHIKV/DENV dual targeting antiviral intron constructs indicated were each challenged with dengue virus serotype 4, processed, and analyzed as described for Fig 7 and in Materials and Methods. I = Wt C6/36 mosquito cells infected with virus indicated; U = uninfected Wt C6/36 mosquito cells; ΔC/Dv1 = ΔCHIKV/DENVv1; ΔD/Cv1 = ΔDENV/CHIKVv1; ΔC/Dv2 = ΔCHIKV/DENVv2; ΔD/Cv2 = ΔDENV/CHIKVv2.
(TIF)
Clonal Ae. albopictus C6/36 cells transformed with the anti-CHIKV, anti-DENV or anti-CHIKV/DENV dual targeting antiviral intron constructs indicated were each mock infected, processed, and analyzed as described for Fig 7 and in Materials and Methods. I = Wt C6/36 mosquito cells infected with virus indicated; U = uninfected Wt C6/36 mosquito cells; ΔC/Dv1 = ΔCHIKV/DENVv1; ΔD/Cv1 = ΔDENV/CHIKVv1; ΔC/Dv2 = ΔCHIKV/DENVv2; ΔD/Cv2 = ΔDENV/CHIKVv2.
(TIF)
The ribonucleotide sequences of each antiviral group I intron are shown. The left column lists the viruses targeted by the dual targeting introns containing respective targeting sequences shown. Targeting sequences specific for each virus are indicated. See methods for description of assembly. CHIKV = Chikungunya virus targeting sequences; DENV = Dengue virus targeting sequences; EGS = external guide sequence; IGS = internal guide sequence; BL = bulge loop; TSD = trans splicing domain; P10 = P10 helix.
(TIF)
Listed are the forward and reverse primer sets used to produce the PCR fragments of anti-CHIKV/DENV introns, negative controls, and the ΔN Bax 3’ exon for plasmid insertion. Restriction sites used are indicated by lowercase nucleic acids. See Materials and Methods for description of vector construct assembly.
(TIF)
Data Availability Statement
All relevant data are within the paper and its Supporting Information files.