

Abstract
Combining 2-formylphenylboronic acid with 4-hydrazinylbenzoic acid in neutral aqueous solution at low, equimolar concentrations of the reagents results in a single, stable product, a 1,2-dihydro-1-hydroxy-2,3,1-benzodiazaborine, in a matter of minutes with no side products. Application of this reaction to protein conjugation demonstrates that the reaction is orthogonal to protein functional groups, and the resulting conjugate withstands SDS-PAGE analysis. This reaction should be particularly useful for couplings that must be performed with low concentrations of reagents under physiologically compatible conditions.
Bioorthogonal reactions occur under conditions that are compatible with a biological environment, with exogenous functional groups that form a covalent bond with one another without reacting with endogenous reactive functional groups.1 Owing to their utility in site-specific bioconjugation reactions, a variety of classes of reagents are widely used and discovery of new reactions is a very active area of research.2-6 A remaining challenge for many of these reactions is that they occur slowly at physiological pH. High concentrations of one or both of the reagents, catalytic additives, or acidic conditions are often required for these reactions to proceed.7 These conditions may be incompatible with the biological system, uneconomical or infeasible.8
One of the oldest bioorthogonal reactions is that of heteroatom-bonded amines, referred to as “α-effect amines” (hydrazine, hydrazide, and other amine-nitrogen compounds, aminooxy reagents, etc.), with carbonyl functionalities (aldehydes or ketones).9 These reactions occur readily in acidic aqueous solution but tend to be slow at physiological pH. In spite of the reaction characteristics, hydrazone and oxime forming reactions have been used successfully for many types of bioconjugations,10, 11 including construction of drug-antibody conjugates.12, 13
The speed of the conjugation reaction at neutral pH can be significantly increased by nucleophilic catalysis using aromatic amines.14 Large excess of the catalyst is required, however, since imine formation between the carbonyl and the aromatic amine is responsible for the increased overall reaction rate. Intramolecular catalysis of hydrazone and oxime formation has been explored to increase the speed of the hydrazone or oxime-forming reaction without high concentrations of additives.15, 16 We have been interested in intramolecular catalysis of hydrazone formation by modification of aromatic aldehyde electrophiles. We discovered that a phosphate group ortho- to an aldehyde could serve as an intramolecular acid catalyst, enhancing the reaction rate by a factor of ~20 at pH 717. It was of interest to assess whether a Lewis acid similarly situated could catalyze hydrazone formation, which we approach using model compounds.
We have found that an ortho-boronic acid on an aromatic aldehyde accelerates the speed of reaction of the aldehyde with an aromatic hydrazine by at least three orders of magnitude. The resulting product, however, is not the expected hydrazone. Instead, a second dehydration reaction occurs, forming a substituted 1,2-dihydro-1-hydroxy-2,3,1-benzodiazaborine as the final product(Figure 1).
Figure 1.
Structures of salicyaldehyde (Sal, 1), p-hydrazinylbenzoic acid (HBA, 2), the hydrazone of Sal and HBA (3), 2-formylphenylboronic acid (2fPBA, 4), and the product of 2fPBA and HBA (5)
Model compounds selected for study were salicylaldehyde (Sal, 1), 4-hydrazinylbenzoic acid (HBA, 2) and 2-formylphenylboronic acid (fPBA, 4). Sal was chosen as the aromatic aldehyde because we have previously performed a number of kinetic and structural studies of hydrazones formed with ortho-hydroxybenzaldehydes.18-20 The progress of the reaction HBA with fPBA or with Sal was monitored by absorption difference spectroscopy. Initially, the reactions were performed under pseudo-first order conditions (325 μM aldehyde + 32.5 μM HBA) and data points were collected every 5 minutes. Under these conditions, hydrazone formation between Sal and HBA (3) was complete in about 3 hours. (ESI, Figure S1) Surprisingly, the reaction between fPBA and HBA was complete before the first data point was collected.
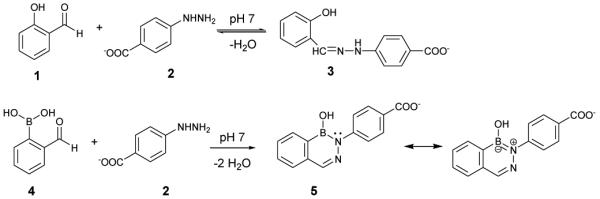
It was therefore necessary to use much lower concentrations of fPBA and HBA to measure the reaction kinetics. Figure 2 shows a typical plot of absorption difference spectra as a function of time collected under second order conditions (5 μM fPBA, 5 μM HBA). In the hydrazone-forming reaction between Sal and HBA, there is an increase in the low energy absorption band as the reaction progresses (ESI, Figure S1). In the reaction of fPBA and HBA, however, there is an rapid increase in the low energy region of the absorption spectrum that decreases over time and a peak at higher energy, which increases and shifts to higher energy over time.
Figure 2.
Absorption difference spectra of fPBA and HBA in 0.1 M phosphate buffer, pH 7, (5 μM each, final concentration) as a function of time. Note that the absorptivity of the low energy band initially increases and then decreases over the course of the reaction.
These data are consistent with an initial, very fast second order reaction between fPBA and HBA, in which a hydrazone is formed. Hydrazone formation is immediately followed by a unimolecular dehydration reaction to form the final product, a substituted 1,2-dihydro-1-hydroxy-2,3,1-benzodiazaborine (DAB). The changes in the absorption spectrum as a function of time provide qualitative support for this idea. The immediate appearance of a lower energy band in the difference spectrum is characteristic of hydrazone formation. It is known that the absorption spectra of hydrazones of aromatic aldehydes are red-shifted compared to those of the parent aldehyde.21 Loss of this low energy band over time is accompanied by an increase in a higher energy region of the spectrum is consistent with a conjugated system becoming an aromatic molecule. This observation is consistent with the absorption properties of known DABs that resemble the aromatic heterocycle isoquinoline, which has an absorption maximum near 300 nm.22 The absorption maximum of the final product of fPBA and HBA reaction is ~305 nm (Figure S2, ESI).
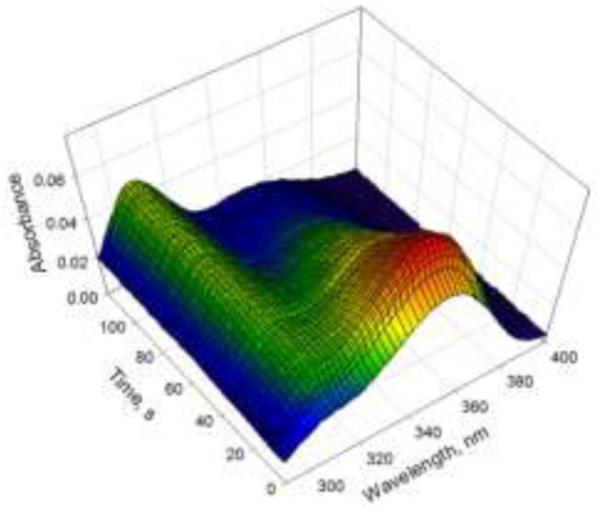
Further evidence that the second step of the reaction is unimolecular was found by collecting kinetic data at varying concentrations of the two components. The decrease in absorption at 350 nm as a function of time was essentially invariant with concentration, as expected for a unimolecular reaction. (ESI, Figure S3) In order to obtain parameters for both steps of the reaction, subsequent experiments were performed under pseudo-first order conditions. The data were fit to a kinetic model of two sequential first order reactions. The rate constant for the second step of the reaction was consistently 0.014 – 0.016 s−1 at 25 °C. The second order rate constant, calculated from the pseudo-first order rate constant for the first step of the reaction, is in the range of ~103 M−1s−1 (~1500 – 2500 M−1s−1, four different experiments). There is more error in this value owing to the speed of the reaction. Figure 3 shows that a significant portion of the second order reaction is complete before data collection could commence. Alternative methods of data collection are required to get precise rate constants, and these experiments are currently underway.
Figure 3.
Kinetic data collected under pseudo-first order conditions. Absorption difference spectra of 50 μM HBA + 2.5 μM fPBA were collected as a function of time. Data were fit to two sequential first order reactions using Olis GlobalWorks software. A plot of the change in absorption at 350 nm vs time is shown (red points), which is proportional to the concentration of the intermediate hydrazone as a function of time. The dashed line is the theoretical curve for k1 = 0.114 s−1 (pseudo-first order rate constant for the formation of the hydrazone) and k2 = 0.015 s−1 (first order rate constant for formation of the DAB), which were obtained from the global fit of the spectral data.
The presence of the boronic acid ortho- to the aldehyde therefore increases the rate of hydrazone formation by about three orders of magnitude. This is similar to the rate enhancement observed for oxime formation with o-formyl phenylboronic acids.23 A significant difference in this system is its ability to undergo a second dehydration reaction and form a stable aromatic heterocycle, a DAB, as the final product.
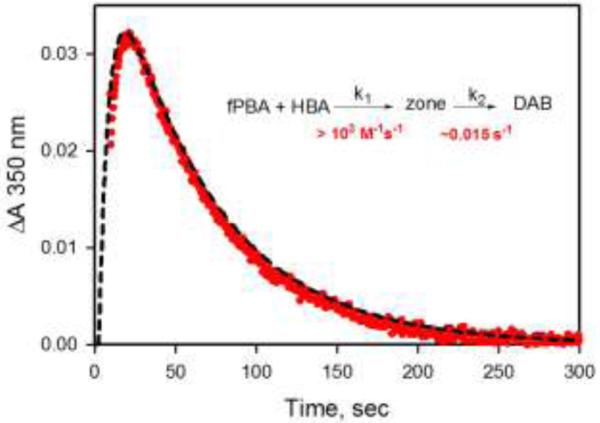
DABs are a little known group of aromatic heterocyclic molecules that contain a boron atom and two nitrogen atoms. Gronowitz, Dewar, Dougherty and others reasoned that the 10-electron system of a DAB fused to an aromatic or heteroaromatic ring should be planar and fully conjugated if the lone pair on the nitrogen atom interacted with the empty orbital on the boron atom.24-26 These compounds, first prepared in the 1960’s, have been prepared by typical synthetic means (organic solvent, acid catalyst, heat, and/or high concentrations of reagents).27-30 To confirm that this DAB can be formed in neutral aqueous solution, 5 was prepared by mixing fPBA with HBA in water at a concentration of about 100 mM. A white precipitate formed, which was isolated and dissolved in DMSO-d6 for NMR. The compound was characterized by 1H, 13C and 11B NMR and mass spectrometry (ESI). In a second experiment, 2 mM solutions of fPBA and HBA in water containing 10% D2O were mixed in a 1:1 ratio. The 1H NMR spectrum of this product conforms to that of the molecule in DMSO-d6. (ESI)
Owing to the aromatic character of the boron heterocycle, the product of this reaction would be expected to be more stable than a corresponding hydrazone. We have found that 5 is highly stable in neutral aqueous solution. There is essentially no decomposition of the molecule in pH 7 buffer for a period of one month. Figure 4A shows the proton NMR spectrum of 5 that results immediately after mixing the solutions of fPBA and HBA in pH 7 buffer. Figure 4B is a spectrum of the same solution after 30 days at room temperature.
Figure 4.
Proton NMR spectrum of 5 in immediately after mixing fPBA and HBA to a final concentration of 2.5 mM each in 0.1 M phosphate buffer, pH 7.0, containing 10% D2O.
A criterion for a reaction to be employed in a biological setting is that the reagents are orthogonal to endogenous functional groups. Cal et al. have shown that fPBA forms a stable iminoboronate with lysine residue that is reversible. The interaction has been exploited for modification of lysine residues in proteins with fPBA-containing molecules.31, 32 We have performed a few preliminary experiments to determine whether endogenous nucleophiles might interfere with the reaction. No effect on reaction kinetics was observed in the presence of millimolar concentrations of lysine, glutathione or sucrose (ESI).
Application of this reaction for bioorthogonal couplings was assessed using a model system. A fluorescent 2-formylphenylboronic acid ester (6, Figure 5) was synthesized as described in ESI. No attempt was made to deprotect the molecule prior to use in the biological coupling reaction. Many boronic acid esters hydrolyze readily in aqueous solution33 and therefore should also be suitable precursors to DABs. This hypothesis was confirmed using the pinacol ester of 2fPBA. The esterified 2fPBA was allowed to react with HBA and the reaction was monitored by absorption difference spectroscopy. The results were comparable with those of the boronic acid (ESI, Figure S3).
Figure 5.
Coumarin-fPBA ester conjugate (6), NHS-ester for labeling BSA with HBA (7)
Next, HBA was covalently attached to bovine serum albumin (BSA) through a hydroxysuccinimidyl ester derivative of the molecule (7). Probe 6 was allowed to react with unmodified BSA or with BSA-hydrazine for 15 min at room temperature at a molar ratio of 1:3 (BSA:probe 6) in 10 mM phosphate buffer (pH 7.0), after which small molecules were removed by rapid gel filtratration. The hydrazine-containing protein retained the fluorophore, while the unmodified protein did not (Figure 6). Figure 7 shows these samples after SDS-PAGE analysis. It is seen in figures 6 and 7 that the fluorophore covalently attaches to the hydrazine functionalized BSA, thereby demonstrating that this chemistry is amenable to bioconjugation processes. Additionally, the DAB remains intact through the high pH of the resolving gel and the acidic pH of the destaining solution, indicating that the aromatic heterocycle is sufficiently stable to allow for downstream analysis of a biomolecule conjugate.
Figure 6.
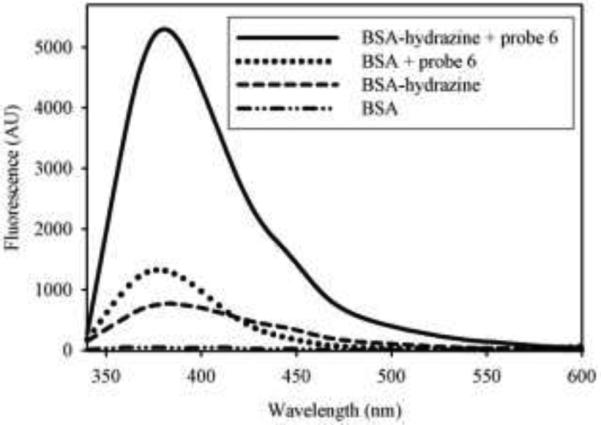
Emission spectra of BSA-hydrazine and BSA reacted with or without probe 6 excited at 320 nm. BSA or BSA-hydrazine (final concentration 25 μM) was allowed to react with or without probe 6 (final concentration 75 μM) at room temperature for 15 min. Excess probe was removed by rapid gel filtration chromatography and emission spectra of equal concentration of each protein sample were obtained.
Figure 7.
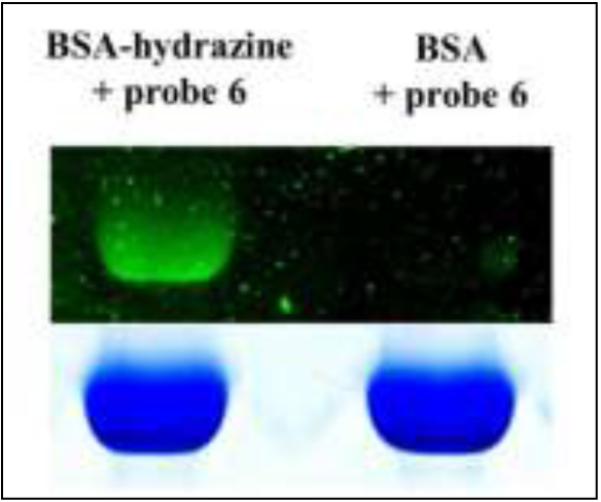
SDS-PAGE of BSA or BSA-hydrazine after reaction with probe 6.
In conclusion, the reaction of an aromatic hydrazine, 4-hydrazinylbenzoic acid, with ortho-formylphenylboronic acid in aqueous solution at pH 7 is remarkably fast, but the product of the reaction is not the expected hydrazone. Instead, the hydrazone is presumably an intermediate in formation of the final product, which is an aromatic heterocycle. The reaction occurs in dilute aqueous solution and forms a single product, which is stable in neutral aqueous solution over a period of weeks. The reaction is orthogonal to protein functional groups, and the product of the reaction is stable for weeks at pH 7 and is stable to SDS-PAGE analysis. The fast kinetics of the reaction at micromolar concentrations, the ability to form a single product at 1:1 molar ratio, and the stability of the phenyl-substituted DAB suggests that coupling reactions of this type will have a broad range of applications, especially in biological systems .
Supplementary Material
Acknowledgments
‡This work was supported by NIH Grant R15 GM-093941 and by a special grant from the Harpur College Dean’s Office, Binghamton University. The Regional NMR Facility (600 MHz instrument) at Binghamton University is supported by NSF (CHE-0922815). The authors thank Dr. John Lloyd, Natural Products Chemistry Section, NIDDK, NIH, for mass spectrometry analyses, Professor Rebecca Kissling for scientific discussion, Dr. Jürgen T. Schulte for assistance in NMR data acquisition and interpretation, Mr. Han Gu and Ms. Samantha L. Grieco for technical assistance and Mr. David Tuttle for photographing the gels.
Footnotes
Electronic Supplementary Information (ESI) available: [experimental procedures, supporting data and NMR spectra]. See DOI: 10.1039/x0xx00000x
References
- 1.Sletten EM, Bertozzi CR. Angew. Chem. Int. Ed. Engl. 2009;48:6974–6998. doi: 10.1002/anie.200900942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Agarwal P, Bertozzi CR. Bioconjugate Chem. 2015;26:176–192. doi: 10.1021/bc5004982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Waengler C, Schirrmacher R, Bartenstein P, Waengler B. Curr. Med. Chem. 2010;17:1092–1116. doi: 10.2174/092986710790820615. [DOI] [PubMed] [Google Scholar]
- 4.Thirumurugan P, Matosiuk D, Jozwiak K. Chem. Rev. 2013;113:4905–4979. doi: 10.1021/cr200409f. [DOI] [PubMed] [Google Scholar]
- 5.McKay CS, Finn MG. Chem. Biol. 2014;21:1075–1101. doi: 10.1016/j.chembiol.2014.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Debets MF, van Hest JC, Rutjes FP. Org Biomol Chem. 2013;11:6439–6455. doi: 10.1039/c3ob41329b. [DOI] [PubMed] [Google Scholar]
- 7.Kalia J, Raines RT. Curr. Org. Chem. 2010;14:138–147. doi: 10.2174/138527210790069839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Saito F, Noda H, Bode JW. ACS Chem. Biol. 2015;10:1026–1033. doi: 10.1021/cb5006728. [DOI] [PubMed] [Google Scholar]
- 9.Lim RKV, Lin Q. Chemical communications (Cambridge, England) 2010;46:1589–1600. doi: 10.1039/b925931g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ulrich S, Boturyn D, Marra A, Renaudet O, Dumy P. Chemistry – A European Journal. 2014;20:34–41. doi: 10.1002/chem.201302426. [DOI] [PubMed] [Google Scholar]
- 11.Algar WR, Prasuhn DE, Stewart MH, Jennings TL, Blanco-Canosa JB, Dawson PE, Medintz IL. Bioconjugate Chem. 2011;22:825–858. doi: 10.1021/bc200065z. [DOI] [PubMed] [Google Scholar]
- 12.Axup JY, Bajjuri KM, Ritland M, Hutchins BM, Kim CH, Kazane SA, Halder R, Forsyth JS, Santidrian AF, Stafin K, Lu Y, Tran H, Seller AJ, Biroc SL, Szydlik A, Pinkstaff JK, Tian F, Sinha SC, Felding-Habermann B, Smider VV, Schultz PG. Proceedings of the National Academy of Sciences. 2012;109:16101–16106. doi: 10.1073/pnas.1211023109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gordon MR, Canakci M, Li L, Zhuang J, Osborne B, Thayumanavan S. Bioconjugate Chem. 2015 doi: 10.1021/acs.bioconjchem.5b00399. DOI: 10.1021/acs.bioconjchem.5b00399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dirksen A, Hackeng TM, Dawson PE. Angewandte Chemie-International Edition. 2006;45:7581–7584. doi: 10.1002/anie.200602877. [DOI] [PubMed] [Google Scholar]
- 15.Crisalli P, Kool ET. Org. Lett. 2013;15:1646–1649. doi: 10.1021/ol400427x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang X, Canary JW. Bioconjugate Chem. 2012;23:2329–2334. doi: 10.1021/bc300430k. [DOI] [PubMed] [Google Scholar]
- 17.Dilek O, Sorrentino AM, Bane S. unpublished work.
- 18.Blanden AR, Mukherjee K, Dilek O, Loew M, Bane SL. Bioconjugate Chem. 2011;22:1954–1961. doi: 10.1021/bc2001566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Banerjee A, Panosian TD, Mukherjee K, Ravindra R, Gal S, Sackett DL, Bane S. ACS Chem. Biol. 2010;5:777–785. doi: 10.1021/cb100060v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dilek O, Bane SL. J. Fluoresc. 2011;21:347–354. doi: 10.1007/s10895-010-0723-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hinman RL. J. Ord. Chem. 1960;25:1775–1778. [Google Scholar]
- 22.Dewar MJS, Dougherty RC. J. Am. Chem. Soc. 1964;86:433–436. [Google Scholar]
- 23.Schmidt P, Stress C, Gillingham D. Chem. Sci. 2015;6:3329–3333. doi: 10.1039/c5sc00921a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dewar MJS, Dougherty RC. J. Am. Chem. Soc. 1962;84:2648–2649. [Google Scholar]
- 25.Gronowitz S, Bugge A. Acta Chem. Scand. 1965;19:1271–1285. [Google Scholar]
- 26.Bosdet MJD, Piers WE. Can. J. Chem. 2012;90:8–29. [Google Scholar]
- 27.Tschampel P, Snyder HR. J. Org. Chem. 1964;29:2168–2172. [Google Scholar]
- 28.Groziak MP, Chen L, Yi L, Robinson PD. J. Am. Chem. Soc. 1997;119:7817–7826. [Google Scholar]
- 29.Grassberger MA, Turnowsky F, Hildebrandt J. J. Med. Chem. 1984;27:947–954. doi: 10.1021/jm00374a003. [DOI] [PubMed] [Google Scholar]
- 30.Yang J, Johnson BJ, Letourneau AA, Vogels CM, Decken A, Baerlocher FJ, Westcott SA. Aust. J. Chem. 2015;68:366–372. [Google Scholar]
- 31.Cal PM, Vicente JB, Pires E, Coelho AV, Veiros LF, Cordeiro C, Gois PM. J. Am. Chem. Soc. 2012;134:10299–10305. doi: 10.1021/ja303436y. [DOI] [PubMed] [Google Scholar]
- 32.Cal PMSD, Frade RFM, Cordeiro C, Gois PMP. Chemistry – A European Journal. 2015;21:8182–8187. doi: 10.1002/chem.201500127. [DOI] [PubMed] [Google Scholar]
- 33.Achilli C, Ciana A, Fagnoni M, Balduini C, Minetti G. Cent. Eur. J. Chem. 2013;11:137–139. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.