

Abstract
The nitrosation of cysteamine (H2NCH2CH2SH) to produce cysteamine-S-nitrosothiol (CANO) was studied in slightly acidic medium by using nitrous acid prepared in situ. The stoichiometry of the reaction was H2NCH2CH2SH + HNO2 → H2NCH2CH2SNO + H2O. On prolonged standing, the nitrosothiol decomposed quantitatively to yield the disulfide, cystamine: 2H2NCH2CH2SNO → H2NCH2CH2S–SCH2CH2NH2 + 2NO. NO2 and N2O3 are not the primary nitrosating agents, since their precursor (NO) was not detected during the nitrosation process. The reaction is first order in nitrous acid, thus implicating it as the major nitrosating agent in mildly acidic pH conditions. Acid catalyzes nitrosation after nitrous acid has saturated, implicating the protonated nitrous acid species, the nitrosonium cation (NO+) as a contributing nitrosating species in highly acidic environments. The acid catalysis at constant nitrous acid concentrations suggests that the nitrosonium cation nitrosates at a much higher rate than nitrous acid. Bimolecular rate constants for the nitrosation of cysteamine by nitrous acid and by the nitrosonium cation were deduced to be 17.9 ± 1.5 (mol/L)−1 s−1 and 6.7 × 104 (mol/L)−1 s−1, respectively. Both Cu(I) and Cu(II) ions were effective catalysts for the formation and decomposition of the cysteamine nitrosothiol. Cu(II) ions could catalyze the nitrosation of cysteamine in neutral conditions, whereas Cu(I) could only catalyze in acidic conditions. Transnitrosation kinetics of CANO with glutathione showed the formation of cystamine and the mixed disulfide with no formation of oxidized glutathione (GSSG). The nitrosation reaction was satisfactorily simulated by a simple reaction scheme involving eight reactions.
Keywords: nitric oxide, nitrosation, thiols, cysteamine, kinetics
Introduction
Nitric oxide (NO) is a simple gaseous molecule with fascinating biological functions.1 It is one of the most versatile bioactive molecules ever identified. Unlike other free radicals, it is relatively stable, but becomes unstable in biological systems due to its reaction with oxygen, superoxide anion (O2•−), and haem proteins.2 Nitrite (NO2−) is the major end product of its reaction with oxygen. Nitrite can also release NO on acidification and by the reductase activity of the enzyme, xanthine oxidoreductase, at pH conditions lower than 6.0.3–5
Nitric oxide has been the focus of many research groups in recent years, probably because of the realization that it is synthesized by mammalian cells and can act both as a physiological messenger and as a cytotoxic agent.6 It is an essential physiological signaling molecule necessary for cell-to-cell communication and it is also involved in the regulation of gene expression and modification of sexual and aggressive behavior.7,8 It is actively involved in smooth muscle relaxation due to its ability to activate guanosine 3′,5′-cyclic monophosphate (cGMP) in smooth muscle tissue.9,10 The role of nitric oxide in the physiological system at any given time significantly depends on its concentration and its environment. It is known to perform most of its beneficial roles, such as vasorelaxation11,12 and neurotransmission,13,14 at low concentrations, on the order of 5–10 nmol/L.15 At this concentration range it cannot effectively compete with superoxide dismutase (SOD) for O2•−.
NO also readily reacts with O2•− to produce the highly damaging and genotoxic oxidant (peroxynitrite, ONOO−):
| [R1] |
The bimolecular rate constant for rxn. [R1] is 6.7 × 109 (mol/L)−1 s−1.15,16 Superoxide anion radical concentrations in the physiological environment are in the order of 4–10 μmol/L.15 When NO concentrations are in the micromolar range, rxn. [R1], which produces the deleterious peroxynitrite, becomes competitive with the deactivating reaction that removes superoxide anion radical.
Excessive production of nitric oxide has thus been linked to so many cytotoxic activities such as septic shock and liver injury.17,18 It is necessary to have processes that can reduce NO concentrations to beneficial levels in the physiological environment. Such processes would include its reaction with thiols to form S-nitrosothiols (RSNOs). For example, RSNOs have been shown to improve end-organ recovery in models of ischemia–reperfusion injury in the heart and liver.19,20 Biological nitrosothiol formation is a post-translational modification of biological thiols that is subsequently linked to all those functions that are attributed to nitric oxide.21 Possible roles of S-nitrosation include the regulation of apoptosis through glyceraldehyde-3-phosphate dehydrogenase modification22 as well as involvement in the pathogenesis of Parkinson’s disease.23 There are several studies on nitrosothiol formation by Williams,24–33 but these studies dwell on formations and characterizations and not so much on the mechanisms and kinetics. Despite the potential benefits of the relevance of the mechanistic basis of nitrosations (both S- and N-nitrosations),34 very few studies have been directed at elucidating these mechanisms.
Cysteamine (2-aminoethanethiol) is a very important physiologically active β-aminothiol. It is produced in vivo by degradation of coenzyme A. Coenzyme A is a cofactor in many enzymatic pathways, which include fatty acid oxidation, heme synthesis, synthesis of the neurotransmitter acetylcholine, and amino acid catabolism.35 Cysteamine is medically known as Cystagon and can be used as oral therapy for the prevention of hypothyroidism and as a growth enhancer in patients with nephropathic cystinosis.36 Cysteamine and its disulfide (cystamine) can also be used in topical eye drops to dissolve corneal cystine crystals.37,38 It has also been shown to be an effective molecule against hepatotoxicity caused by excessive production of nitric oxide.17
Although a series of studies have been performed on the metabolism of this thiol and its metabolites with respect to oxidation by reactive oxygen species,39–41 nothing has been done on the elucidation of its mechanism of nitrosation by NO and (or) NO-derived metabolites (N2O3 and NO+). Its nitrosation is well-known and has been reported in several manuscripts,42,43 but the exact mechanistic details are still unknown. We report herein on the detailed kinetics and mechanism of the nitrosation of cysteamine. The nitrosating agent is produced in situ, by premixing H+ and NO2 and utilizing a double-mixing stopped-flow spectrophotometer. This manuscript also reports on detailed product analysis as well as on transnitrosation kinetics. The most prevalent thiol-based antioxidant in the physiological environment is glutathione. It is envisaged that, in any discussion of S-nitrosation, glutathione would be a major player.
Experimental
Materials
Cysteamine hydrochloride (CA, 2-aminoethanethiol hydrochloride) 98%, cystamine, cysteine (99+%), sodium perchlorate (Acros), glutathione, sodium nitrite, sodium chloride, perchloric acid (72%), ferrous sulfate (Fisher Scientific), cuprous chloride, and cupric chloride (GSF chemicals) were used as purchased. Stock solutions of CA and nitrite were prepared just before use. Reagent solutions were prepared with distilled and deionized water from a Barnstead Sybron Corporation water purification unit. Inductively coupled plasma mass spectrometry (ICP–MS) was used to show that our aqueous reaction medium did not contain enough transition-metal ions to affect the overall reaction kinetics and mechanism The highest metal ion concentration was cadmium at 1.5 ppb followed by lead at 0.43 ppb. Copper was virtually undetectable.
Methods
All experiments were carried out at 25.0 ± 0.1 °C and at a constant ionic strength of 1.0 mol/L (NaClO4). The progress of the nitrosation reaction was monitored spectrophotometrically by following the absorbance of S-nitrosocysteamine (CANO) at its experimentally determined absorption peak of 545 nm, where an absorptivity coefficient of 16.0 ± 0.1 (mol/L)−1 cm−1 was evaluated. Although CANO has a second absorption peak at 333 nm, with a much higher absorptivity coefficient of 536.0 (mol/L)−1 cm−1, its maximum absorption at 545 nm was used owing to the lack of interference from the absorption peaks of nitrogen species (NO2−, NO+, and HNO2) at this absorption wavelength (see Fig. 1).
Fig. 1.
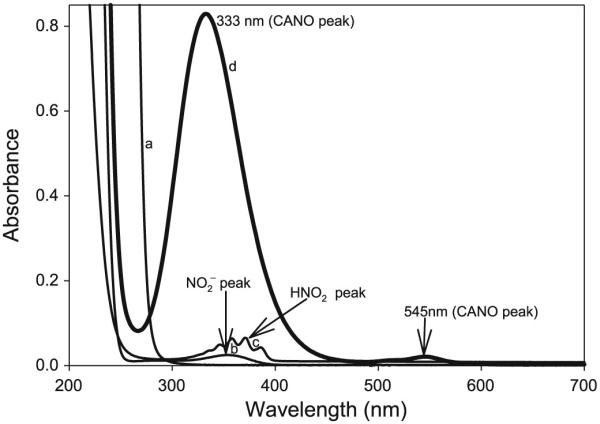
UV–vis spectral scan of reactants 0.01 mol/L cysteamine hydrochloride (CA, a), 0.001 mol/L NaNO2− (b), 0.001 mol/L HNO2 (c), and product 0.001 mol/L S-nitrosocysteamine (CANO, d).
Product identification and verification was achieved by complementary techniques: UV–vis spectrophotometry, HPLC, quadrupole time-of-flight mass spectrometry (TOF-MS), and electron paramagnetic resonance (EPR) spectrometry. Kinetics measurements for fast reactions were performed on a Hi-Tech Scientific double-mixing SF61-DX2 stopped-flow spectrophotometer. For slower reactions, a PerkinElmer Lambda 25 UV–vis spectrophotometer was used.
Stoichiometric determinations were performed by mixing excess perchloric acid and nitrite with a fixed amount of CA such that CA was the limiting reagent. The reaction was allowed to proceed to completion while rapidly scanning the reacting solution between 200 and 700 nm. The concentration of CANO was deduced from its maximum absorption at 545 nm.
HPLC technique
The HPLC system utilized a Shimadzu Prominence (Columbia, Maryland) SPD-M10A VP diode array detector, dual LC 600 pumps, LPT-6B LC–PC interface controller, SIL-10AD auto injector, and Class-VP chromatography data system software. All samples were loaded on a reversed phase Discovery 5 μm C18 column (Supelco, Bellefonte, Pennsylvania). They were run isocractically at 0.15% trifluoroacetic acid (TFA) / H2O to 10% methanol / H2O, and CANO was detected at 545 nm (for selective scans) with maximum spectral scan mode employed to detect all eluents. A flow rate of 1 mL/min was maintained. All solutions for HPLC analysis were made with Milli-Q Millipore purified water and filtered with Whatman Polypropylene 0.45 μm pore-size filter devices before injection (10 μL) into the column using the auto injector. To curb the interaction of the protonated amines on the analytes with the silanol groups on the stationary phase (which was causing tailing of peaks), the sodium salt of 1-octanesulfonic acid (0.005 mol/L) was incorporated into the aqueous mobile phase as an HPLC ion-pairing agent. This was sufficient to neutralize the protonated amines and produce good resolution while eliminating peak tailing.
TOF-MS
Mass spectra were acquired on a Micromass QTOF-II (Waters Corporation, Millford, MA) quadrupole time-of-flight mass spectrometer. Analytes were dissolved in 50:50 acetonitrile / water – 1% formic acid mixture, and analyte ions were generated by positive-mode electrospray ionization (ESI) at a capillary voltage of 2.8 kV and a flow rate of 5 μL/min. The source block was maintained at 80 °C and the nitrogen desolvation gas was maintained at 150 °C and at a flow rate of 400 L/h. Data were visualized and analyzed with the Micromass MassLynx 4.0 software suite for Windows XP (Water Corporation, Millford, Massachusetts).
EPR Measurements
A Bruker e-scan EPR spectrometer (X-band) was used for the detection of radicals and to confirm the release of NO upon the decomposition of CANO. Detection of NO was through the use of the NO-specific spin trap, aci-nitroethane.44 The following settings were used for a typical run: microwave power, 19.91 mW; modulation amplitude, 1.41 G; receiver gain, 448; time constant, 10.24 ms; sweep width, 100 G; and frequency, 9.78 GHz. All measurements were taken at room temperature.
Results
The major reaction studied was a simple nitrosation of cysteamine through in situ production of nitrous acid as the nitrosating agent. The yield of CANO varied, based on the order of mixing reagents. The highest yield, which gave quantitative formation of CANO, was obtained by premixing nitrite with acid and incubating the solution for approximately 1 s before subsequently reacting this nitrous acid with cysteamine in a third reagent stream of the double-mixing stopped-flow spectrophotometer. The value of the absorptivity coefficient of CANO at 545 nm adopted for this study of 16.0 mol/L−1 cm−1 was derived from this mixing order. Premixing CA with acid before the addition of nitrite gave a lower yield of CANO. The available protons are reduced by the protonation of CA.
| [R2] |
This subsequently reduces the amount of the immediately available nitrosating agent, HNO2.
Stoichiometry and product analysis
The stoichiometry, in conditions of excess acid and nitrite over CA, was strictly 1:1.
| [R3] |
Stoichiometry rxn. [R3] is attained within a minute of mixing the reagents at high acid concentrations, and a little longer in lower acid concentrations. High acid concentrations rapidly form the product (CANO), which also rapidly decomposes. A resolvable mass spectrum is taken at low acid concentrations, and this generally will show both the reagent (CA) and the single product (CANO). This mass spectrum is shown in Fig. 2A. There are no other peaks that can be discerned from the reaction mixture, apart from those belonging to the reagent and the product. Incubation of the product of nitrosation overnight shows a final conversion of the CANO into the disulfide, cystamine (H2NCH2CH2S–SCH2CH2NH2, Fig. 2B).
Fig. 2.

Mass spectrometry analysis of the product of nitrosation of cysteamine hydrochloride (CA) showing (A) formation of S-nitrosocysteamine (CANO), m/z = 107.9999; (B) product of decomposition of CANO after 24 h. Cystamine, a disulfide of CA was produced with a strong peak, m/z = 153.0608.
| [R4] |
Nitric oxide can be trapped and observed by EPR techniques through the use of a nitroethane trap.44 The aci-anion of this trap, formed in basic environments, generates a long-lived spin adduct with NO, which is detectable and quantifiable by its distinct EPR spectrum pattern and g value.
| [R5] |
This adduct ([CH3C(NO)(NO2)]•2−) is EPR-active. No peaks were obtained in the EPR spectrum when the nitroethane trap was utilized during the formation of CANO, indicating that NO is not released during the S-nitrosation and that it is not a major nitrosation agent in this mechanism. Upon use of the trap, however, during the decomposition of CANO, a strong EPR spectrum indicating the presence of NO was obtained, proving stoichiometry [R4]. Figure 3 shows a series of EPR spectra that proves the presence of NO in the decomposition of CANO. The first three spectra show that, on their own, nitroethane, CA, and NO2− cannot generate any active peaks that can be attributed to NO. In excess concentrations of the trapping reagent, heights of the spectral peaks can be used, after a standardization, as a quantitative measure of the available NO. NO has a very poor solubility in water, and so the technique is limited only to low NO concentrations.
Fig. 3.

Electron paramagnetic resonance (EPR) spectra of the NO radical generated during the decomposition of S-nitrosocysteamine (CANO) using 0.5 mol/L nitroethane (NE) in 1.0 mol/L NaOH solution as the spin trap. (A) 0.5 mol/L NE, (B) 0.01 mol/L NO2−, (C) 0.04 mol/L cysteamine hydrochloride (CA), (D) [CANO] = 0.01 mol/L, (E) [CANO] = 0.02 mol/L, (F) [CANO] = 0.03 mol/L, and (G) [CANO] = 0.04 mol/L.
Reaction kinetics
The reaction has a very simple dependence on cysteamine concentrations (see Figs. 4A and 4B). There is a linear first-order dependence on the rate of nitrosation with CA concentrations with an intercept kinetically indistinguishable from zero. The reaction kinetics are more reproducible in the format utilized in Fig. 4 in which concentrations of acid and nitrite are equal and in overwhelming excess over CA concentrations. The effect of nitrite is also simple and first-order (see Figs. 5) when nitrite concentrations do not exceed the initial acid concentrations. Effectively, under these conditions, all N(III) species in the reaction environment are in the form of protonated HNO2. Acid effects are much more complex and are dependent on the ratio of initial acid to nitrite concentrations. The nitrosation response to acid differs, depending on whether initial proton (H3O+) concentrations exceed initial nitrite concentrations. The most important parameter affecting the rate of nitrosation is the concentration of HNO2. A set of kinetics traces that encompass a proton concentration variation, which ranges from concentrations that are lower than nitrite to those that exceed, was performed. A simple plot of added acid to the rate of nitrosation shown in Fig. 6A clearly displays a discontinuity at the point where acid exceeds nitrite concentrations. By utilizing the dissociation constant of HNO2, the final concentrations of H3O+, NO2−, and HNO2 can be calculated for each set of initial concentrations. These data are shown in Table 1 for the input concentrations used for the set of kinetics experiments. Close examination of the data in Table 1 shows that the kinetics of the reaction are vastly different depending on whether initial acid concentrations exceed initial nitrite concentrations. When nitrite is in excess (the first four readings), there is a sharp nonlinear increase in the rate of nitrosation with nitrous acid concentrations (Fig. 6B), and a saturation in rate with respect to the actual proton concentrations despite the recalculation of the concentrations undertaken in Table 1. Figure 6B would strongly indicate second-order kinetics in nitrous acid, and a plot of nitrosation rate versus nitrous acid to the second power is linear (plot not shown). Table 1 shows that, in the series of data plotted for Fig. 6B, nitrous acid concentrations and excess proton concentrations increase while nitrite concentrations decrease, and the final observed rate of nitrosation is derived from these three species concentrations and not isolated to nitrous acid. At acid concentrations that exceed initial nitrite concentrations, there is a linear dependence in rate on acid concentrations (Fig. 6C). Note that a saturation would have been achieved in nitrous acid concentrations as would be expected in this format in which the nitrous acid saturates at 50 mmol/L. The increase in nitrosation rate with an increase in acid after saturation of HNO2 indicates that other nitrosants apart from HNO2 exist in the reaction environment.
Fig. 4.

(A) Absorbance traces showing the effect of varying cysteamine hydrochloride (CA) concentrations. The reaction displays first-order kinetics in CA. [NO2−]0 = 0.10 mol/L; [H+]0 = 0.10 mol/L; and (a) [CA]0 = 0.005 mol/L; (b) [CA]0 = 0.006 mol/L; (c) [CA]0 = 0.007 mol/L; (d) [CA]0 = 0.008 mol/L; (e) [CA]0 = 0.009 mol/L; and (f) [CA]0 = 0.010 mol/L. (B) Initial rate plot of the data in Fig. 4A. The plot shows the strong first-order dependence of the rate of formation of S-nitrosocysteamine (CANO) on CA, with an intercept kinetically indistinguishable from zero.
Fig. 5.
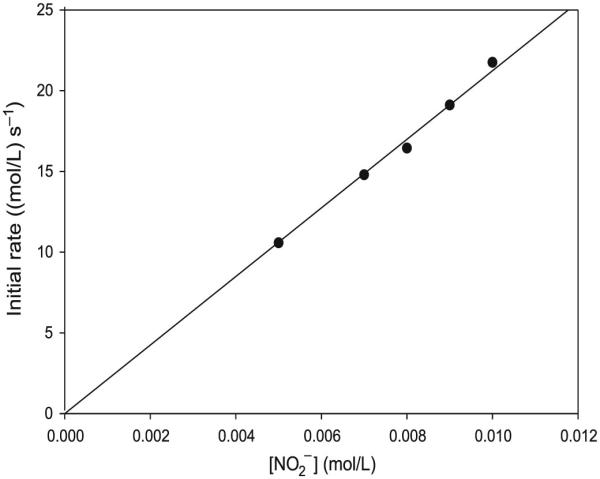
Initial rate plot from the data derived from varying nitrite ion concentrations in excess acid over nitrite. [CA]0 = 0.10 mol/L; [H+]0 = 0.10 mol/L; and [NO2−]0 was varied from 0.005 to 0.010 mol/L. The plot shows strong first-order dependence on initial rate of formation of S-nitrosocysteamine (CANO) on nitrite. CA, cysteamine hydrochloride.
Fig. 6.

(A) Plot of the raw kinetics data for the effect of added acid on the rate of nitrosation.[CA]0 = 0.05 mol/L; [NO2−]0 = 0.05 mol/L; and [H+]0 was varied between 0.01 and 0.08 mol/L. For the first four data points, [NO2−]0 > [H+]0. The linear potion of the plot represents excess proton concentrations over nitrite. Acid concentrations plotted here are those experimentally added and not calculated. (B) Plot of initial rate versus nitrous acid concentrations of the experimental data in Fig. 6A and calculated in Table 1. This plot involves the region at which initial nitrite concentrations exceed acid concentrations. (C) Plot showing the linear dependence of the rate of formation of S-nitrosocysteamine (CANO) on [H3O+] in high acid concentrations, where initial acid concentrations exceed nitrite concentrations. Nitrous acid concentrations are nearly constant in this range (see Table 1).
Table 1.
Input species concentrations and final reagent concentrations for acid dependence experiments (units of mol/L).
| [H+]added | [NO2−]added | [HNO2]initial | [NO2−]final | [HNO2]final | [H3O+]final |
|---|---|---|---|---|---|
| 0.01 | 0.05 | 0.01 | 4.01×10−2 | 9.86×10−3 | 1.38×10−4 |
| 0.02 | 0.05 | 0.02 | 3.04×10−2 | 1.97×10−2 | 3.63×10−4 |
| 0.03 | 0.05 | 0.03 | 2.08×10−2 | 2.92×10−2 | 7.90×10−4 |
| 0.04 | 0.05 | 0.04 | 1.18×10−2 | 3.82×10−2 | 1.82×10−3 |
| 0.05 | 0.05 | 0.05 | 5.03×10−3 | 4.50×10−2 | 5.03×10−3 |
| 0.06 | 0.05 | 0.05 | 2.22×10−3 | 4.78×10−2 | 1.22×10−2 |
| 0.07 | 0.05 | 0.05 | 1.07×10−3 | 4.89×10−2 | 2.11×10−2 |
| 0.08 | 0.05 | 0.05 | 8.93×10−4 | 4.91×10−2 | 3.09×10−2 |
Effect of copper
Copper has been implicated in many reactions involving organosulfur compounds.45–47 Cu(I) is known to be much more active than Cu(II). It is easier to assay for Cu(II) and the addition of Cu(II) to organosulfur reactions involves its initial conversion to the more active Cu(I). Figure 7A shows that addition of Cu(II) rapidly increases the nitrosation reaction. Cu(II) ions, however, also catalyze the decomposition of CANO. A plot of the rate of nitrosation versus Cu(II) concentrations shows the sigmoidal rate increase followed by an abrupt saturation, which is typical of catalytic mechanisms (figure not shown). No nitrosation occurs in the presence of nitrite without acid (which is necessary to generate the HNO2). Figure 7B, however, shows that, even in the absence of acid, Cu(II) ions can still effect the nitrosation of CA. Cu(I) ions, however, could not effect nitrosation in the absence of acid. Figure 7C shows the catalytic effect of Cu(II) ions on the decomposition of CANO. CANO was prepared in situ from two feed streams of the double-mixing stopped flow ensemble. Cu2+ ions derived from cupric chloride were next added from a third feed stream after complete production of CANO. All feed streams were buffered at pH 7.4. The two control experiments (traces a and b) show the slow decomposition expected from CANO solutions at pH 7.4. Trace b was treated with EDTA to sequester all metal ions in the reaction solution. This trace is identical to the one without EDTA (trace a), confirming the absence of metal ions in reagent solution, which can affect the decomposition kinetics. Addition of micromolar quantities of Cu(II) ions effects a big acceleration in the rate of decomposition. Higher Cu(II) concentrations give a strong autocatalytic decay of the nitrosothiol (trace f in Fig. 7C). The catalytic effect of Cu(II) ions on the decomposition can also be evaluated through the rate of production of nitric oxide, which is known to be the major product, together with the disulfide, of nitrosothiol decompositions. Figure 7D shows EPR spectra using the nitroethane trap, of the enhanced production of NO with addition of Cu(II) ions. All three spectra were taken after the same time lapse from CANO incubation, and thus the peak heights of the NO-induced spectra can be correlated with the extent of reaction.
Fig. 7.

(A) Absorbance traces showing the effect of varying Cu2+ concentrations on the rate of formation of CANO. There is a progressive increase in the rate of S-nitrosocysteamine (CANO) formation with an increase in Cu2+ concentration. [CA]0 = [NO2−]0 = [H ]0 = 0.05 mol/L; and (a) [Cu2+]0 = 0.00 mol/L, (b) [Cu2+]0 = 5 μmol/L, (c) [Cu2+]0 = 15 μmol/L, (d) [Cu2+]0 = 100 μmol/L, (e) [Cu2+]0 = 1 mmol/L, and (f) [Cu2+]0 = 2.5 mmol/L. CA, cysteamine hydrochloride. (B) Absorbance traces showing the formation of CANO via a reaction of CA, nitrite, and copper(II) without acid. The amount of CANO formed increases with an increase in Cu2+ concentration. No formation of CANO was observed with Cu+. The result shows the catalytic effect of Cu2+ on the rate of formation of S-nitrosothiol (RSNO). [CA]0 = [NO2 ]0 = [H ]0 = 0.05 mol/L; and (a) [Cu2+] = 0.0025 mol/L, (b) [Cu2+] = 0.0030 mol/L, (c) [Cu2+] = 0.0035 mol/L, (d) [Cu2+] = 0.0040 mol/L, (e) [Cu2+] = 0.0045 mol/L, and (f) [Cu2+] = 0.0050 mol/L. (C) Effect of Cu2+ on the stability of CANO in a pH 7.4 phosphate buffer. This reaction involves the reduction of Cu2+ to Cu+, which causes the decomposition of CANO. [CANO]0 = 0.028 mol/L; (a) [Cu2+]0 = 0.00 mol/L (pure CANO), (b) [Cu2+]0 = 0.00 mol/L (pure CANO in 0.00001 mol/L EDTA), (c) [Cu2+]0 = 5 μmol/L, (d) [Cu2+]0 = 10 μmol/L, (e) [Cu2+]0 = 20 μmol/L, and (f) [Cu2+]0 = 30 μmol/L. (D) Electron paramagnetic resonance (EPR) spectra of the NO radical showing the catalytic effect of Cu2+ on the rate of decomposition of CANO. The intensity of the spectra increases with an increase in Cu2+ concentration. [CANO]0 = 0.01 mol/L; (a) [Cu2+]0 = 0.00, (b) [Cu2+]0 = 1.0 × 10−4 mol/L, and (c) [Cu2+]0 = 2.0 × 10−4 mol/L.
Transnitrosation
The most abundant thiol in the physiological environment is glutathione (GSH). A pertinent reaction to study would be the interaction of any nitrosothiol formed in the physiological environment with glutathione. There is a crucial question about the lifetimes of the nitrosothiols formed. If thiols are carriers of NO from the point of production to the point of its usage, the nitrosothiols formed should be stable enough and have a long enough lifetime to enable them to perform this activity. Spectrophotometric study of transnitrosation is rendered complex by the fact that many nitrosothiols seem to absorb around the same wavelength and with similar absorptivity coefficients. Figure 8A shows a series of superimposed spectra of four well-known nitrosothiols, which show nearly identical spectra. Figure 8B shows the spectrophotometric characterization of possible transnitrosation in mixtures of CANO and GSH. GSNO has a higher absorptivity coefficient than CANO at the same wavelength,48 and this explains the initial increase in absorbance on addition of GSH to CANO. LC–MS data have shown that the reaction mixture can transiently (before attainment of the equilibrium mixture) contain any of at least six components at varying times and concentrations: CANO, S-nitrosoglutathione (GSNO), GSH, CA– CA (cystamine), GSSG (oxidized glutathione), and CA–GS (mixed disulfide). The rate of transnitrosation will be difficult to evaluate owing to the presence of the mixed disulfide. Figure 8B shows that, relatively, CANO is more stable than GSNO because of the rapid rate of consumption of GSNO upon its formation as compared with the much slower CANO decomposition with or without EDTA. Figure 9 shows a GC–MS spectrum of mixtures of CANO and GSH. This involved a 3:5 ratio of CANO to GSH, which is also trace f in Fig. 8B. The remarkable aspect of this spectrum is the absence of the GSSG, whose precursor is GSNO. Thus, GSNO, when formed, rapidly decomposes by reacting with CA to form the mixed disulfide and releasing NO. The cystamine formed and observed in the spectrum is derived from the normal decomposition of CANO (see rxn. [R10], vide infra).
Fig. 8.

(A) Superimposed spectra of four well-known nitrosothiols (a) CysNO, (b) S-nitrosocysteamine (CANO), (c) S-nitrosoglutathione (GSNO), and (d) nitrosothiol of penicillamine (SNAP). Three of them nearly have the same ε at 545 nm. The observed separation in the absorbances of these three at 545 nm is due to staggering of the time lag before acquiring the spectrum. (B) The effect of glutathione (GSH) on the stability of CANO in a pH 7.4 phosphate buffer. All experimental traces have [CA]0 = 0.03 mol/L, [NO2−]0 = 0.03 mol/L, and [H+]0 = 0.05 mol/L. (a) [GSH] = 0, (b) [GSH] = 0, (c) [GSH] = 0.01 mol/L, (d) [GSH] = 0.02 mol/L, (e) [GSH] = 0.03 mol/L, and (f) [GSH] = 0.05 mol/L. Traces a and b are controls. Trace a has no EDTA. Trace b is the same as trace a with 10 μmol/L of EDTA. There is no significant difference between traces a and b, indicating that the water used for preparing reagent solutions did not contain enough trace metal ions to affect the kinetics. Ten microlitres of EDTA was added to
Fig. 9.

A GC–MS spectrum of a 3:5 ratio of S-nitrosocysteamine (CANO) to glutathione (GSH); trace f in Fig. 8B. Final products were predominantly the mixed disulfide and cysteamine with no evidence of oxidized glutathione (GSSG). CA, cysteamine hydrochloride.
Mechanism
The overall rate of nitrosation is dependent on the rate of formation and accumulation of the nitrosating agents. A number of possibilities have been suggested, and these include HNO2, NO, NO2, N2O3, and the nitrosonium cation, +NO. Some nitrosants cannot exist under certain conditions; for example, the +NO cannot exist at high pH49 conditions and NO2 cannot be produced in strictly anaerobic conditions. Figure 10 shows two nitrosation reactions at equivalent conditions except that one was run in solutions that had been degassed with argon and immediately capped. Aerobic environments can form NO2 from NO and subsequently N2O3 will also be formed.50
Fig. 10.

Evaluating the effect of oxygen on the rate of nitrosation of cysteamine. The solutions purged with argon (trace a) give a very slightly lower rate of nitrosation, indicating a small and insignifant contribution of nitrosation by N2O3. [CA]0 = 0.05 mol/L; [H+]0 = 0.08 mol/L; and [NO2−]0 = 0.07 mol/L. CA, cysteamine hydrochloride.
| [R6] |
| [R7] |
Both NO2 and N2O3 are potent nitrosating agents.29 There is a slightly higher rate of nitrosation in aerobic environments when compared with those in anaerobic environments. This is a reflection of the low dissolved oxygen concentrations available in aqueous solutions at approximately 2 × 10−4 mol/L. Thus, although formation for these nitrosants is favored, their viability is hampered by the low concentrations of oxygen as well as NO, which is only produced during the decomposition of the nitrosothiol. NO was not detected during the formation of CANO. In the aqueous environment utilized for these experiments, reaction of aqueous N2O3 to nitrous acid would be overwhelmingly dominant. This is a rapid equilibrium reaction that favors the weak acid, HNO2.
| [R8] |
Early studies by Bunton and Steadman51 had erroneously estimated a value for KH−1 = 0.20 (mol/L)−1, which would indicate appreciable amounts of N2O3(aq) over nitrous acid. A more accurate evaluation by Markovits et al.52 gavea more realistic value of KH−1 = 3.03 × 10−3 (SD 0.23 × 10−3) (mol/L)−1. According to Turney,53 appreciable amounts of N2O3 can only exist in highly acidic environments, in the region of 5 mol/L perchloric acid. Such conditions are not applicable for the physiological environment. Most nitrosation studies in aerobic NO environments neglect the contribution to nitrosation by NO2 and only concentrate on NO and N2O3. NO2 concentration will always be negligibly low because of rxn. [R6], which is two orders of magnitude greater than the rate of nitrosation by NO2 (1.1 × 109 (mol/L)−1 s−1 vs 2.0 × 107 (mol/L)−1 s−1, respectively),54,55 is hampered by the low concentrations of oxygen available. In the presence of NO, it is more unstable with respect to N2O3.
All nitrosation kinetics show first-order dependence in both thiol and nitrite with a complex dependence on acid. Data in Figs. 4 and 5 show this strong first-order dependence on CA and on nitrite. In conditions in which acid concentrations are less than initial nitrite concentrations, the effect of nitrite is more difficult to interpret, since a change in its concentrations concomitantly induces a change in both acid and HNO2 concentrations, and thus its sole effect cannot be isolated. First-order kinetics in nitrite strongly suggest that the major nitrosant at the beginning of the reaction is HNO2, since there is a direct correlation between nitrite concentrations and HNO2, especially when there is consistently excess acid over nitrite. After performing calculations for the relevant reactive species in the reaction medium (Table 1), the plot of the initial rate of nitrosation versus nitrous acid concentrations shows a parabolic dependence, which erroneously suggests second-order kinetics in HNO2 (Fig. 6B) for the region in which initial nitrite concentrations exceed acid. As mentioned in the Results section, the data in Table 1 show that HNO2 is not changing in isolation. Both nitrite and acid are also concomitantly changing, and so the effect observed in rate is not solely attributable to changes in HNO2 concentrations. When acid is in excess over nitrite, the amount of HNO2 saturates, but as can be seen in Fig. 6C, the rate keeps increasing linearly with an increase in acid, indicating the presence of multiple nitrosating agents apart from HNO2. The simplest rate law that can explain this nitrosation rate involves +NO and HNO2. +NO can be formed from the protonation of HNO2:
| [R9] |
(H(OH)N=O+ is a hydrated nitrosonium cation, +NO + H2O).
The overall rate of reaction, using these two nitrosants gives
| [1] |
where [N(III)]T is the total concentrations of nitrous acid and nitrite as calculated in Table 1 (columns 4 and 5) and [H+] is the calculated acid concentration in column 6 of Table 1. Ka is the acid dissociation constant of nitrous acid, Kb is the equilibrium for the protonation of nitrous acid (rxn. [R9]), k1 is the bimolecular rate constant for the nitrosation of CA by HNO2, and k2 is the bimolecular rate constant for the nitrosation by +NO. Ka was taken as 5.62 × 10−4 (mol/L)−1 and, thus, at high acid concentrations, a plot of Rate/[N(III)]T [CA] vs [H+] should give a straight line with slope k2Kb and intercept k1. This type of plot only applies to conditions of excess acid over nitrite (Fig. 6C). One can derive values for k1 and product k2Kb from these series of plots. A value of k1 = 17.9 ± 1.5 (mol/L)−1 s−1 was derived from a series of several complementary experiments. k2 was deduced as 6.70 × 104 (mol/L)−1 s−1 after assuming a Kb value of 3.16 ×10−2 (mol/L)−1.
Decomposition of CANO
RSNOs differ greatly in their stabilities, and most of them have never been isolated in solid form. Homolysis of the S–N bond occurs both thermally and photochemically, but, generally, these processes are very slow at room temperature and in the absence of radiation of the appropriate wavelength. In vivo decomposition of RSNOs is likely to proceed through other pathways apart from these two. The presence of trace amounts of Cu(II) or Cu(I) can efficiently catalyze RSNOs to disulfide and nitric oxide (see Figs. 7A–7D).
| [R10] |
The characterization of the S–N bond is crucial in determining the stability of RSNOs, and, unfortunately, very little work has been performed in this area. The little crystallographic data available on isolable RSNOs indicate that the S–N bond is weak, sterically unhindered, elongated at between 0.17 and 0.189 nm,56 and exists as either the cis (syn) or trans (anti) conformers. These conformers are nearly isoenergetic but separated by a relatively high activation barrier of ~11 kcal mol−1 (1 cal = 4.184 J). CANO is a primary S-nitrosothiol, with primary and secondary nitrosothiols known to be kinetically unstable to decomposition when compared with tertiary nitrosothiols.57,58 S-Nitroso-N-acetylpenicillamine is the most well-known stable tertiary nitrosothiol.59 The mechanism of decomposition of nitrosothiols was believed to proceed through the homolytic cleavage of this weak S–N bond. Computed S–N bond dissociation energies for nitrosothiols show very little variation, however, between primary, secondary, and tertiary nitrosothiols, resulting in very similar homolysis rates of reaction at elevated temperatures for all nitrosothiols. The computed activation parameters for thermal homolysis to occur are prohibitively high for this to be a dominant pathway under normal biological conditions. For example, assuming an approximate S–N bond energy of 31 kcal,58 a rough calculation will indicate that if decomposition was to proceed solely through homolysis, the half-life of a typical nitrosothiol would be in the region of years instead of the minutes to hours that are observed.60 This analysis does not support the work reported by Roy et al.61 in which they established homolysis as the major decomposition pathway. They also managed to trap the thiyl radical, RS·. Close examination of their reaction conditions show high trap concentrations (0.2 mol/L DMPO) and nitrosothiol (0.1 mol/L). Such conditions would catch even very low concentrations of thiyl radical, but would not necessarily indicate that this was the dominant pathway and whether it was derived solely from homolytic cleavage of the S–N bond. Nitrosothiol decomposition rates in the absence of metal ions are known to be mostly zero order57 and this is inconsistent with an S–N bond homolysis-driven decomposition,62 which would have been expected to be first order. In the presence of excess thiol, however, decomposition of nitrosothiols proceeds via a first-order decay process.59 The absence of first-order decay kinetics in the decomposition of CANO also rules out homolysis as a dominant pathway. For a series of nitrosothiols, the homolysis-dependent decomposition rates are not dependent on the bulkiness of the substituent groups attached to the nitrosothiols, which seems to enhance the calculations that showed no discernible difference in the S–N bond energies irrespective of whether the nitrosothiol was primary, secondary, or tertiary.63 In aerated solutions, the decay was autocatalytic, thus implicating N2O3 as the autocatalytic propagating species (see rxns. [R6] and [R7]). Nitrosothiol decompositions were much slower in solutions in which NO was not allowed to escape, implicating NO as a possible retardant of nitrosothiol decomposition.25 This can be explained by the recombination of the thiyl radical with NO in the reaction cage after initial cleavage. Elimination of NO from the medium by bubbling air forces rxn. [R11] to the right.
| [R11] |
No mechanism has yet been proposed for the implication of N2O3 in the autocatalytic mechanism. One can assume that N2O3 should assist in the cleavage of the S–N bond, thus accelerating the rate of homolytic decomposition of RSNOs.
In aerated solutions, NO then undergoes rxns. [R6] and [R7] to yield N2O3.
| [R12] |
The incorporation of the NO2− group in the nitrosothiol would weaken the S–N bond, leading to its easy cleavage and regeneration of the N2O3 molecule, which will further catalyze the decomposition.
| [R13] |
The sequence of rxns. [R11]–[R13] will deliver autocatalysis in N2O3, since rxn. [R13] produces N2O3, which recycles back into rxn. [R12].
Recent results have suggested a novel pathway for the decomposition of nitrosothiols that involves an initial internal rearrangement to form an N-nitrosation product.64 Typical nitrosothiol decomposition products after such an internal rearrangement would include thiiranes and 2-hydroxy-mercaptans. This mechanism has been used successfully to explain the production of a small but significant quantity of 2-hydroxy-3-mercaptopropionic acid (apart from the dominant product cystine) in the decomposition of S-nitrosocysteine. The GC–MS spectrum shown in Fig. 2, however, gives a dominant signal for dimeric cystamine, with very little evidence of any other significant products, indicating very little initial N-transnitrosation.
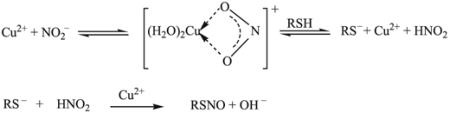
The copper-catalyzed decomposition of nitrosothiols has been well-documented.65–67 The varying rates of nitrosothiol decompositions reported by different research groups can be traced possibly to adventitious metal ion catalysis from the water used in preparing reagent solutions. Even in our case, with the maximum metal ion concentrations of 0.43 ppb in Pb2+, small differences can be observed in EDTA-laced reaction solutions and those that are not (see traces a and b in Fig. 7C). Nitrosothiol decompositions have also implicated other metal ions and not just specifically copper.66 Although Cu(II) ions are used to catalyze nitrosothiol decomposition, the active reagent is Cu(I). In this catalytic mechanism, Cu redox cycles between the +2 and +1 oxidation states. Only trace amounts of Cu(I) are needed to effect the catalysis, and these can be generated from the reduction of Cu(II) ions from minute amounts of thiolate anions that exist in equilibrium with the nitrosothiol.
| [R14] |
| [R15] |
| [R16] |
| [R17] |
The cascade of rxns. [R14]–[R17] continues until the nitrosothiol is depleted and converted to the disulfide. The complex formed in rxn. [R16] has been justified through density functional theory studies that report that Cu(I) binds more strongly to the S center than to the N center of the nitrosothiol.65 This preferential binding weakens and lengthens the S–N bond, thus facilitating its cleavage. It is difficult to extrapolate this proposed mechanism into the physiological environment, but some experimental results have shown that protein-bound Cu2+ catalyzed nitric oxide generation from nitrosothiols, but not with the same efficiency as the free hydrated ion.68 Further studies are needed to evaluate the efficiency of this catalysis, especially in copper-based enzymes.
Effect of Cu(II) on the nitrosation of cysteamine (Fig. 7B)
The fact that Cu(II) ions can assist in nitrosating CA from nitrite in the absence of acid while Cu(I) ions are unable to effect this can be explained by recognizing that nitrosation is an electrophilic process, where NO effectively attaches as the (formally) positive charge, while eliminating a positively charged proton. Cu(II), in this case, acts by complexing to the nitrite and forming a nitrosating species that will release the Cu(II) ions after nitrosation.
Further experiments on the effect of nitrous acid
Since nitrous acid is the major nitrosation species, another series of experiments were undertaken in which the effect of nitrous acid could be unambiguously determined. For the data shown in Fig. 11A, nitrite and acid were kept at equimolar concentrations and varied with that constraint. Final proton and nitrite concentrations after dissociation are also equal for electroneutrality (see Table 2). Thus, if our simple mechanism is plausible, a plot of nitrous acid versus initial nitrosation rate should be linear with the expected slight tailing at either extreme of the nitrous acid concentrations owing to nonlinear generation of the free acid derived from the quadratic equation. Using the same rate law as eq. [1], a value of k1 can be derived and compared with values obtained from the Fig. 6C plot. Kinetics constants derived from the Fig. 11B plot are not expected to be as accurate as those from Fig. 6C, but should deliver values close to those from high acid environments. For the case of nitrous acid as the nitrosating agent (too low a concentration of excess H3O+ to effect equilibrium of rxn. [R9]), data derived from Fig. 11B delivered an upper limit rate constant for k1 that was higher than that derived from Fig. 6C-type data, but well within the error expected for such a crude extrapolation. Other sets of kinetics data were derived in which CA concentrations were varied while keeping the nitrous acid concentrations constant. Varying nitrous acid concentrations were used and, for each, a CA dependence was plotted (data not shown). These were straight lines through the origin and with the slope determined by the concentration of nitrous acid and the free protons existing in solution after the dissociation of nitrous acid. For these kinetics data, eq. [1] had to be evaluated completely, since Ka ≈ [H3O+] and no valid approximation could be made. The values of k1 and k2Kb were utilized in eq. [1] to recalculate initial rates and slopes with respect to CA concentrations. These values were utilized to check the accuracy of the kinetics constants derived from high-acid-dependent experiments. It was noted that, within experimental error, these values checked and were generally robust.
Fig. 11.

(A) Evaluation of unambigous nitrous acid dependence by employing [H+]0 = [NO2−]0 and varying both at the same equimolar concentrations. [CA]0 fixed at 0.25 mol/L. (a) [H+]0 = [NO2−]0 = 0.005 mol/L, (b) [H+]0 = [NO2−]0 = 0.010 mol/L, (c) [H+]0 = [NO2−]0 = 0.015 mol/L, (d) [H+]0 = [NO2−]0 = 0.020 mol/L, (e) [H+]0 = [NO2−]0 = 0.025 mol/L, (f) [H+]0 = [NO2−]0 = 0.030 mol/L, and (g) [H+]0 = [NO2−]0 = 0.035 mol/L. (B) Plot of initial rate of nitrosation versus initial nitrous acid concentrations as calculated in Table 2 and derived from the data in Fig. 11A. This plot is linear, as opposed to Fig. 6C.
Table 2.
Input species concentration and final reagent concentrations for nitrous acid dependence experiments with [H+]0 = [NO2−]0.
| [H+]added | [NO2−]added | [HNO2]initial | [H3O+]final | [NO2−]final | [HNO2]final |
|---|---|---|---|---|---|
| 5.00×10−3 | 5.00×10−3 | 5.00×10−3 | 1.42×10−3 | 1.42×10−3 | 3.58×10−3 |
| 1.00×10−2 | 1.00×10−2 | 1.00×10−2 | 2.11×10−3 | 2.11×10−3 | 7.89×10−3 |
| 1.50×10−2 | 1.50×10−2 | 1.50×10−2 | 2.64×10−3 | 2.64×10−3 | 1.24×10−2 |
| 2.00×10−2 | 2.00×10−2 | 2.00×10−2 | 3.08×10−3 | 3.08×10−3 | 1.69×10−2 |
| 2.50×10−2 | 2.50×10−2 | 2.50×10−2 | 3.48×10−3 | 3.48×10−3 | 2.15×10−2 |
| 3.00×10−2 | 3.00×10−2 | 3.00×10−2 | 3.83×10−3 | 3.83×10−3 | 2.62×10−2 |
| 3.50×10−2 | 3.50×10−2 | 3.50×10−2 | 4.16×10−3 | 4.16×10−3 | 3.08×10−2 |
Simulations
A small set of simple reactions was utilized to simulate the nitrosation kinetics. This set is shown in Table 3. At low acid concentrations, the nitrosation is dominated by HNO2 as the nitrosant, and as acid concentrations are increased, both nitrous acid and the nitrosonium cation are involved in the nitrosation. Even though Table 3 includes nitrosation by N2O3, the lack of oxygen (approximately kept constant at 2.0 × 10−4 mol/L) and its most favored and rapid reaction to nitrous acid made the nitrosation by N2O3 insignificant. N2O3 is the dehydrated form of HNO2 (rxn. [R8]), while reaction conditions were in an overwhelming excess of water. Its insignificant concentration ensured that the simulations were insensitive to value of the bimolecular nitrosation constant for N2O3 (kM8). Figure 12B clearly shows that the concentration of N2O3 never rises to a level where it would be significant. The rate constants adopted for rxn. [M1] (Table 3) were not crucial for as long as they were fast (in both directions) and were connected by Ka. Increasing the forward and reverse rate constants while maintaining Ka only slowed the simulations, but did not alter the simulation results. Reaction [M2] (Table 3) was also a rapid hydration reaction that strongly favored HNO2, thus rapidly depleting any N2O3 formed or merely keeping these concentrations depressed should the tandem rxns. [R6] + [R7] have been added to the mechanism. These were not utilized for this simulation because EPR experiments had not detected NO at the beginning of the reaction and had only detected NO during the decomposition of the nitrosothiol. There are no reliable thermodynamics parameters for rxn. [M3] (Table 3), since this would be heavily dependent on the pH of the medium. For the purposes of this simulation and some of our previous estimates, the equilibrium constant for rxn. [M3] was set at 3.16 × 10−2 (mol/L)−1. Kinetics constants for rxns. [M4] and [M5] (Table 3) were derived from this study. In the absence of significant concentrations of N2O3, the simulations were insensitive to the kinetics constants utilized for rxns. [M6] and [M8] (Table 3). Reaction [M7] (Table 3) was assumed to be slow and, in the absence of Cu(II) ions or other adventitious ions, this is indeed so (see Figs. 7A and 7C). The simulations are shown in Fig. 12A, which gives a reasonably good fit from such a crude mechanism. Figure 12B is provided to show the concentration variation of some of the species that could not be experimentally determined. Of note is that N2O3 concentrations are depressed throughout the lifetime of the nitrosation. Even though +NO is also depressed in concentration, this can be explained by its high rate of nitrosation as soon as it is formed and an equilibrium that favors its hydration back to HNO2 once the thiol is depleted.
Table 3.
The set of reactions used for simulating the nitrosation of cysteamine. RSH, cysteamine.
| Reaction No. |
Reaction | k f | k r |
|---|---|---|---|
| M1 | H+ + NO2− ⇌ HNO2 | 1.10×109 | 6.18×105 |
| M2 | 2HNO2 ⇌ N2O3 + H2O | 1.0×102 | 1.0×108 |
| M3 | HNO2 + H+ ⇌ +N=O + H2O | 3.16×105 | 1.00×107 |
| M4 | RSH + HNO2 → RSNO + H2O | 17.9 | ~0 |
| M5 | RSH + +N=O ⇌ RSNO + H+ | 6.7×104 | 1.0×10−3 |
| M6 | N2O3 ⇌ NO + NO2 | 8.0×109 | 6.0×108 |
| M7 | 2RSNO ⇌ RSSR + 2NO | 4.0×10−3 | ~0 |
| M8 | RSH + N2O3 → RSNO + HNO2 | 5.0×103 | ~0 |
Note: kf, forward rate constant; kr, reverse rate constant.
Fig. 12.

(A) Simulation of the short set of reactions shown in Table 3. Solid lines are experimental data and the symbols indicate simulations. The mechanism is dominated by the value of the bimolecular rate constant for the nitrosation of cysteamine hydrochloride (CA) by HNO2 (k1). [CA]0 = [NO2−]0 = 0.05 mol/L. (B) Results from the modeling that delivered simulations of the two traces shown in Fig. 12A. These simulations show the concentration variations of those species that could not be experimentally monitored. Neither N2O3 nor NO+ rise to any significant concentration levels.
Conclusion
This detailed kinetics study has shown that, even though there are several possible nitrosating agents in solution, in the physiological environment, with its slightly basic pH and low oxygen concentrations, the major nitrosant is HNO2. Furthermore, the lifetime of CANO is not long enough to be able to be effectively used as a carrier of NO. GSH, which has been touted as a possible carrier of NO, appears to have a shorter lifetime than CANO. However, because of its overwhelming concentration in the physiological environment it might still be a major carrier of NO when it is compared with other thiols. Protein thiols, it appears, would be the best carriers by either intra- or inter-molecular transfer of NO through protein nitrosothionylation through exposed and accessible cysteine groups.69
Acknowledgements
This work was supported by Grant No. CHE 1053611 from the National Science Foundation (R.H.S.) and partly from Grant No. R01 EB2044 from the National Institutes of Health (R.S.).The findings and conclusions in this report are those of the authors and do not necessarily represent the views of the National Institute for Occupational Safety and Health.
Contributor Information
Moshood K. Morakinyo, Department of Chemistry, Portland State University, Portland, OR 97207-0751, USA
Itai Chipinda, Allergy and Clinical Immunology Branch, Health Effects Laboratory Division, National Institute of Occupational Safety and Health, Centers for Disease Control and Prevention, 1095 Willowdale Road, Morgantown, WV 26505, USA.
Justin Hettick, Allergy and Clinical Immunology Branch, Health Effects Laboratory Division, National Institute of Occupational Safety and Health, Centers for Disease Control and Prevention, 1095 Willowdale Road, Morgantown, WV 26505, USA.
Paul D. Siegel, Allergy and Clinical Immunology Branch, Health Effects Laboratory Division, National Institute of Occupational Safety and Health, Centers for Disease Control and Prevention, 1095 Willowdale Road, Morgantown, WV 26505, USA
Jonathan Abramson, Department of Physics, Portland State University, Portland, OR 97207-0751, USA.
Robert Strongin, Department of Chemistry, Portland State University, Portland, OR 97207-0751, USA.
Bice S. Martincigh, School of Chemistry, University of KwaZulu-Natal Westville Campus, Private Bag X54001, Durban 4000, Republic of South Africa
Reuben H. Simoyi, Department of Chemistry, Portland State University, Portland, OR 97207-0751, USA; School of Chemistry, University of KwaZulu-Natal Westville Campus, Private Bag X54001, Durban 4000, Republic of South Africa
References
- (1).Thomas DD, Ridnour LA, Isenberg JS, Flores-Santana W, Switzer CH, Donzelli S, Hussain P, Vecoli C, Paolocci N, Ambs S, Colton CA, Harris CC, Roberts DD, Wink DA. Free Radic. Biol. Med. 2008;45(1):18. doi: 10.1016/j.freeradbiomed.2008.03.020. doi:10.1016/j.freeradbiomed.2008.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Möller MN, Li Q, Vitturi DA, Robinson JM, Lancaster JR, Jr, Denicola A. Chem. Res. Toxicol. 2007;20(4):709. doi: 10.1021/tx700010h. doi:10.1021/tx700010h. [DOI] [PubMed] [Google Scholar]
- (3).Li H, Samouilov A, Liu X, Zweier JL. J. Biol. Chem. 2001;276(27):24482. doi: 10.1074/jbc.M011648200. doi:10.1074/jbc.M011648200. [DOI] [PubMed] [Google Scholar]
- (4).Zweier JL, Li H, Samouilov A, Liu X. Nitric Oxide. 2010;22(2):83. doi: 10.1016/j.niox.2009.12.004. doi:10.1016/j.niox.2009.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Godber BL, Doel JJ, Sapkota GP, Blake DR, Stevens CR, Eisenthal R, Harrison R. J. Biol. Chem. 2000;275(11):7757. doi: 10.1074/jbc.275.11.7757. doi:10.1074/jbc.275.11.7757. [DOI] [PubMed] [Google Scholar]
- (6).Lincoln J, Hoyle CHV, Burnstock G. Nitric Oxide in Health and Disease. Cambridge University Press; Cambridge: 1997. [Google Scholar]
- (7).Williams DLH. Nitrosation Reactions and the Chemistry of Nitric Oxide. Elsevier; London: 2004. Nitric Oxide in Biological Systems; p. 187. [Google Scholar]
- (8).Priviero FBM, Leite R, Webb RC, Teixeira CE. Acta Pharmacol. Sin. 2007;28(6):751. doi: 10.1111/j.1745-7254.2007.00584.x. doi:10.1111/j.1745-7254. 2007.00584.x. [DOI] [PubMed] [Google Scholar]
- (9).Nakane M. Clin. Chem. Lab. Med. 2003;41(7):865. doi: 10.1515/CCLM.2003.131. doi:10. 1515/CCLM.2003.131. [DOI] [PubMed] [Google Scholar]
- (10).Stuart-Smith K, Warner DO, Jones KA. Eur. J. Pharmacol. 1998;341(2–3):225. doi: 10.1016/s0014-2999(97)01455-6. doi:10.1016/S0014-2999 (97)01455-6. [DOI] [PubMed] [Google Scholar]
- (11).Newsholme P, Homem De Bittencourt PI, Hagan C, De Vito G, Murphy C, Krause MS. Clin. Sci. (Lond.) 2010;118(5):341. doi: 10.1042/CS20090433. doi:10.1042/CS20090433. [DOI] [PubMed] [Google Scholar]
- (12).Nahrevanian H. Braz. J. Infect. Dis. 2009;13:440. doi: 10.1590/s1413-86702009000600010. [DOI] [PubMed] [Google Scholar]
- (13).Jadhav V, Jabre A, Chen M-F, Lee T-J. Stroke. 2009;40(1):261. doi: 10.1161/STROKEAHA.108.516104. doi:10.1161/STROKEAHA.108.516104. [DOI] [PubMed] [Google Scholar]
- (14).De Felice M, Porreca F. Cephalalgia. 2009;29(12):1277. doi: 10.1111/j.1468-2982.2009.01873.x. doi:10.1111/j.1468-2982.2009.01873.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Beckman JS, Koppenol WH. Am. J. Physiol. Cell Physiol. 1996;40:C1424. doi: 10.1152/ajpcell.1996.271.5.C1424. [DOI] [PubMed] [Google Scholar]
- (16).Huie RE, Padmaja S. Free Radic. Res. Commun. 1993;18(4):195. doi: 10.3109/10715769309145868. doi:10.3109/10715769309145868. [DOI] [PubMed] [Google Scholar]
- (17).Ozaki T, Kaibori M, Matsui K, Tokuhara K, Tanaka H, Kamiyama Y, Nishizawa M, Ito S, Okumura T. JPEN J. Parenter. Enteral Nutr. 2007;31(5):366. doi: 10.1177/0148607107031005366. doi:10.1177/ 0148607107031005366. [DOI] [PubMed] [Google Scholar]
- (18).Cauwels A. Kidney Int. 2007;72(5):557. doi: 10.1038/sj.ki.5002340. doi:10.1038/sj.ki. 5002340. [DOI] [PubMed] [Google Scholar]
- (19).Glantzounis GK, Rocks SA, Sheth H, Knight I, Salacinski HJ, Davidson BR, Winyard PG, Seifalian A. Free Radic. Biol. Med. 2007;42(6):882. doi: 10.1016/j.freeradbiomed.2006.12.020. doi:10.1016/j. freeradbiomed.2006.12.020. [DOI] [PubMed] [Google Scholar]
- (20).Chen YY, ; Chu HM, Pan KT, Teng CH, Wang DL, Wang AH, Khoo KH, Meng TC. J. Biol. Chem. 2008;283(50):35265. doi: 10.1074/jbc.M805287200. doi:10.1074/jbc.M805287200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Wang K, Zhang W, Xian M, Hou YC, Chen XC, Cheng JP, Wang PG. Curr. Med. Chem. 2000;7:821. doi: 10.2174/0929867003374633. [DOI] [PubMed] [Google Scholar]
- (22).Hara MR, Agrawal N, Kim SF, Cascio MB, Fujimuro M, Ozeki Y, Takahashi M, Cheah JH, Tankou SK, Hester LD, Ferris CD, Hayward SD, Snyder SH, Sawa A. Nat. Cell Biol. 2005;7(7):665. doi: 10.1038/ncb1268. doi:10.1038/ ncb1268. [DOI] [PubMed] [Google Scholar]
- (23).Yao DD, Gu ZZ, Nakamura T, Shi ZQ, Ma YL, Gaston B, Palmer LA, Rockenstein EM, Zhang ZH, Masliah E, Uehara T, Lipton SA. Proc. Natl. Acad. Sci. U.S.A. 2004;101(29):10810. doi: 10.1073/pnas.0404161101. doi:10.1073/pnas.0404161101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Askew SC, Barnett DJ, Mcaninly J, Williams DLH. J. Chem. Soc. Perkin Trans. 1995;2:741. [Google Scholar]
- (25).Beloso PH, Williams DLH. Chem. Commun. (Camb.) 1997;(1):89. doi:10.1039/a607228c. [Google Scholar]
- (26).Dicks AP, Li E, Munro AP, Swift HR, Williams DLH. Can. J. Chem. 1998;76:789. doi:10.1139/v98-062. [Google Scholar]
- (27).Dicks AP, Beloso PH, Williams DLH. J. Chem. Soc. Perkin Trans. 2. 1997:1429. [Google Scholar]
- (28).Holmes AJ, Williams DLH. J. Chem. Soc. Perkin Trans. 2. 2000:1639. [Google Scholar]
- (29).Noble DR, Williams DLH. J. Chem. Soc. Perkin Trans. 2. 2002:1834. [Google Scholar]
- (30).Williams DLH. Acc. Chem. Res. 1999;32(10):869. doi:10. 1021/ar9800439. [Google Scholar]
- (31).Williams DLH. Nitric Oxide. 1997;1(6):522. doi: 10.1006/niox.1997.0159. doi:10.1006/ niox.1997.0159. [DOI] [PubMed] [Google Scholar]
- (32).Williams DLH. Chem. Commun. (Camb.) 1996;(10):1085. doi:10.1039/cc9960001085. [Google Scholar]
- (33).Williams DLH. Transition Met. Chem. 1996;21(2):189. doi:10.1007/BF00136555. [Google Scholar]
- (34).Zamora R, Matthys KE, Herman AG. Eur. J. Pharmacol. 1997;321(1):87. doi: 10.1016/s0014-2999(96)00918-1. doi:10.1016/S0014-2999(96)00918-1. [DOI] [PubMed] [Google Scholar]
- (35).Kelly GS. Altern. Med. Rev. 1997 [Google Scholar]
- (36).Gahl WA. Eur. J. Pediatr. 2003 doi: 10.1007/s00431-003-1349-x. doi:10. 1007/s00431-003-1349-x. [DOI] [PubMed] [Google Scholar]
- (37).Iwata F, Kuehl EM, Reed GF, Mccain LM, Gahl WA, Kaiser-Kupfer MI. Mol. Genet. Metab. 1998;64(4):237. doi: 10.1006/mgme.1998.2725. doi:10.1006/mgme.1998.2725. [DOI] [PubMed] [Google Scholar]
- (38).Skovby F, Kleta R, Anikster Y, Christensen R, Gahl WA. Am. J. Hum. Genet. 2002 [Google Scholar]
- (39).Chanakira A, Chikwana E, Peyton D, Simoyi R. Can. J. Chem. 2006;84(1):49. doi:10.1139/v05-263. [Google Scholar]
- (40).Martincigh BS, Mundoma C, Simoyi RH. J. Phys. Chem. 1998;102(48):9838. doi:10.1021/jp982575c. [Google Scholar]
- (41).Simoyi RH, Streete K, Mundoma C, Olojo R. South African J. Chem. (Suid-Africaanse Tydskrif Vir Chemie) 2002;55:136. [Google Scholar]
- (42).Liu X, Gillespie JS, Martin W. Br. J. Pharmacol. 1994;111:1287. doi: 10.1111/j.1476-5381.1994.tb14885.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Whiteside WM, Sears DN, Young PR, Rubin DB. Radiat. Res. 2002;157(5):578. doi: 10.1667/0033-7587(2002)157[0578:possnc]2.0.co;2. doi:10.1667/0033-7587(2002) 157[0578:POSSNC]2.0.CO;2. [DOI] [PubMed] [Google Scholar]
- (44).Reszka KJ, Bilski P, Chignell CF. Nitric Oxide. 2004;10(2):53. doi: 10.1016/j.niox.2004.03.001. doi:10.1016/j.niox.2004.03.001. [DOI] [PubMed] [Google Scholar]
- (45).Choi JS, Yoon NM. J. Org. Chem. 1995;60(11):3266. doi:10.1021/jo00116a001. [Google Scholar]
- (46).Zhu BZ, Antholine WE, Frei B. Free Radic. Biol. Med. 2002;32(12):1333. doi: 10.1016/s0891-5849(02)00847-x. doi:10.1016/S0891-5849(02)00847-X. [DOI] [PubMed] [Google Scholar]
- (47).Reed CJ, Douglas KT. Biochem. Biophys. Res. Commun. 1989;162(3):1111. doi: 10.1016/0006-291x(89)90788-2. doi:10.1016/0006-291X(89)90788-2. [DOI] [PubMed] [Google Scholar]
- (48).Williams DLH. Nitrosation Reactions and the Chemistry of Nitric Oxide. Elsevier: London: 2004. Synthesis, Properties and Reactions of S-Nitrosothiols. p 137. [Google Scholar]
- (49).Chipinda I, Simoyi RH. J. Phys. Chem. B. 2006;110(10):5052. doi: 10.1021/jp0531107. doi:10.1021/jp0531107. [DOI] [PubMed] [Google Scholar]
- (50).Jourd’heuil D, Jourd’heuil FL, Feelisch M. J. Biol. Chem. 2003;278(18):15720. doi: 10.1074/jbc.M300203200. doi:10.1074/jbc.M300203200. [DOI] [PubMed] [Google Scholar]
- (51).Bunton CA, Stedman G. J. Chem. Soc. 1958 doi:10. 1039/jr9580002440. [Google Scholar]
- (52).Markovits GY, Schwartz SE, Newman L. Inorg. Chem. 1981;20(2):445. doi:10.1021/ic50216a025. [Google Scholar]
- (53).Turney TA. J. Chem. Soc. 1960 doi:10.1039/ jr9600004263. [Google Scholar]
- (54).Huie RE. Toxicology. 1994;89(3):193. doi: 10.1016/0300-483x(94)90098-1. doi:10.1016/0300-483X(94)90098-1. [DOI] [PubMed] [Google Scholar]
- (55).Ford E, Hughes MN, Wardman P. Free Radic. Biol. Med. 2002;32(12):1314. doi: 10.1016/s0891-5849(02)00850-x. doi:10.1016/S0891-5849(02)00850-X. [DOI] [PubMed] [Google Scholar]
- (56).Timerghazin QK, Peslherbe GH, English AM. Phys. Chem. Chem. Phys. 2008;10(11):1532. doi: 10.1039/b715025c. doi:10.1039/ b715025c. [DOI] [PubMed] [Google Scholar]
- (57).Stamler JS, Toone EJ. Curr. Opin. Chem. Biol. 2002;6(6):779. doi: 10.1016/s1367-5931(02)00383-6. doi:10.1016/S1367-5931(02)00383-6. [DOI] [PubMed] [Google Scholar]
- (58).Grossi L, Montevecchi PC, Strazzari S. J. Am. Chem. Soc. 2001;123(20):4853. doi: 10.1021/ja005761g. doi:10.1021/ja005761g. [DOI] [PubMed] [Google Scholar]
- (59).Bartberger MD, Mannion JD, Powell SC, Stamler JS, Houk KN, Toone EJ. J. Am. Chem. Soc. 2001;123(36):8868. doi: 10.1021/ja0109390. doi:10.1021/ja0109390. [DOI] [PubMed] [Google Scholar]
- (60).Baciu C, Gauld JW. J. Phys. Chem. A. 2003;107(46):9946. doi:10.1021/jp035205j. [Google Scholar]
- (61).Roy B, du Moulinet d’Hardemare A, Fontecave M. J. Org. Chem. 1994;59(23):7019. doi:10.1021/jo00102a028. [Google Scholar]
- (62).Grossi L, Montevecchi PC. J. Org. Chem. 2002;67(24):8625. doi: 10.1021/jo026154+. doi:10.1021/jo026154+. [DOI] [PubMed] [Google Scholar]
- (63).Field L, Dilts RV, Ravichandran R, Lenhert PG, Carnahan GE. J. Chem. Soc. Chem. Commun. 1978 doi:10.1039/c39780000249. [Google Scholar]
- (64).Peterson LA, Wagener T, Sies H, Stahl W. Chem. Res. Toxicol. 2007;20(5):721. doi: 10.1021/tx700095u. doi:10.1021/tx700095u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (65).Baciu C, Cho KB, Gauld JW. J. Phys. Chem. B. 2005;109(4):1334. doi: 10.1021/jp0443759. doi:10.1021/jp0443759. [DOI] [PubMed] [Google Scholar]
- (66).McAninly J, Williams DLH, Askew SC, Butler AR, Russell C. J. Chem. Soc. Chem. Commun. 1993;(23):1758. doi:10.1039/c39930001758. [Google Scholar]
- (67).Singh RJ, Hogg N, Joseph J, Kalyanaraman B. J. Biol. Chem. 1996;271(31):18596. doi: 10.1074/jbc.271.31.18596. doi:10.1074/jbc.271.31.18596. [DOI] [PubMed] [Google Scholar]
- (68).Dicks AP, Williams DLH. Chem. Biol. 1996;3(8):655. doi: 10.1016/s1074-5521(96)90133-7. doi:10.1016/S1074-5521(96)90133-7. [DOI] [PubMed] [Google Scholar]
- (69).Foster MW, Hess DT, Stamler JS. Trends Mol. Med. 2009;15(9):391. doi: 10.1016/j.molmed.2009.06.007. doi:10.1016/j.molmed.2009.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]