

Abstract
Airway smooth muscle (ASM) is a key target cell in allergen-induced asthma known to contribute to airway hyperresponsiveness (AHR) and chronic airway remodeling. Changes in ASM calcium homeostasis have been shown to contribute to AHR although the mechanisms and Ca2+ signal effectors are incompletely understood. In the present study we tested the function of ASM multifunctional protein kinase Ca2+/calmodulin-dependent kinase II (CaMKII) isoforms CaMKIIδ and CaMKIIγ in allergen-induced AHR and airway remodeling in vivo. Using a murine model of atopic asthma we demonstrate CaMKIIδ protein is up-regulated in ASM derived from ovalbumin (OVA)-treated animals compared to controls. A genetic approach to conditionally knockout smooth muscle CaMKIIδ and CaMKIIγ in separate Cre-loxp systems was validated, and using this loss-of function approach, the function of these CaMKII isoforms was tested in ovalbumin (OVA)-induced airway remodeling and AHR. OVA treatment in control mice had no effect on ASM remodeling in this model of AHR and CaMKIIδ knockouts had no independent effects on ASM content. However, at 1day post final OVA challenge, OVA-induced AHR were eliminated in the CaMKIIδ knockouts. OVA-induced peribronchial inflammation and bronchoalveolar lavage fluid (BALF) levels of the Th2 cytokine IL-13 were significantly decreased in the CaMKIIδ knockouts. Unexpectedly, we found increased peribronchial eosinophils in the smooth muscle CaMKIIδ knockouts compared to control animals at 1 day post final challenge, suggesting that lack of ASM CaMKIIδ delays the progression of AHR rather than inhibiting it. Indeed, when AHR was determined at 7 day post final OVA challenge, CaMKIIδ knockouts showed robust AHR while AHR was fully resolved in OVA-challenged control mice. These in vivo studies demonstrate a role for smooth muscle CaMKIIδ in promoting airway inflammation and AHR and suggest a complex signaling role for CaMKIIδ in regulating ASM function. These studies confirm the diverse roles of ASM cells as immune effectors that control AHR and calls for further studies into CaMKIIδ-mediated signaling in ASM cells during disease.
Introduction
Airway smooth muscle (ASM), a major structural component of the airway, is strongly implicated in the pathophysiology of atopic asthma. Drug therapies targeting ASM contractile activity, or bronchial thermoplasty targeting ASM viability, decrease airway hyperresponsiveness (AHR) and improve symptoms in asthmatics [10,11]. Still, there is considerable debate over the exact role(s) ASM plays in the pathophysiology of asthma [17,39].
Smooth muscle cells, including ASM, exhibit phenotypic modulation in response to environmental cues, switching between a differentiated, quiescent contractile phenotype and a “synthetic” phenotype which is motile and proliferative [19,37]. Although important during development and physiological adaptations to stress and injury, unregulated smooth muscle phenotypic changes may also promote disease pathologies including airway remodeling associated with chronic asthma [38,58]. Recently, in vitro studies in ASM cells have shown these cells respond to stimulation with cytokines, such as IL-1β and TNF-α, by expressing inflammatory marker proteins including intercellular adhesion molecule 1 (ICAM-1) and vascular cell adhesion molecule 1 (VCAM-1) [2,30]. Furthermore, ASM has been shown to secrete various pro-inflammatory cytokines including granulocyte-macrophage colony-stimulating factor (GM-CSF) [45], IL-6, IL-11 [14], IL-5, interferon gamma (IFN-γ) [18], as well as chemokines, including RANTES (regulated on activation, normal T cell expressed) and eotaxin [7,25]. These studies provide evidence that ASM may acquire a pro-inflammatory phenotype leading to both recruitment and adhesion of inflammatory cells to sites of allergen-induced mucosal injury [42,53]. However, there is little direct in vivo evidence for this important concept, as in vivo model systems to detect and manipulate the interplay between activated inflammatory cells and asthmatic ASM are lacking.
Changes in calcium homeostasis in ASM cells exposed to inflammatory factors promote allergen-induced airway responses [55]. For example, we recently demonstrated stromal interaction molecule 1 (STIM1) and Ca2+ release activated Ca2+ channel protein 1(Orai1), two proteins involved in store- operated Ca2+ entry (SOCE) [16] are up-regulated in a murine model of allergen induced asthma. Silencing expression of these proteins inhibited PDGF- induced Ca2+ entry into ASM cells and Ca2+ release- activated Ca2+ (CRAC) current [51]. One ubiquitously expressed family of effector proteins regulated by intracellular Ca2+ signals is multifunctional Ca2+/calmodulin-dependent protein kinase II (CaMKII). CaMKII is a multi-gene family with CaMKIIδ and CaMKIIγ (Camk2d and Camk2g genes, respectively) being the predominant isoforms expressed in vascular smooth muscle [20]. Recent studies in human asthmatics and a murine model of asthma linked increases in oxidation and activation of CaMKII to NF-κB-mediated transcription of inflammatory factors in airway epithelial cells. Using general pharmacological inhibitors of CaMKII administered intranasally, allergen induced AHR and airway remodeling was prevented confirming the role of this kinase in atopic asthma. [44] However, the expression and function of specific CaMKII isozymes in ASM physiology and pathophysiology is poorly characterized.
In this study, we test the hypothesis that CaMKIIδ regulates ASM function in allergen-induced AHR. A murine model of ovalbumin (OVA)-induced AHR was used which recapitulates aspects of the human inflammatory response in asthmatics, including goblet cell hyperplasia, epithelial cell hypertrophy, elevated IgE, AHR and overall airway inflammation [35]. We show for the first time up-regulation of CaMKIIδ protein expression in ASM in response to OVA sensitization and challenge in allergen-induced AHR in vivo. Mice with smooth muscle deletion of CaMKIIδ were produced by breeding animals with floxed Camk2d alleles [5] with those harboring a Transgelin (SM22α) promoter driving a Cre recombinase transgene [26]. In conditional CaMKIIδ knockout animals, allergen induced AHR was delayed. This was a CaMKII isoform specific phenomenon as OVA-induced AHR in conditional knockout mice of CaMKIIγ was similar to controls. OVA-induced inflammatory responses were attenuated in CaMKIIδ knockouts as assessed by extent of peribronchial infiltrate and BALF concentration of the inflammatory cytokine IL-13. These studies for the first time implicate a function for ASM CaMKIIδ in promoting allergen-induced AHR in vivo, and suggest a role for this kinase in promoting a pro-inflammatory ASM phenotype.
Materials and Methods
Mouse Models
Tagln;Cre reporter animals were created by crossing transgenics expressing Cre recombinase under control of a Transgelin (SM22α) promoter [26] (B6.Cg-Tg(Tagln-cre)1Her/J; Jackson labs) with a reporter line (“mTmG”) expressing either membrane localized tdTomato or GFP (B6.129(Cg)-Gt(ROSA) 26Sortm4(ACTB-tdTomato-EGFP)luo/J; Jackson Labs). CaMKIIδ and CaMKIIγ smooth muscle knockout mice were produced by breeding mixed background B6;129;FVB mice harboring floxed Camk2 alleles with the Tagln;Cre transgenics. Camk2γf/f [6]and Camk2δf/f founders [5] were provided by Drs. Johannes Backs (Univ. of Heidelberg) and Eric Olsen, (UT Southwestern). All animal procedures were carried out with protocols approved by the Albany Medical Center Institutional Animal Care and Use Committee.
Sensitization/challenge Protocol
3–4 week old males were sensitized by intraperitoneal injection of ovalbumin (OVA; 25 μg) plus aluminum hydroxide adjuvant (2 mg) in 0.5 ml sterile physiological salt solution (PSS) weekly for 3 weeks. The IP injections were followed by challenge with OVA (100 μg in 25 μl PSS intranasal) daily for 10 days [51]. Controls received PSS. Mice were studied using the forced oscillation techniques and/or euthanized by overdose of sodium pentobarbital (100 μg/g IP) for tissue harvesting 24 hr post-final challenge.
Airway mechanics
Mice were analyzed 1 day or 7 days post-final challenge. Animals were anesthetized by i.p. injection with xylaxine (0.01 mg/g) and ketamine (0.1 mg/g). Animals were tracheotomized and connected to a computer controlled small animal mechanical ventilator, part of the FlexiVent operating system (SCIREQ, Montreal, PQ, Canada). The animals were then paralyzed with an i.p. injection of pancuronium bromide (0.8 mg/kg). Measurements were taken after administration of increasing aerosolized doses of methacholine (Mch): 0mg/ml, 3.8mg/ml, 7.5mg/ml, 15mg/ml, 30mg/ml, 60mg/ml and 100mg/ml. Pre-programmed single frequency snapshot perturbation was executed by the FlexiWare7 software (SCIREQ). The software inputs resulting data into a mathematical single compartment model and outputs systemic airway resistance (Rrs) values at each concentration of Mch. For all perturbations a coefficient of determination (COD) of 0.90 was determined to be the limit of the lower range for measurement acceptance.
BALF IL-13 analysis
Bronchoalveolar lavage fluid (0.75 ml PSS) was collected from euthanized mice and centrifuged (5,000 rpm, 1 min). Interleukin-13 (IL-13) was quantified by enzyme-linked immunoassay (ELISA; R&D Systems, Minneapolis, MN).
Immunoblotting
Tracheobronchial tissue was harvested, epithelium mechanically removed, and lysates prepared as described previously [20]. Western blotting was by standard methods. Antibodies used for the study included rabbit anti-peptide antibodies specific for the C-terminus in CaMKIIδ2 PSCIPNGKENFSGSTSLWQNI and CaMKIIγ (aa567LNVHYHCSGAPAAPL581), as previously described[20,48]. GAPDH (Sigma) was used as a loading control. Secondary HRP-conjugated anti-rabbit IgG (GE Healthcare) chemiluminescent signals (SuperSignal, West Pico) were imaged and quantified with a Fuji LAS4000 Imaging Station.
Immunohistochemistry
Tissue analysis of Tagln-Cre Reporter Mice
Lungs were harvested, embedded in OCT, and sectioned for imaging of lower airway fluorescence. Imaging of trachealis (cleaned of epithelium) was on a glass bottom culture dish (MatTek) in HBSS (Cellgro). Confocal fluorescence microscopy was carried out on a Zeiss LSM 510META.
Smooth muscle area analysis
Smooth muscle area was quantified by IHC staining using a primary antibody specific for smooth muscle alpha actin (α-SMA; Abcam) using Vectastain Elite ABC Kit (Vector Laboratories) for detection. Smooth muscle area was quantified from 40× images using the ImageJ Color Deconvolution plugin to separate out Nova Red initiated stain. Images were thresholded to remove background and the area of signal was determined. Length of basement membrane was determined using ImageJ freehand line tool to normalize smooth muscle area measurement to airway size.
Airway Inflammation Analysis
Lungs were harvested from euthanized animals and fixed overnight in 10% buffered formalin (Fisher Scientific, Fair Lawn, New Jersey), dehydrated and embedded in paraffin blocks. 5 μm sections were cut and recovered on Superfrost Plus slides (Fisher Scientific, Fair Lawn, New Jersey). Sections were stained with hematoxylin and eosin (H&E) (Thermo Scientific). Lower airways (~100–200 μm in diameter) were examined at 20× magnification (100× magnification for infiltrate assessment) on an Olympus BX51 microscope. Quantification of the extent of peribronchial infiltrate was determined by a measurement adapted from a previous publication [13]. ImageJ freehand line tool was used to trace the total length of the airway lumen. The length of the lumen covered by infiltrate was then assessed and a percentage of airway covered by infiltrate was determined. Peribronchial infiltrate depth was quantified using the ImageJ straight line tool. Each airway was analyzed to locate the area where the infiltrate was the deepest, and the length of this infiltrate was then quantified.
Infiltrate Characterization
Paraffin lung sections obtained from processed lungs as described above were used to determine the number of peribronchial: eosinophils using rat anti-mouse eosinophil antibody (anti-MPB- a generous gift from Dr. Jamie Lee), neutrophils and macrophages using mouse anti-Ly-6C Ly-6G antibody (BD Pharmingen), this antibody recognizes both leukocytes. Distinction between the two leukocytes was determined by assessing nuclear morphology, Ly6C+/6G+ cells with a polymorphonucleaus were counted as neutrophils while Ly6C+/6G+ cells with a uniform nuclear morphology were counted as a macrophage. Lymphocytes were detected using the T- lymphocyte antibody (anti-CD3, abcam). Data represents leukocyte per unit area. Sections were cleared and rehydrated as described above. Heat- mediated antigen retrieval was carried out when staining for lymphocytes, macrophages and neutrophils using the Citric Acid-EDTA Buffer. When staining for eosinophils antigen retrieval was carried out using pepsin (0.5% in 5mM HCl) at room temperature. Slides were washed in 1× PBS then endogenous peroxidase activity was quenched by incubating samples in 3% H202 for 20 minutes at room temperature. Lungs were then stained with Vectastain ABC Kit (vector Laboratories) with Nova Red chromagen. Sections were further counterstained with hematoxylin (Thermo Scientific).
Immunofluorescence staining
10% buffered formalin fixed lungs were cyroprotected by incubation with serial 0.1M glycine washes at (4°C) for 1 hr and stored in 0.6 M sucrose (4°C) overnight, embedded in optimal cutting temperature (OCT) embedding compound (Tissue-Tek) and snap frozen in liquid nitrogen. 10 μm frozen sections were rinsed in 1× PBS and permeabilized with pre-cooled acetone at 4°C. Tissue was blocked with buffer containing: 1× PBS, 5% goat serum (MP Biomedicals), 0.5% fish gelatin and 0.1% TritonX-100 (Sigma). Staining of lung sections was performed by direct fluorescence using the following antibodies: α-smooth muscle actin (α-SMA) antibody conjugated to Cy3 (Sigma), and indirect fluorescence using an antibody specific for the c-terminus of CaMKIIδ [48], SM-MHC (Biomedical Technologies), α-SMA antibody (abcam). Appropriate secondary antibodies were used including anti-rabbit secondary antibodies (488 nm, 647nm; Molecular Probes) in order to detect signal. Sections were mounted with aqueous mounting medium Fluoromount-G (SouthernBiotech) or Vectshield with dapi (Vector). Confocal microscopy was carried out on processed sections with a Zeiss laser scanning microscope (LSM) 510META at 63× magnification.
ASMC dispersion and culture
Mouse ASMCs were obtained from the tracheobronchial tissue of airways derived from 9 week old male δf/f; smCre- or δf/f; smCre+ animals. Following the removal of epithelial cells ASM cells were enzymatically dispersed and cultured in DMEM- Ham’s F-12 with 10% fetal bovine serum. The mouse ASMCs were maintained C in a 5% CO2-95% humidity atmospheric environment. Cells were split twice weekly and media was changed every 3 days. Experiments were carried out on passages 3–7.
Inflammatory Cocktail Treatment
δf/f; smCre- and δf/f; smCre+ ASMCs were treated with either 0.4% FBS or with an inflammatory cocktail consisting of IL-1β (20ng/ml), TNF-α (20ng/ml) and IFNγ (200u/ml) in 0.4% FBS for 6 hours. After 6 hours total RNA was extracted as described below.
Quantitative PCR
Total RNA was extracted from mouse airway smooth muscle by homogenization in Trizol (Invitrogen, Carlsbad, CA) and first-strand cDNA synthesized using QuantiTect Reverse Transcription (Qiagen, Valencia, CA). Quantitative RT-PCR was performed on Mx3000P QPCR System (Agilient, Santa Clara, CA) using iQ SYBR Green supermix (Bio-Rad, Hercules, CA). The primers for mouse targets genes and internal controls were as follows: camk2d forward 5′-GAC GAG TAT CAG CTC TTT GAG G-3′, and reverse, 5′-GTT TCT GAT GGT CCC TAG CAG-3′; camk2g forward 5′-CGA CTA CCA GCT TTT CGA GG-3′, and reverse, 5′-GCC TCT CGT TCT AGT TTC TGA TG-3′; GAPDH forward 5′-TCC ATG ACA ACT TTG GCA TTG -3′, and reverse, 5′-TCA CGC CAC AGC TTT CCA-3′, eotaxin forward 5′-GCTCCATCCCAACTTCCTG -3′, and reverse 5′-AGATCTCTTTGCCCAACCTG 3′; MMP9 forward 5′-GATCCCCAGAGCGTCATTC-3′, and reverse 5′-CCACCTTGTTCACCTCATTTTG-3′; IL-33 forward 5′-TTCCAACTCCAAGATTTCCCC-3′ and reverse 5′-CAGAACGGAGTCTCATGCAG-3′.
Statistical Analysis
All data are expressed as means ± SEM. Statistical tests were performed using GraphPad Prism 4 software and are reported in Figure legends.
RESULTS
CaMKIIδ is up-regulated in allergen induced asthma
Mice were sensitized by intraperitoneal (i.p.) injection of ovalbumin (OVA) and adjuvant aluminum hydroxide, once a week for three consecutive weeks followed by daily challenge with intranasal OVA for 10 days (Figure 1A; adapted from Schramm et al 2004[47] ). One day post-final challenge, hematoxylin and eosin (H&E) staining confirmed intense peribronchial cellular infiltrate and mucosal hypertrophy indicative of a strong inflammatory response in OVA-treated animals compared to PSS treated controls (Figure 1B). Airway dynamics were assessed using the forced oscillation technique (FlexiVent, SCIREQ). Using systemic airway resistance (Rrs) as the endpoint, OVA-treated animals were found hyperresponsive to increased concentrations of the contractile agonist methacholine compared to control animals (Figure 1C), a characteristic response of allergic airways to contractile agonists. T helper 2 cells (Th2) are known to drive the inflammatory response in asthmatic airways and one major pro-inflammatory cytokine secreted by these cells is interleukin 13 (IL-13) [12,57]. IL-13 levels in bronchoalveolar lavage fluid (BALF) measured by ELISA were found to be markedly increased in OVA-challenged animals compared to controls (Figure 1D).
Figure 1. Characterization of acute murine model of atopic asthma.

(A.) Mice were sensitized with 3 weekly intraperitoneal (IP) injections of 25 μg ovalbumin (OVA) and 2 mg of AlOH adjuvant (Adj). Control animals received IP injections of PSS. One week after the third injection animals were challenged by intranasal administration of either 100 μg of OVA or PSS (Sal) once a day for 10 days. (B.) H&E staining of lower airways from a saline (Sal) and OVA sensitized/challenged animal 24 hr post-final challenge. Arrow points to area of dense peribronchial inflammatory infiltrate. Images taken with 20× objective. Images are representative of n=3 animals per condition and 10–25 airways per animal. (C.) Systemic airway resistance (Rrs) in response to increasing concentrations of methacholine was quantified 24 hr post-final challenge using the forced oscillation technique (FlexiVent, SCIREQ). Values means ± SEM; ***P<0.001, **P<0.01, by repeated measures two-way ANOVA with Bonferroni post-test. (D.) Th2 cytokine IL-13 levels were quantified by ELISA in BALF 24 hrs post- final challenge. Values are means ± SEM,*P< 0.05, by Student t- test.
Expression of Ca2+-signaling related proteins including SOCE channel proteins STIM1 and Orai1 [51] and sarco-endoplasmic reticulum Ca2+- ATPase (SERCA) [31] are known to be modulated in asthmatic ASM. As an intracellular mediator of Ca2+ signaling, CaMKII expression in ASM was determined by Western blotting tracheobronchial smooth muscle extracts with antibodies that specifically distinguish the C-terminus of CaMKIIδ and γ isoforms [20,48]. CaMKIIδ protein was up-regulated 1.5-fold in OVA-treated animals as compared to controls (Figure 2A and 2B) with a corresponding small but significant decrease in the expression of CaMKIIγ in asthmatic airways (Figure 2C and D). Indirect immunofluorescence confirmed CaMKIIδ expression in lower airways from both control and asthmatic animals and co-localization with ASM as detected by staining with α-smooth muscle actin (Figure 2E).
Figure 2. CaMKIIδ protein is up-regulated in acute murine model of atopic asthma.

Tracheobronchial lysates from OVA challenged and control (Sal) mice were immunoblotted with isoform- specific antibodies recognizing the c-terminus of CaMKIIδ (A.) or CaMKIIγ (C.). Lower panels quantify ECL signals from 5 mice/group (B. & D.). Values shown as means + SEM; *P<0.05 by Student t-test. (E.) Indirect immunofluorescence localization of CaMKIIδ protein (green) and smooth muscle α-actin (sm-α actin; red) in the lower airways from control (Sal) and OVA-challenged animals. Images are taken at 63× magnification.
Genetic deletion of CaMKIIδ Expression in ASM
To determine the function of ASM CaMKIIδ in atopic asthma, conditional knockout mice were developed by breeding mixed background mice harboring Camk2d floxed alleles [5] with transgenics containing Cre recombinase under control of a 2.8kb Transgelin (SM22α) promoter (B6.Cg-Tg(Tagln-cre)1Her/J) [26]. Previous work using alternative versions of the SM22α promoter were inconclusive regarding its activity in bronchial and tracheal smooth muscle [27,34]. In order to localize expression of Tagln-Cre in this system, the transgenics were crossed to a double fluorescent reporter line expressing membrane targeted tdTomato (an RFP mutant) or membrane targeted EGFP (“mTmG” mice), depending on Cre recombinase activity. In this system, mT is constitutively expressed while Cre activity deletes the floxed mT allele and puts in frame the EGFP allele. In the airways of Tagln-Cre(−);mTmG animals EGFP was not detected in the lower airway smooth muscle (Figure 3A). In the double Tagln-Cre;mTmG transgenic, EGFP fluorescence was detected submucosally in the walls of lower airways and pulmonary blood vessels (Figure 3B), consistent with localization in lower ASM and vascular smooth muscle. Compared to expression levels of mG in Cre(−) animals (Figure 3C), mG expression was found to be stronger in tracheobronchial tissue from Tagln-Cre(+) animals (Figure 3D). These results confirm activity of Tagln-Cre throughout the airway tree of the transgenics.
Figure 3. Expression of SM22α-Cre recombinase and conditional CaMKIIδ Knockout.
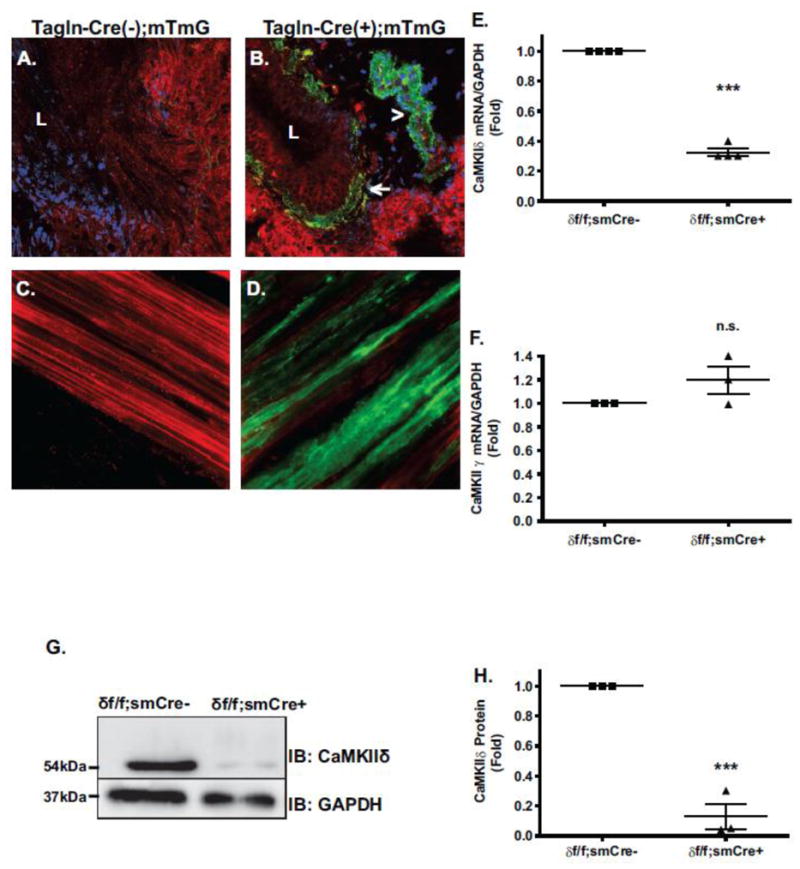
Lung sections were imaged by confocal fluorescence microscopy with 40× objective. SM22α-Cre expression is marked by membrane localized EGFP (mG) expression (green fluorescence channel); all other cells express membrane localized rtTomato (mT), an RFP mutant (red channel). Images show merged channels. (A.) Lower airway section from control mTmG;SM22α-Cre (−/−) animal. “L” signifies airway lumen. (B.) Lower airway section from mTmG;SM22α-Cre (+/−) animal. SM22α-Cre expression is localized in the walls of both lower airways (arrow) and blood vessels (arrowhead), consistent with ASM and VSM expression, respectively. (C.) Live whole mount of trachealis from control mT/mG; SM22α-Cre(−/−) mouse. (D.) Live whole mount of trachealis from experimental mT/mG; SM22α Cre (+/−) mouse showing SM22α –Cre expression(green) in ASM layer. (E.) qPCR analysis of CaMKIIδ mRNA and (F.) CaMKIIγ mRNA in tracheobronchial tissue derived from SM22α –Cre(+/−);Camk2d (f/f) smooth muscle conditional knockouts (δ f/f; smCre+) and littermate controls (δ f/f; smCre-). Values are means + SEM n= 4; ***P>0.001, (n.s.) not significant, by Student t- test. (G.) Representative Western blot of CaMKIIδ in passaged cells derived from tracheobronchial smooth muscle tissue of δf/f; smCre- and δf/f; smCre+ animals with an antibody specific for the CaMKIIδ2 c-terminus. (H.) Quantification of ECL signals. Values are means + SEM n= 3, ***P= 0.0005 using Student t- test.
Given evidence of Tagln-Cre activity in ASM, Camk2d (f/f) mice were bred with Tagln-Cre (+/−) to produce conditional smooth muscle CaMKIIδ knockouts and littermate controls. The extent of CaMKIIδ knockout was evaluated by qPCR of CaMKIIδ mRNA in tracheobronchial layer extracts containing predominantly ASM. CaMKIIδ expression decreased by 68% in the Cre+ animals (Figure 3E), with no compensatory increases in CaMKIIγ expression (Figure 3F). In lysates from enzyme dispersed cells from tracheobronchial tissue, CaMKIIδ knockout was nearly complete at the protein level (Figure 3G and 3H). Based on these results, we concluded that Tagln-Cre activity efficiently deleted CaMKIIδ in ASM cells with a residual low expression most likely originating from contaminating cell types in the complex tracheobronchial tissue.
Allergen- induced AHR is ameliorated in smooth muscle conditional CaMKIIδ KO mice but not CaMKIIγ KO mice
In order to evaluate function of smooth muscle CaMKIIδ in allergen-induced AHR, systemic airway resistance (Rrs) was assessed using the Flexivent system in OVA-challenged CaMKIIδ knockout mice (δf/f; smCre+) and littermate controls (δf/f; smCre-). Conditional deletion of smooth muscle CaMKIIδ had no effect on baseline responsiveness to methacholine in saline-treated controls (Figure 4A, solid lines), suggesting no intrinsic changes in contractility in the conditional knockouts. Remarkably, the characteristic OVA-induced hyperresponsiveness to methacholine observed in control mice was completely absent in δf/f; smCre+ mice lacking smooth muscle CaMKIIδ (Figure 4A). Parallel studies were conducted in mice with smooth muscle conditional knockout of CaMKIIγ isoform expression. OVA-induced AHR (Rrs) were unaffected in the CaMKIIγ knockouts, indicating an isoform-specific function for smooth muscle CaMKIIδ in promoting OVA-induced AHR (Figure 4B).
Figure 4. Effects of smooth muscle CaMKIIδ deletion on AHR induction in OVA challenged mice is CaMKII isoform specific.

(A.) Systemic airway resistance (Rrs) was quantified using the forced oscillation technique (FlexiVent, SCIREQ) 24 hr post-final challenge in control δf/f; smCre- animals (black lines), smooth muscle CaMKIIδ knockouts (δf/f; smCre+; red lines). OVA challenged animals are signified by dashed lines and Saline controls by solid lines. Values are means ± SEM, n=4–7. +++p<0.001 between δf/f; smCre+;OVA and littermate δf/f; smCre-;OVA controls. *** p<0.001 between δf/f; smCre-;OVA and δf/f; smCre-;OVA; Sal by repeated measures two-way ANOVA with Bonferroni post-test. (B.) Systemic airway resistance (Rrs) was assessed 24 hours post final challenge in control γf/f; smCre- animals (black lines) and smooth muscle CaMKIIγ knockouts (γf/f; smCre+, red lines). OVA challenged animals are signified by dashed lines and Saline controls by solid lines. Values are means ± SEM; n= number of animals.+p<0.05, +++p<0.001 between γf/f; smCre+;OVA and littermate γf/f; smCre-;OVA controls. ***p<0.001 between γf/f; smCre-;OVA and γf/f; smCre-;OVA; Sal. Statistics are repeated measures two-way ANOVA with Bonferroni post-test.
Allergen- induced AHR is independent of ASM remodeling or alterations in contractile proteins
We showed previously that increased expression of CaMKIIδ promotes vascular smooth muscle proliferation and migration in vitro [20,33] and vascular remodeling in response to balloon angioplasty-induced injury in vivo [21]. In this model of OVA-induced AHR, no increases in ASM area were observed in control OVA-treated δf/f; smCre- mice compared to saline controls (Figure 5A) and smooth muscle deletion of CaMKIIδ had no effect on either baseline ASM content in saline treated animals, or on ASM content in OVA-treated animals (Figure 5B). Similarly, no OVA-induced changes in protein expression of the smooth muscle specific contractile marker, smooth muscle myosin heavy chain (SM-MHC) were observed (Figure 5C) suggesting a lack of effect of CaMKIIδ deletion on ASM contractile phenotype. From these results we conclude that AHR in this specific OVA-challenge model is independent of ASM remodeling and smooth muscle CaMKIIδ deletion has no effect on baseline ASM structure or contractile markers.
Figure 5. Smooth muscle CaMKIIδ deletion does not affect ASM remodeling or contractile phenotype.

(A.) Representative lung sections from Saline (A1) and OVA (A2) challenged control animals (δf/f; smCre-) were analyzed for α-SM actin staining using immunohistochemistry. Black arrows indicate ASM, arrowheads indicate VSM in blood vessels. Images taken at 40× magnification. (B.) Smooth muscle area was quantified in controls (δf/f; smCre-) and smooth muscle CaMKIIδ knockouts (δf/f; smCre+) using ImageJ de-convolution plugin and normalized by airway circumference of basement membrane (BM). Values are means ± SEM; n=number of animals; 18 airways from each animal were averaged. There were no statistical differences between groups using two-way ANOVA with a Bonferroni post-hoc test. (C.) Lung sections were imaged by confocal microscopy with 40× objective. Lung sections harvested 24 hours after final challenge were stained with SM-MHC (Green) using indirect immunofluorescence. Qualitative assessment of SM-MHC protein expression in the lower airway of representative lung sections shows minimal changes in the extent of expression in saline challenged δf/f; smCre- animals, saline challenged δf/f; smCre+, OVA challenged δf/f; smCre- and OVA challenged δf/f; smCre+ animals.
Allergen-induced inflammatory response is attenuated in smooth muscle CaMKIIδ knockout mice
To identify the effect of smooth muscle CaMKIIδ deletion on the process of acute inflammatory cell infiltration [24], lung sections were examined by H&E staining (Figure 6A). Extent of peribronchial inflammation (circumference covered) in response to OVA challenge was significantly decreased by 49% in smooth muscle CaMKIIδ knockouts (δf/f; smCre+) compared to littermate controls (δf/f; smCre-) (Figure 6B). As a separate index, the depth of peribronchial infiltrate was also reduced to a similar extent in CaMKIIδ knockout compared to control animals (Figure 6C). Consistent with decreased extent of inflammatory cell infiltrate, IL-13 concentrations in BALF was reduced by about 70% in allergen-challenged CaMKIIδ knockouts compared to littermate controls (Figure 6D).
Figure 6. Smooth muscle CaMKIIδ deletion inhibits OVA-induced airway inflammation.
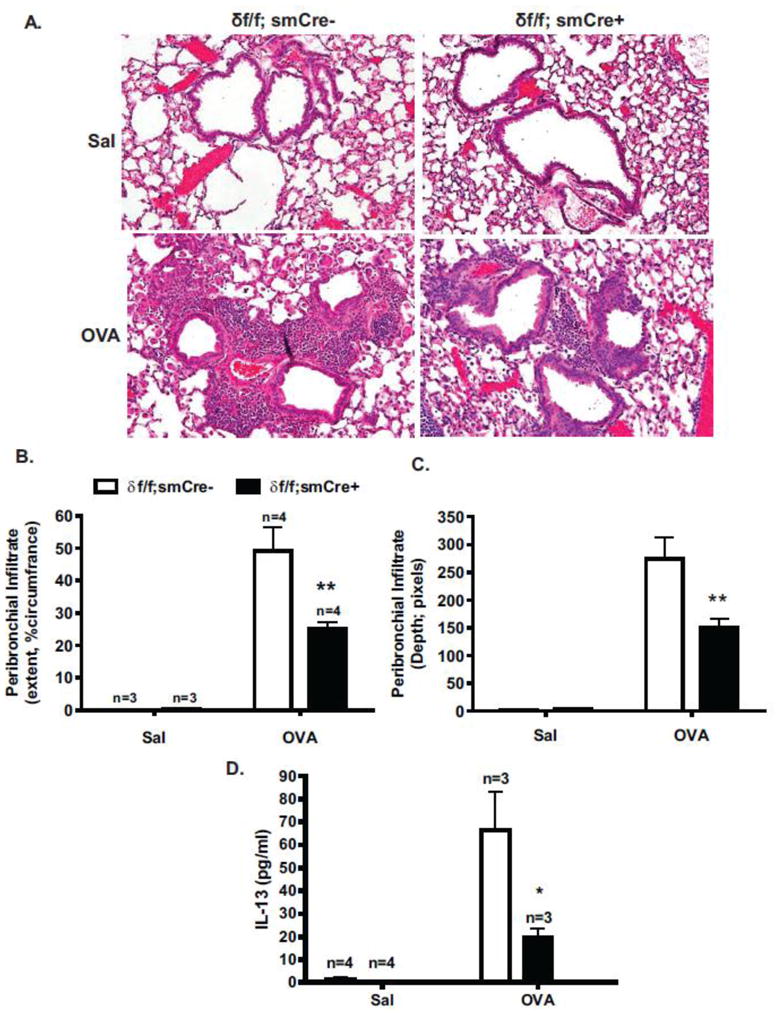
Lung sections from control (δf/f; smCre-) and smooth muscle CaMKIIδ knockout (δf/f; smCre+) saline (Sal) and OVA treated mice were stained with H&E. Images taken with a 20× objective. Peribronchial inflammation was scored as a function of airway circumference covered (B.) and depth from the airway wall (C.). 10–25 airways from 3–4 animals were analyzed. Values are means ± SEM, **P< 0.01. Statistical analysis was determined using two-way ANOVA with a Bonferroni post-hoc test. (D.) BALF was collected 24 hr post-final challenge and Th2 cytokine IL-13 levels measured by ELISA in smooth muscle CaMKIIδ knockout (δf/f; smCre+) mice (closed bars) and littermate (δf/f; smCre-) controls (open bars). Groups were challenged with control Saline (Sal) or ovalbumin (OVA) as indicated. Values are means ± SEM, n= number of animals, *P< 0.05 using two-way ANOVA with a Bonferroni post-hoc test.
Leukocyte specific antibodies were used to stain eosinophils, lymphocytes, neutrophils and macrophages in serial lung sections derived from lungs harvested 1 day after final challenge (Figure 7A–D). Areas of dense peribronchial infiltrate were examined using a high powered objective (100x) and the relative number of specific leukocytes per unit area was determined after examining the entire image (Figure 7E). We observed a decrease in the content of lymphocytes and macrophages in CaMKIIδ knockout animals as compared to OVA-challenged wild type animals, while the occurrence of intact eosinophils was significantly higher in the peribronchial space of airways from OVA-challenged CaMKIIδ knockouts compared to OVA-challenged controls. Eosinophils have been strongly linked to initiating airway remodeling in allergic asthma [56]. The presence of this granulocyte is transient however, upon secretion of their granular contents these cells are cleared from the airway during the resolution phase of injury [15,32]. Our results showing higher eosinophil count in OVA-challenged CaMKIIδ knockouts suggest that the inflammatory response in OVA-challenged δf/f; smCre+ animals is delayed rather than prevented. Indeed, at the later time point of 7 days after final OVA challenge (instead of 1 day post final OVA challenge as depicted in Figures 1 and 4), allergen-induced AHR was resolved in OVA-treated control δf/f; smCre- animals, while OVA-treated δf/f; smCre+ knockouts were hyperrespponsive to methacholine treatment. (Figure 7F).
Figure 7. Characterization of inflammatory response in OVA challenged wild type and smooth muscle CaMKIIδ knockout animals.
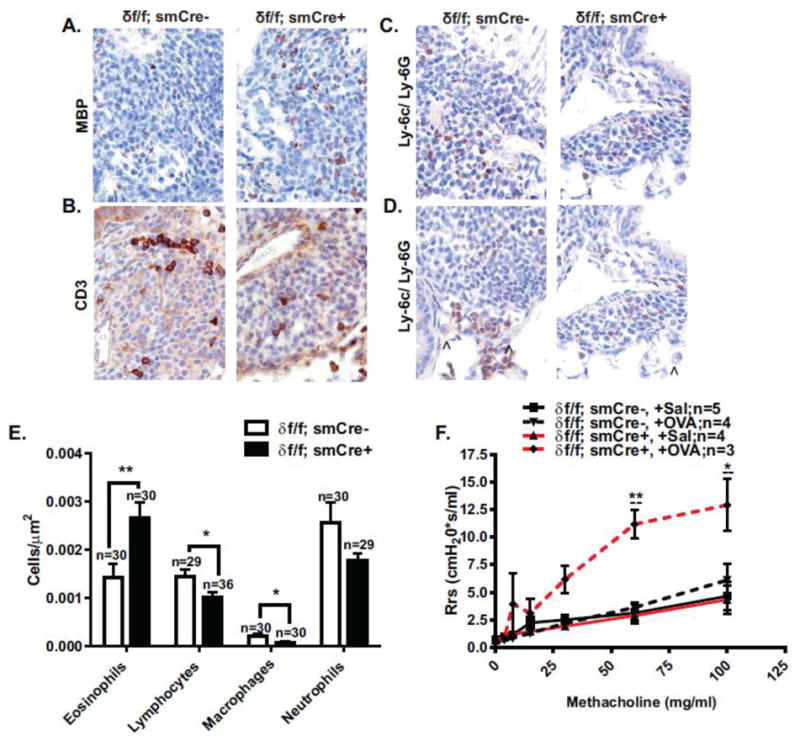
Immunohistochemical assessment of peribronchial inflammatory cell composition was carried out on lung sections derived from OVA challenged wild type (δf/f; smCre-) and smooth muscle CaMKIIδ knockout (δf/f; smCre+) animals 24 hours after final challenge. Immunoperoxidase staining (brown) was carried out on representative sections in order to detect number of (A.) eosinophils (MBP), (B.) lymphocytes (CD3), (C.) neutrophils (Ly-6C/Ly-6G) and (D.) macrophages (Ly-6C/Ly-6G). Sections were counterstained with nuclear stain hematoxylin. Arrowheads point to macrophages. (E.) Number of cells per μm2 in OVA challenged smooth muscle CaMKIIδ knockout (δf/f; smCre+) mice (closed bars) and littermate (δf/f; smCre-) controls (open bars) are depicted. Values are means ± SEM, n= number of animals, *P< 0.05, **P<0.01 using a Student-t test. (F.) Systemic airway resistance (Rrs) was quantified using the forced oscillation technique (FlexiVent, SCIREQ) 7 days post-final challenge in control δf/f; smCre- animals (black lines), smooth muscle CaMKIIδ knockouts (δf/f; smCre+; red lines). OVA challenged animals are signified by dashed lines and Saline controls by solid lines. Values are means ± SEM, n=3–5. * p<0.05, **p<0.01 between δf/f; smCre+;OVA and littermate δf/f; smCre-;OVA controls, --p<0.01,δ f/f; smCre+;OVA and δf/f; smCre+; Sal between by repeated measures two-way ANOVA with Bonferroni post-test.
CaMKIIδ regulates cytokine dependent up-regulation of IL-33 in cultured ASM cells
ASM themselves have been shown to initiate a pro- inflammatory transcriptional profile when stimulated by various inflammatory factors, secreting growth factors and adhesion molecules, further propagating the inflammatory cascade. Recently CaMKII has been linked to the activation of this profile in cultured human ASM cells [30]. To identify if ASM CaMKIIδ regulates the transcription of pro-inflammatory cytokines, we assessed the level of mRNA expression of the eosinophil chemokine eotaxin, the matrix metallopeptidase 9 (MMP9) and the recently identified IL-1 family member, interleukin -33 (IL-33). IL-33 has been shown to be up-regulated in asthmatic airways[41]. This cytokine promotes the differentiation of T helper (Th) and innate lymphoid type 2 cells (ILC2) into a Th2 phenotype, inducing the secretion of pro-inflammatory cytokine IL-13 [46]. ASM cells from wild type and CaMKIIδ knockout were treated with an inflammatory cytokine cocktail consisting of IL-1β, TNF-α and IFN-γ for 6 hours and the extent of eotaxin, MMP9 and IL-33 mRNA induction was quantified by qPCR (Figure 8). IL-33 mRNA levels were attenuated by 60% in ASM cells derived from CaMKIIδ knockout animals as compared to ASM cells from control animals. There were no significant differences in induction of eotaxin or MMP9 expression between the two cell lines.
Figure 8. In vitro Regulation of IL-33 mRNA expression by ASM CaMKIIδ.
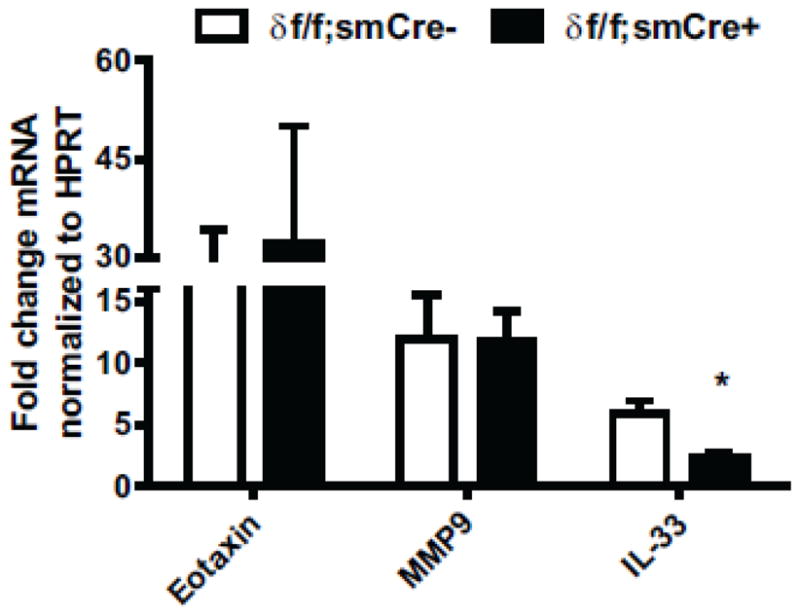
qPCR analysis of Eotaxin, Mmp9 and IL-33 mRNA expression in cultured cells derived from the tracheobronchial tissue of smooth muscle conditional knockout (δ f/f; smCre+) and littermate controls (δ f/f; smCre-) animals treated with TNF-α (20ng/ml), IL-1β (20ng/ml) and IFN-γ (200 u/ml). Expression levels were normalized to HPRT and represented values depict fold induction over corresponding non-treated cells as represented by means + SEM n= 3; *p<0.05, by Student t-test.
Discussion
Airway smooth muscle (ASM) cells are both a target and source of inflammatory mediators that promote allergen-induced AHR and remodeling [3,4]. Ca2+ and oxidative signaling through the multi-functional serine/threonine protein kinase, CaMKII, has been shown to promote inflammatory responses in various cell types, including cardiomyocytes [29]. The present study for the first time implicates a specific function for the CaMKIIδ isoform in promoting ASM pro-inflammatory function in vivo in a murine model of allergen-induced AHR. Recently, CaMKII oxidation and activity was shown to be increased in airway epithelium in bronchial biopsy specimens from severe asthmatic patients as well as in a murine model of atopic asthma [44]. Inhibition of CaMK activity using molecular/genetic approaches or by inhaled KN-93, a CaMKII inhibitor [52], strongly attenuated AHR and inflammation in the mouse model. While that study highlighted the role of CaMKII as a mediator and therapeutic target in atopic asthma, it did not examine CaMKII isoform-specific functions and was focused on CaMKII activity and function in airway epithelium. Together, these in vivo studies emphasize the role of CaMKII in the pathophysiology of atopic asthma and highlight the complex interplay of Ca2+ and ROS signaling pathways and airway/immune cell interactions in the promotion of the asthmatic phenotype.
Four differentially expressed genes (α, β, δ, γ) encode CaMKII [54] which is expressed in vivo as a large heteromultimeric holoenzyme composed of 12 protein kinase subunits [22]. We determined that CaMKIIδ and γ isoforms were primarily expressed in differentiated ASM, similar expression patterns in differentiated VSM [20]. Significantly, ovalbumin sensitization and challenge resulted in induced CaMKIIδ up-regulation relative to CaMKIIγ in tracheal smooth muscle, correlating with marked AHR and extensive airway inflammation. Smooth muscle-specific CaMKII knockout mouse models were employed in this study. The lineage tracing approach was first used to verify expression of Tagln;Cre in ASM surrounding both large conducting airways and lower airways. After verification of Cre expression in peripheral ASM we were able to use these animals to test the function of specific CaMKII isoforms in the induction of allergen induced AHR. Although application of conditional smooth muscle knockout mouse models have been widely applied in studies of vascular smooth muscle (VSM) biology and pathophysiology, only one other set of studies has been reported using a similar approach with a smooth muscle myosin heavy chain (Myh11);Cre line to study ASM biology and function in vivo. Employing this novel transgenic system, we determined that OVA-induced AHR and airway inflammation were absent in mice with smooth muscle conditional CaMKIIδ knockout. This effect was specific for the CaMKIIδ isoform as conditional deletion of CaMKIIγ had no significant effects on OVA-induced AHR, indicating non-equivalent functions of closely related CaMKIIδ and γ isoforms in this system.
Smooth muscle CaMKIIδ knockout had no effect on baseline methacholine reactivity in saline-treated animals, arguing for a lack of CaMKIIδ effect on intrinsic ASM contractility. However, OVA-induced AHR, as defined by increased Systemic Airway Resistance (Rrs), failed to develop in CaMKIIδ knockout animals. The absence of OVA induced alterations in ASM remodeling or changes in contractile protein expression in this model led us to focus our attention on the OVA-induced inflammatory response as a potential target for the “protective” effect on AHR observed in CaMKIIδ knockout mice. Total OVA-induced inflammatory burden was significantly reduced in the smooth muscle CaMKIIδ knockout animals as assessed histomorphometrically by the extent of inflammatory infiltrate, and by BALF quantification of the classic Th2 cytokine IL-13. Although the mechanisms by which CaMKIIδ deletion in smooth muscle decreases the net inflammatory response remain to be determined, it is possible that signaling via this kinase promotes transcription or release of pro-inflammatory cytokines and/or chemokines from ASM [30]. In vitro studies have emphasized cytokine-induced pro-inflammatory functions of ASM resulting in production of leukocyte adhesion molecules such as ICAM-1 or VCAM-1 and/or cytokines (e.g. IL-6, IL-8) and chemokines (e.g. RANTES, eotaxin-1) [1,9,23,36,43,49] and limited pharmacological studies support a function for CaMKII mediated signaling in promoting these effects [30]. In this study, in vitro treatment of ASM cells with an inflammatory cytokine cocktail promoted transcription of the pro-inflammatory cytokine IL-33. This up-regulation of IL-33 expression is dependent on smooth muscle CaMKIIδ as IL-33 expression was attenuated in CaMKIIδ knockout cells. This finding supports a role of ASM CaMKIIδ in promoting the inflammatory phenotype.
Inhibition of CaMKII activity has recently been shown to attenuate transcription of eotaxin in airway epithelia, with corresponding decreases in BALF content of eosinophils [44]. Our findings of increased peribronchial eosinophils in the smooth muscle CaMKIIδ knockouts may indicate compromised transmigration of these cells across the airway wall, a process driven by eotaxin and/or related factors produced by activated airway epithelia and ASM cells. Eosinophils have been found to further promote the Th2 mediated response perpetuating the inflammatory response [50], potentially explaining the decreased BALF Th2 cytokine, IL-13 concentrations, inflammatory infiltrate and ultimately AHR in allergen challenged CaMKIIδ knockout animals.
Our respiratory mechanics studies at the later time point (7 days) support the hypothesis that ASM CaMKIIδ plays a complex role in the airways during allergen exposure. Clearly, CaMKIIδ up-regulation in ASM has a pathological role in promoting or accelerating allergen induced AHR. However, OVA-challenged CaMKIIδ knockout animals are hyperresponsive after exposure to methacholine at 7 day after final OVA challenge whilst at this time point we observed resolution of AHR in OVA challenged wild type animals. It is unclear whether at these later time points ASM CaMKIIδ plays a protective and necessary physiological role by helping in the resolution of airway injury and further experiments are needed to resolve this issue. This idea is supported by the larger number of intact eosinophils in airway sections derived from OVA-challenged CaMKIIδ knockout as compared to OVA-challenged wild type animals. Future studies identifying the molecular mechanisms involved in a potential dual role of ASM CaMKIIδ in promoting both inflammation and resolution of airway inflammation could further advance our understanding of the dynamic and complex process of airway inflammation.
We have previously defined selective increases in CaMKIIδ protein expression in arterial vascular smooth muscle (VSM) in response to balloon angioplasty injury [20]. One limitation of current approaches for studying ASM function using either the Tagln;Cre or Myh11;Cre transgenics, is expression of Cre recombinase in lung vasculature [8,26]. Given the close physical relationship between lung airways and blood vessels, and the fact that the vascular wall is a barrier to circulating leukocytes, we cannot rule out a contribution of CaMKIIδ in VSM as a contributing factor to any observed effects on OVA-induced leukocyte trafficking and inflammation. In VSM, CaMKIIδ promotes proliferation, migration, and ultimately vascular wall remodeling in response to injury [20,21,28]. An interesting possibility that remains to be tested is that CaMKIIδ up-regulation in VSM promotes a pro-inflammatory function similar to that indicated by the present studies in ASM in the context of an atopic asthma model. Airway remodeling resulting from increased ASM growth and proliferation is a key feature of chronic asthma in humans and source of hyperreactivity [40,59]. Whether or not allergen induced up-regulation of ASM CaMKIIδ protein expression promotes airway remodeling will require additional studies using an alternative model that manifests with significant increases in ASM proliferation, hypertrophy, and wall thickness.
Acknowledgments
The authors greatly acknowledge the following research support: AMC’s Candice Weir Fund, NIH grant HL097111 (to MT), NIH grant HL049426 (to HAS), Albany Medical Center fellowship and stipend support (AMS). We would also like to acknowledge receipt of generous gifts including founder mice carry floxed CaMKIIδ and CaMKIIγ alleles which were kindly provided by Dr. Johannes Backs (Heidelberg) and Dr. Eric Olson (UTSW) and the eosinophil specific MPB antibody, provided by Dr. Jamie Lee (Mayo Clinic Scottsdale, Arizona). We also thank Xiaolan Ding, Miao Jiang and Diane Singer for their general animal and cell culture support. To Dr. Margarida Barroso for her assistance and insight into immunohistochemical and microscopy techniques. The Singer, Trebak and Jourd’heuil lab for the exchange of ideas and experiences on the 1st floor of the ME building at AMC.
Reference List
- 1.Ammit AJ, Moir LM, Oliver BG, Hughes JM, Alkhouri H, Ge Q, Burgess JK, Black JL, Roth M. Effect of IL-6 trans-signaling on the pro-remodeling phenotype of airway smooth muscle. Am J Physiol Lung Cell Mol Physiol. 2007;292:L199–L206. doi: 10.1152/ajplung.00230.2006. [DOI] [PubMed] [Google Scholar]
- 2.Amrani Y, Lazaar AL, Hoffman R, Amin K, Ousmer S, Panettieri RA., Jr Activation of p55 tumor necrosis factor-alpha receptor-1 coupled to tumor necrosis factor receptor-associated factor 2 stimulates intercellular adhesion molecule-1 expression by modulating a thapsigargin-sensitive pathway in human tracheal smooth muscle cells. Mol Pharmacol. 2000;58:237–245. doi: 10.1124/mol.58.1.237. [DOI] [PubMed] [Google Scholar]
- 3.Amrani Y, Panettieri RA. Airway smooth muscle: contraction and beyond. Int J Biochem Cell Biol. 2003;35:272–276. doi: 10.1016/s1357-2725(02)00259-5. [DOI] [PubMed] [Google Scholar]
- 4.An SS, Bai TR, Bates JH, Black JL, Brown RH, Brusasco V, Chitano P, Deng L, Dowell M, Eidelman DH, Fabry B, et al. Airway smooth muscle dynamics: a common pathway of airway obstruction in asthma. Eur Respir J. 2007;29:834–860. doi: 10.1183/09031936.00112606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Backs J, Backs T, Neef S, Kreusser MM, Lehmann LH, Patrick DM, Grueter CE, Qi X, Richardson JA, Hill JA, Katus HA, et al. The delta isoform of CaM kinase II is required for pathological cardiac hypertrophy and remodeling after pressure overload. Proc Natl Acad Sci U S A. 2009;106:2342–2347. doi: 10.1073/pnas.0813013106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Backs J, Stein P, Backs T, Duncan FE, Grueter CE, McAnally J, Qi X, Schultz RM, Olson EN. The gamma isoform of CaM kinase II controls mouse egg activation by regulating cell cycle resumption. Proc Natl Acad Sci U S A. 2010;107:81–86. doi: 10.1073/pnas.0912658106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Berkman N, Krishnan VL, Gilbey T, Newton R, O’Connor B, Barnes PJ, Chung KF. Expression of RANTES mRNA and protein in airways of patients with mild asthma. Am J Respir Crit Care Med. 1996;154:1804–1811. doi: 10.1164/ajrccm.154.6.8970374. [DOI] [PubMed] [Google Scholar]
- 8.Camoretti-Mercado B, Forsythe SM, LeBeau MM, Espinosa R, III, Vieira JE, Halayko AJ, Willadsen S, Kurtz B, Ober C, Evans GA, Thweatt R, et al. Expression and cytogenetic localization of the human SM22 gene (TAGLN) Genomics. 1998;49:452–457. doi: 10.1006/geno.1998.5267. [DOI] [PubMed] [Google Scholar]
- 9.Catley MC, Sukkar MB, Chung KF, Jaffee B, Liao SM, Coyle AJ, Haddad E, Barnes PJ, Newton R. Validation of the anti-inflammatory properties of small-molecule IkappaB Kinase (IKK)-2 inhibitors by comparison with adenoviral-mediated delivery of dominant-negative IKK1 and IKK2 in human airways smooth muscle. Mol Pharmacol. 2006;70:697–705. doi: 10.1124/mol.106.023150. [DOI] [PubMed] [Google Scholar]
- 10.Cazzola M, Page CP, Rogliani P, Matera MG. beta2-agonist therapy in lung disease. Am J Respir Crit Care Med. 2013;187:690–696. doi: 10.1164/rccm.201209-1739PP. [DOI] [PubMed] [Google Scholar]
- 11.Cox G, Thomson NC, Rubin AS, Niven RM, Corris PA, Siersted HC, Olivenstein R, Pavord ID, McCormack D, Chaudhuri R, Miller JD, et al. Asthma control during the year after bronchial thermoplasty. N Engl J Med. 2007;356:1327–1337. doi: 10.1056/NEJMoa064707. [DOI] [PubMed] [Google Scholar]
- 12.Del Prete GF, De CM, D’Elios MM, Maestrelli P, Ricci M, Fabbri L, Romagnani S. Allergen exposure induces the activation of allergen-specific Th2 cells in the airway mucosa of patients with allergic respiratory disorders. Eur J Immunol. 1993;23:1445–1449. doi: 10.1002/eji.1830230707. [DOI] [PubMed] [Google Scholar]
- 13.Doherty TA, Soroosh P, Broide DH, Croft M. CD4+ cells are required for chronic eosinophilic lung inflammation but not airway remodeling. Am J Physiol Lung Cell Mol Physiol. 2009;296:L229–L235. doi: 10.1152/ajplung.90543.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Elias JA, Wu Y, Zheng T, Panettieri R. Cytokine- and virus-stimulated airway smooth muscle cells produce IL-11 and other IL-6-type cytokines. Am J Physiol. 1997;273:L648–L655. doi: 10.1152/ajplung.1997.273.3.L648. [DOI] [PubMed] [Google Scholar]
- 15.Erjefalt JS, Persson CG. New aspects of degranulation and fates of airway mucosal eosinophils. Am J Respir Crit Care Med. 2000;161:2074–2085. doi: 10.1164/ajrccm.161.6.9906085. [DOI] [PubMed] [Google Scholar]
- 16.Frischauf I, Schindl R, Derler I, Bergsmann J, Fahrner M, Romanin C. The STIM/Orai coupling machinery. Channels (Austin ) 2008;2:261–268. doi: 10.4161/chan.2.4.6705. [DOI] [PubMed] [Google Scholar]
- 17.Gunst SJ, Panettieri RA., Jr Last Word on Point: Alterations in airway smooth muscle phenotype do cause airway hyperresponsiveness in asthma. J Appl Physiol. 2012;113:847. doi: 10.1152/japplphysiol.00714.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hakonarson H, Maskeri N, Carter C, Grunstein MM. Regulation of TH1- and TH2-type cytokine expression and action in atopic asthmatic sensitized airway smooth muscle. J Clin Invest. 1999;103:1077–1087. doi: 10.1172/JCI5809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Halayko AJ, Solway J. Molecular mechanisms of phenotypic plasticity in smooth muscle cells. J Appl Physiol. 2001;90:358–368. doi: 10.1152/jappl.2001.90.1.358. [DOI] [PubMed] [Google Scholar]
- 20.House SJ, Ginnan RG, Armstrong SE, Singer HA. Calcium/calmodulin-dependent protein kinase II-delta isoform regulation of vascular smooth muscle cell proliferation. Am J Physiol Cell Physiol. 2007;292:C2276–C2287. doi: 10.1152/ajpcell.00606.2006. [DOI] [PubMed] [Google Scholar]
- 21.House SJ, Singer HA. CaMKII-delta isoform regulation of neointima formation after vascular injury. Arterioscler Thromb Vasc Biol. 2008;28:441–447. doi: 10.1161/ATVBAHA.107.156810. [DOI] [PubMed] [Google Scholar]
- 22.Hudmon A, Schulman H. Structure-function of the multifunctional Ca2+/calmodulin-dependent protein kinase II. Biochem J. 2002;364:593–611. doi: 10.1042/BJ20020228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jarai G, Sukkar M, Garrett S, Duroudier N, Westwick J, Adcock I, Chung KF. Effects of interleukin-1beta, interleukin-13 and transforming growth factor-beta on gene expression in human airway smooth muscle using gene microarrays. Eur J Pharmacol. 2004;497:255–265. doi: 10.1016/j.ejphar.2004.06.055. [DOI] [PubMed] [Google Scholar]
- 24.Kips JC. The relation between morphologic and functional airway changes in bronchial asthma. Verh K Acad Geneeskd Belg. 2003;65:247–265. [PubMed] [Google Scholar]
- 25.Lamkhioued B, Renzi PM, bi-Younes S, Garcia-Zepada EA, Allakhverdi Z, Ghaffar O, Rothenberg MD, Luster AD, Hamid Q. Increased expression of eotaxin in bronchoalveolar lavage and airways of asthmatics contributes to the chemotaxis of eosinophils to the site of inflammation. J Immunol. 1997;159:4593–4601. [PubMed] [Google Scholar]
- 26.Lepore JJ, Cheng L, Min LM, Mericko PA, Morrisey EE, Parmacek MS. High-efficiency somatic mutagenesis in smooth muscle cells and cardiac myocytes in SM22alpha-Cre transgenic mice. Genesis. 2005;41:179–184. doi: 10.1002/gene.20112. [DOI] [PubMed] [Google Scholar]
- 27.Li L, Miano JM, Mercer B, Olson EN. Expression of the SM22alpha promoter in transgenic mice provides evidence for distinct transcriptional regulatory programs in vascular and visceral smooth muscle cells. J Cell Biol. 1996;132:849–859. doi: 10.1083/jcb.132.5.849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li W, Li H, Sanders PN, Mohler PJ, Backs J, Olson EN, Anderson ME, Grumbach IM. The multifunctional Ca2+/calmodulin-dependent kinase II delta (CaMKIIdelta) controls neointimaformation after carotid ligation and vascular smooth muscle cell proliferation through cell cycle regulation by p21. J Biol Chem. 2011;286:7990–7999. doi: 10.1074/jbc.M110.163006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ling H, Gray CB, Zambon AC, Grimm M, Gu Y, Dalton N, Purcell NH, Peterson K, Brown JH. Ca2+/Calmodulin-dependent protein kinase II delta mediates myocardial ischemia/reperfusion injury through nuclear factor-kappaB. Circ Res. 2013;112:935–944. doi: 10.1161/CIRCRESAHA.112.276915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Luo SF, Chang CC, Lee IT, Lee CW, Lin WN, Lin CC, Yang CM. Activation of ROS/NF-kappaB and Ca2+/CaM kinase II are necessary for VCAM-1 induction in IL-1beta-treated human tracheal smooth muscle cells. Toxicol Appl Pharmacol. 2009;237:8–21. doi: 10.1016/j.taap.2009.02.025. [DOI] [PubMed] [Google Scholar]
- 31.Mahn K, Hirst SJ, Ying S, Holt MR, Lavender P, Ojo OO, Siew L, Simcock DE, McVicker CG, Kanabar V, Snetkov VA, et al. Diminished sarco/endoplasmic reticulum Ca2+ ATPase (SERCA) expression contributes to airway remodelling in bronchial asthma. Proc Natl Acad Sci U S A. 2009;106:10775–10780. doi: 10.1073/pnas.0902295106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Malm-Erjefalt M, Greiff L, Ankerst J, Andersson M, Wallengren J, Cardell LO, Rak S, Persson CG, Erjefalt JS. Circulating eosinophils in asthma, allergic rhinitis, and atopic dermatitis lack morphological signs of degranulation. Clin Exp Allergy. 2005;35:1334–1340. doi: 10.1111/j.1365-2222.2005.02335.x. [DOI] [PubMed] [Google Scholar]
- 33.Mercure MZ, Ginnan R, Singer HA. CaM kinase II delta2-dependent regulation of vascular smooth muscle cell polarization and migration. Am J Physiol Cell Physiol. 2008;294:C1465–C1475. doi: 10.1152/ajpcell.90638.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Moessler H, Mericskay M, Li Z, Nagl S, Paulin D, Small JV. The SM 22 promoter directs tissue-specific expression in arterial but not in venous or visceral smooth muscle cells in transgenic mice. Development. 1996;122:2415–2425. doi: 10.1242/dev.122.8.2415. [DOI] [PubMed] [Google Scholar]
- 35.Nials AT, Uddin S. Mouse models of allergic asthma: acute and chronic allergen challenge. Dis Model Mech. 2008;1:213–220. doi: 10.1242/dmm.000323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Odaka M, Matsukura S, Kuga H, Kokubu F, Kasama T, Kurokawa M, Kawaguchi M, Ieki K, Suzuki S, Watanabe S, Homma T, et al. Differential regulation of chemokine expression by Th1 and Th2 cytokines and mechanisms of eotaxin/CCL-11 expression in human airway smooth muscle cells. Int Arch Allergy Immunol. 2007;143(Suppl 1):84–88. doi: 10.1159/000101412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Owens GK. Molecular control of vascular smooth muscle cell differentiation. Acta Physiol Scand. 1998;164:623–635. doi: 10.1111/j.1365-201x.1998.tb10706.x. [DOI] [PubMed] [Google Scholar]
- 38.Owens GK, Kumar MS, Wamhoff BR. Molecular regulation of vascular smooth muscle cell differentiation in development and disease. Physiol Rev. 2004;84:767–801. doi: 10.1152/physrev.00041.2003. [DOI] [PubMed] [Google Scholar]
- 39.Pare PD, Mitzner W. Last Word on Counterpoint: Alterations in airway smooth muscle phenotype do not cause airway hyperresponsiveness in asthma. J Appl Physiol. 2012;113:848. doi: 10.1152/japplphysiol.00779.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pepe C, Foley S, Shannon J, Lemiere C, Olivenstein R, Ernst P, Ludwig MS, Martin JG, Hamid Q. Differences in airway remodeling between subjects with severe and moderate asthma. J Allergy Clin Immunol. 2005;116:544–549. doi: 10.1016/j.jaci.2005.06.011. [DOI] [PubMed] [Google Scholar]
- 41.Prefontaine D, Lajoie-Kadoch S, Foley S, Audusseau S, Olivenstein R, Halayko AJ, Lemiere C, Martin JG, Hamid Q. Increased expression of IL-33 in severe asthma: evidence of expression by airway smooth muscle cells. J Immunol. 2009;183:5094–5103. doi: 10.4049/jimmunol.0802387. [DOI] [PubMed] [Google Scholar]
- 42.Ramos-Barbon D, Fraga-Iriso R, Brienza NS, Montero-Martinez C, Verea-Hernando H, Olivenstein R, Lemiere C, Ernst P, Hamid QA, Martin JG. T Cells localize with proliferating smooth muscle alpha-actin+ cell compartments in asthma. Am J Respir Crit Care Med. 2010;182:317–324. doi: 10.1164/rccm.200905-0745OC. [DOI] [PubMed] [Google Scholar]
- 43.Robins S, Roussel L, Schachter A, Risse PA, Mogas AK, Olivenstein R, Martin JG, Hamid Q, Rousseau S. Steroid-insensitive ERK1/2 activity drives CXCL8 synthesis and neutrophilia by airway smooth muscle. Am J Respir Cell Mol Biol. 2011;45:984–990. doi: 10.1165/rcmb.2010-0450OC. [DOI] [PubMed] [Google Scholar]
- 44.Sanders PN, Koval OM, Jaffer OA, Prasad AM, Businga TR, Scott JA, Hayden PJ, Luczak ED, Dickey DD, Allamargot C, Olivier AK, et al. CaMKII Is Essential for the Proasthmatic Effects of Oxidation. Sci Transl Med. 2013;5:195ra97. doi: 10.1126/scitranslmed.3006135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Saunders MA, Mitchell JA, Seldon PM, Yacoub MH, Barnes PJ, Giembycz MA, Belvisi MG. Release of granulocyte-macrophage colony stimulating factor by human cultured airway smooth muscle cells: suppression by dexamethasone. Br J Pharmacol. 1997;120:545–546. doi: 10.1038/sj.bjp.0700998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Schmitz J, Owyang A, Oldham E, Song Y, Murphy E, McClanahan TK, Zurawski G, Moshrefi M, Qin J, Li X, Gorman DM, et al. IL-33, an interleukin-1-like cytokine that signals via the IL-1 receptor-related protein ST2 and induces T helper type 2-associated cytokines. Immunity. 2005;23:479–490. doi: 10.1016/j.immuni.2005.09.015. [DOI] [PubMed] [Google Scholar]
- 47.Schramm CM, Puddington L, Wu C, Guernsey L, Gharaee-Kermani M, Phan SH, Thrall RS. Chronic inhaled ovalbumin exposure induces antigen-dependent but not antigen-specific inhalational tolerance in a murine model of allergic airway disease. Am J Pathol. 2004;164:295–304. doi: 10.1016/S0002-9440(10)63119-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Schworer CM, Rothblum LI, Thekkumkara TJ, Singer HA. Identification of novel isoforms of the delta subunit of Ca2+/calmodulin-dependent protein kinase II. Differential expression in rat brain and aorta. J Biol Chem. 1993;268:14443–14449. [PubMed] [Google Scholar]
- 49.Shan L, Redhu NS, Saleh A, Halayko AJ, Chakir J, Gounni AS. Thymic stromal lymphopoietin receptor-mediated IL-6 and CC/CXC chemokines expression in human airway smooth muscle cells: role of MAPKs (ERK1/2, p38, and JNK) and STAT3 pathways. J Immunol. 2010;184:7134–7143. doi: 10.4049/jimmunol.0902515. [DOI] [PubMed] [Google Scholar]
- 50.Shi HZ, Humbles A, Gerard C, Jin Z, Weller PF. Lymph node trafficking and antigen presentation by endobronchial eosinophils. J Clin Invest. 2000;105:945–953. doi: 10.1172/JCI8945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Spinelli AM, Gonzalez-Cobos JC, Zhang X, Motiani RK, Rowan S, Zhang W, Garrett J, Vincent PA, Matrougui K, Singer HA, Trebak M. Airway smooth muscle STIM1 and Orai1 are upregulated in asthmatic mice and mediate PDGF-activated SOCE, CRAC currents, proliferation, and migration. Pflugers Arch. 2012;464:481–492. doi: 10.1007/s00424-012-1160-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sumi M, Kiuchi K, Ishikawa T, Ishii A, Hagiwara M, Nagatsu T, Hidaka H. The newly synthesized selective Ca2+/calmodulin dependent protein kinase II inhibitor KN-93 reduces dopamine contents in PC12h cells. Biochem Biophys Res Commun. 1991;181:968–975. doi: 10.1016/0006-291x(91)92031-e. [DOI] [PubMed] [Google Scholar]
- 53.Tan X, Alrashdan YA, Alkhouri H, Oliver BG, Armour CL, Hughes JM. Airway smooth muscle CXCR3 ligand production: regulation by JAK-STAT1 and intracellular Ca(2)(+) Am J Physiol Lung Cell Mol Physiol. 2013;304:L790–L802. doi: 10.1152/ajplung.00356.2012. [DOI] [PubMed] [Google Scholar]
- 54.Tombes RM, Faison MO, Turbeville JM. Organization and evolution of multifunctional Ca(2+)/CaM-dependent protein kinase genes. Gene. 2003;322:17–31. doi: 10.1016/j.gene.2003.08.023. [DOI] [PubMed] [Google Scholar]
- 55.Trian T, Benard G, Begueret H, Rossignol R, Girodet PO, Ghosh D, Ousova O, Vernejoux JM, Marthan R, Tunon-de-Lara JM, Berger P. Bronchial smooth muscle remodeling involves calcium-dependent enhanced mitochondrial biogenesis in asthma. J Exp Med. 2007;204:3173–3181. doi: 10.1084/jem.20070956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Venge P. The eosinophil and airway remodelling in asthma. Clin Respir J. 2010;4(Suppl 1):15–19. doi: 10.1111/j.1752-699X.2010.00192.x. [DOI] [PubMed] [Google Scholar]
- 57.Walker C, Bode E, Boer L, Hansel TT, Blaser K, Virchow JC., Jr Allergic and nonallergic asthmatics have distinct patterns of T-cell activation and cytokine production in peripheral blood and bronchoalveolar lavage. Am Rev Respir Dis. 1992;146:109–115. doi: 10.1164/ajrccm/146.1.109. [DOI] [PubMed] [Google Scholar]
- 58.Wright DB, Trian T, Siddiqui S, Pascoe CD, Johnson JR, Dekkers BG, Dakshinamurti S, Bagchi R, Burgess JK, Kanabar V, Ojo OO. Phenotype modulation of airway smooth muscle in asthma. Pulm Pharmacol Ther. 2013;26:42–49. doi: 10.1016/j.pupt.2012.08.005. [DOI] [PubMed] [Google Scholar]
- 59.Zuyderduyn S, Sukkar MB, Fust A, Dhaliwal S, Burgess JK. Treating asthma means treating airway smooth muscle cells. Eur Respir J. 2008;32:265–274. doi: 10.1183/09031936.00051407. [DOI] [PubMed] [Google Scholar]