

Abstract
Mismatch repair (MMR) deficiency gives rise to cisplatin resistance and can lead to poor prognosis in cancers. Various models have been proposed to explain this low level of resistance caused due to loss of MMR proteins. We have shown that MMR proteins are required to maintain cisplatin interstrand cross-links (ICLs) on the DNA leading to increased cellular sensitivity. In our previous studies, we have shown that BER processing of the cisplatin ICLs is mutagenic. Polymerase β (Polβ) can generate mismatches which leads to the activation and the recruitment of mismatch repair proteins. In this paper, we distinguished between the requirement of different downstream MMR proteins for maintaining cisplatin sensitivity. We show that the MutSα (MSH2-MSH6) heterocomplex is required to maintain cisplatin sensitivity, whereas the Mutsβ complex has no effect. These results can be correlated with the increased repair of cisplatin ICLs and ICL induced DNA double strand breaks (DSBs) in the resistant cells. Moreover, we show that MLH1 proficient cells displayed a cisplatin sensitive phenotype when compared with the MLH1 deficient cells and the ATPase activity of MLH1 is essential to mediate this effect. Based on these results, we propose that MutSα as well as the downstream MMR pathway proteins are essential to maintain a cisplatin sensitive phenotype as a consequence of processing Polβ induced mismatches at sites flanking cisplatin ICLs.
Keywords: BER, cisplatin, MMR, resistance, ATPase
1.1 Introduction
The DNA mismatch repair (MMR) system which is involved in the post replicative repair of mismatches plays a crucial role in the maintenance of genomic stability [1]. In addition to the recognition of mismatches, MMR proteins have also been involved in the recognition and processing of DNA damage inflicted by a number of chemotherapeutic agents like cisplatin, carboplatin, alkylating agents and 5-Fluorouracil [2–6]. The MMR pathway is composed of recognition proteins with MSH2 as a common partner in two heterocomplexes namely MutSα (MSH2-MSH6) and MutSβ (MSH2MSH3) [7]. MutSα is required for the repair of base-base mismatches and one base pair insertion deletion loops (IDLs). On the other hand, MutSβ carries out the repair of IDLs with single or multiple base pairs. The mismatch recognition step is followed by the recruitment of downstream MMR proteins including MutLα (MLH1-PMS2), Exonuclease I, DNA polymerase δ and DNA ligase.
MMR has also been shown to participate in the DNA damage response after treatment with certain chemotherapeutic agents. Loss of MMR proteins has been associated with resistance to a number of anti-cancer agents (e.g., alkylating agents and cisplatin) [5, 6]. Various models have been proposed for the possible role of MMR in maintaining drug sensitivity. Adducts formed by alkylating agents can result in the generation of mismatched bases. It has been suggested that MMR proteins take part in futile cycles of repair of these mismatches in the daughter strand. The resulting strand breaks signal apoptosis and loss of this function can give rise to drug resistance [8–10]. In addition, MMR proteins have been shown to directly signal the DNA damage, eventually resulting in cell death [3, 11]. These studies, however, did not differentiate between the different types of DNA adducts that are formed by cisplatin, namely intrastrand adducts which are formed within the same DNA strand versus interstrand crosslinks (ICLs) which are formed between adjoining strands of DNA. MutSα proteins have been shown to recognize cisplatin intrastrand adducts [10]. In addition, MutSβ was found to be one of the proteins that interact with cisplatin ICLs [12]. However, the exact role of MSH3 in the processing of cisplatin adducts has not been clearly evaluated.
Recent studies have shown that MSH3 is required for the repair of DNA double strand breaks (DSBs) induced during cisplatin and oxaliplatin treatment [13, 14]. Thus, the MMR pathway has been shown to be required for the sensitization of colorectal cancer cells to cisplatin and oxaliplatin, and this effect is believed to be independent of the canonical MMR processing. However, other studies have shown that MSH3 proficient cells, which were more resistant to chemotherapy, expressed higher levels of NER proteins which could explain the reason for increased resistance [15]. Thus, the exact role of MSH3 in modulating platinum cytotoxicity remains to be determined.
In our previous studies, we have shown that loss of base excision repair (BER) and MMR proteins gives rise to resistance to cisplatin and these two pathways take part in the same mechanistic pathway to mediate cisplatin sensitivity [16, 17]. In the absence of these proteins, increased repair of cisplatin ICLs was observed which leads to decreased cellular cytotoxicity. We also showed that this mechanism is dependent upon the low fidelity of DNA polymerase β (Polβ), which leads to mis-incorporation of bases and generation of mismatches at sites flanking a cisplatin ICL. This mismatch in turn activates the MMR pathway. In this report, we distinguish between the requirement of different downstream MMR proteins to mediate this effect, and we show that in contrast to previous studies, there is a clear distinction between the initial MMR recognition heterocomplexes. MutSα is required to maintain cisplatin sensitivity while MutSβ plays no role at least in breast cancer cell lines and mouse embryonic fibroblasts in mediating cisplatin cytotoxicity. Moreover, we show that the ATPase activity of MLH1 is required for maintaining a cisplatin sensitive phenotype highlighting the importance of the MMR pathway in the non-productive processing of cisplatin ICLs and not just shielding of the DNA damage by MutSα.
1.2 Materials and Methods
1.2.1 Chemicals and Antibodies
Cisplatin, oxaliplatin and myricetin were purchased from Sigma-Aldrich. All other chemicals and reagents were from standard suppliers. Antibodies directed against MSH3, MSH6 and MLH1 were from BD Pharmigen and α-tubulin was from Sigma-Aldrich. For the stock preparation, cisplatin and oxaliplatin were diluted in 1X PBS and vortexed vigorously until the drug dissolved completely. The stock concentration was 1 mM. Cisplatin was prepared fresh before each experiment. Stock solutions for oxaliplatin were stored at −80 °C for up to 6 months and thawed at room temperature (RT) when needed.
1.2.2 Cell lines
The human breast adenocarcinoma MDA-MB-231 cells were grown in RPMI 1640 containing 10% FBS and geneticin (700μg/ml). MDA-MB-231 Polβ knockdown cells (Polβ lentiviral shRNA) were grown in the presence of 0.5μg/mL puromycin. The development and characterization of the MDAMB-231/Polβ-KD cells were described previously [18]. MLH1-null HCT116 cells were used for complementation with wtMLH1 and its S44L and S44P ATPase mutants. A site-specific mutagenesis was performed using the QuikChange multi site-directed mutagenesis kit from Stratagene. MLH1 constructs were inserted into the pQCXIN retroviral vector. Infected cells with stable expression of the vector sequences were selected in the presence of geneticin [19]. The HCT116 cells were grown in DMEM F-12 media with 10% FBS, antibiotics and 600 μg/ml of geneticin. The DLD-1 and DLD-1 + chr 2 cells were kindly provided by Dr. Thomas Kunkel (NIH) and were maintained in DMEM F-12 media with 10% FBS, antibiotics. The chromosome complemented DLD-1 + chr 2 cells were maintained in 400 μg/ml of geneticin.
1.2.3 shRNA transfection
Mission shRNA plasmid bacterial stocks directed against human MSH6 and MSH3 were obtained from Sigma Aldrich. The plasmid DNA was purified using a plasmid purification maxi prep kit from Qiagen. Lentiviral particles were packaged using 293FT cells with the help of 3rd generation packaging plasmids PMD2G, PMDLG/RRE and PRSV/RRE. Lipofectamine 2000 reagent (Invitrogen) was used for the transfection of the plasmid DNA. The media was changed after 24 hrs of transfection. The viral particles were harvested 48 hr and 72 hr after the transfection by centrifugation followed by filtration through 0.2 micron filters. The viral stocks were stored as aliquots at −80°C for future use. At the time of the experiment, the viral stocks were used along with polybrene (Sigma Aldrich) for the knock down of proteins of interest. Cells were harvested at the 72 hr timepoint post transduction to check for protein and transcript expression.
1.2.4 siRNA transfection
ON-TARGET plus SMART pool siRNAs specific for human MSH3 and MLH1 were purchased from Dharmacon RNAi technologies, Thermo Scientific. The non-targeting control siRNA was used as a control for non-specific effects. siRNA transfection was carried out as per the manufacturer’s protocol. Briefly, the cells were plated in 6 well plates in the antibiotic free media. At the time of transfection, the cell density was maintained at 60–70% and two transfections were done with an interval of 24 hrs. Dharmafect transfection reagent 1 and 4 were used for MEFs and MDA-MB-231 cells, respectively. The cells were harvested at 48 and 72 hr timepoints after transfection for the detection of protein and transcript expression.
1.2.5 Western blot analysis
Cells were harvested at 96, 120 and 144 hrs after the infection, washed with PBS and lysed in lysis buffer (10 mM Tris pH 8.0, 120 mM NaCl, 0.5% NP-40, 1mM EDTA) containing protease inhibitors (0.5 M phenyl methyl sulphonyl fluoride PMSF, 1mg/`ml Leupeptin and 1 mg/ml pepstatin A). The proteins were separated on 8% SDSpolyacrylamide gels and transferred onto Immobilon-P transfer membranes (Millipore). After blocking (2% non-fat dry milk), the membranes were probed with primary antibodies recognizing human MLH1, with α-tubulin as a loading control. The membranes were incubated with appropriate secondary antibodies and the signal was detected by using Enhanced chemiluminescence detection system.
1.2.6 Real time PCR for the measurement of transcript levels
At indicated post-transfection time points, cells were harvested and pelleted. RNA was isolated using TRIzol reagent (Invitrogen) by standard procedures. The total RNA was reverse transcribed using MMLV reverse transcriptase enzyme (Invitrogen) as per the manufacturer’s protocol. The transcript levels were quantified using iQ SYBR green supermix (Bio-Rad) in iCycler iQ System, with GAPDH as an endogenous control. The percent transcript knockdown was determined from 2^−∆∆CT values as previously described [17].
1.2.7 Colony survival assay
Cells (~500) were treated with increasing concentrations of cisplatin for 2 hr. After treatment, fresh medium was added and the cells were allowed to grow for 7–14 days. Colonies were fixed with 95% methanol and stained with 0.2 % crystal violet. Colonies with ≥50 cells were counted and colony survival was expressed as the ratio of the average number of colonies in drug treated cells versus control cells × 100. The experiment was done in triplicates for each drug concentration.
1.2.8 Alkaline comet assay
Alkaline comet assay was used to analyze the repair of cisplatin ICLs as described [20, 21]. Cell suspensions (~10000 cells) were embedded on a microscopic slide, lysed and incubated in ice-cold alkaline solution for 20 min to allow the DNA to unwind. Electrophoresis was carried out for 30 min at 28 V, 300 mA. Slides were neutralized and stained with SYBR green (Invitrogen). The comets were scored using a Nikon epifluorescence microscope. At least fifty cells were analyzed per slide using Komet Assay Software 5.5F (Kinetic Imaging, Liverpool, UK). The data was expressed as the percentage of crosslinks that remained at that particular time point normalized to 100% at 0 hr post treatment. This time point corresponds to 2 hr post cisplatin treatment.
1.2.9 Immunofluorescence
Double-strand break (DSB) repair was assessed by monitoring the nuclear γ-H2AX foci by immunofluorescence. Cells were fixed using 4% paraformaldehyde, permeabilized and probed with monoclonal anti γ-H2AX antibody (1:500, Millipore). The images were visualized using a Nikon Eclipse T2000-U microscope. Foci were counted in at least 200 cells at each time point per condition in each cell line and results are expressed as % γH2AX foci positive nuclei.
1.2.10 ELISA
The rate of repair of cisplatin intrastrand adducts was assessed by ELISA using a specific monoclonal antibody against cisplatinintrastrand adducts (ICR4, kindly provided by Michael J. Tilby, University of Newcastle, UK). Cells were treated with cisplatin for 2 hrs and were harvested at 0–72 hr timepoints. Genomic DNA was isolated using QIAGEN DNeasy blood and tissue kit. The DNA was coated on 96 well Profoldin DNA binding ELISA plates and probed with ICR4 antibody. The plates were washed to remove any unbound antibody followed by incubation with HRP conjugated goat anti-rat secondary antibody (Calbiochem). After addition of TMB (1 step ultra TMB-ELISA, Thermo Scientific), the reaction was stopped with 2M sulfuric acid and absorbance was measured at 450 nm (Spectramax M5 plate reader, Molecular Devices). The % intrastrand adducts were calculated using OD 450 nm reading. The % of adducts at 0 hr time point were normalized to 100% intrastrand adducts in each cell line.
1.2.11 MTS assay
Cells (~10,000) were plated in 96 well plates and were treated with cisplatin for 2 hrs. The media was changed to complete media and the cells were allowed to incubate for 72 hrs after the treatment. MTS assay was performed as per the manufacturer’s protocol. (Promega). Briefly, the MTS dye was added to the plate, followed by incubation for 2–3 hrs at 37°C. The absorbance was measured at 490 nm and the survival was expressed by normalizing untreated cells to 100%. The experiment was done in triplicate for each drug concentration.
1.3 Results
1.3.1 Requirement of different MMR recognition heterocomplexes to maintain cisplatin sensitivity
To differentiate between the role of MutSα and MutSβ in mediating cisplatin sensitivity, colony survival assays were performed. Using wild type and Polβ deficient MDA-MB-231 cells, we knocked down MSH3 or MSH6 using shRNA directed against these proteins. The knockdown efficiency was analyzed at both protein and transcript levels using western blot analysis and real time PCR, respectively (Supplementary Figure S1). The level of knockdown was found to be 80–90 % compared to the controls. In addition we also checked for the expression of MSH3, MSH6 and MSH2 in absence of MSH3 and/or MSH6 (Supplementary Figures 2 A–F). Using clonogenicity assays, we found that MSH6 deficient cells were resistant to cisplatin as compared to the wild type cells (Figure 1A). Loss of Polβ also gave rise to cisplatin resistance, which is consistent with our previous results [16,17]. However, down regulation of MSH6 in Polβ deficient cells did not give rise to any additive increase in the degree of resistance reinforcing our previous observations of an epistatic relationship between BER and MMR proteins in mediating cisplatin cytotoxicity. In support of these studies, we also performed colony survival assays using MSH6 deficient DLD-1 and MSH6 proficient DLD-1 + chr 2 cells after cisplatin treatment. MSH6 deficient cells displayed a cisplatin resistant phenotype compared to MSH6 proficient cells indicating that MSH6 is required to mediate cisplatin sensitivity (Figure 1B). The expression of MSH2, MSH3 and MSH6 in these cells is shown in Supplementary Figure 3. In addition, we also performed clonogenic assays using MSH3 knockdown cells, and we observed that down regulation of MSH3 had minimal effect on cisplatin cytotoxicity. Similar to the wild type cells, MSH3 deficient cells maintained a cisplatin sensitive phenotype (Figure 1C). Comparable results were observed in wild type mouse embryonic fibroblasts and in Polβ null cells no additional cisplatin resistance was observed following MSH3 knockdown (Supplementary Figure S4). This data is consistent with MSH3 having no role in mediating cisplatin sensitivity. Also, knockdown of both MSH3 and MSH6 together gave rise to a ~ 2 fold resistant phenotype in response to cisplatin similar to MSH6 knockdown and MSH6 depleted cells alone (Figure 1D). These data indicate that MSH6 is required for mediating sensitivity whereas, MSH3 does not influence cisplatin cytotoxicity at least in the cell lines used in this study. These data along with our previous studies showing loss of MSH2 results in cisplatin resistance implicate MutSα in mediating cisplatin efficacy and not MutSβ [17].
Figure 1. Cisplatin cytotoxicity in MSH6 and MSH3 knockdown cells.
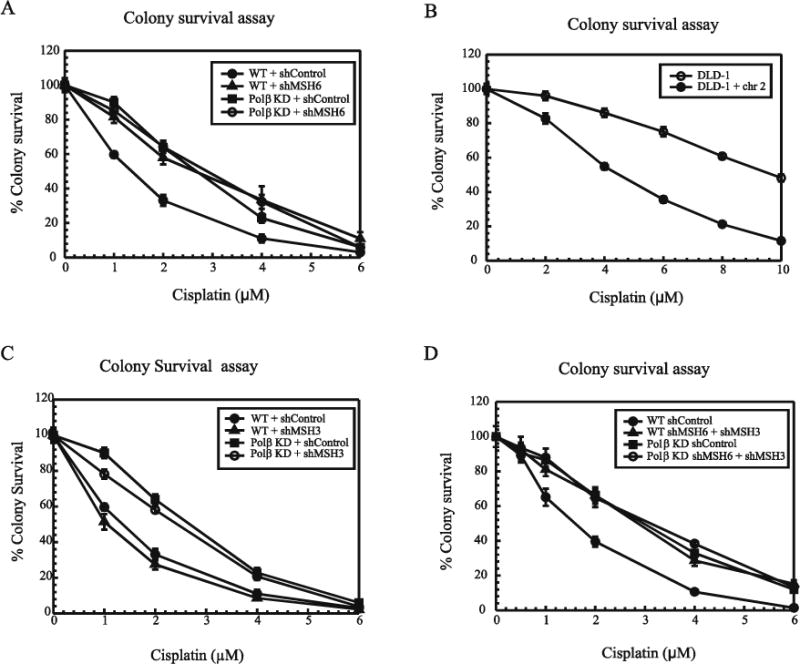
(A) Colony survival assay in MDA-MB-231 wildtype (WT) cells or following MSH6 knockdown: Control (closed circles), MSH6 (open triangles), Polβ (closed squares) and Polβ MSH6 KD (open circles). (B) Colony survival assay in DLD-1 (open circles) and DLD-1 + chr2 cells (closed circles) (C) Colony survival assay in MDA-MB-231 cells following MSH3 knockdown: Control (closed circles), MSH3 (open triangles), Polβ (closed squares) and Polβ MSH3 KD (open circles). (D) Colony survival assay in MDA-MB-231 cells following a double knockdown of MSH3 and MSH6: WT control (closed circles), MSH3 +MSH6 KD in WT cells (open triangles), Polβ KD control (closed squares) and MSH3 +MSH6 KD in Polβ KD cells (open circles). shRNA transfected cells were treated with increasing doses of cisplatin and cytotoxicity was determined by colony survival assay as described in Materials and Methods. Results are represented as mean ± SD from 3 independent experiments.
Previously, we have shown that BER and MMR processing of ICLs is dependent upon the unique structure produced by the cisplatin ICL. This distorted structure is unique to cisplatin with respect to the distortion surrounding the ICL. For this reason, we studied cell viability in response to oxaliplatin in MSH3 and MSH6 knockdown cells using MTS assays (Supplementary Figure S5A and S5B). Cell survival studies showed no difference in the oxaliplatin cytotoxicity highlighting the fact that the role of MMR proteins in modulating chemo sensitivity is specific to cisplatin and carboplatin. In support of these studies, Zdraveski et al have shown that the oxaliplatin adducts are not recognized by MMR proteins [22].
1.3.2 Differential role of MMR recognition proteins in cisplatin ICL repair
Enhanced DNA repair has been shown to be a major mechanism for the development of cisplatin resistance [23]. MMR proteins have been shown to bind to the cisplatin GG intrastrand adducts [9, 10]. In our previous studies, we have shown that down-regulation of MSH2, which is a common partner in MutSα and MutSβ heterocomplexes, does not influence the rate of repair of cisplatin intrastrand adducts [17]. Thus, based on our previous observations, we did not expect MSH3 and MSH6 to be involved in the repair of cisplatin intrastrand adducts. As the repair of intrastrand adducts was expected to be unaffected, we hypothesized that the rate of repair of cisplatin ICLs influences cisplatin cytotoxicity. For this reason, we studied cisplatin ICL repair using modified alkaline comet assay in MSH3 or MSH6 knockdown cells. The alkaline comet assay is modified using hydrogen peroxide to generate a fixed level of DNA strand breaks [17, 24]. During electrophoresis, these strand breaks migrate to form a comet tail.
However, presence of ICLs in DNA hinders the migration of DNA strands giving rise to a shorter tail. The comparison between the olive tail moments of H2O2 alone treated and H2O2 plus cisplatin treated cells allows to calculate % of ICLs remaining in the DNA.
Using this assay, we observed increased repair of cisplatin ICLs over the time course of 0–72 hr in all cell types with no difference in the repair rate up to the 24 hr time point. At the 48 hr and 72 hr time point, however, we observed increased repair of cisplatin ICLs in Polβ deficient cells consistent with our previous studies [16, 17]. MSH6 knockdown in both WT and Polβ deficient cells showed increased repair of cisplatin ICLs as compared to WT cells treated with control shRNA suggesting the involvement of MSH6 in cisplatin ICL processing (Figure 2A). Moreover, we did not see any additive increase in the repair capacity in the absence of both MSH6 and Polβ. In addition, we also performed similar assays using MSH3 shRNA and in contrast to MSH6 knockdown cells, following MSH3 knockdown, we did not see any change in the repair rate of cisplatin ICLs in these cells (Figure 2B). It should be noted that the modified alkaline comet assay used in this report is sensitive to the initial unhooking of the ICL from the double stranded DNA and therefore, this assay can represent the involvement of MMR proteins in the initial stages of ICL repair. Thus, these data suggest that MSH6 interferes with or inhibits the repair of cisplatin ICLs whereas MSH3 does not participate in the cisplatin ICL processing which leads to similar rates of ICL repair in WT and MSH3 knockdown cells. Moreover, increased repair rates have been shown to be one of the mechanisms of cisplatin resistance and poor prognosis and these data correlate well with the cisplatin cytotoxicity profile after down regulation of these proteins (Figure 1).
Figure 2.
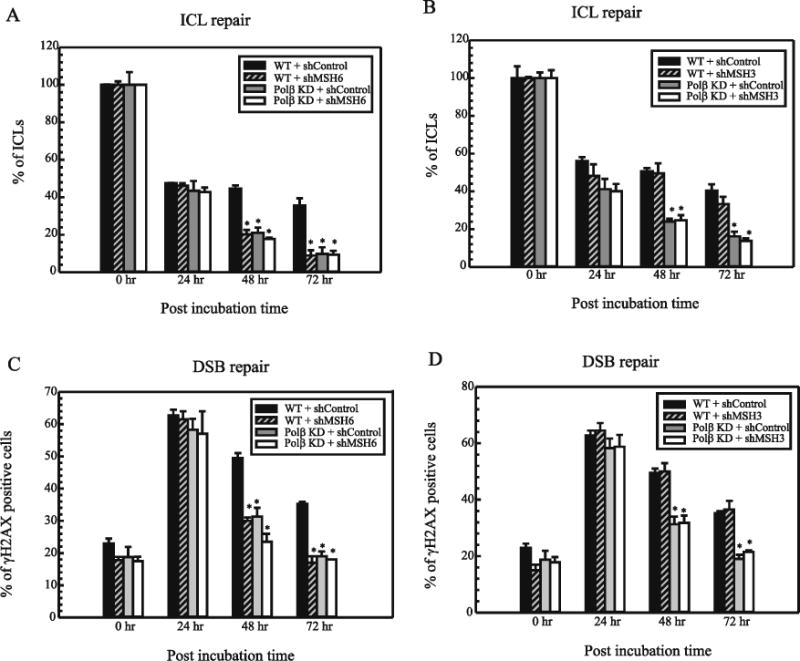
Repair of cisplatin ICLs in MDA-MB-231 cells. (A) MSH6 KD (B) MSH3 KD in MDA-MB-231. Cells were treated with cisplatin for 2 hrs and comet assays were performed as described in Materials and Methods at different time intervals (0, 24, 48 and 72 hr) to assess ICL levels. The data was collected using komet 5.5 software. The percentage of ICLs present at each time point was calculated using olive tail moments. Results are represented as mean ± SD of three independent experiments. Statistical analysis was performed by student’s t test and comparisons are made between wildtype and proficient cells vs deficient cells. NS – non-significant; * - P< 0.01. Repair of cisplatin ICL induced DSBs in MDA-MB-231 (C) MSH6 KD (D) MSH3 KD. Cells were treated with cisplatin for 2 hrs and immunofluorescence was performed as described in Materials and Methods at different time intervals (0, 24, 48 and 72 hr). A minimum of 200 cells were analyzed for each time point. The percentage of γH2AX foci positive cells at each time point was calculated. Results are represented as mean ± SD of three independent experiments. Statistical analysis was performed by student’s t test and comparisons are made between wildtype and proficient cells vs deficientcells. NS–nonsignificant;*-P<0.05.
Furthermore, cisplatin ICL processing leads to the generation of DNA DSBs. The ATM kinase recognizes the DSBs and causes phosphorylation of a histone variant H2AX at serine 139. This causes recruitment of downstream DSB repair proteins. The phosphorylated γH2AX proteins form distinct nuclear foci and can be studied using an immunofluorescence assay. In this assay, we observed induction of γH2AX foci and a decrease in γH2AX positive cells from 24 hr to 72 hr time points. The Polβ deficient cells showed decreased percentage of foci positive cells at 48 hr and 72 hr as compared to WT cells consistent with our previous report [16]. However, MSH6 knockdown cells also showed decreased percentage of foci positive cells and therefore, increased repair of cisplatin induced DSBs as compared to the WT cells (Figure 2C). These data validate the inhibitory role of MSH6 in the processing of cisplatin ICLs. However, in contrast to some of the other studies done in colorectal cancer cells lines [13, 14], MSH3 knockdown cells did not show any change in the phosphorylation status of γH2AX when compared to the WT breast adenocarcinoma cell lines used in this study (Figure 2D). These results suggest that unlike MSH6, loss of MSH3 does not affect the repair of cisplatin ICLs and likely not the repair rate of cisplatin ICL induced DSBs.
1.3.3 MutL homolog 1 plays a key role in maintaining cisplatin sensitivity
MLH1 is the human homologue of the E. coli MMR gene MutL. The MMR pathway involves recognition of a base mismatch or insertion/deletion loop by a MutS homolog followed by recruitment of a MutLα heterodimeric complex consisting of MLH1 and PMS2 [7]. To understand the importance of MLH1 in mediating cisplatin cytotoxicity, we knocked down MLH1 using siRNA in MDA-MB-231 cell lines. The knock down efficiency was found to be 80–90% (Supplementary Figure S6A, S6B). Colony survival assays were performed to address the effect of MLH1 down-regulation on the cell viability in response to cisplatin (Figure 3A). MLH1 knock down in WT cells showed ~2 fold resistance to cisplatin as compared to the control cells. However, MLH1 knock down in Polβ deficient cells did not give rise to any additional increase in cisplatin resistance indicating an overlapping role of these two proteins in the same mechanistic pathway to mediate cisplatin sensitivity. To understand the mechanism of resistance, we checked for the effect of MLH1 knockdown on the repair rates of cisplatin DNA adducts. We performed enzyme linked immunosorbent assay using a monoclonal antibody specific for cisplatin GG adduct, which is a major intrastrand adduct formed by cisplatin [16, 17]. Knockdown of BER and MMR showed no difference in the repair of cisplatin intrastrand adducts indicating that these pathways do not influence the repair rate of cisplatin GG adducts (Figure 3B). As cisplatin intrastrand adduct repair was unaffected, we checked for the repair of cisplatin ICLs. Modified alkaline comet assay was used to evaluate the rate of ICL repair from 0–72 hr after cisplatin treatment. Down-regulation of Polβ showed decreased percentage of ICLs at 48 hr and 72 hr time points. MLH1 has shown to be required for signaling DNA damage in response to psoralen crosslinks [25]. In these studies using cisplatin, depletion of MLH1 in the WT as well as Polβ deficient cells resulted in increased repair of cisplatin ICLs (Figure 3C). In addition, similar results were observed in an immunofluorescence assay where MLH1 KD cells showed increased repair of cisplatin ICL induced DSBs as compared to the WT cells (Figure 3D). These results indicate that increased repair of cisplatin ICLs accounts for the cisplatin resistant phenotype seen in these cells. Furthermore, we observed similar levels of ICL repair in the absence of both BER and MMR pathways as compared to the knockdown of MMR and BER alone. These data suggest an epistatic relationship of these two pathways in the mediating cisplatin sensitivity.
Figure 3. Effect of MLH1 knockdown on cisplatin cytotoxicity and repair.
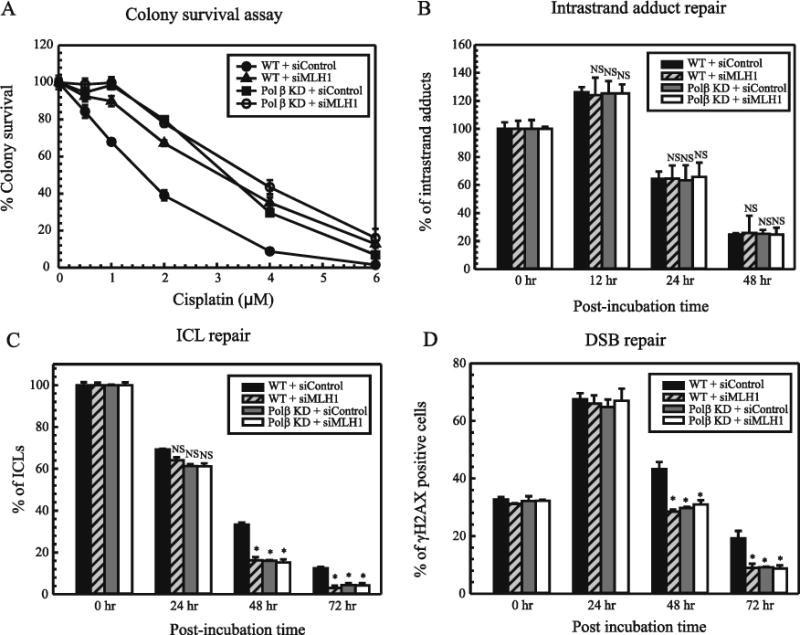
(A) Colony survival assays in WT and Polβ deficient MDA-MB-231 cells with MLH1 KD. (B) ELISA for cisplatin GG intrastrand adduct repair in MLH1 KD MDA-MB-231 cells. Cells were treated with cisplatin for 2 hrs and genomic DNA was extracted at the indicated time points. ELISA was performed using an antibody specific for cisplatin GG intrastrand adduct and the percentage of GG adducts remaining was calculated as described. Results are represented as mean ± SD of three independent experiments. Statistical analysis was performed by student’s t test and comparisons are made between wildtype and proficient cells vs deficient cells. NS – non-significant (C) Cisplatin ICL DNA repair using alkaline comet assay performed in WT and Pol β deficient MDA-MB231 cells as described previously. (D) Immunofluorescence assay for γH2AX assessing DSB repair in WT, MLH1 KD and Polβ deficient MDA-MB-231 cells.
1.3.4 ATPase activity of MLH1 is essential for cellular sensitivity to cisplatin
We have shown that BER and MMR play an epistatic role in mediating cisplatin sensitivity [17]. This overlapping role is dependent upon BER processing and the error prone nature of Polβ to generate a mismatch flanking a cisplatin ICL, thereby leading to activation of the MMR pathway. Our results indicate that the complete knockdown of MLH1 gives rise to a cisplatin resistant phenotype, suggesting that a functional downstream MMR pathway is essential to maintain cellular sensitivity to cisplatin. Next, to confirm the involvement of MMR and the actual processing of the mismatch we utilized human colon cancer cell lines that are deficient in the MLH1 ATPase activity (Figure 4A). The HCT116 cells are deficient in MLH1 and MSH3. These cells were reconstituted with WT MLH1, S44L and S44F ATPase mutant MLH1. Owing to a point mutation that affects the serine 44 residue in the ATPase domain, these cells are deficient in MMR activity. We utilized these cells lines to differentiate between the effects of MLH1 binding to the MSH complexes at the mismatch as opposed to the role of MLH1 in actual MMR processing of the mismatch.
Figure 4. MLH1 ATPase mutants in mediating cisplatin cytotoxicity.
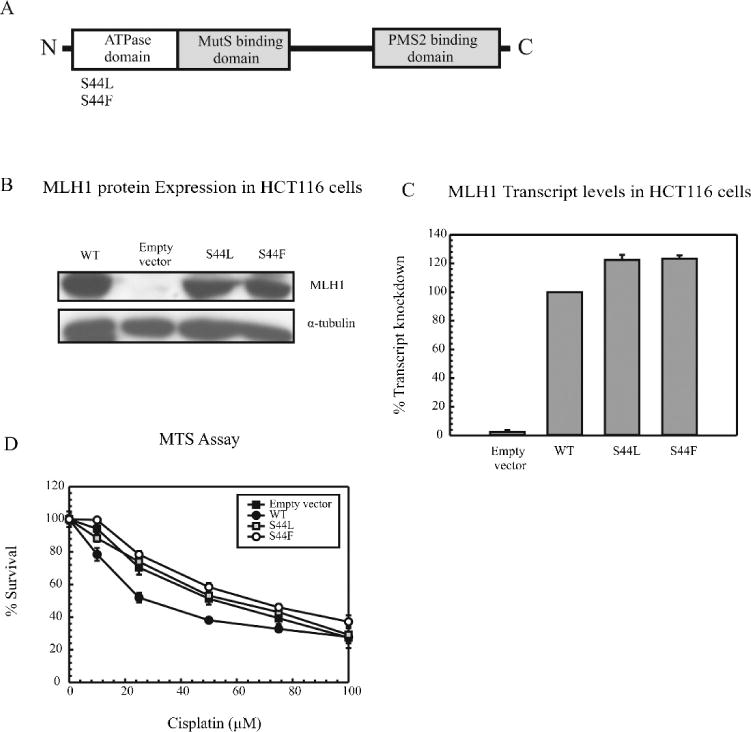
(A) Schematic of MLH1 ATPase mutant, (B) Western blot analysis and (C) real time PCR were performed to check the expression levels of MLH1 protein and transcript. (D) MTS assays were performed in HCT116 cells using increasing concentrations of cisplatin for 2 hrs. WT cells (closed circles), Empty vector (closed squares), S44L cells (open squares) and S44F cells (open circles). Results are represented as mean ± SE from 3 independent experiments.
We studied the expression of these mutants and it was found to be similar at both the protein and the transcript level (Figure 4B and 4C). After that, we checked the effect of these mutations on cisplatin cytotoxicity using an MTS assay. The MLH1 deficient empty vector cells showed a cisplatin resistant phenotype as compared to the MLH1 proficient WT cells consistent with our previous results. Moreover, The MLH1 ATPase mutant cells also showed a cisplatin resistant phenotype indicating the importance of the ATPase activity of MLH1 in mediating cisplatin sensitivity (Figure 4D). In contrast, using mouse embryonic fibroblasts, previous studies have shown that loss of ATPase activity still maintains sensitivity to cisplatin, highlighting the genetic basis for the direct DNA damage signaling by MMR proteins [26, 27]. Our studies in human cells, however, showed that this activity is essential for cellular sensitivity, indicating that different ATPase mutations can have a differential effect on cisplatin cytotoxicity. Furthermore, we did not see any significant difference in the survival profiles of these cells in response to oxaliplatin (Supplementary Figure S5C). These results suggest that these effects are specific to cisplatin and can be attributed to the unique structure produced by cisplatin ICLs.
To understand the mechanism of resistance and to appreciate the events taking place near the ICLs, we studied how these ATPase mutations affect the repair of cisplatin ICLs. We used a modified alkaline comet assay to study the repair rates of ICLs up to the 72 hr time point after cisplatin treatment. MLH1 deficient empty vector cells showed increased repair of cisplatin ICLs at the 48 hr and 72 hr time point. Moreover, the MLH1 ATPase mutant cells also showed increased repair of cisplatin ICLs at both the 48 hr and 72 hr time points as compared to the MLH1 proficient WT cells (Figure 5A). In addition, in an immunofluorescence assay MLH1 deficient empty vector cells and MLH1 ATPase mutant cells also showed increased repair of cisplatin ICL induced DSBs as compared to the WT MLH1 proficient cells (Figure 5B). These results indicate that the ATPase activity of MLH1 is essential to maintain cisplatin ICLs in the DNA and therefore, this increased repair can be correlated with the resistant phenotype observed in the cell survival studies. Next, we wanted to assess whether MSH6 and MLH1 work within the same pathway to mediate cisplatin cytotoxicity. Hence, we knocked down MLH1 in MSH6 deficient DLD-1 cells and MSH6 proficient DLD-1 + chr 2 cells and assessed the degree of protein knock down (Supplementary Figure S6C). We did not see any additional change in the degree of resistance in the absence of both MSH6 and MLH1 as compared to the knockdown of MSH6 and MLH1 alone (Figure 5C). These results suggest that MLH1 is recruited downstream of MSH6 and an entire MMR pathway is essential for mediating cisplatin sensitivity.
Figure 5. Repair of cisplatin ICLs in HCT116 MLH1 ATPase mutant cells.
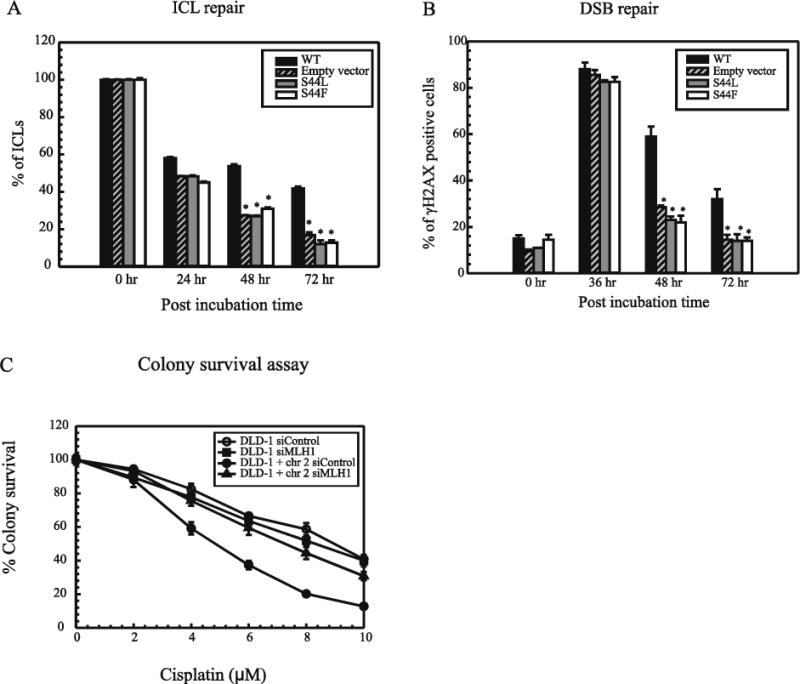
(A) Alkaline comet assay was performed as previously described to assess cisplatin ICL DNA repair. (B) Repair of DSBs in HCT116 cells was measured using an immunofluorescence assay with an antibody specific for γH2AX as previously described Materials and Methods. (C) MLH1 was knocked down in DLD-1 and DLD-1 + chr 2 cells using siRNA transfection: DLD-1 control (open circles), MLH1 KD in DLD-1 cells (closed squares), DLD-1 + chr 2 control (closed circles) and MLH1 KD in DLD-1 + chr 2 (open triangles). Colony survival assays were performed as described previously.
We hypothesize that this role of MLH1 is dependent on the generation of a mismatch due to BER processing. To understand whether MMR and BER pathways play an overlapping role in these ATPase mutant cells, we inhibited BER in HCT116 cells using myricetin. Myricetin is a small molecule inhibitor of the repair function of apurinic/apyridiminic endonuclease 1 (APE1) (Supplementary Figure S7). Pretreatment with myricetin gave rise to a cisplatin resistant phenotype in MLH1 proficient WT cells, consistent with our previous findings. However, when we used MLH1 deficient empty vector cells and MLH1 ATPase mutant cells, both of which are already resistant to cisplatin, myricetin pretreatment did not give rise to any additive increase in the degree of resistance. These results suggest that MMR processing occurs downstream of BER and these two pathways play an epistatic role in mediating cisplatin cytotoxicity. In conclusion, our results suggest that downstream MMR pathway activation plays a critical role in mediating cisplatin cytotoxicity. In addition, the ATPase activity is essential for the processing of the mismatch near the ICL region and this MMR processing hinders the productive repair of cisplatin specific ICLs and thus, gives rise to a cisplatin sensitive phenotype.
1.4 Discussion
The role of MutS and MutL homologues in the repair of cisplatin ICLs has been previously studied using psoralen ICLs [25, 28]. However, the role of individual MMR proteins in the processing of cisplatin ICLs remains to be determined. Cisplatin forms ICLs that have a unique structure compared to other platinum agents as well as other cross-linking agents. As a consequence, cisplatin treatment leads to the recruitment of different DNA repair proteins and initiation of different DNA repair pathways. Previously, we have shown a novel mechanism of crosstalk between BER and MMR pathways in response to cisplatin ICLs. The cisplatin ICL structure involves the flipping out of the cytosine residues, making these structures prone to undergo oxidative deamination. These deamination events can lead to the conversion of cytosine residues to uracil in the DNA flanking the cisplatin ICL. Uracil acts as a substrate for the initiation of the BER pathway. BER processing adjacent to the cisplatin ICLs involves error prone DNA synthesis by Polβ [16]. The low fidelity nature of Polβ can generate mismatches that can lead to activation of the MMR pathway [16, 17]. We have shown the increased recruitment of the MMR protein MSH2 on BER processed cisplatin ICL DNA substrates as compared to unprocessed substrates [17]. The aim of this work was to understand the requirement of downstream MMR proteins in mediating cisplatin sensitivity. It was anticipated that this information will help us to further understand the crosstalk between the BER and MMR pathways near the cisplatin ICL site. Furthermore, it will help us to elucidate the role of MMR proteins in the complex process of cisplatin ICL DNA repair. Repair of ICLs is a complicated process and the exact mechanism of cisplatin ICL repair and how different repair proteins play a role is still not completely understood. From a clinical point of view, increased repair of ICLs has been shown to be associated with resistance to chemotherapy drugs and poor prognosis of cancer [29, 30]. Therefore, understanding this mechanism of cisplatin ICL repair and evaluating the role of different proteins in these pathways becomes clinically relevant.
Recently, it was shown that MSH3 is involved in the repair of cisplatin ICL induced DNA DSBs [13, 14]. In contrast, Vaismann et al have shown that MSH3 deficiency does not have any significant effect on cisplatin or oxaliplatin cytotoxicity [31]. Consistent with these studies, when we knocked down MSH3 in breast adenocarcinoma cell lines, we did not see any effect on cisplatin or oxaliplatin cytotoxicity indicating that MSH3 does not influence platinum cytotoxicity at least in the cell lines used in this study. These observations suggest that MSH3 deficient cells should still retain sensitivity to cisplatin-based chemotherapy. Depletion of MSH3 in Polβ deficient cells did not give rise to a cisplatin sensitive phenotype indicating that MSH3 is not essential and does not play any significant role in maintaining cisplatin sensitivity. In addition, MSH6 knockdown cells showed a cisplatin resistant phenotype consistent with the literature [31, 32]. Furthermore, MSH6 down-regulation in Polβ knock down cells did not give rise to any additive increase in cisplatin resistance, indicating that these proteins play an overlapping mechanistic role and are required to maintain cisplatin sensitivity.
In accordance with the cell survival studies, we wanted to elucidate the mechanism of cisplatin resistance in these cells. Increased repair of adducts from the DNA has been shown to be one of the major mechanisms of cisplatin resistance [23, 33]. MSH2 forms a common partner in MutSα (MSH2-MSH6) and MutSβ (MSH2-MSH3) heterocomplexes. Previously, we have shown that lack of MSH2 does not affect the repair rate of cisplatin intrastrand adducts [17]. For this reason, we did not expect MSH3 and MSH6 to affect cisplatin intrastrand adduct repair. As the repair of intrastrand adducts remains unaffected, we checked the repair kinetics of cisplatin ICLs. In MSH3 knockdown cells, we did not see any change in the repair rate of cisplatin ICLs as well as ICL-induced DSBs. Zhu and Lippard have shown that MutSβ is one of the proteins that bind specifically to cisplatin ICLs [12], indicating that the MSH2-MSH3 complex might play some role in the repair of cisplatin ICLs. However, based on our observations, MSH3 is not essential, as lack of MSH3 did not affect cellular response to cisplatin within our experimental conditions. Our previous studies show that Pol β driven mutagenesis is responsible for generation of a mismatch near ICL sites. As MutSα complex i.e. MSH2-MSH6 are required to process a single base mismatch, differential recognition patterns of MutSα and MutSβ might be responsible for differential requirement of MSH6 and MSH3 in mediating cisplatin cytotoxicity. MSH6 deficiency has been known to cause cisplatin resistance [31]. Various models have been proposed to explain the resistance mechanism. There have been studies that link futile cycles of mismatch repair of cisplatin intrastrand adduct with increased sensitivity [10]. Some other studies have shown that MSH6 is essential to signal apoptosis. Yang et al have shown that MSH6 T1219D missense mutation causes the loss of mismatch repair function while retaining ability to signal apoptotic response followed by treatment with DNA damaging agents [32]. As compared to MSH6 knockout cells these mutants remain sensitive to cisplatin indicating that MMR proteins play a vital role in sensing and signaling the cell death. These studies have been attributed to all kinds of adducts formed by cisplatin overall. However, we found that MSH6 knockdown resulted in the increased repair of cisplatin ICLs and ICL induced DNA double strand breaks. It will be interesting to study whether MSH6 T1219D mutation has any effect on cisplatin ICL repair. Biochemical understanding of whether this mutant protein can sense the mismatch and whether it stays bound to the damaged site can provide useful information for the sensitive phenotype displayed by these cells in response to cisplatin. In conclusion, our results suggest that the drug sensitizing effects of MMR proteins are dependent on the processing of cisplatin ICLs and not intrastrand adducts. Also, MutSα and not MutSβ is required to maintain a cisplatin sensitive phenotype. MutSα and MutSβ recognize and process different mismatch repair substrates [34]. This difference in substrate recognition of MutSα and MutSβ may be responsible for their differential role in mediating cisplatin cytotoxicity. Single base mismatches generated by Polβ driven mutagenesis might be responsible for the recruitment of MutSα near the ICL sites and its role in maintaining a cisplatin sensitive phenotype.
Inactivation of MLH1 has been shown to give rise to cisplatin resistance [26]. In our studies, knockdown of MLH1 in Polβ deficient cells did not give rise to any additional increase in the degree of resistance, suggesting that these two proteins are involved in the same mechanism to mediate cisplatin cytotoxicity. The difference in the sensitivities could be correlated with the difference in the repair rates of cisplatin ICLs and ICL induced DSBs. These results suggest that following MutSα binding to a Polβ induced mismatch at a cisplatin ICL site, the downstream MMR processing events are required to maintain cisplatin sensitivity. We hypothesized that the MMR pathway tries to repair the mismatch that is generated due to error prone DNA synthesis by Polβ at a cisplatin ICL. This mismatch repairing ability is dependent upon the ATPase domain of MLH1, and this activity is essential for the initiation of the repair processes that recruit downstream DNA repair proteins and facilitate the removal of the mismatch. To understand the importance of these processes in response to cisplatin, we utilized MLH1 ATPase mutant cell lines. MLH1 missense mutations form about 65% of all the MMR mutations associated with HNPCC syndrome [35]. Many of these mutations occur in the ATPase domain of this protein giving rise to MMR deficiency and a high level of microsatellite instability [36]. S44F mutations in the ATPase domain have frequently been associated with the HNPCC syndrome [35]. S44 is a critical amino acid residue for MLH1 function as 18 out of 19 possible substitutions gave a loss of MMR activity [36]. This residue has no known biochemical function but it is located in the conserved ATP binding motif I. Mutations in this region are predicted to dislocate a conserved glutamic acid residue (E34) which is important for ATP binding and hydrolysis [36]. In cell lines used in these studies, these mutations did not have any significant effect on the level of mutant MLH1 protein. The absence of ATPase activity led to a cisplatin resistant phenotype, indicating that the mismatch repairing ability is essential to maintain a cisplatin sensitive phenotype. Previous studies have shown that the MLH1 G67R mutation still retains drug sensitivity despite having a defect in the repair capacity [26]. The G67R MLH1 mutation is located within the ATP binding motif II and also results in compromised MMR [26, 41]. Unlike S44F, however, the G67R substitution decreases stability of MLH1, reduces its nuclear accumulation and appears to disrupt its interactions with the PMS2 endonuclease in human cells [41]. The studies with G67R pointed toward a role for MLH1 in the activation of processes that directly signal apoptosis. The futile cycle model involves the futile repair of the mismatch generated in response to alkylating agents, which gives rise to strand breaks ultimately leading to cell death [8]. In our studies, the MLH1 deficient and MLH1 ATPase mutant cells showed a resistant phenotype in response to the alkylating agent MNNG, where the expression of MGMT in the ATPase mutant cells was comparable to that in the WT cells (data not shown). On the other hand, the second model involves the role of MMR proteins as DNA damage sensors to directly signal apoptosis in response to alkylating and platinating agents [11, 26, 27, 32, 37]. PMS2 binds to MLH1 and forms MutLα. This complex is involved in all MMR processes. In response to cisplatin treatment, PMS2 has been shown to interact with p73 to signal apoptosis [38, 39]. ATP binding to MLH1 has been shown to enhance the interaction between MLH1 and PMS2 [40]. Consistent with other studies [41], the S44 ATPase mutations in MLH1 did not have any effect on the stability of PMS2 (Supplementary Figure S8A) suggesting that the signaling function of PMS2, if any, remains unaffected. Our studies suggest that the MMR protein binding near the cisplatin ICL site blocks the productive repair of cisplatin ICLs and in turn results in a sensitive phenotype. The ATPase mutant MLH1 protein lacks the repair capacity but retains its function as a DNA damage sensor. Cell survival studies in the ATPase mutant cell lines show that this activity is essential for mediating cisplatin cytotoxicity. This would predict that either MLH1 does not have any DNA damage signaling role in response to cisplatin ICLs or ATPase mutants are not sequestered at the damage site long enough to trigger DNA damage signaling as they are unable to repair the damage. These are possibilities that can be examined in future studies. The current study shows that MLH1 ATPase S44L and S44F mutations behave differently as compared to the MLH1 ATPase G67R mutation studied before, suggesting that the different MLH1 ATPase mutations respond differently to cisplatin therapy.
Initially we set out to differentiate between the futile repair cycle model and the direct DNA damage-signaling model involving MMR proteins in response to cisplatin ICLs. However, one caveat here is that the specific domains in the MMR proteins that are required for sensing and signaling DNA damage are still unclear. Therefore, we still cannot make any clear conclusions about the role of direct signaling by MMR proteins in the development of cisplatin resistance. However, considering that the MMR deficient ATPase mutants showed increased cisplatin resistance, the mismatch repair ability and MMR processing near the cisplatin ICL sites appears to play a crucial role in maintaining cisplatin sensitivity. When we looked at how these mutations affect ICL repair by using an alkaline comet assay, we found that these mutant cells as well as MLH1 null cells showed increased repair of cisplatin ICLs and ICL-induced DSBs. These results suggest that the repair activities by MMR proteins or the MMR processing near the ICL are essential to maintain ICLs on the DNA and therefore, to maintain a cisplatin sensitive phenotype. This increased repair of ICLs can be due to reduced binding of the MLH1 mutant proteins at these sites due to an inability to process and repair the mismatch. This, in turn, results in increased repair of cisplatin ICLs through an enhanced accessibility of NER and HR proteins to the ICL site. On the other hand, the increased repair of ICLs could also be due to increased expression of NER and HR proteins involved in the repair of ICLs. In accordance, some recent studies have shown that the increased resistance in the MSH3 proficient cells is the result of increased expression of XPF and ERCC1 proteins known to be involved in ICL repair [15]. In addition, various studies have shown that increased expression of these proteins is responsible for the development of cisplatin and oxaliplatin resistance [42, 43]. To rule this out, we assessed the expression of XPF and ERCC1 proteins for a period of 0 to 72 hr after cisplatin treatment in MLH1 ATPase mutant cells. We did not see any change in the expression levels of these proteins in cisplatin treated (Supplementary Figure S8B) or in untreated cells (data not shown), which suggests that enhanced HR and NER protein expression is not responsible for the cisplatin resistance observed. In addition, we also checked for expression of XPF in the absence of MSH3 after cisplatin treatment. We did not see any significant change in the expression patterns of XPF followed by transient MSH3 knockdown (Supplementary Figure S9). These studies indicate that as the MMR proteins process the Polβ induced mismatches near the cisplatin ICL site, they block productive ICL repair and give rise to cisplatin sensitivity. In turn, the lack of MMR proteins provides enhanced accessibility to cisplatin ICL DNA repair proteins, which results in increased repair of cisplatin ICLs and development of cisplatin resistance.
Supplementary Material
Highlights.
MSH6 deficiency gives rise to increased ICL repair and cisplatin resistance.
MSH3 deficient cells remain sensitive to cisplatin.
Lack of MLH1 leads to enhanced ICL repair and cisplatin resistance.
ATPase activity of MLH1 is required to maintain a cisplatin sensitive phenotype.
A functional MMR pathway is essential to maintain cisplatin sensitivity.
Acknowledgments
Financial support: The National Institutes of Health (GM088249) to SMP, (ES008786) to AZ and (CA148629) to RWS.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of interest:
None declared. RWS is a scientific consultant for Trevigen, Inc.
The authors’ state that there are no conflicts of. The work is not submitted to any other journal.
References
- 1.Iyer RR, Pluciennik A, Burdett V, Modrich PL. DNA mismatch repair: Functions and mechanisms. Chemical Reviews. 2006;106:302–323. doi: 10.1021/cr0404794. [DOI] [PubMed] [Google Scholar]
- 2.Fink D, Zheng H, Nebel S, Norris PS, Aebi S, Lin TP, Nehme A, Christen RD, Haas M, MacLeod CL, Howell SB. In vitro and in vivo resistance to cisplatin in cells that have lost DNA mismatch repair. Cancer Research. 1997;57:1841–1845. [PubMed] [Google Scholar]
- 3.Wang JYJ, Edelmann W. Mismatch repair proteins as sensors of alkylation DNA damage. Cancer Cell. 2006;9:417–418. doi: 10.1016/j.ccr.2006.05.013. [DOI] [PubMed] [Google Scholar]
- 4.Pani E, Stojic L, El-Shemerly M, Jiricny J, Ferrari S. Mismatch repair status and the response of human cells to cisplatin. Cell Cycle. 2007;6:1796–1802. doi: 10.4161/cc.6.14.4472. [DOI] [PubMed] [Google Scholar]
- 5.Fink D, Nebel S, Aebi S, Zheng H, Cenni B, Nehme A, Christen RD, Howell SB. The role of DNA mismatch repair in platinum drug resistance. Cancer Research. 1996;56:4881–4886. [PubMed] [Google Scholar]
- 6.Fink D, Aebi S, Howell SB. The role of DNA mismatch repair in drug resistance, Clinical. Cancer Research. 1998;4:1–6. [PubMed] [Google Scholar]
- 7.Jiricny J. The multifaceted mismatch-repair system. Nature Reviews Molecular Cell Biology. 2006;7:335–346. doi: 10.1038/nrm1907. [DOI] [PubMed] [Google Scholar]
- 8.Mojas N, Lopes M, Jiricny J. Mismatch repair-dependent processing of methylation damage gives rise to persistent single-stranded gaps in newly replicated DNA. Genes Dev. 2007;21:3342–3355. doi: 10.1101/gad.455407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yamada M, ORegan E, Brown R, Karran P. Selective recognition of a cisplatinDNA adduct by human mismatch repair proteins. Nucleic Acids Research. 1997;25:491–495. doi: 10.1093/nar/25.3.491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mello JA, Acharya S, Fishel R, Essigmann JM. The mismatch-repair protein hMSH2 binds selectively to DNA adducts of the anticancer drug cisplatin. Chemistry & Biology. 1996;3:579–589. doi: 10.1016/s1074-5521(96)90149-0. [DOI] [PubMed] [Google Scholar]
- 11.Yoshioka KI, Yoshioka Y, Hsieh P. ATR kinase activation mediated by MutS alpha and MutL alpha in response to cytotoxic O-6-methylguanine adducts. Molecular Cell. 2006;22:501–510. doi: 10.1016/j.molcel.2006.04.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhu G, Lippard SJ. Photoaffinity Labeling Reveals Nuclear Proteins That Uniquely Recognize Cisplatin-DNA Interstrand Cross-Links. Biochemistry. 2009;48:4916–4925. doi: 10.1021/bi900389b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Takahashi M, Koi M, Balaguer F, Boland C, Goel A. MSH3 Mediates Sensitization of Colorectal Cancer Cells to Cisplatin, Oxaliplatin, and a Poly(ADP-ribose) Polymerase Inhibitor. Journal of Biological Chemistry. 2011;286 doi: 10.1074/jbc.M110.198804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Park JM, Huang S, Tougeron D, Sinicrope FA. MSH3 Mismatch Repair Protein Regulates Sensitivity to Cytotoxic Drugs and a Histone Deacetylase Inhibitor in Human Colon Carcinoma Cells. Plos One. 2013;8:e65369. doi: 10.1371/journal.pone.0065369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tentori L, Muzi A, Dorio AS, Dolci S, Campolo F, Vernole P, Lacal PM, Praz F, Graziani G. MSH3 expression does not influence the sensitivity of colon cancer HCT116 cell line to oxaliplatin and poly(ADP-ribose) polymerase (PARP) inhibitor as monotherapy or in combination. Cancer Chemother Pharmacol. 2013;72:117–125. doi: 10.1007/s00280-013-2175-0. [DOI] [PubMed] [Google Scholar]
- 16.Kothandapani A, Dangeti VSMN, Brown AR, Banze LA, Wang XH, Sobol RW, Patrick SM. Novel Role of Base Excision Repair in Mediating Cisplatin Cytotoxicity. Journal of Biological Chemistry. 2011;286:14564–14574. doi: 10.1074/jbc.M111.225375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kothandapani A, Sawant A, Dangeti VSMN, Sobol RW, Patrick SM. Epistatic role of base excision repair and mismatch repair pathways in mediating cisplatin cytotoxicity. Nucleic Acids Research. 2013;41:7332–7343. doi: 10.1093/nar/gkt479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Trivedi RN, Wang XH, Jelezcova E, Goellner EM, Tang JB, Sobol RW. Human methyl purine DNA glycosylase and DNA polymerase beta expression collectively predict sensitivity to temozolomide. Molecular Pharmacology. 2008;74:505–516. doi: 10.1124/mol.108.045112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Reynolds MF, Peterson-Roth EC, Bespalov IA, Johnston T, Gurel VM, Menard HL, Zhitkovich A. Rapid DNA Double-Strand Breaks Resulting from Processing of Cr-DNA Cross-Links by Both MutS Dimers. Cancer Research. 2009;69:1071–1079. doi: 10.1158/0008-5472.CAN-08-2306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Arora S, Kothandapani A, Tillison K, Kalman-Maltese V, Patrick SM. Downregulation of XPF-ERCC1 enhances cisplatin efficacy in cancer cells. Dna Repair. 2010;9:745–753. doi: 10.1016/j.dnarep.2010.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.De Silva IU, Mchugh PJ, Clingen PH, Hartley JA. Defects in interstrand cross-link uncoupling do not account for the extreme sensitivity of ERCC1 and XPF cells to cisplatin. Nucleic Acids Research. 2002;30:3848–3856. doi: 10.1093/nar/gkf479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zdraveski ZZ, Mello JA, Farinelli CK, Essigmann JM, Marinus MG. MutS preferentially recognizes cisplatin- over oxaliplatin-modified DNA. Journal of Biological Chemistry. 2002;277:1255–1260. doi: 10.1074/jbc.M105382200. [DOI] [PubMed] [Google Scholar]
- 23.Eastman A, Schulte N. Enhanced Dna-Repair As A Mechanism of Resistance to Cis-Diamminedichloroplatinum(Ii) Biochemistry. 1988;27:4730–4734. doi: 10.1021/bi00413a022. [DOI] [PubMed] [Google Scholar]
- 24.De Silva IU, Mchugh PJ, Clingen PH, Hartley JA. Defects in interstrand cross-link uncoupling do not account for the extreme sensitivity of ERCC1 and XPF cells to cisplatin. Nucleic Acids Res. 2002;30:3848–3856. doi: 10.1093/nar/gkf479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wu Q, Vasquez KM. Human MLH1 Protein Participates in Genomic Damage Checkpoint Signaling in Response to DNA Interstrand Crosslinks, while MSH2 Functions in DNA Repair. Plos Genetics. 2008;4 doi: 10.1371/journal.pgen.1000189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Avdievich E, Reiss C, Scherer SJ, Zhang Y, Maier SM, Jin B, Hou H, Jr, Rosenwald A, Riedmiller H, Kucherlapati R, Cohen PE, Edelmann W, Kneitz B. Distinct effects of the recurrent Mlh1(G67R) mutation on MMR functions, cancer, and meiosis. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:4247–4252. doi: 10.1073/pnas.0800276105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lin DP, Wang YX, Scherer SJ, Clark AB, Yang K, Avdievich E, Jin B, Werling U, Parris T, Kurihara N, Umar A, Kucherlapati R, Lipkin M, Kunkel TA, Edelmann W. An Msh2 point mutation uncouples DNA mismatch repair and apoptosis. Cancer Research. 2004;64:517–522. doi: 10.1158/0008-5472.can-03-2957. [DOI] [PubMed] [Google Scholar]
- 28.Wu Q, Christensen LA, Legerski RJ, Vasquez KM. Mismatch repair participates in error-free processing of DNA interstrand crosslinks in human cells. Embo Reports. 2005;6:551–556. doi: 10.1038/sj.embor.7400418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wynne P, Newton C, Ledermann J, Olaitan A, Mould T, Hartley J. Enhanced repair of DNA interstrand crosslinking in ovarian cancer cells from patients following treatment with platinum-based chemotherapy. British Journal of Cancer. 2007;97:927–933. doi: 10.1038/sj.bjc.6603973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Clingen P, Wu J, Miller J, Mistry N, Chin F, Wynne P, Prise K, Hartley J. Histone H2AX phosphorylation as a molecular pharmacological marker for DNA interstrand crosslink cancer chemotherapy. Biochemical Pharmacology. 2008;76:19–27. doi: 10.1016/j.bcp.2008.03.025. [DOI] [PubMed] [Google Scholar]
- 31.Vaisman A, Varchenko M, Umar A, Kunkel TA, Risinger JI, Barrett JC, Hamilton TC, Chaney SG. The role of hMLH1, hMSH3, and hMSH6 defects in cisplatin and oxaliplatin resistance: Correlation with replicative bypass of platinum-DNA adducts. Cancer Research. 1998;58:3579–3585. [PubMed] [Google Scholar]
- 32.Yang GZ, Scherer SJ, Shell SS, Yang K, Kim M, Lipkin M, Kucherlapati R, Kolodner RD, Edelmann W. Dominant effects of an Msh6 missense mutation on DNA repair and cancer susceptibility. Cancer Cell. 2004;6:139–150. doi: 10.1016/j.ccr.2004.06.024. [DOI] [PubMed] [Google Scholar]
- 33.Parker RJ, Eastman A, Bostick-Bruton F, Reed E. Acquired cisplatin resistance in human ovarian cancer cells is associated with enhanced repair of cisplatinDNA lesions and reduced drug accumulation. J Clin Invest. 1991;87:772–777. doi: 10.1172/JCI115080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Genschel J, Littman SJ, Drummond JT, Modrich P. Isolation of MutSbeta from human cells and comparison of the mismatch repair specificities of MutSbeta and MutSalpha. J Biol Chem. 1998;273:19895–19901. doi: 10.1074/jbc.273.31.19895. [DOI] [PubMed] [Google Scholar]
- 35.Bronner CE, Baker SM, Morrison PT, Warren G, Smith LG, Lescoe MK, Kane M, Earabino C, Lipford J, Lindblom A, Tannergard P, Bollag RJ, Godwin AR, Ward DC, Nordenskjold M, Fishel R, Kolodner R, Liskay RM. Mutation in the Dna Mismatch Repair Gene Homolog Hmlh1 Is Associated with Hereditary Nonpolyposis Colon-Cancer. Nature. 1994;368:258–261. doi: 10.1038/368258a0. [DOI] [PubMed] [Google Scholar]
- 36.Ellison AR, Lofing J, Bitter GA. Human MutL homolog (MLH1) function in DNA mismatch repair: a prospective screen for missense mutations in the ATPase domain. Nucleic Acids Research. 2004;32:5321–5338. doi: 10.1093/nar/gkh855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Topping RP, Wilkinson JC, Scarpinato KD. Mismatch Repair Protein Deficiency Compromises Cisplatin-induced Apoptotic Signaling. Journal of Biological Chemistry. 2009;284:14029–14039. doi: 10.1074/jbc.M809303200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shimodaira H, Yoshioka-Yamashita A, Kolodner RD, Wang JYJ. Interaction of mismatch repair protein PMS2 and the p53-related transcription factor p73 in apoptosis response to cisplatin. Proceedings of the National Academy of Sciences of the United States of America. 2003;100:2420–2425. doi: 10.1073/pnas.0438031100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Marinovic-Terzic I, Yoshioka-Yamashita A, Shimodaira H, Avdievich E, Hunton IC, Kolodner RD, Edelmann W, Wang JY. Apoptotic function of human PMS2 compromised by the nonsynonymous single-nucleotide polymorphic variant R20Q. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:13993–13998. doi: 10.1073/pnas.0806435105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tomer G, Buermeyer AB, Nguyen MM, Liskay RM. Contribution of human mlh1 and pms2 ATPase activities to DNA mismatch repair. J Biol Chem. 2002;277:21801–21809. doi: 10.1074/jbc.M111342200. [DOI] [PubMed] [Google Scholar]
- 41.Raevaara TE, Korhonen MK, Lohi H, Hampel H, Lynch E, Lonnqvist KE, Holinski-Feder E, Sutter C, McKinnon W, Duraisamy S, Gerdes AM, Peltomaki P, Kohonen-Ccorish M, Mangold E, Macrae F, Greenblatt M, de la Chapelle A, Nystrom M. Functional significance and clinical phenotype of nontruncating mismatch repair variants of MLH1. Gastroenterology. 2005;129:537–549. doi: 10.1016/j.gastro.2005.06.005. [DOI] [PubMed] [Google Scholar]
- 42.Usanova S, Piee-Staffa A, Sied U, Thomale J, Schneider A, Kaina B, Koeberle B. Cisplatin sensitivity of testis tumour cells is due to deficiency in interstrandcrosslink repair and low ERCC1-XPF expression. Molecular Cancer. 2010;9 doi: 10.1186/1476-4598-9-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Arora S, Kothandapani A, Tillison K, Kalman-Maltese V, Patrick SM. Downregulation of XPF-ERCC1 enhances cisplatin efficacy in cancer cells. Dna Repair. 2010;9:745–753. doi: 10.1016/j.dnarep.2010.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.