

Abstract
Preventing breast cancer will require the development of targeted strategies that can effectively block disease progression. Tamoxifen and aromatase inhibitors are effective in addressing estrogen receptor-positive (ER+) breast cancer development, but estrogen receptor-negative (ER−) breast cancer remains an unmet challenge due to gaps in pathobiological understanding. In this study, we used reverse phase protein array (RPPA) to identify activation of Src kinase as an early signaling alteration in premalignant breast lesions of women who did not respond to tamoxifen, a widely used ER antagonist for hormonal therapy of breast cancer. Src kinase blockade with the small molecule inhibitor saracatinib prevented the disorganized 3D growth of ER− mammary epithelial cells in vitro and delayed the development of pre-malignant lesions and tumors in vivo in mouse models developing HER2+ and ER− mammary tumors, extending tumor-free and overall survival. Mechanistic investigations revealed that Src blockade reduced glucose metabolism as a result of an inhibition in ERK1/2-MNK1-eIF4E-mediated cap-dependent translation of c-Myc and transcription of the glucose transporter GLUT1, thereby limiting energy available for cell growth. Taken together, our results provide a sound rationale to target Src pathways in premalignant breast lesions to limit the development of breast cancers.
Keywords: Estrogen receptor negative (ER−) breast cancer, cancer prevention, glucose metabolism
Introduction
Despite recent advances in effective treatment, including targeted therapies, many women still die of breast cancer (1). Ultimately, the most effective way to reduce breast cancer mortality is disease prevention (2). In large-scale prevention trials, tamoxifen and aromatase inhibitors reduced the incidence of estrogen receptor-positive (ER+) breast cancer in high-risk women by approximately 50% (3,4). However, prevention of estrogen receptor negative (ER−) and tamoxifen-resistant (TamR) breast cancer remains an overarching unmet demand. ER− breast cancers account for approximately 30% of total breast cancers (5), of which epidermal growth factor receptor 2-positive (HER2+)/ER− and triple negative breast cancer subtypes account for approximately 10–15% and 15–20% of total breast cancers, respectively (5,6). Here, we explored strategies to prevent ER− breast cancer in an HER2+/ER− mammary tumor model.
One of the molecular mechanisms of ER loss in ER− breast cancer is constitutive proteolysis of ERα via Src activation (7). Moreover, Src promotes ER phosphorylation leading to increased proliferation and tamoxifen resistance in breast cancer cells (8,9). ER− breast cancer cells have been found to be more dependent on Src activation than ER+ breast cancer cells and noncancerous breast cells, as Src silencing increased ER− breast cancer cell death (10). In TamR breast cancer cells, Src activity is upregulated and associated with a more aggressive phenotype (11). These findings indicate that Src activation is a key signaling event driving ER− and TamR breast cancer progression and suggest that targeting Src may prevent ER− breast cancer.
Src-targeting agents, such as the small molecule tyrosine kinase inhibitors dasatinib, bosutinib, and saracatinib, have been extensively tested in the clinic for treatment of metastatic breast cancer (12); however, they have never been explored for cancer prevention purposes. Given the benefits associated with targeted cancer therapies, e.g. better tolerance and lower cost of small molecule inhibitors relative to chemotherapy (13), we postulated that low-dose Src-targeting agents may be an effective and well-tolerated option for prevention of ER− breast cancer. Dasatinib and bosutinib are active against a broad spectrum of kinases, whereas saracatinib is a potent and more selective Src inhibitor (13), making it a good candidate for proof-of-concept prevention studies. Additionally, saracatinib used in combination with fulvestrant (ER antagonist), circumvented anti-estrogen resistance in ER+ breast and ovarian cancer preclinical models (14,15). Given the lack of low-toxicity targeted agents for ER− breast cancer prevention (16), saracatinib at lower doses may possess unrecognized potential for breast cancer prevention.
Metabolic dysregulations have been associated with increased breast cancer risk (17). Pre-malignant and neoplastic cells exhibit increased demand for energy and nutrients to proliferate and survive (18,19). Increased glucose uptake enables the generation of building blocks for dysregulated cellular growth which facilitates cancer initiation (18). In tumor cells, c-Myc (Myc) plays an important role in regulating glycolysis and glutaminolysis (20). Endocrine-resistant breast cancer cells overexpress Myc which correlates with increased dependency on glucose and glutamine, and these cells could survive on glutamine upon glucose deprivation (21). Notably, ER− primary breast tumors had an increased uptake of 18F-fluorodeoxyglucose (a glucose analog) and express higher levels of glucose transporter 1 (GLUT1) than ER+ tumors (22,23). However, the role of glucose metabolic dysregulation in the early stages of cancer is unclear and the role of Src in regulating glucose metabolism in cancer cells is not well studied.
In this study, we set out to tackle the challenges of preventing HER2+ and ER− breast cancer. We found that Src activation was a key signaling alteration during early stage cancer initiation and that Src inhibition suppressed cap-dependent translation of Myc, reduced GLUT1 transcription and glucose uptake in premalignant ER− mammary epithelial cells (MECs), consequently inhibiting cell proliferation and ER− mammary tumor initiation and development. These preclinical findings provide a strong scientific foundation of using Src-inhibitors for preventing, at least this subtype, of ER− breast cancer.
Materials and Methods
Patient samples
Patient sample collection was carried out in accordance with institutional review board-approved protocol at the Duke University Medical Center. “High-risk patient” includes women with greater than 20% lifetime risk of developing breast cancer, as assessed by at least one of the criteria: a) prior biopsy containing atypia, DCIS, or LCIS, b) known or suspected to have BRCA mutations, or c) first-degree family member with premenopausal breast cancer. No BRCA mutation carriers were included in this study. The presence of atypia in random periareolar fine needle aspiration (RPFNA) has been used as a surrogate marker to track cytological response to chemoprevention agents. Informed consent was obtained prior to enrolling women in the study. In the following analysis, ER status was not determined in any of the materials collected from patients.
Cells and vectors
The ER− mammary epithelial cells, MCF10A and MCF12A were obtained from American Type Culture Collection. The pLKO.1-based shRNAs for Src were purchased from Sigma-Aldrich and the Src mutant (Y527F) construct from Addgene. shRNAs for GLUT1 were obtained from MD Anderson Cancer Center’s shRNA core facility.
Amplex Red Glucose Assay Kit
The Amplex Red glucose assay kit was used (Life Technologies) following previously published protocol (24). Briefly, 1.3 × 104 cells were plated in poly-HEMA coated 96 wells. After 24 hours, media was collected and diluted 1:4000 in water. The amount of glucose in the media was then determined using the Amplex Red Assay according to the manufacturer’s instructions. Glucose uptake was analyzed by subtracting the amount of glucose in each sample from the total amount of glucose in the media (without cells). The data represents experiments from three independent replicates. To examine the effect of saracatinib on glucose uptake, cells pretreated with either vehicle or saracatinib for 3 days were seeded to perform the assay.
ER− Mammary Tumor Prevention Studies
Mouse experiments were performed in accordance with approved protocols from the Institutional Animal Care and Use Committee of MD Anderson Cancer Center. Female MMTV-neu mice were treated with either vehicle (0.5% hydroxypropyl methylcellulose with tween-80) or saracatinib by oral gavage once daily for 6 days a week. Tumor sizes were measured twice a week. Tumor-free survival was defined as the time from date of birth to the first appearance of a palpable mammary tumor at least 100 mm3 in size. The fourth pair of normal looking mammary fat pads (MFPs) were isolated from these mice at 32 weeks of age. For histological analyses non-serial sections thought-out the MFPs were analyzed. Another cohort was set up using female MMTV-Neu* mice. These mice were treated either with vehicle or saracatinib. Tumor-free and overall survivals were monitored.
Statistical analyses
Quantitative results were analyzed either by one-way ANOVA (multiple groups) or t-test (2 groups). Differences with P < 0.05 (2-sided) were considered statistically significant. *, P < 0.05, **, P < 0.01 and ***, P < 0.001. For patient samples, Wilcoxon rank-sums test was used. Tumor-free and overall survival analyses were performed using the Kaplan-Meier Wilcoxon test. Bars represent means ± SEM.
Results
Elevated Src expression in premalignant breast lesions of women who did not respond to tamoxifen
To develop effective prevention strategy for ER antagonist-unresponsive breast cancer, we sought to identify targetable molecular signature in premalignant lesions of women who did not respond to tamoxifen and were at a higher risk of developing tamoxifen-unresponsive breast cancer. Eighteen high-risk women with greater than 20% lifetime risk of developing breast cancer were given tamoxifen (20 mg, PO qd) for cancer prevention. After 6–12 months of treatment, women who experienced a disappearance of atypia or did not progress to develop atypical lesions were classified as tamoxifen-sensitive (Tam-S, n = 12), and those who had persistent atypical lesions or developed atypical lesions were classified as tamoxifen-non responder (Tam-NR, n = 6). Next, reverse phase protein array (RPPA) was performed in duplicate from a total of 22 Tam-S and Tam-NR random periareolar fine needle aspiration (RPFNA) samples (8 bilaterally and 14 unilaterally). Six of the biomarkers (c-Src, E-cadherin, phospho-Bad-S136, phospho-Bcl2-S70, phospho-IκB-S32/36, and phospho-P70S6K-T412) were significantly increased in Tam-NR compared with Tam-S samples (Fig. 1A). Among these, c-Src (Src) is a readily targetable molecule as Src inhibitors have shown efficacy in clinical trials for treatment of late stage cancers (12) and Src acts as a key node of multiple cancer cell signaling pathways (12,25). Additionally, analysis of the Total Cancer Proteome Atlas (TCPA) breast tumor dataset (26) revealed higher phospho-Src-Y416 and total Src levels in ER− breast tumors than ER+ breast tumors (Supplementary Fig. S1A). Furthermore, increased phospho-Src-Y416 was detected in both HER2-enriched and basal-like ER− breast tumors than ER+ luminal type breast tumors, although higher total Src expression was detected only in basal-like breast tumors compared to luminal type breast tumors (Supplementary Fig. S1B and S1C). Therefore, we investigated whether Src activation plays an important role in ER− breast cancer initiation and may be a feasible target for prevention/intervention of ER− breast cancer.
Figure 1.
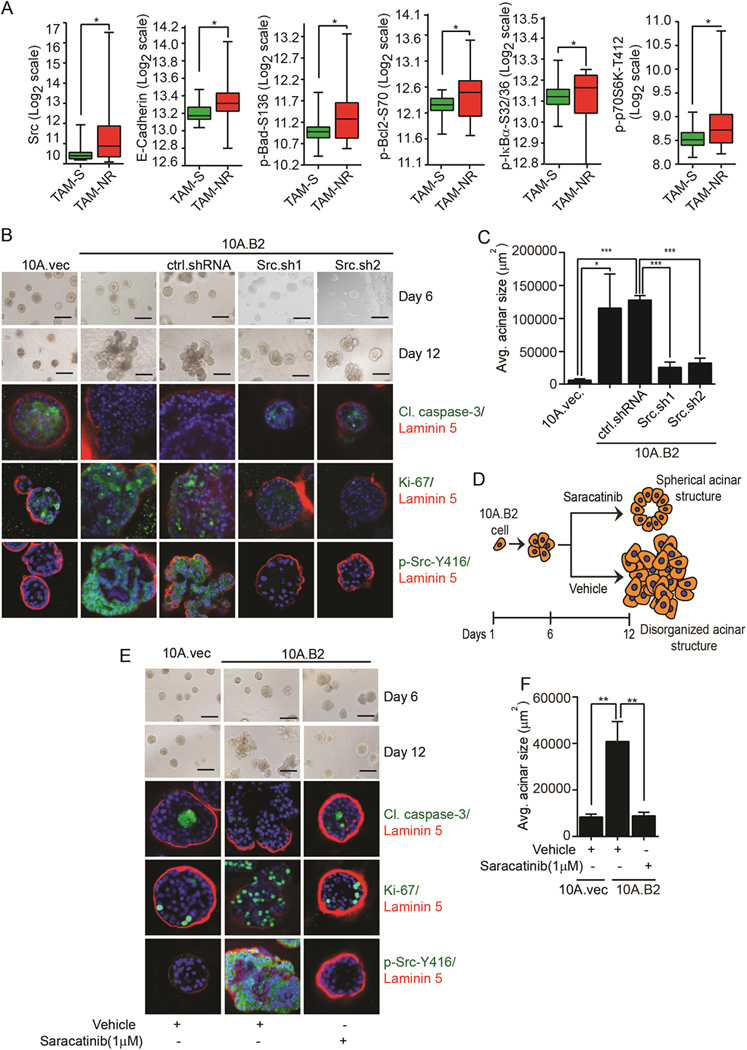
Elevated Src expression in Tam-NR premalignant lesions from patients and effects of targeting Src on disorganized acini formation of ER−, Src-activated MECs. A, Protein markers with statistically significant differences in their central tendencies between the Tam-S and Tam-NR groups. B, Phase-contrast images of 10A.vec, 10A.B2, ctrl.shRNA, and Src.sh clones of 10A.B2 cells in 3D culture. IF images showing cleaved caspase-3, Ki-67, phospho-Src-Y416, laminin 5, and DAPI staining in acini. C, Quantification of average (avg.) acinar size. D, Schematic showing the effect of vehicle and saracatinib on the acinar growth of HER2-overexpressing MECs in the prevention setting. E, Phase-contrast images of 10A.vec, vehicle- and saracatinib-treated 10A.B2 cells. IF images showing cleaved caspase-3, Ki-67, phospho-Src-Y416, laminin 5, and DAPI staining in acini. F, Quantification of avg. acinar size (scale bar = 200 µm).
Targeting Src prevents disorganized growth of ER− MECs
Because HER2-overexpressing MECs exhibit higher Src activation (27), to determine the role of Src in ER− premalignant MECs growth, we generated ER− MCF10A and MCF12A MECs stable clones harboring HER2-overexpression (10A.B2 and 12A.B2) or control vectors (10A.vec and 12A.vec) as in vitro models (27) (Supplementary Fig. S1D). In 3D culture, 10A.B2 and 12A.B2 cells form noninvasive disorganized acinar structures with filled lumen due to increased proliferation and reduced apoptosis compared to the control cells (Fig. 1B, second column from left and Supplementary Fig. S1G, middle column). These acinar structures mimic ductal carcinoma in situ (DCIS) in patients (27) and can be used for testing therapeutics (28). On the contrary, the vector control cells form spherical acinar structures with a hollow lumen that mimic normal mammary glands in vivo (Fig. 1B and Supplementary Fig. S1G, left columns) (29). To determine the role of Src in the disorganized acinar growth of 10A.B2 cells, we knocked down Src in 10A.B2 cells (Supplementary Fig. S1E). In 3D culture, 10A.B2 cells and control shRNA (ctrl.shRNA)-expressing 10A.B2 cells formed disorganized acinar structures, whereas Src knock down (Src.sh) resulted in smaller spherical acini structurally similar to those of the 10A.vec cells (Fig. 1B and 1C). Staining of markers for apoptosis (cleaved caspase-3), proliferation (Ki-67), and basement membrane (laminin 5) showed that acini formed by 10A.B2.Src.sh cells exhibited fewer proliferating cells and more apoptotic cells than those of 10A.B2.ctrl.shRNA cells (Fig. 1B and 1C). These data demonstrated that Src is required for apoptosis-resistance, MEC proliferation, and the disorganized acinar growth of 10A.B2 cells in 3D culture. Next, we tested the Src inhibitor saracatinib for prevention of the growth of disorganized acini in Src-activated MECs (Fig. 1D). We first confirmed that saracatinib indeed inhibited activation of Src and its down-stream targets, such as phospho-FAK-Y576, phospho-P130 Cas-Y410, and phospho-Paxillin-Y118 in both 10A.B2 and 12A.B2 cells in 3D cultures (Supplementary Fig. S1F). We then treated 10A.B2 and 12A.B2 cells with either vehicle or saracatinib (1 µM) on day 6 of 3D culture, when there was no significant difference in acinar growth of 10A.B2 and 12A.B2 cells compared to their vector control cells (Fig. 1D). Saracatinib indeed prevented the disorganized acinar growth of 10A.B2 and 12A.B2 cells, resulting in smaller spherical acinar structures with hollow lumina similar to those of vector control cells (Fig. 1E, 1F, Supplementary Fig. S1G and S1H). Saracatinib also induced apoptosis and inhibited proliferation of acini, as shown by increased cleaved caspase-3, and decreased Ki-67 and MCM2 (Fig. 1E, Supplementary Fig. S1G and S1I).
Targeting Src delays ER− mammary tumor development in mice
We next examined the effect of saracatinib on preventing ER− mammary tumors in the MMTV-neu 202 Mul/J (denoted as MMTV-neu, neu is the rat homologue of human ErbB2/HER2) mouse model (30). MMTV-neu mouse overexpresses wild-type HER2 in the mammary gland and progressively develop ER− mammary intraepithelial neoplasia (MIN) (~10–18 weeks) and invasive ductal carcinomas (IDC, ~30 weeks onwards) lesions (data not shown). As the well tolerated saracatinib dose used for cancer treatment in clinical trials is 175 mg/day ((31), equivalent to 33.2 mg/kg in mouse), we used saracatinib at a lower dose (25 mg/kg, n=20) for treating MMTV-neu mice and gave vehicle to control group (n=20) and monitored the mice for tumor-free survival. All mice in the vehicle group developed mammary tumors by 260 days of age, when 9 out of 20 mice in the saracatinib-treated group remained tumor-free (Fig. 2A). Compared with the vehicle group, saracatinib significantly increased tumor-free (vehicle; T(50) = 211 days, saracatinib; T(50) = 249 days) and overall (vehicle; T(50) = 258 days, saracatinib; T(50) = 285 days) survival of these mice (Fig. 2A and Supplementary Fig. S2A) without obvious toxicity to mice ((32) and data not shown). Saracatinib significantly reduced the development of hyperplastic, MIN, and IDC lesions (Fig. 2B). Immunohistochemical (IHC) staining confirmed that saracatinib treatment inhibited Src activity in MFPs (Supplementary Fig. 2C).
Figure 2.
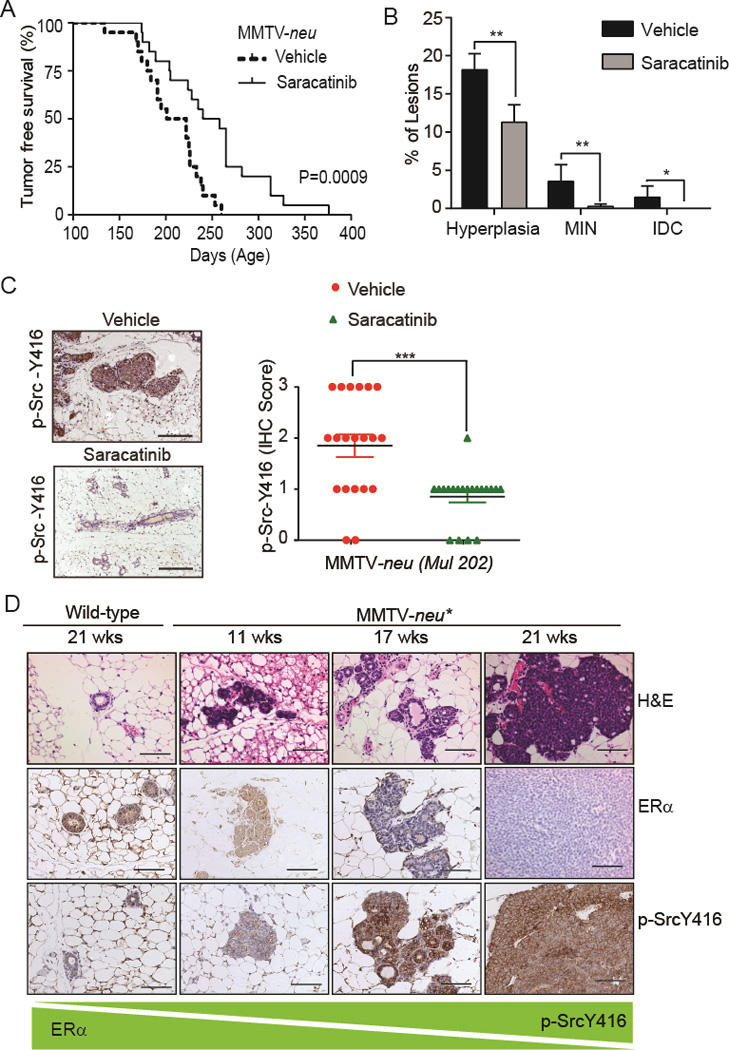
Targeting Src delays ER− mammary tumor development in mice. A&B, Kaplan-Meier tumor-free survival curve and percentage of hyperplastic, MIN, and IDC lesions in MFPs of MMTV-neu mice treated with either vehicle (n=20) or saracatinib (n=20). C, IHC images and scores of phospho-Src-Y416 in MFPs of vehicle- (n=20) and saracatinib- (n=20) treated MMTV-neu mice. D, H&E and IHC staining of ERα and phospho-SrcY416 in MFPs of wild-type and MMTV-neu* mice at different ages (hyperplasia at 11 wks, MIN at 17 wks, and IDC at 21 wks). Student’s t-test was used to calculate statistical significance. Differences with P < 0.05 were considered statistically significant. *, P < 0.05, **, P < 0.01.
To monitor mammary tumor progression from hyperplasia to MIN to IDC during saracatinib treatment in real-time, we implemented the high resolution micro-endoscopy (HRME), an innovative imaging technology, which has been used as a diagnostic tool to distinguish normal tissue from benign and neoplastic tissues in head and neck and cervical cancers (33,34). Fresh MFPs from 32 weeks old vehicle- and saracatinib-treated mice were imaged to detect pre-malignant lesions using HRME with or without structured illumination (SI), a method of rejecting out-of-focus light to improve image contrast in thick tissue samples. The images of HRME with SI (HRME-SI) were co-registered with images obtained from confocal microscopy and H&E staining for comparison of resolutions (Supplementary Fig. S2B). The quality of images from HRME-SI, which could be applied in real time in living animals through a biopsy needle, was comparable with that of confocal microscopy and H&E staining. The HRME-SI detected MIN lesions in the vehicle-treated MMTV-neu mice, whereas only hyperplastic lesions were detected in the saracatinib-treated mice (Supplementary Fig. S2B).
We also tested the effect of saracatinib in MMTV-neu NDL 2–5 (referred to as MMTV-neu*) mouse model expressing the activated neu as female MMTV-neu* mice rapidly develop ER− mammary tumors (35). Specifically, MMTV-neu* female mice develop hyperplastic lesions between 8–11 weeks, MIN lesions between 11–19 weeks, and IDC lesions between 19–26 weeks of age (Fig. 2D). IHC staining of MFP biopsies showed that MMTV-neu* mice developed atypia and MIN lesions with a gradual loss of ER and increase in Src activation as disease progressed to mammary tumors (Fig. 2D).
Given the appearance of MIN lesions between 11–19 weeks and an enhanced Src activation (Fig. 2D) in MIN lesions of MMTV-neu* mice, we tested saracatinib for prevention of the development of ER− IDC in this model by starting treatment in 10-week-old female mice. We gave saracatinib (25 mg/kg, n=20) to MMTV-neu* mice and used vehicle in control group (n=20). Compared with vehicle treatment, saracatinib significantly increased the median tumor-free [vehicle; T(50) = 155 days, saracatinib; T(50) = 176 days] and overall [vehicle; T(50) = 176 days, saracatinib; T(50) = 196 days] survival of mice (Supplementary Fig. S2C and S2D). To determine whether saracatinib inhibited Src activation and the appearance of hyperplastic, MIN and IDC lesions, the MFPs were collected at 21 weeks of age from vehicle- and saracatinib-treated mice when there was abundance of hyperplastic and MIN lesions but less than 10% IDC lesions. Indeed, low-dose saracatinib treatment reduced Src activation (Supplementary Fig. S2E). MFPs from saracatinib-treated mice exhibited significantly reduced number of IDC lesions and fewer MIN lesions than vehicle-treated mice (Supplementary Fig. S2F). Altogether, these data clearly demonstrated that saracatinib delayed initiation and progression of premalignant lesions, and increased both tumor-free and overall survival in two mouse models of ER− and Src-activated mammary tumors.
Src regulates glucose metabolism and GLUT1 expression
Since metabolic alterations are prominent in early stage cancers (17), to gain mechanistic insights into the delayed initiation and progression of premalignant lesions by targeting Src, we performed metabolomic profiling of MFPs of vehicle- and saracatinib-treated MMTV-neu* mice. Metabolite set enrichment analysis showed that starch and sucrose metabolism, the pentose phosphate pathway, and glycolysis were among the most significantly downregulated pathways in response to Src inhibition (Supplementary Fig. S3A). Glucose 6-phoshate and fructose 6-phosphate were among the most inhibited metabolites in response to saracatinib treatment (Supplementary Fig. S3B and S3C). Given that glucose 6-phosphate is the common metabolite of these pathways, we hypothesized that Src inhibition may impact on the availability of glucose to premalignant cells, which would in turn affect the synthesis of downstream metabolites of the aforementioned pathways. Therefore, we compared glucose uptake by 10A.B2 and 12A.B2 cells to their corresponding control cells with or without saracatinib treatment. Compared with vector control cells, 10A.B2 and 12A.B2 MECs had a significant increase in glucose uptake, which was inhibited by saracatinib treatment (Fig. 3A and 3B). Furthermore, knocking down Src (Supplementary Fig. S1E and S3D) also significantly reduced the glucose uptake compared with control shRNA transfected cells (Fig. 3C and 3D). These data indicate that Src plays an essential role in regulating glucose uptake.
Figure 3.
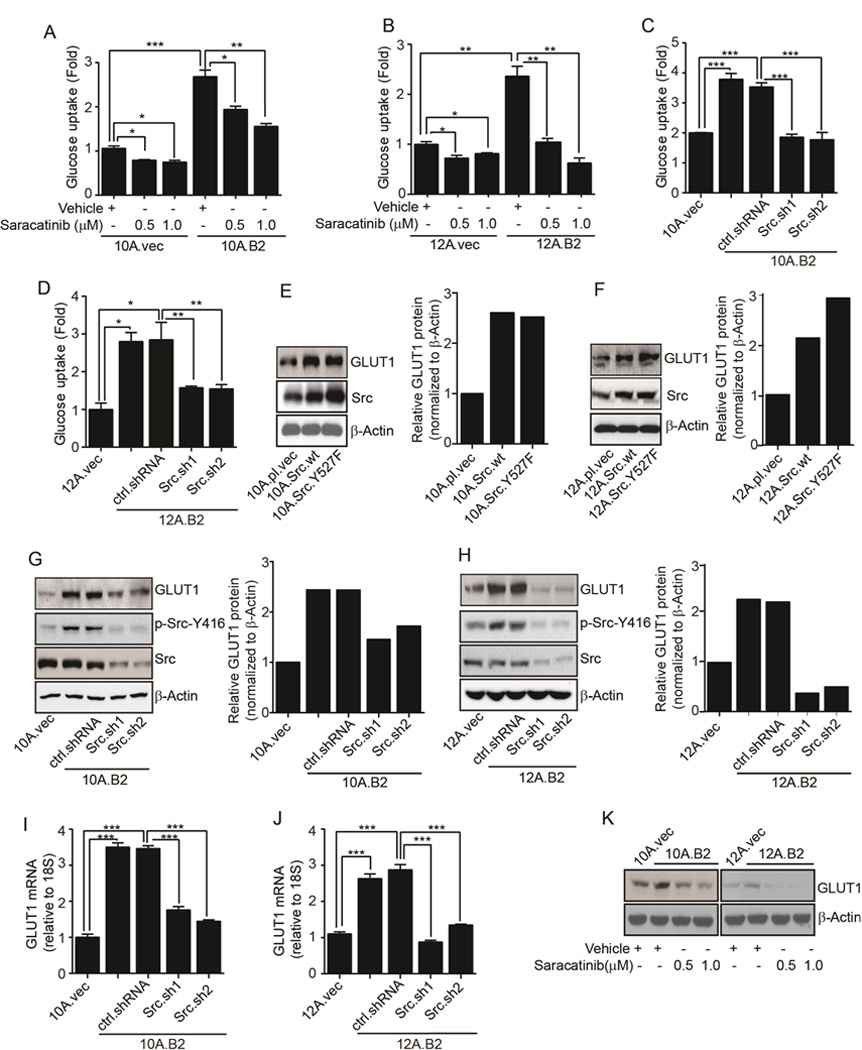
Src regulates glucose uptake and GLUT1 expression. A & B, Glucose uptake in vector control, 10A.B2, and 12A.B2 cells treated with vehicle or saracatinib. C & D, Glucose uptake in vector control, ctrl.shRNA-, and Src.sh-transfected 10A.B2 and 12A.B2 cells. E&F, Immunoblots and respective quantifications showing GLUT1 and Src expression in vector-, Src wt-, and SrcY527F-expressing MCF10A and MCF12A cells in 3D culture. G&H, Immunoblots and respective quantifications showing GLUT1, phospho-Src-Y416, and Src in vector control, 10A.B2, 12A.B2, ctrl.shRNA-, and Src.sh-clones of 10A.B2 and 12A.B2 cells grown in 3D culture. I&J, qRT-PCR showing GLUT1 in vector control, 10A.B2, 12A.B2, ctrl.shRNA and Src.sh clones of 10A.B2 and 12A.B2 cells grown in 3D culture. K, Immunoblot showing GLUT1 in vehicle- and saracatinib-treated 10A.B2 and 12A.B2 cells grown in 3D culture.
Transport of glucose across the plasma membrane of mammalian cells is carried out by glucose transporter (GLUT) proteins that are widely dysregulated in many cancers (36). Mammary glands predominantly express GLUT1 which also facilitates transport of mannose and galactose in addition to glucose (37). Particularly, GLUT1 is expressed at much higher levels compared with other members of the GLUT family in mammary tumors of the MMTV-neu mouse model (38). To investigate the effect of Src on GLUTs, we established Src wild-type (wt)-expressing (10A.Src.wt and 12A.Src.wt) and constitutively active Src.Y527F-expressing (10A.Src.Y527F and 12A.Src.Y527F) stable sublines from MCF10A and MCF12A cells, respectively. We examined the expression of GLUT1–5 and GLUT12, since altered expression of GLUT1–5 has been reported in many tumors and GLUT12 is highly expressed in breast tumors (39). 10A.Src.wt and 10A.Src.Y527F sublines had significantly increased GLUT1 expression than 10A.pl.vec whereas no significant changes were observed in GLUT2/3/4/5/12 (Supplementary Fig. S3E). 12A.Src.wt and 12A.Src.Y527 sublines also exhibited higher expression of GLUT1 compared to control (Supplementary Fig. S3F). Src.wt and SrcY527F-overexpressing sublines of MCF10A and MCF12A exhibited higher GLUT1 protein compared to their respective vector controls (Fig. 3E and 3F). Conversely, 10A.B2.Src.sh and 12A.B2.Src.sh cells had significantly reduced GLUT1 protein and mRNA levels compared with control shRNA transfected cells (Fig. 3G–J). Likewise, saracatinib also reduced GLUT1 protein and mRNA expression in 10A.B2 and 12A.B2 cells (Fig. 3K, Supplementary Fig. S3G, and S3H). These data indicated that Src activation increased glucose uptake, at least partly, by upregulating GLUT1 expression. Additionally, we examined the effect of saracatinib on the level of glycolytic enzymes. Saracatinib moderately inhibited HKII and LDHA expressions without changing PFK-1 (Supplementary Fig. S3I), indicating that some glycolytic enzymes were affected by Src inhibition but not as dramatic as GLUT1.
High GLUT1 expression is required for disorganized acinar growth of ER− and Src- activated MECs
To determine whether GLUT1 is critical for glucose uptake in ER− and Src-activated MECs, we stably knocked down GLUT1 in 10A.B2 cells (10A.B2.GLUT1.sh) (Fig. 4A). Clearly, 10A.B2.GLUT1.sh cells had reduced glucose uptake compared with control cells (Fig. 4B), indicating that GLUT1 is the major glucose transporter responsible for glucose uptake in 10A.B2 cells. Remarkably, 10A.B2.GLUT1.sh stable cells did not form disorganized acinar structures as 10A.B2 and control shRNA transfected 10A.B2 cells did in 3D culture (Fig. 4C, top, and 4D). Moreover, immunofluorescence (IF) staining showed that silencing GLUT1 resulted in increased apoptosis and decreased proliferation in acini of 10A.B2 cells (Fig. 4C, lower panels). These data indicated that GLUT1 plays an important role in disorganized acinar growth of ER−, Src-activated 10A.B2 cells by increasing glucose uptake to provide energy.
Figure 4.
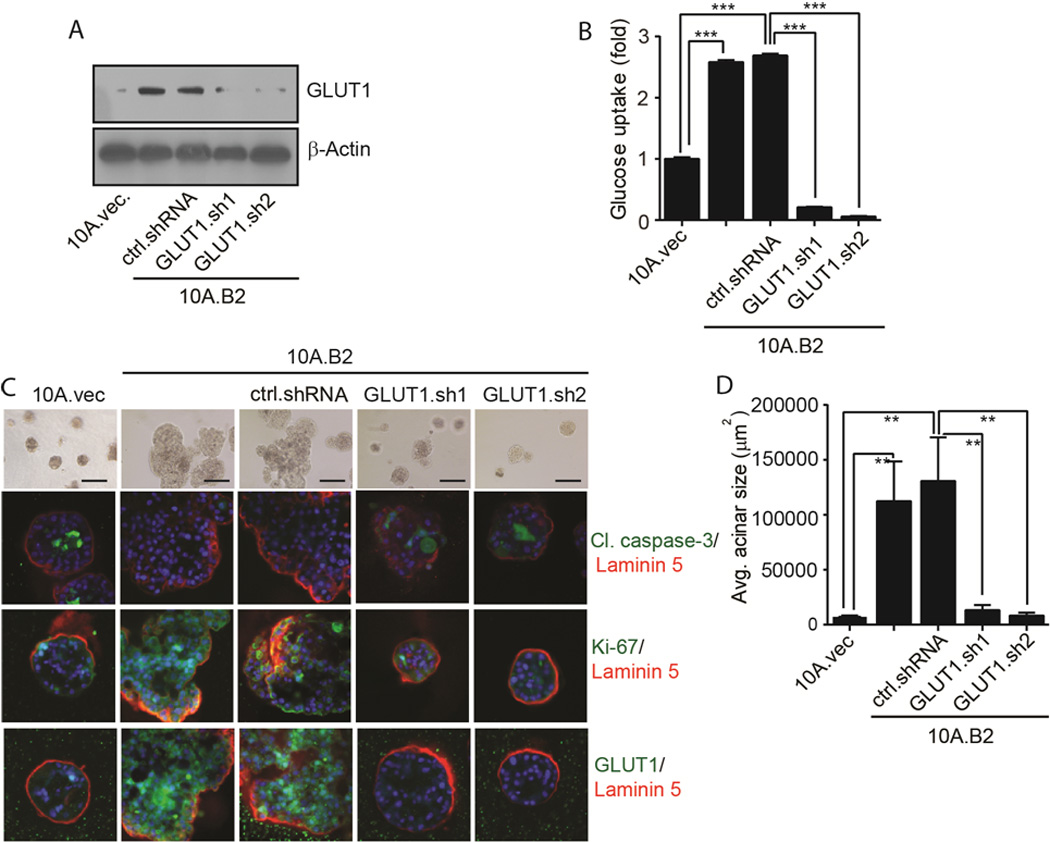
High GLUT1 expression is required for disorganized acinar growth of ER− and Src- activated MECs. A, Immunoblot showing GLUT1 in 10A.vec, 10A.B2, ctrl.shRNA- and GLUT1.sh clones of 10A.B2 cells. B, Glucose uptake in 10A.vec, 10A.B2, ctrl.shRNA and GLUT1.sh clones of 10A.B2 cells. C, Phase-contrast of 10A.vec, 10A.B2, 10A.B2.ctrl.shRNA, and 10A.B2.GLUT1.sh cells acini. IF images showing cleaved caspase-3, Ki-67, GLUT1, laminin 5, and DAPI staining in acini. D, Quantification of avg. acinar size (scale bar = 200 µm).
Src upregulates GLUT1 expression through increasing c-Myc protein synthesis
To gain mechanistic insights on how Src inhibition reduced GLUT1 expression and glucose uptake (Fig. 3), we treated 10A.B2 cells grown in 3D culture with either vehicle or saracatinib and performed RPPA. Among many detected alterations, c-Myc was one of the top 3 transcriptional regulators that were downregulated by saracatinib treatment (Supplementary Table S1). Myc downregulation by saracatinib was validated by immunoblotting in both 10A.B2 and 12A.B2 cells (Fig. 5A). As Myc had been reported to transcriptionally upregulate GLUT1 expression in Rat-1 fibroblasts (40), we examined whether Myc regulated GLUT1 expression in 10A.B2 and 12A.B2 cells. Myc silencing by siRNA resulted in decreased GLUT1 at both the protein and mRNA levels in 10A.B2 and 12A.B2 cells (Fig. 5B–D). As the data clearly demonstrated that Myc regulates GLUT1 expression in these MECs, we further investigated how Src inhibition impacted Myc expression. Although saracatinib inhibited Myc protein levels of 10A.B2 and 12A.B2 cells, it did not inhibit Myc mRNA expression (Fig. 5A and Supplementary Fig. S4A). Myc protein expression was also dramatically reduced in 3D-cultured 10A.B2.Src.sh and 12A.B2.Src.sh cells compared with control shRNA transfected cells (Fig. 5E). Additionally, Src.wt- and Src.Y527F-transfected MCF10A and MCF12A cells showed increased Myc protein expression compared to vector control cells but no significant changes in Myc mRNA levels (Fig. 5F and Supplementary Fig. S4B). These data indicated that Src regulates Myc at the protein level. Furthermore, Myc protein expression significantly correlated with phospho-Src-Y416 level, among human breast tumors in the TCPA dataset (Supplementary Fig. S4C).
Figure 5.
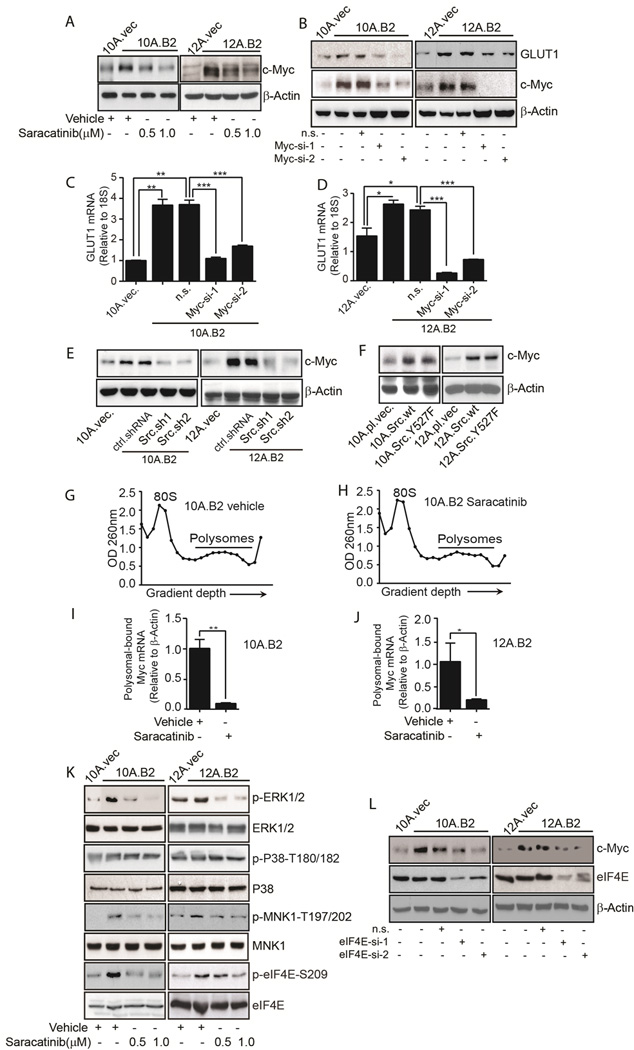
Src upregulates GLUT1 expression by increasing Myc protein synthesis. A, Immunoblot shows Myc expression in vehicle- or saracatinib-treated 10A.B2 and 12A.B2 cells. B, Immunoblots showing GLUT1 and Myc in vector control, 10A.B2, 12A.B2, non-silencing (n.s.), and Myc siRNA-transfected 10A.B2 and 12A.B2 cells. C & D, qRT-PCR shows GLUT1 mRNA in vector control, 10A.B2, 12A.B2, n.s.- and Myc siRNA-transfected 10A.B2 and 12AB2 cells. E, Immunoblot showing Myc in 10A.vec, 12A.vec, 10A.B2, 12A.B2, ctrl.shRNA and Src.sh clones of 10A.B2 and 12AB2 cells. F, Immunoblot showing Myc in vector-, Src wt-, and SrcY527F-expressing MCF10A and MCF12A cells. G & H, ODs at 260 nm are shown as a function of gradient depth for 10A.B2 cells treated with vehicle or saracatinib. I & J qRT-PCR measuring Myc mRNA in the polysomal fractions of vehicle- or saracatinib-treated 10A.B2 and 12A.B2 cells. K, 10A.B2 and 12A.B2 cells were treated either with vehicle or saracatinib and immunoblotting was conducted. L, Immunoblots showing Myc and eIF4E in vector control, 10A.B2, 12A.B2, n.s.-, and eIF4E-siRNA transfected 10A.B2 and 12AB2 cells.
The combination of protein synthesis and stability determine steady state protein levels (41). To examine whether Src inhibition affects Myc protein stability, vehicle- or saracatinib-treated 12A.B2 MECs were treated with cycloheximide to block protein synthesis and cell lysates were collected at different treatment times (0–60 mins.) followed by Myc immunoblotting. Similar rates of Myc protein degradation were detected between vehicle- and saracatinib-treated cells (Supplementary Fig. S4D), indicating that Src inhibition did not reduce the stability of Myc protein.
To determine the effect of Src inhibition on Myc protein synthesis, polysome fractions were collected from vehicle- and saracatinib-treated 10A.B2 and 12A.B2 cells. Saracatinib did not significantly change the overall polysome profile compared with vehicle controls (comparison of Fig. 5G with 5H and Supplementary Fig. S4E with S4F), indicating that Src inhibition did not impact global protein synthesis. However, saracatinib drastically attenuated polysomal recruitment of Myc mRNA compared to vehicle (Fig. 5I, 5J, Supplementary Fig. S4G, and S4H), indicating that saracatinib inhibited Myc protein translation in these MECs.
Src increases Myc protein by enhancing cap-dependent translation
The synthesis of many oncogenic proteins is promoted by cap-dependent translation which is regulated by phosphorylated eIF4E (42–44). The eIF4E is phosphorylated at serine 209 by MAPK signal-integrating kinases 1 and 2 (MNK1 and MNK2) (45). MNK2 is constitutively active, whereas MNK1 is activated by external stimuli via the extracellular signal-regulated kinase (ERK) and p38 mitogen activated protein kinase (MAPK) (45). To investigate whether saracatinib inhibited Myc translation by inhibiting cap-dependent translation, we compared phospho-eIF4E-S209 and its upstream kinases in vehicle- or saracatinib-treated 10A.B2 and 12A.B2 cells. Saracatinib suppressed phospho-eIF4E-S209, and its upstream kinases phospho-MNK1-T197/202 and phospho-ERK1/2-T202/Y204, but not phospho-p38 MAPK-T180/182, suggesting that Src affects cap-dependent translation via the ERK1/2-MNK1-eIF4E pathway (Fig. 5K). Furthermore, eIF4E-silenced 10A.B2 and 12A.B2 cells showed decreased levels of Myc protein, confirming cap-dependent translation of Myc in these cells (Fig. 5L). Thus, saracatinib reduced cap-dependent translation of Myc by inhibiting the ERK1/2-MNK1-eIF4E pathway.
Saracatinib inhibits Myc and GLUT1 expression in mouse models
Next, we set out to determine if the above findings from 3D-cultured MECs recapitulated the effects of targeting Src with saracatinib in mouse models. MFPs collected from vehicle- and saracatinib-treated MMTV-neu and MMTV-neu* mice were compared for Ki-67, Myc, and GLUT1 expression by IHC staining. Compared to vehicle-treated mice, Ki-67 in MFPs from saracatinib-treated mice was significantly reduced (Fig. 6A and Supplementary Fig. S5A), consistent with the delayed tumor onset and progression observed in these mice (Fig. 2A, Supplementary Fig. S2A, S2C, and S2D). Myc was significantly reduced in MFPs of saracatinib-treated mice (Fig. 6B and Supplementary Fig. S5B) with a corresponding reduction of GLUT1 expression (Fig. 6C and Supplementary Fig. S5C). Together, these in vivo findings were consistent with the Src inhibition-mediated responses in 3D-cultured 10A.B2 and 12A.B2 cells, indicating that targeting Src with saracatinib inhibited Myc expression, resulting in reduced GLUT1 expression, consequently impeding glucose metabolism and ER−, Src-activated mammary tumor initiation and progression.
Figure 6.
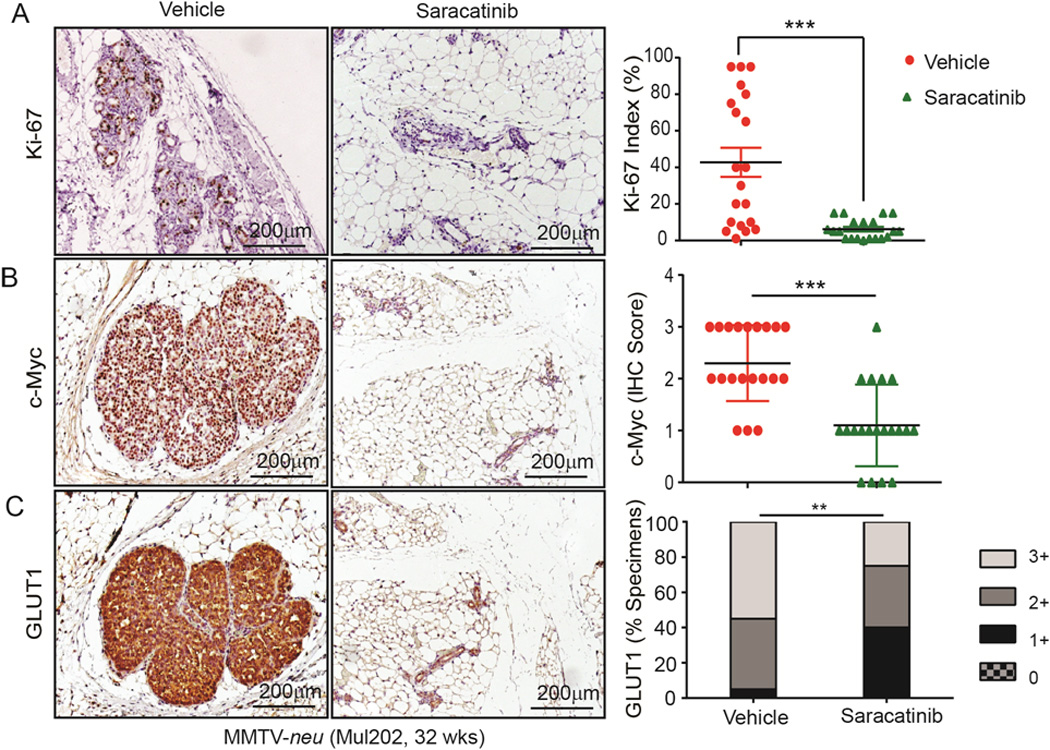
Saracatinib inhibits proliferation and Myc and GLUT1 expression in MMTV-neu mouse model. A–C, IHC staining for (A) Ki-67, (B) Myc, (C) and GLUT1 and their respective intensity scores in MFPs of vehicle- and saracatinib-treated MMTV-neu mice.
Discussion
In spite of recent advances in treatment, breast cancer accounts for an estimated 29% (232,670) of new cancer cases, highest among all female-related cancers, and 15% (40,000) of cancer deaths per year (1). Effective prevention strategies are needed to reduce breast cancer deaths. The development of agents to prevent tamoxifen-unresponsive and ER− breast cancer requires better understanding of the critical molecular alterations driving early lesion (atypia) progression towards breast cancer. In this study, we found that Src activation is a targetable molecular alteration in early-stage Tam-NR atypical lesions of women at higher risk of developing tamoxifen-unresponsive breast cancer. Here we show that saracatinib effectively targets Src and prevents the disorganized acinar growth of HER2-overexpressing, Src-activated, and ER− MECs in 3D culture, a condition mimicking the growth of premalignant lesions in early-stage breast disease. Importantly, saracatinib markedly prevented the development of premalignant lesions and delayed tumor onset in two ER− mammary tumor mouse models with HER2-induced Src activation. These strong preclinical data have led to the recent initiation of a clinical trial using a Src inhibitor for secondary prevention of ER− breast cancer at MD Anderson Cancer Center (ClinicalTrials.gov identifier: NCT01471106).
Previous RPPA analysis in primary breast tumors showed significantly higher Src expression in ERα- and progesterone receptor-negative (PR-) tumors than in ERα+ and/or PR+ tumors, and ERα and Src expression were inversely correlated in all tumors (46). Deletion of Src in epithelial cells delayed polyomavirus middle-T antigen (PyVmT)-driven ER− mammary tumorigenesis (47). Recently, dasatinib was shown to delay mammary tumor onset driven by activated HER2 and PTEN-loss (48). These studies in advanced breast cancer and mammary tumor models support our findings that Src plays an important role in ER− breast cancer progression. However, our current study represents the first discovery that Src activation is a readily targetable event in women who are at high risk of developing tamoxifen-unresponsive breast cancer, and that Src inhibition effectively prevents the progression of ER− early breast lesions.
Cancer metabolism is a recently recognized hallmark of cancer, as chronic and uncontrolled cell proliferation often also involves dysregulation of energy metabolism (49). Recently, clinical trials implementing caloric restriction (CR) and exercise have shown promising results for the prevention of breast cancer in high-risk women (50). Because of the behavioral difficulty in maintaining a CR diet for a long period of time, currently there is a focus on identifying and developing agents that could complement, synergize, or mimic the anticancer effects of CR (51). Our metabolomic data showed for the first time that in an ER− breast cancer model, Src regulates carbohydrate metabolism, partly by regulating glucose uptake. Our data revealed that Src activation increases glucose uptake via upregulating GLUT1 and that saracatinib inhibits glucose uptake by downregulating GLUT1, indicating that targeting Src could help to achieve the goals of CR by reducing glucose uptake.
Previous microarray analysis showed that genes associated with increased glucose metabolism significantly overlap with those of an ER− molecular phenotype (22) suggesting that increased glucose metabolism contributes to ER− breast cancer development. In this study, we were surprised to find that GLUT1 silencing alone prevented the abnormal acinar growth of ER− 10A.B2 cells, indicating that GLUT1 is required for disorganized acinar growth. Our data provided direct evidence of the critical role of glucose metabolism in dysregulated growth of ER− MECs. Mechanistically, our data showed that Src-mediated GLUT1 upregulation is Myc-dependent. We revealed that Src promotes cap-dependent translation of Myc protein via activation of the ERK1/2-MNK1-eIF4E pathway (Fig. 7). Although Src was found to increase cap-dependent translation of β-catenin and hypoxia inducible factor 1-alpha (HIF1α) (42,43), our data demonstrated for the first time that Src enhances the translation of Myc, a strong oncogene activated by various mitogenic signals. Consistent with our in-depth mechanistic findings in vitro, saracatinib also significantly inhibited Src activation and Myc and GLUT1 expression in mouse models. Together, our data present proof of concept that targeting glucose metabolism can be a promising approach for future prevention strategies of ER− breast cancer.
Figure 7.
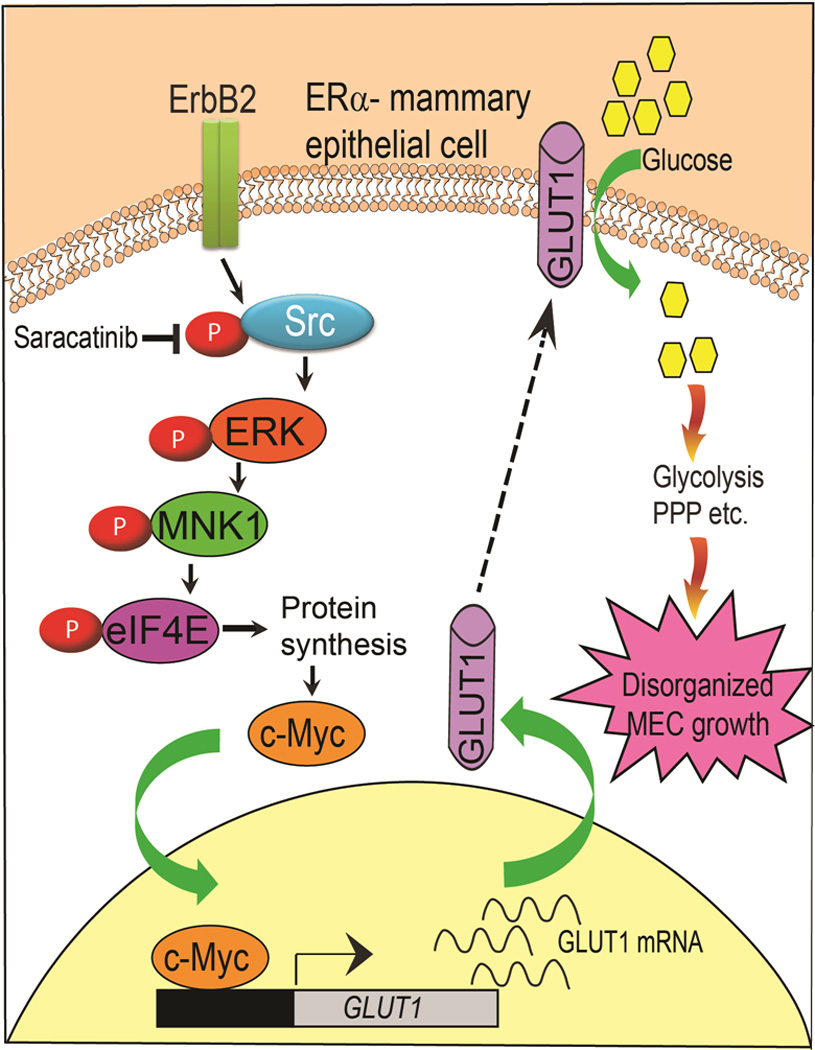
Schematic showing that Src promotes cap-dependent translation of Myc protein via activation of ERK1/2-MNK1-eIF4E pathway leading to GLUT1 expression, which in turn enhances glucose uptake by premalignant cells by controlling several metabolic pathways, such as glycolysis and the pentose phosphate pathway that contribute to premalignant cells growth.
The detection of premalignant lesions at the early stages of cancer development allows for potential early management. In this study, we applied HRME-SI, imaging technology to monitor mammary tumor progression (Supplementary Fig. S2B). Since HRME-SI offers several advantages over traditional measures, e.g. low cost, real time imaging, ease of interpretation at the point-of-care, and visualization of cellular and architectural features (52–54), we have begun testing the HRME-SI imaging technology in human breast cancer surgical specimens, aiming at bring the pilot imaging strategies to the clinic in the near future.
In summary, we identified Src activation as a targetable alteration for prevention of ER− breast cancer and demonstrated that targeting Src with saracatinib delayed ER− mammary tumor initiation and progression. The prevention strategies developed in this study can be immediately implemented to test a wide range of cancer prevention agents. Our approach here could be ultimately developed to achieve a significant reduction in mortality from ER− breast cancer.
Supplementary Material
Acknowledgments
We thank AstraZeneca for providing us saracatinib, S. S. Joshi for suggestions, S. K. Rehman and F. J. Lowery for comments on the manuscript.
Grant Support: This work was supported by Susan G. Komen Breast Cancer Foundation promise grant KG091020 (D.Y.), PO1-CA099031 project 4 (D.Y.), RO1-CA112567-06 (D.Y.), MDACC Duncan Family Institute Fund (D.Y.), US Department of Defense Breast Cancer Research Program postdoctoral fellowship W81XWH-11-1-0004 (S.J.), and MD Anderson Cancer Center Support Grant CA016672. Dr. D. Yu is the Hubert L. & Olive Stringer Distinguished Chair in Basic Science at MDACC.
Footnotes
Conflicts of Interest: None
Author’s Contributions
Conception and design: S. Jain and D. Yu.
Development of methodology: S. Jain, V.L. Seewaldt, R. Richards-Kortum, B. Arun, and D. Yu.
Acquisition of data (provision of animals and facilities, enrollment and management of patients): S. Jain, X. Wang, C-C. Chang, C. Ibarra-Drendall, H. Wang, Q. Zhang, S.W. Brady, P. Li, H. Zhao, J. Dobbs, M. Kyrish, T. S. Tkaczyk, A. Ambrose, C. Sistrunk, V.L. Seewaldt, and D. Yu
Analysis and interpretation of data: S. Jain, X. Wang, S.W. Brady, V.L. Seewaldt and D. Yu
Writing, review, and/or revision of the manuscript: S. Jain, X. Wang, D. Yu, V.L. Seewaldt, R. Richards-Kortum, B. Arun, and W. Jia.
Administrative, technical, or material support (i.e., reporting or organizing data, constructing databases): W. Jia, D. Yu,
References
- 1.Siegel R, Ma J, Zou Z, Jemal A. Cancer statistics, 2014. CA: a cancer journal for clinicians. 2014;64:9–29. doi: 10.3322/caac.21208. [DOI] [PubMed] [Google Scholar]
- 2.Cazzaniga M, Bonanni B. Breast cancer chemoprevention: old and new approaches. Journal of biomedicine & biotechnology. 2012;2012:985620. doi: 10.1155/2012/985620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fisher B, Costantino JP, Wickerham DL, Cecchini RS, Cronin WM, Robidoux A, et al. Tamoxifen for the prevention of breast cancer: current status of the National Surgical Adjuvant Breast and Bowel Project P-1 study. Journal of the National Cancer Institute. 2005;97:1652–1662. doi: 10.1093/jnci/dji372. [DOI] [PubMed] [Google Scholar]
- 4.Yue W, Wang JP, Li Y, Bocchinfuso WP, Korach KS, Devanesan PD, et al. Tamoxifen versus aromatase inhibitors for breast cancer prevention. Clinical cancer research : an official journal of the American Association for Cancer Research. 2005;11:925s–930s. [PubMed] [Google Scholar]
- 5.Carey LA, Perou CM, Livasy CA, Dressler LG, Cowan D, Conway K, et al. Race, breast cancer subtypes, and survival in the Carolina Breast Cancer Study. JAMA : the journal of the American Medical Association. 2006;295:2492–2502. doi: 10.1001/jama.295.21.2492. [DOI] [PubMed] [Google Scholar]
- 6.Dolle JM, Daling JR, White E, Brinton LA, Doody DR, Porter PL, et al. Risk factors for triple-negative breast cancer in women under the age of 45 years. Cancer epidemiology, biomarkers & prevention : a publication of the American Association for Cancer Research, cosponsored by the American Society of Preventive Oncology. 2009;18:1157–1166. doi: 10.1158/1055-9965.EPI-08-1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhou W, Slingerland JM. Links between oestrogen receptor activation and proteolysis: relevance to hormone-regulated cancer therapy. Nature reviews Cancer. 2013;14:26–38. doi: 10.1038/nrc3622. [DOI] [PubMed] [Google Scholar]
- 8.Castoria G, Giovannelli P, Lombardi M, De Rosa C, Giraldi T, de Falco A, et al. Tyrosine phosphorylation of estradiol receptor by Src regulates its hormone-dependent nuclear export and cell cycle progression in breast cancer cells. Oncogene. 2012;31:4868–4877. doi: 10.1038/onc.2011.642. [DOI] [PubMed] [Google Scholar]
- 9.Likhite VS, Stossi F, Kim K, Katzenellenbogen BS, Katzenellenbogen JA. Kinase-specific phosphorylation of the estrogen receptor changes receptor interactions with ligand, deoxyribonucleic acid, and coregulators associated with alterations in estrogen and tamoxifen activity. Molecular endocrinology. 2006;20:3120–3132. doi: 10.1210/me.2006-0068. [DOI] [PubMed] [Google Scholar]
- 10.Zheng X, Resnick RJ, Shalloway D. Apoptosis of estrogen-receptor negative breast cancer and colon cancer cell lines by PTP alpha and src RNAi. International journal of cancer Journal international du cancer. 2008;122:1999–2007. doi: 10.1002/ijc.23321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hiscox S, Morgan L, Green TP, Barrow D, Gee J, Nicholson RI. Elevated Src activity promotes cellular invasion and motility in tamoxifen resistant breast cancer cells. Breast cancer research and treatment. 2006;97:263–274. doi: 10.1007/s10549-005-9120-9. [DOI] [PubMed] [Google Scholar]
- 12.Puls LN, Eadens M, Messersmith W. Current status of SRC inhibitors in solid tumor malignancies. The oncologist. 2011;16:566–578. doi: 10.1634/theoncologist.2010-0408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gerber DE. Targeted therapies: a new generation of cancer treatments. American family physician. 2008;77:311–319. [PubMed] [Google Scholar]
- 14.Simpkins F, Hevia-Paez P, Sun J, Ullmer W, Gilbert CA, da Silva T, et al. Src Inhibition with saracatinib reverses fulvestrant resistance in ER-positive ovarian cancer models in vitro and in vivo. Clinical cancer research : an official journal of the American Association for Cancer Research. 2012;18:5911–5923. doi: 10.1158/1078-0432.CCR-12-1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chen Y, Alvarez EA, Azzam D, Wander SA, Guggisberg N, Jorda M, et al. Combined Src and ER blockade impairs human breast cancer proliferation in vitro and in vivo. Breast cancer research and treatment. 2011;128:69–78. doi: 10.1007/s10549-010-1024-7. [DOI] [PubMed] [Google Scholar]
- 16.den Hollander P, Savage MI, Brown PH. Targeted Therapy for Breast Cancer Prevention. Frontiers in oncology. 2013;3:250. doi: 10.3389/fonc.2013.00250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ibarra-Drendall C, Dietze EC, Seewaldt VL. Metabolic Syndrome and Breast Cancer Risk: Is There a Role for Metformin? Current breast cancer reports. 2011;3:142–150. doi: 10.1007/s12609-011-0050-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gillies RJ, Robey I, Gatenby RA. Causes and consequences of increased glucose metabolism of cancers. Journal of nuclear medicine : official publication, Society of Nuclear Medicine. 2008;49(Suppl 2):24S–42S. doi: 10.2967/jnumed.107.047258. [DOI] [PubMed] [Google Scholar]
- 19.Dang CV. Links between metabolism and cancer. Genes & development. 2012;26:877–890. doi: 10.1101/gad.189365.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dang CV, Le A, Gao P. MYC-induced cancer cell energy metabolism and therapeutic opportunities. Clinical cancer research : an official journal of the American Association for Cancer Research. 2009;15:6479–6483. doi: 10.1158/1078-0432.CCR-09-0889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shajahan-Haq AN, Cook KL, Schwartz-Roberts JL, Eltayeb AE, Demas DM, Warri AM, et al. MYC regulates the unfolded protein response and glucose and glutamine uptake in endocrine resistant breast cancer. Molecular cancer. 2014;13:239. doi: 10.1186/1476-4598-13-239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Osborne JR, Port E, Gonen M, Doane A, Yeung H, Gerald W, et al. 18F-FDG PET of locally invasive breast cancer and association of estrogen receptor status with standardized uptake value: microarray and immunohistochemical analysis. Journal of nuclear medicine : official publication, Society of Nuclear Medicine. 2010;51:543–550. doi: 10.2967/jnumed.108.060459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hussein YR, Bandyopadhyay S, Semaan A, Ahmed Q, Albashiti B, Jazaerly T, et al. Glut-1 Expression Correlates with Basal-like Breast Cancer. Translational oncology. 2011;4:321–327. doi: 10.1593/tlo.11256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Schafer ZT, Grassian AR, Song L, Jiang Z, Gerhart-Hines Z, Irie HY, et al. Antioxidant and oncogene rescue of metabolic defects caused by loss of matrix attachment. Nature. 2009;461:109–113. doi: 10.1038/nature08268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhang S, Huang WC, Li P, Guo H, Poh SB, Brady SW, et al. Combating trastuzumab resistance by targeting SRC, a common node downstream of multiple resistance pathways. Nature medicine. 2011;17:461–469. doi: 10.1038/nm.2309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li J, Lu Y, Akbani R, Ju Z, Roebuck PL, Liu W, et al. TCPA: a resource for cancer functional proteomics data. Nature methods. 2013;10:1046–1047. doi: 10.1038/nmeth.2650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lu J, Guo H, Treekitkarnmongkol W, Li P, Zhang J, Shi B, et al. 14-3-3zeta Cooperates with ErbB2 to promote ductal carcinoma in situ progression to invasive breast cancer by inducing epithelial-mesenchymal transition. Cancer cell. 2009;16:195–207. doi: 10.1016/j.ccr.2009.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Debnath J, Muthuswamy SK, Brugge JS. Morphogenesis and oncogenesis of MCF-10A mammary epithelial acini grown in three-dimensional basement membrane cultures. Methods. 2003;30:256–268. doi: 10.1016/s1046-2023(03)00032-x. [DOI] [PubMed] [Google Scholar]
- 29.Debnath J, Brugge JS. Modelling glandular epithelial cancers in three-dimensional cultures. Nature reviews Cancer. 2005;5:675–688. doi: 10.1038/nrc1695. [DOI] [PubMed] [Google Scholar]
- 30.Guy CT, Webster MA, Schaller M, Parsons TJ, Cardiff RD, Muller WJ. Expression of the neu protooncogene in the mammary epithelium of transgenic mice induces metastatic disease. Proceedings of the National Academy of Sciences of the United States of America. 1992;89:10578–10582. doi: 10.1073/pnas.89.22.10578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Baselga J, Cervantes A, Martinelli E, Chirivella I, Hoekman K, Hurwitz HI, et al. Phase I safety, pharmacokinetics, and inhibition of SRC activity study of saracatinib in patients with solid tumors. Clinical cancer research : an official journal of the American Association for Cancer Research. 2010;16:4876–4883. doi: 10.1158/1078-0432.CCR-10-0748. [DOI] [PubMed] [Google Scholar]
- 32.Liu KJ, He JH, Su XD, Sim HM, Xie JD, Chen XG, et al. Saracatinib (AZD0530) is a potent modulator of ABCB1-mediated multidrug resistance in vitro and in vivo. International journal of cancer Journal international du cancer. 2013;132:224–235. doi: 10.1002/ijc.27649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Vila PM, Park CW, Pierce MC, Goldstein GH, Levy L, Gurudutt VV, et al. Discrimination of benign and neoplastic mucosa with a high-resolution microendoscope (HRME) in head and neck cancer. Annals of surgical oncology. 2012;19:3534–3539. doi: 10.1245/s10434-012-2351-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Quinn MK, Bubi TC, Pierce MC, Kayembe MK, Ramogola-Masire D, Richards-Kortum R. High-resolution microendoscopy for the detection of cervical neoplasia in low-resource settings. PloS one. 2012;7:e44924. doi: 10.1371/journal.pone.0044924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Siegel PM, Ryan ED, Cardiff RD, Muller WJ. Elevated expression of activated forms of Neu/ErbB-2 and ErbB-3 are involved in the induction of mammary tumors in transgenic mice: implications for human breast cancer. The EMBO journal. 1999;18:2149–2164. doi: 10.1093/emboj/18.8.2149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Macheda ML, Rogers S, Best JD. Molecular and cellular regulation of glucose transporter (GLUT) proteins in cancer. Journal of cellular physiology. 2005;202:654–662. doi: 10.1002/jcp.20166. [DOI] [PubMed] [Google Scholar]
- 37.Zhao FQ. Biology of glucose transport in the mammary gland. Journal of mammary gland biology and neoplasia. 2014;19:3–17. doi: 10.1007/s10911-013-9310-8. [DOI] [PubMed] [Google Scholar]
- 38.Young CD, Lewis AS, Rudolph MC, Ruehle MD, Jackman MR, Yun UJ, et al. Modulation of glucose transporter 1 (GLUT1) expression levels alters mouse mammary tumor cell growth in vitro and in vivo. PloS one. 2011;6:e23205. doi: 10.1371/journal.pone.0023205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pujol-Gimenez J, de Heredia FP, Idoate MA, Airley R, Lostao MP, Evans AR. Could GLUT12 be a Potential Therapeutic Target in Cancer Treatment? A Preliminary Report. Journal of Cancer. 2015;6:139–143. doi: 10.7150/jca.10429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Osthus RC, Shim H, Kim S, Li Q, Reddy R, Mukherjee M, et al. Deregulation of glucose transporter 1 and glycolytic gene expression by c-Myc. The Journal of biological chemistry. 2000;275:21797–21800. doi: 10.1074/jbc.C000023200. [DOI] [PubMed] [Google Scholar]
- 41.Kristensen AR, Gsponer J, Foster LJ. Protein synthesis rate is the predominant regulator of protein expression during differentiation. Molecular systems biology. 2013;9:689. doi: 10.1038/msb.2013.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Karni R, Dor Y, Keshet E, Meyuhas O, Levitzki A. Activated pp60c-Src leads to elevated hypoxia-inducible factor (HIF)-1alpha expression under normoxia. J Biol Chem. 2002;277:42919–42925. doi: 10.1074/jbc.M206141200. [DOI] [PubMed] [Google Scholar]
- 43.Karni R, Gus Y, Dor Y, Meyuhas O, Levitzki A. Active Src elevates the expression of beta-catenin by enhancement of cap-dependent translation. Mol Cell Biol. 2005;25:5031–5039. doi: 10.1128/MCB.25.12.5031-5039.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.De Benedetti A, Graff JR. eIF-4E expression and its role in malignancies and metastases. Oncogene. 2004;23:3189–3199. doi: 10.1038/sj.onc.1207545. [DOI] [PubMed] [Google Scholar]
- 45.Topisirovic I, Sonenberg N. mRNA translation and energy metabolism in cancer: the role of the MAPK and mTORC1 pathways. Cold Spring Harbor symposia on quantitative biology. 2011;76:355–367. doi: 10.1101/sqb.2011.76.010785. [DOI] [PubMed] [Google Scholar]
- 46.Chu I, Arnaout A, Loiseau S, Sun J, Seth A, McMahon C, et al. Src promotes estrogen-dependent estrogen receptor alpha proteolysis in human breast cancer. The Journal of clinical investigation. 2007;117:2205–2215. doi: 10.1172/JCI21739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Marcotte R, Smith HW, Sanguin-Gendreau V, McDonough RV, Muller WJ. Mammary epithelial-specific disruption of c-Src impairs cell cycle progression and tumorigenesis. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:2808–2813. doi: 10.1073/pnas.1018861108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Karim SA, Creedon H, Patel H, Carragher NO, Morton JP, Muller WJ, et al. Dasatinib inhibits mammary tumour development in a genetically engineered mouse model. The Journal of pathology. 2013;230:430–440. doi: 10.1002/path.4202. [DOI] [PubMed] [Google Scholar]
- 49.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 50.Imayama I, Ulrich CM, Alfano CM, Wang C, Xiao L, Wener MH, et al. Effects of a caloric restriction weight loss diet and exercise on inflammatory biomarkers in overweight/obese postmenopausal women: a randomized controlled trial. Cancer research. 2012;72:2314–2326. doi: 10.1158/0008-5472.CAN-11-3092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hursting SD, Dunlap SM, Ford NA, Hursting MJ, Lashinger LM. Calorie restriction and cancer prevention: a mechanistic perspective. Cancer & metabolism. 2013;1:10. doi: 10.1186/2049-3002-1-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Houssami N, Hayes DF. Review of preoperative magnetic resonance imaging (MRI) in breast cancer: should MRI be performed on all women with newly diagnosed, early stage breast cancer? CA: a cancer journal for clinicians. 2009;59:290–302. doi: 10.3322/caac.20028. [DOI] [PubMed] [Google Scholar]
- 53.Independent UKPoBCS. The benefits and harms of breast cancer screening: an independent review. Lancet. 2012;380:1778–1786. doi: 10.1016/S0140-6736(12)61611-0. [DOI] [PubMed] [Google Scholar]
- 54.Kyrish M, Dobbs J, Jain S, Wang X, Yu D, Richards-Kortum R, et al. Needle-based fluorescence endomicroscopy via structured illumination with a plastic, achromatic objective. Journal of biomedical optics. 2013;18:096003. doi: 10.1117/1.JBO.18.9.096003. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.