

Abstract
There are two major types of ocular neovascularization that affect the retina, retinal neovascularization (NV) and subretinal or choroidal NV. Retinal NV occurs in a group of diseases referred to as ischemic retinopathies in which damage to retinal vessels results in retinal ischemia. Most prevalent of these are diabetic retinopathy and retinal vein occlusions. Subretinal and choroidal NV occur in diseases of the outer retina and Bruch’s membrane, the most prevalent of which is age-related macular degeneration. Numerous studies in mouse models have helped to elucidate the molecular pathogenesis underlying retinal, subretinal, and choroidal NV. There is considerable overlap because the precipitating event in each is stabilization of hypoxia inducible factor-1 (HIF-1) which leads to upregulation of several hypoxia-regulated gene products, including vascular endothelial growth factor (VEGF), angiopoietin 2, vascular endothelial-protein tyrosine phosphatase (VE-PTP), and several others. Stimulation of VEGF signaling and suppression of Tie2 by angiopoietin 2 and VE-PTP are critical for sprouting of retinal, subretinal, and choroidal NV, with perturbation of Bruch’s membrane also needed for the latter. Additional HIF-1-regulated gene products cause further stimulation of the NV. It is difficult to model macular edema in animals and therefore proof-of-concept clinical trials were done and demonstrated that VEGF plays a central role and that suppression of Tie2 is also important. Neutralization of VEGF is currently the first line therapy for all of the above disease processes, but new treatments directed at some of the other molecular targets, particularly stabilization of Tie2, are likely to provide additional benefit for subretinal/choroidal NV and macular edema. In addition, the chronicity of these diseases as well as the implication of VEGF as a cause of retinal nonperfusion and progression of background diabetic retinopathy make sustained delivery approaches for VEGF antagonists a priority.
Keywords: Angiogenesis, age-related macular degeneration, diabetic retinopathy, hypoxia-inducible factor-1, vascular endothelial growth factor, vascular endothelial-protein tyrosine phosphatase, TIE2, angiopoietins, platelet-derived growth factor
1.1 Retinal blood supply
The retina is supplied by the retinal and choroidal vasculatures. The central retinal artery enters the eye through the optic nerve, branches on the surface of the nerve, and sends progressively branching arterioles along the surface of the retina to its anterior border. The surface arterioles send numerous penetrating branches to form the intermediate and deep capillary beds which supply the inner 2/3 of the retina. Retinal capillaries drain into retinal venules that retrace the path of the arterioles into progressively larger branch veins that enter the central retinal vein that exits the eye through the optic nerve. The endothelial cells of retinal vessels have tight junctions and specialized vesicular transport that limit efflux of plasma and its components into the interstitial space of the retina constituting the inner blood-retinal barrier. The outer 1/3 of the retina consisting of the outer nuclear layer containing photoreceptor cell bodies, photoreceptor inner segments, and photoreceptor outer segments is completely devoid of blood vessels (Figure 1). Erythrocytes coursing through blood vessels contain hemoglobin which absorbs light and so the absence of blood vessels immediately anterior to photoreceptor outer segments is an important adaptation for visual function, but presents a unique challenge to the supply of oxygen and nutrients to photoreceptors. The challenge is heightened by the high metabolic activity of photoreceptors. The choroidal circulation surmounts this challenge because it has very high blood flow and forms the choriocapillaris, a dense plexus of fenestrated capillaries that allow plasma to bath the retinal pigmented epithelium (RPE) which contains apical tight junctions and specialized vesicular transport constituting the outer blood-retinal barrier.
Figure 1. Schematic showing the vascular supply of the retina.

The retinal arteries branch to form the superficial capillary bed near the surface of the retina and send penetrating branches to form the intermediate and deep capillaries. Retinal vessels supply the inner two-thirds of the retina with oxygen and nutrients. The outer third of the retina which consists of photoreceptor outer and inner segments and cells bodies is avascular. It receives oxygen and nutrients from the choroidal circulation. Large choroidal vessels branch and become progressively smaller until they form the choriocapillaris which is fenestrated and allows plasma to pool along Bruch’s membrane. The RPE, which has barrier characteristics prevents fluid from entering the outer retina but allows oxygen and nutrients to enter.
2.1 Retinal and choroidal vascular diseases
The unique blood supply of the retina is an elegant solution to the challenges rendered by the need to eliminate blood vessels immediately anterior to photoreceptor outer segments, but is also the source of vulnerabilities. It requires a high flow vascular system in close proximity to the avascular outer retina ending in fenestrated capillaries allowing plasma free access to the RPE which has both epithelial and endothelial functions. The distance over which oxygen and nutrients must travel is greater than that in most tissues. The situation is further complicated by the need to maintain a narrow range of ionic concentrations in the extracellular space of retinal neurons so that neurotransmission is not perturbed. This requires RPE and retinal vascular endothelial cells to maintain their highly specialized barrier characteristics in order for normal retinal function to be maintained. Since the demands on RPE and retinal vascular cells are high, they have high vulnerability to disease and consequences of their dysfunction are large. Pathologies that affect retinal endothelial cells are classified as retinal vascular diseases and some pathologies that affect the RPE result in choroidal vascular diseases.
2.2 Retinal vascular diseases
Pathologic processes that damage retinal vessels can result in vessel closure and hence retinal ischemia and/or compromise of barrier characteristics of retinal endothelial cells resulting in excessive vascular leakage. Severe retinal ischemia results in retinal neovascularization (NV) which grows into the vitreous cavity and can cause traction retinal detachment. Diseases in which this occurs are referred to as ischemic retinopathies and include diabetic retinopathy, retinal vein occlusions (RVOs), retinopathy of prematurity (ROP), and sickle cell retinopathy. The mechanism by which hyperglycemia leads to closure of retinal vessels and retinal ischemia is uncertain and because years of hyperglycemia are required, it has not been possible to produce an animal model of proliferative diabetic retinopathy. However, the demonstration that in new born kittens, high levels of inspired oxygen cause retinal vessel closure and retinal NV helped to elucidate the mechanism of ROP and provide a template for reliable models of ischemic retinopathy (Patz et al., 1953). This model has been reproduced in dogs, rats, and mice. Due to the relatively low cost of mice and the advanced understanding of mouse genetics compared to other species, the mouse model of oxygen-induced ischemic retinopathy (Smith et al., 1994) has been widely used to elucidate molecular signals involved in retinal NV. The basis of the molecular pathogenesis is that retinal blood vessels develop after birth in mice, so that initially the retina is relatively hypoxic which contributes to upregulation of the hypoxia-regulated gene product, vascular endothelial growth factor (VEGF), a critical stimulus for retinal vascular development. Placement of neonatal mice into an environment high in oxygen reduces levels of VEGF in the retina, halting vascular development, and causing regression of many of the newly developed blood vessels, because their endothelial cells, unlike the endothelial cells of mature vessels, are dependent upon VEGF for survival (Alon et al., 1995; Stone et al., 1995). When mice are returned to room air, the poorly perfused retina becomes hypoxic causing excessive production of VEGF which overshoots the normal level and stimulates retinal NV (Pierce et al., 1996). Figure 2A shows an ocular section through a normal mouse retina stained with Griffonia simplicifolia (GSA) lectin that selective stains vascular cells. The superficial capillaries are seen adjacent to ganglion cells near the surface of the retina, intermediate capillaries are near the inner border of the inner nuclear layer and the deep capillaries are along the outer border of the inner nuclear layer. In the retinas of mice with oxygen-induced ischemic retinopathy, the capillaries within the retina are dilated and neovascularization is present on the surface of the retina extending into the vitreous cavity (Figure 2B). Retinal NV and hyaloid vessels stain more intensely with GSA than normal retinal vessels, so that they are selectively stained by brief incubations of retinal flat mounts with FITC-labeled GSA (Figure 3)(Shen et al., 2014). This is advantageous for quantification of the area of NV by image analysis because the software recognizes both retinal NV and hyaloid vessels and the area of the hyaloid vessels is easily subtracted.
Figure 2. Ocular sections from mouse models of ocular neovascularization (NV).

(A) A normal mouse retina histochemically stained with Griffonia simplicifolia lectin that selectively stains vascular cells and counter-stained to show other retinal cells illustrates the 3 capillary beds of the retina, the superficial capillaries near the surface, the intermediate capillaries at the inner border of the inner nuclear layer, and the deep capillaries at the outer border of the inner nuclear layer. There are also some penetrating vessels showing connections or part of connections between the beds.
(B) An ocular section from a mouse with oxygen-induced ischemic retinopathy shows dilation of retinal vessels making it easier to see connections between the three capillary beds. There is retinal NV on the surface of the retina extending into the vitreous cavity (arrows).
(C) This is an ocular section from the eye of a rho/VEGF transgenic mouse in which the rhodopsin promoter drives expression of VEGF in photoreceptors. These mice sprout new vessels from the deep capillaries of the retina resulting in new vessels that penetrate through the outer nuclear layer of the retina into the subretinal space. This is a model of retinal angiomatous proliferation which occurs in patients with neovascular age-related macular degeneration.
(D) This is an ocular section from a mouse with choroidal NV after laser-induced rupture of Bruch’s membrane. The arrow shows a vessel extending from the choroid through the rupture in Bruch’s membrane and retinal pigmented epithelium into the subretinal space where it connects to a large convoluted network of new vessels, many of which are cut in cross section to show their lumens (astericks). The superior margin of the choroidal NV that borders the photoreceptors is shown by the arrowheads.
Figure 3. Selective staining of neovascularization (NV) in retina of mouse with oxygen-induced ischemic retinopathy.
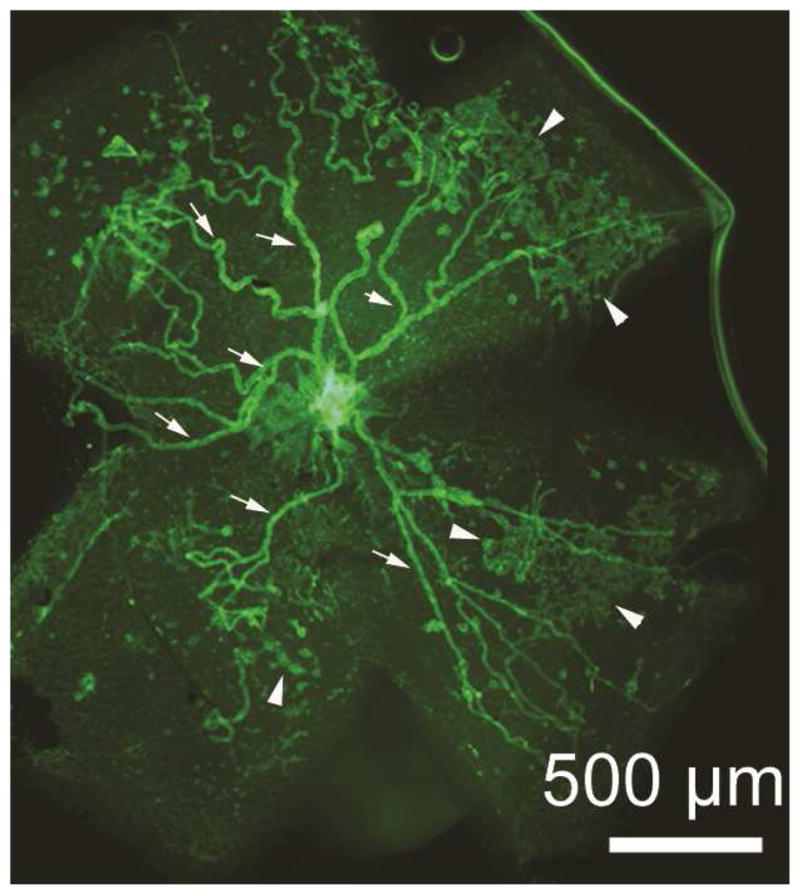
A retinal from a mouse with ischemic retinopathy was stained with FITC-labeled Griffonia simplicifolia lectin for 20 minutes which stains retinal NV (arrowheads) and hyaloid vessels (arrows) but not the pre-existent retinal vessels which facilitates quantification of NV by image analysis. After radial cuts, the retina was flat-mounted allowing visualization of the entire retina and measurement of all retinal NV.
Inhibition of VEGF suppresses retinal NV indicating that high levels of VEGF are necessary for its occurrence and maintenance (Aiello et al., 1995). To determine if high levels of VEGF are sufficient to cause retinal NV, rho/VEGF transgenic mice were generated in which the rhodopsin promoter was used to drive expression of VEGF165 in photoreceptors (Okamoto et al., 1997). In rho/VEGF mice, NV sprouted from the deep capillary bed of the retina (Figure 2C), but not from the intermediate or deep capillaries, indicating that another critical stimulus for retinal NV might be constitutively expressed in the vicinity of the deep capillary bed, but not the superficial or intermediate capillaries. This difference between the capillary beds within the retina indicates a very important principle in vascular biology. A vasoactive protein may have different effects on different vascular beds because the microenvironment may differ in important ways. For this reason, mechanisms involved in different types of NV may differ. There are similarities and differences involved in the mechanisms of retinal and choroidal NV, and they have some similarities and greater differences with NV in neoplastic tissues (reviewed in (Campochiaro, 2006)). It is useful to discuss the contribution of various proteins to retinal and choroidal NV together to point out similarities and differences.
2.3 Choroidal vascular diseases
There is a great deal of interdependence between photoreceptors, RPE cells, and the choriocapillaris. The RPE promotes a vascular environment along its basal surface and an avascular environment along its apical surface. Vascular endothelial cells require survival factors that can originate from extracellular matrix, surrounding cells, or plasma. Vascular endothelial growth factor (VEGF) is an important mediator of paracrine and endocrine trophic support for endothelial cells (Alon et al., 1995). Fenestrated endothelial cells are particularly dependent upon VEGF (Inai et al., 2004; Kamba et al., 2005) and the choriocapillaris is no exception. Development of the choriocapillaris requires RPE-derived VEGF (Mameros et al., 2005) and maintenance of the choriocapillaris after development requires production of soluble isoforms of VEGF by the RPE (Saint-Geniez et al., 2009). Conditional knockout of all VEGF-A isoforms in RPE cells of adult mice results in rapid death of the choriocapillaris followed by death of the RPE and photoreceptors (Kurihara et al., 2012). RPE cells are polarized cells and under normal circumstances secrete VEGF only from their basolateral surface which supports the choriocapillaris. The basal lamina of the RPE is the innermost layer of Bruch’s membrane, a complex extracellular matrix structure that provides mechanical support, physical separation from the choriocapillaris, and important signals for maintenance of polarization. When in culture, RPE cells must be seated on preformed basal lamina to form polarized monolayers or they must secrete their own resulting in gradual polarization over time (Campochiaro and Hackett, 1993; Davis et al., 1995). Abnormalities of Bruch’s membrane cause RPE dysfunction; diffuse thickening of Bruch’s membrane and focal deposits called drusen occur in patients with age-related macular degeneration (AMD) and large drusen increase risk of both forms of advanced AMD, choroidal neovascularization (CNV) and geographic atrophy (GA) (Age-Related, 2001; Klein et al., 2008). Cultured RPE cells grown on matrix containing components of drusen produce increased levels VEGF (Mousa et al., 1999). However, transgenic mice with inducible expression of VEGF in RPE cells do not develop CNV, demonstrating that high expression of VEGF by RPE cells is not sufficient to cause CNV (Oshima et al., 2004b). Several diseases in which CNV occurs have abnormalities and/or defects in Bruch’s membrane including AMD, pathologic myopia, pseudoxanthoma elasticum with angioid streaks, and Sorsby Dystrophy. Sorsby Dystrophy is due to a mutation in Timp3 which causes deposits of aggregated TIMP3 in Bruch’s membrane and is complicated by severe CNV and degeneration of the RPE and photoreceptors (Weber et al., 1994). Rupture of Bruch’s membrane with laser photocoagulation in primates, rats, or mice results in CNV (Dobi et al., 1989; Ryan, 1982; Tobe et al., 1998b). Seven or 14 days after rupture of Bruch’s membrane, ocular sections stained with GSA show vessels growing from the choroid into the subretinal space and collection of fluid under the retina (Figure 3D). Antagonists of VEGF strongly suppressed CNV in the mouse model suggesting that VEGF plays an important role in CNV just like it does in retinal NV (Kwak et al., 2000). This was confirmed by the demonstration that ranibizumab suppresses CNV in primates with rupture of Bruch’s membrane (Kryzstolik et al., 2002) and that aflibercept (VEGF-trap) suppresses CNV in the mouse model of CNV (Saishin et al., 2003). These preclinical studies have been confirmed in large multicenter clinical trials in patients with NVAMD demonstrating that results in the primate and mouse models can be predictive of results in humans (Brown et al., 2006; Heier et al., 2012; Rosenfeld et al., 2006). The mouse model is now widely used to screen drugs for potential therapeutic activity for NVAMD.
Transgenic rho/VEGF mice demonstrated that increased levels of VEGF in the outer retina without any perturbation of Bruch’s membrane fails to cause CNV (Okamoto et al., 1997; Tobe et al., 1998a). However, the increased expression of VEGF in photoreceptors caused sprouting of NV from the deep capillary bed of the retina (Figure 2C). This provides a model of retinal angiomatous proliferation, which occurs in 30% of patients with NVAMD (Yannuzzi et al., 2001). Why does NV grow from the deep capillary bed, but not the intermediate or superficial capillary beds of the retina? Is it because the level of VEGF expression in rho/VEGF mice provides concentrations of VEGF sufficient to stimulate NV at the relatively nearby deep capillaries, but not the more remote intermediate and deep capillary beds? Experimental observations have suggested against this possibility; sustained release of VEGF165 in the vitreous cavity of primates caused severe vascular leakage and macular edema, but no NV (Ozaki et al., 1997) and intravitreous injection of an adenoviral vector expressing VEGF165 in adult mice resulted in high expression of VEGF at the surface of the retina, but no sprouting of NV from the superficial capillaries (Oshima et al., 2004c). In rho/VEGF mice, VEGF expression begins at postnatal day (P) 7 and sprouts of NV occur from the deep capillaries by P10. A second hypothesis was that another gene product is required in addition to VEGF for NV to occur, and that gene product is only expressed in the region of the deep capillary bed at P10. To test this hypothesis, double transgenic mice with doxycycline-inducible expression of VEGF were generated so that the timing of VEGF expression could be controlled (Ohno-Matsui et al., 2002). Two driver lines were utilized, one in which the reverse tetracycline transactivator (rtTA) was coupled to the opsin promoter (rtTA/opsin mice) and another in which the rtTA was coupled to the interphotoreceptor retinoid binding protein (IRBP) promoter (rtTA/IRBP mice) and each of these lines was crossed with transgenics that expressed the tetracycline response element (TRE) coupled to cDNA coding for VEGF165 (TRE/VEGF mice) to generated rtTA/opsin-TRE/VEGF and rtTA/IRBP-TRE/VEGF (shortened to Tet/opsin/VEGF and Tet/IRBP/VEGF) double transgenic mice. Tet/opsin/VEGF mice express VEGF in photoreceptors when doxycycline is given provided it is after P7 when constitutive expression of the opsin promoter begins, but because the IRBP promoter turns on prior to birth, Tet/IRBP/VEGF mice have inducible expression of VEGF even as early as P0. When optimal doses of doxycycline were given to adult Tet/opsin/VEGF mice or Tet/IRBP/VEGF mice, NV sprouted only from the deep capillary bed, but levels of VEGF were substantially higher than those in rho/VEGF mice, resulting in severe vascular leakage and exudative detachments (Figure 4) (Ohno-Matsui et al., 2002). When doxycycline was administered to Tet/IRBP/VEGF mice starting at P0, they developed NV sprouting from the superficial capillaries between P0 and P7, the intermediate capillaries between P4 and P7, and the deep capillaries at P7 and beyond (Oshima et al., 2004a). These time periods correspond to the windows of development for each of the capillary beds, respectively and suggest that at least one other gene product in addition to VEGF is needed for developmental sprouting or pathologic sprouting of new vessels in the retina.
Figure 4. Treatment of Tet/opsin/VEGF double transgenic mice with doxycycline results in exudative retinal detachments that can be imaged by fundus photography.
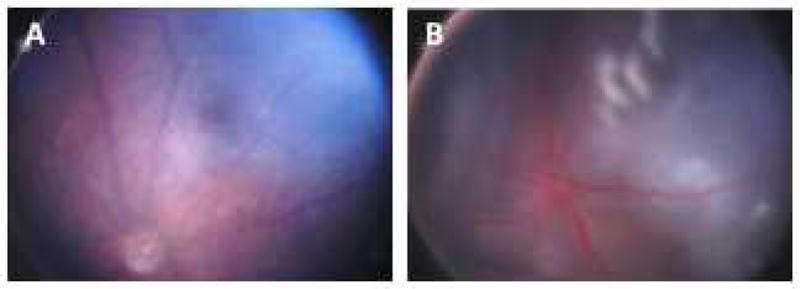
A male Tet/opsin/VEGF double transgenic mouse was given normal drinking water (A) and another was given water containing 2 mg/ml doxycycline in drinking water for 3 days (B). Fundus photography shows a normal retina in the mouse given normal drinking water (A) and an exudative retinal detachment due to severe VEGF-induced vascular leakage in the doxycycline-treated mouse (B). The entire retina is detached giving the retina an opalescent appearance with folds seen superiorly. Also the retinal vessels are dilated giving the vessels a dark red appearance.
3.1 Molecular Signals
3.1.1 VEGF
As is clear from sections 2.2 and 2.3, VEGF plays a critical role in retinal and subretinal/choroidal neovascularization. Its role in macular edema is discussed in sections 5.1.1 and 5.1.2. Recently it has been determined that VEGF has other activities that make it a therapeutic target for early stages of diabetic retinopathy and this is discussed in section 6.1.
It is important to note that there is controversy regarding the safety of intraocular injections of VEGF. Systemic suppression of VEGF in patients with metastatic cancer is complicated by thromboembolic and hemorrhagic events.(Elice et al., 2009; Eremina et al., 2008) Severely compromised endothelial cells overlying atherosclerotic plaque or in regions where vessels are damaged are more dependent upon VEGF for survival than normal endothelial cells. Neutralization of VEGF can result in death of these compromised endothelial cells causing the luminal surface to become thrombogenic and thereby increasing the likelihood of thrombosis. In regions where there is pre-existent damage to the other parts of the vessel wall, endothelial death can promote hemorrhage. It was thought that after intraocular injection of a VEGF-neutralizing protein, there would be not be enough suppression of systemic VEGF levels to increase risk of thrombotic or hemorrhagic events. However, phase 3 trials sponsored by Genentech consistently showed greater numbers of thromboembolic events in patients treated with 0.5mg ranibizumab compared with those treated with 0.3mg ranibizumab or sham. Although the differences were not statistically significant there was enough concern for Genentech to issue a letter warning of the possibility that in patients given intraocular injections of ranibizumab, there might be increased risk of thromboembolic events, particularly in patients with a history of a recent stroke. Patients with diabetes are at higher risk for thromboembolic events than nondiabetic patients and therefore in patients with DME, 0.3mg of ranibizumab was approved rather than 0.5mg. Whether there are difference among VEGF-neutralizing proteins with regard to risk of systemic vascular events after intraocular injection is uncertain, but since systemic suppression of VEGF is greater after intraocular injections of bevacizumab or aflibercept than that seen after intraocular injection of ranibizumab (Avery et al., 2014), they could be associated with a higher risk. Any difference in risk in patients treated with intraocular anti-VEGF agents compared to the baseline risk in the target population is so small that it would take a very large study to demonstrate its existence, and if there are differences among anti-VEGF agents, even larger studies would be needed to demonstrate that. Therefore, there are no data to guide treatment and physicians are left to decide whether they think it is plausible that there is an increased risk of systemic vascular events after intraocular anti-VEGF treatment and whether there is any way to mitigate risk if it exists.
There is also controversy as to whether there is any risk to the retina, RPE, and choroid from intraocular suppression of VEGF. Conditional knock out of VEGF in RPE cells causes atrophy of the choriocapillaris which in turn causes damage to the overlying RPE and retina (Kurihara et al., 2012). However, it is unlikely that intraocular injections of VEGF neutralizing proteins could ever cause sufficient suppression of RPE-derived VEGF to mimic a genetic knockout of VEGF and compromise the choriocapillaris, because there was no damage to the choriocapillaris, RPE, or retina in transgenic mice in which high expression of potent VEGF-neutralizing protein was driven in photoreceptor cells for up to 7 months (Ueno et al., 2008). Whether subretinal injection of an AAV vector that transduces RPE cells to produce high levels of a VEGF-neutralizing protein within the RPE can mimic a genetic knockout of VEGF in RPE is an unanswered question. Macular atrophy occurs in some patients with neovascular AMD treated with intraocular injections of VEGF-neutralizing proteins (Grunwald et al., 2012), but this is not due to damage to retina, RPE, or choriocapillaris from VEGF suppression, because it is never seen in patients with other disease processes treated with anti-VEGF agents. Instead, it occurs in areas of regression of choroidal neovascularization and suggests the hypothesis that in some instances choroidal neovascularization provides oxygen and/or nutrients to compromised RPE cells and that elimination of choroidal neovascularization can result in RPE death and overlying retinal atrophy (Channa et al., 2015). If that hypothesis is correct, then a treatment that causes maturation and stabilization of choroidal neovascularization may be preferable to a treatment that eliminates choroidal neovascularization. However at this time only anti-VEGF agents are available and the visual consequences of macular atrophy during anti-VEGF treatment is uncertain because in many patients with atrophy the fovea is spared and visual acuity remains good (Channa et al., 2015). Therefore it is hazardous to minimize intraocular anti-VEGF injections because of the appearance of extrafoveal macular atrophy since under-treatment tend to promote subretinal fibrosis which may be a greater threat to vision.
3.1.2 Placental growth factor
Placental growth factor (PlGF) specifically activates VEGFR1, whereas VEGF-A activates both VEGFR1 and VEGFR2 (Park et al., 1994). The function of VEGFR1 in endothelial cells is uncertain, but it is also present in bone marrow-derived cells and contributes to recruitment of those cells (Barleon et al., 1996). In retinal and choroidal vascular diseases in which VEGF-A levels are high, it is unlikely that specific neutralization of PlGF would have much effect because VEGF-A also activates VEGFR1. On the other hand, if PlGF levels are high, neutralization of VEGF does not eliminate all stimuli for recruitment of bone marrow-derived cells and whatever they contribute to the disease process. Therefore, it is conceivable that co-suppression of VEGF-A and PlGF could provide benefits over suppression of VEGF-A alone. This could provide an advantage for aflibercept over ranibizumab and bevacizumab, but since aflibercept is a somewhat better antagonist of VEGF-A than ranibizumab or bevacizumab with superior pharmacokinetics it is difficult to know whether any added benefit aflibercept provides in any particular patient is due to those characteristics or its PlGF-neutralizing activity. Therefore, at this time, it is not known if combined suppression of VEGF-A and PlGF provides added benefit over suppression of VEGF-A alone.
3.1.3 The Tie2 Pathway
Tie2 is a tyrosine kinase receptor that is expressed predominantly on vascular endothelial cells (Sato et al., 1995). Angiopoietin 1 (Ang1) binds Tie2 and stimulates phosphorylation which leads to phosphorylation of eNOS, Akt, and ERK (Davis et al., 1996; Kim et al., 2000a). Angiopoietin 2 is a weak agonist that competes with Ang1 for binding and reduces phosphorylation of Tie2 thereby acting as a Tie2 antagonist (Kim et al., 2000b; Maisonpierre et al., 1997; Yuan et al., 2009). Ang2 expression is increased during retinal vascular development or in ischemic retina (Hackett et al., 2000). Heterozygous knockin mice in which angiopoietin 2 gene was replaced by GFP, showed GFP expression along the surface of the retina between P0 and P7 in association with development of the superficial retinal vessels, adjacent to the developing intermediate capillaries between P4 and P7, and in the region of the deep capillary bed at P7 and throughout adulthood. In mice with ischemic retinopathy, GFP was re-expressed at the surface of the retina at P14 in association with sprouting of NV from the superficial capillaries (Hackett et al., 2002). In homozygous GFP knockin mice deficient in Ang2, retinal vascular development failed to occur indicating Ang2 is necessary for developmental sprouting of new vessels in the retina (Gale et al., 2002; Hackett et al., 2002). In adult mice, Ang2 is also necessary for sprouting of NV, because there was no NV when adenoviral-mediated gene transfer was used to express high levels of VEGF at the surface of the retina, but when VEGF and Ang2 were co-expressed, NV grew from the superficial capillaries (Oshima et al., 2004c). Therefore, both Ang2 and VEGF are needed to stimulate NV in the retina, and the reason why NV grows from the deep capillary bed in rho/VEGF mice is because there is constitutive expression of Ang2 in horizontal cells adjacent to the deep capillary bed of the retina (Hackett et al., 2000). This makes remodeling of the deep capillary bed possible. In rd1 mice in which there is a primary rod cell degeneration, the oxygen level in the outer retina is markedly increased (Yu et al., 2000) which down regulates VEGF and due to constitutive expression of Ang2, the reduction in VEGF results in pruning of the deep capillary bed (Yamada et al., 1999). Similarly, wild type mice placed in hyperoxia for several weeks have down-regulation of VEGF associated with constitutive expression of Ang 2 in the region of the deep capillary bed resulting in regression of the deep capillary bed (Yamada et al., 1999). The possible significance of this for humans is that if individuals live in hypoxic environments, such as at high altitudes, the lower levels of oxygen in blood result in relative hypoxia particularly at the border of the outer nuclear and outer plexiform regions, the region of the deep capillary bed. This causes increased production of VEGF which in combination with the constitutive expression of Ang2 in horizontal cells, stimulates growth of the deep capillaries to increase supply of oxygen to the outer nuclear layer. If the same individual relocates to a town at sea-level, environmental oxygen, oxygen in the blood, and tissue oxygen in the region of the deep capillary bed are increased from the previous situation when the individual lived at high altitude. This decreases VEGF expression and due the constitutive expression of Ang2 in horizontal cells, there should be pruning of the deep capillary bed to decrease the tissue oxygenation in the outer retina. In rho/VEGF mice, the production of VEGF cannot be down-regulated by remodeling of the deep capillary bed so that NV grows all the way through the outer nuclear layer into the subretinal space (Okamoto et al., 1997; Tobe et al., 1998a).
To further explore the role of the Tie2 system in retinal and choroidal NV, Tet/opsin/Ang2 mice and Tet/opsin/Ang1 mice with doxycycline-inducible expression of Ang2 or Ang1 in photoreceptors were generated. Induced expression of Ang2 in mice with ischemic retinopathy at the outset of the ischemic period when VEGF levels are high caused a marked increase in retinal NV, while onset of Ang2 expression at P20 when VEGF levels are low, resulted in rapid regression of retinal NV (Oshima et al., 2005). In mice with choroidal NV due to rupture of Bruch’s membrane, VEGF levels are transiently increased and are low 7 days after rupture of Bruch’s membrane; increased expression of Ang2 at day 7 causes rapid regression of choroidal NV. Unlike Ang2, induced expression of Ang1 suppresses retinal or choroidal NV, it never stimulates it nor does it cause regression of NV (Nambu et al., 2004). It also prevents VEGF-induced leakage from retinal vessels and in Tet/opsin/VEGF mice, co-expression of Ang1 and VEGF prevents severe leakage and exudative retinal detachment (Nambu et al., 2005). Therefore, Ang1 makes endothelial cells less responsive to VEGF, while Ang2 makes them more responsive to VEGF; however, if VEGF levels are low, Ang2 promotes regression of NV.
Tie2 is also regulated by a phosphatase, vascular endothelial-protein tyrosine phosphatase (VE-PTP) (Fachinger et al., 1999; Krueger et al., 1990). VE-PTP is physically associated with Tie2 and dephosphorylates it which inactivates it, thereby acting like Ang2 (Yacyshyn et al., 2009). Like Ang2, VE-PTP is increased in hypoxic retina. Antagonism of VE-PTP with an anti-VE-PTP antibody suppresses retinal NV in mice with ischemic retinopathy, suppresses subretinal NV in rho/VEGF mice, and inhibits choroidal NV (Figure 5) (Shen et al., 2014). AKB-9778 is a small molecule, competitive inhibitor of the catalytic activity of VE-PTP that activates Tie2. In cultured endothelial cells, Tie 2 phosphorylation is increased by incubation with AKB-9778 and further increased by co-incubation with AKB-9778 and Ang1 or Ang2. Thus, when VE-PTP is inhibited, Ang2 acts like a Tie2 agonist. Subcutaneous or intraocular injections of AKB-9778 suppress ischemia-induced retinal NV and subretinal NV in rho/VEGF mice (Figure 6). AKB-9778 strongly inhibits NV at Bruch’s membrane rupture sites and has additive activity when combined with aflibercept. AKB-9778 also reduces VEGF-induced leakage from retinal vessels. In Tet/opsin/VEGF mice, subcutaneous injections of AKB-9778 have the same dramatic effect as co-expression of Ang1 by preventing severe vascular leakage and exudative retinal detachments (Shen et al., 2014). In a phase 1B clinical trial in patients with diabetic macular edema (DME), subcutaneous injections of AKB-9778 for 4 weeks substantially reduced edema and improved vision in several patients (Campochiaro et al., 2015). Thus, activation of Tie2 provides a novel therapeutic approach for retinal and choroidal vascular diseases.
Figure 5. Inhibition of vascular endothelial-protein tyrosine phosphatase (VE-PTP) suppresses ocular neovascularization (NV).

At P12, mice with oxygen-induced ischemic retinopathy were given an intravitreous injection in one eye of 0.1, 0.5, or 2 μg of an antibody directed against VE-PTP or 2 μg of IgG isotype control (n≥12 for each). At P17, staining with GSA lectin showed extensive retinal NV (all of the dark green areas are retinal NV) on the surface of the retina in control IgG-injected eyes and significantly less in eyes injected with 2 μg of anti-VE-PTP (top row, *p<0.001 for comparison with IgG control by one-way ANOVA and Bonferroni to adjust for multiple comparisons; bar=500μm). At P15, rho/VEGF transgenic mice were given an intravitreous injection of 0.5 or 2 μg of anti-VE-PTP in one eye and a corresponding dose of control IgG in the fellow eye (n=6 for each). At P21, retinas were stained with GSA lectin and flat mounted with photoreceptor side facing up. All of the dark green spots are subretinal NV and there was significantly less in eyes injected with 0.5 or 2 μg of anti-VE-PTP than corresponding IgG control eyes (middle row, *p=0.01 by unpaired t-test for comparison with fellow eye IgG control; bar=100μm). After laser-induced rupture of Bruch’s membrane in each eye, mice had intravitreous injection of 0.1, 0.5 or 2 μg of anti-VE-PTP in one eye and 2 μg of control IgG or no injection in the other eye (n≥12 for each). After 7 days, eyecups were stained with GSA lectin and flat mounted. The area of choroidal NV was significantly less in eyes injected with 2 μg of anti-VE-PTP compared with control IgG (bottom row, *p<0.001 by one-way ANOVA and Bonferroni, bar=100μm).
Figure 6. Subcutaneous or intraocular injection of AKB-9778 suppresses choroidal and subretinal neovascularization (NV).

After laser-induced rupture of Bruch’s membrane at 3 locations in each eye, mice were given subcutaneous injections of vehicle or 10 or 20 mg/kg AKB-9778 (n=10 for each) twice a day or a single intraocular injection of 1, 3, or 5 μg of AKB9778 in one eye and vehicle in the fellow eye (n=10 for each group). After 7 days, eyecups were stained with FITC-labeled Griffonia simplicifolia (GSA) lectin. Starting at P15, hemizygous rho/VEGF transgenic mice were injected subcutaneously with AKB-9778 (10 mg/kg bid) or vehicle (n=10 for each) or given an intraocular injection of 3 μg of AKB-9778 in one eye and vehicle in the fellow eye (n=10). At P21, retinas were stained with GSA lectin and retinas were flat-mounted. (A) Subcutaneous injection of 10 or 20 mg/kg of AKB-9778 caused significant reduction in choroidal NV (*p=0.03;**p=0.004 for comparison with control by one-way ANOVA and Bonferroni to adjust for multiple comparisons, bar=100μm). (B) Subcutaneous injection of 10 mg/kg of AKB-9778 significantly reduced the area of subretinal NV in rho/VEGF transgenics (p=0.038 for comparison with control by unpaired t-test, bar=100μm). (C) Intraocular injection of 3 or 5 μg, but not 1 μg of AKB-9778 significantly reduced the area of choroidal NV (*p<0.01 for comparison with vehicle control by one-way ANOVA and Bonferroni, bar=100μm). (D) Intraocular injection of 5 μg of AKB-9778 significantly reduced subretinal NV in rho/VEGF transgenic mice (p=0.022 for difference from vehicle control by unpaired t-test, bar=100μm). In an independent experiment, mice had rupture of Bruch’s membrane with laser and then had intraocular injection of 40 μg of the anti-VEGF agent aflibercept or PBS and subcutaneous injections of 20 mg/kg AKB-9778 or PBS twice a day. This resulted in 4 groups of mice with n=19 in each group: control, aflibercept, AKB-9778, or aflibercept + AKB-9778. After 7 days, compared with the area of GSA lectin-stained choroidal NV in control mice, there was a significant reduction in aflibercept- and AKB-9778- treated mice (E, *p<0.01 by ANOVA with Dunnett’s correction). Compared with mice treated with aflibercept or AKB-9778, those treated with the combination had significantly less choroidal NV (**p<0.05 by ANOVA with Dunnett’s correction).
3.1.4 Platelet-derived growth factor-B (PDGF-B)
Transgenic mice in which the rhodopsin promoter drives expression of platelet-derived growth factor-B (PDGF-B) in the retina develop retinal detachments similar to those seen in doxycycline-treated Tet/opsin/VEGF mice (Seo et al., 2000). Initially this was interpreted as traction retinal detachment, but subsequent studies showed that mice that over-express PDGF-B develop NV, while those that over-express PDGF-A have a phenotype in which there is excessive proliferation of glial cells but no NV (Mori et al., 2002a). The expression of PDGF-B is increased by hypoxia, predominantly in vascular endothelial cells and is important for pericyte recruitment; mice deficient in PDGF-B have poor pericyte coverage of retinal vessels which are leaky (Betsholtz, 2004; Lindahl et al., 1997; Lindblom et al., 2003). Selective blockade of PDGF-B causes mild suppression of choroidal NV at Bruch’s membrane rupture sites and has an additive suppressive effect when combined with a VEGF antagonist (Dong et al., 2014). Combined blockade of VEGF and PDGF-B also has a better effect than VEGF blockade alone in prevention of leakage and exudative detachment in Tet/opsin/VEGF mice (Figure 7). This is because the VEGF-stimulated endothelial cells that are activated and proliferating produce high levels of PDGF-B which stimulates pericytes and aggravates vascular leakage and NV (Dong et al., 2014). Thus there is a paracrine interaction between endothelial cells and pericytes that augments leakage and NV, which is the basis for the additive beneficial effect from combined blockade. In patients with neovascular AMD, the combination of ranibizumab with an aptamer that blocks PDGF-B resulted in significantly greater improvement in visual acuity than treatment with ranibizumab alone (Boyer et al., 2009).
Figure 7. Prevention of exudative retinal detachment in Tet/opsin/VEGF double transgenic mice is greater with both anti-PDGF-BB designed ankyrin repeat protein (DARPin) and anti-VEGF-A DARPin than either alone.

Adult male Tet/opsin/VEGF double transgenic mice were given daily subcutaneously injections of 50mg/kg of doxcycline and intraperitoneal injections of PBS as control (a,b), 1mg/kg anti-PDGF-BB DARPin (c,d), 1mg/kg anti-VEGF-A DARPin (e,f), or 1mg/kg of both DARPins (g,h). After 4 days, fundus photographs and fluorescein angiograms showed total bullous retinal detachments in eyes of mice treated with PBS (a,b) or anti-PDGF-BB DARPin (c,d), partial retinal detachments in eyes of mice treated with anti-VEGF-A DARPin (e,f), and little or no retinal detachment in eyes of mice treated with both DARPins (g,h). Ocular sections stained with Hoechst or FITC-Griffonia simplicifolia lectin confirmed total retinal detachments (i) and extensive NV thoughout the outer retina (j) in eyes of mice treated with anti-PDGF-BB DARPin, partial retinal detachments (k) and dilated vessels with NV extending from deep capillaries (l) in eyes of mice treated with anti-VEGF-A DARPin, and no retinal detachments (M) and normal vessels with little or no NV (n) in eyes of mice treated with both DARPins. Grading of the incidence and severity of leakage or retinal detachments (o) showed that eyes from mice treated with both DARPins had significantly less severe grades than those seen in eyes of mice treated with anti-VEGF-A DARPin (x, p=0.0329) or anti-PDGF-BB DARPin (†, p=0.0098) by Wilcoxin test. (p) Tet/Opsin/VEGF mice and C57BL/6 mice (n=5 for each) were treated with doxycycline and after 3 days, the relative of expression of Pdgfb mRNA normalized to cyclophilin A mRNA was significantly greater in Tet/Opsin/VEGF mice (p=0.0002 by unpaired t-test).
3.1.5 Stromal-derived factor-1 (SDF-1)
Like VEGF and PDGF-B, stromal-derived factor-1 (SDF-1) is increased in hypoxic retina primarily in retinal glia (Lima e Silva et al., 2007). The receptor for SDF-1, CXCR4, is located primarily on macrophages and the increased production of SDF-1 in ischemic retina leads to increased recruitment and retention of macrophages which line up along retinal blood vessels and form small caps around sprouts of NV (Shen et al., 2007). They provide paracrine support for the new vessels because both retinal and choroidal NV are reduced by CXCR4 antagonists (Lima e Silva et al., 2007).
3.1.6 Hypoxia-inducible factor-1 (HIF-1)
HIF-1 is a heterodimer made up of HIF-1α and HIF-1β (Semenza, 2000). In normoxic tissue a prolyl hydroxylase dehydroxylates HIF-1α resulting in ubiquitination and degradation. Hypoxia inactivates the prolyl hydroxylase causing HIF-1α to accumulate and bind to HIF-1β to form HIF-1 which translocates to the nucleus and binds to the HRE located in the promoter region of several genes to stimulate their transcription. Genes that are transcriptionally regulated by HIF-1 include VEGF and its receptor VEGFR1, PDGF-B, and its receptor PDGFRβ, SDF-1 and its receptor CXCR4, Ang2, and VE-PTP among several others (Kelly et al., 2003). Thus HIF-1 transcriptionally regulates all of the genes coding for proteins discussed above that have been implicated in retinal and choroidal NV. HIF-1α levels are increased in hypoxic retina and correlate temporally and spatially with increases in VEGF (Ozaki et al., 1999). Mice in which the HRE has been removed from the Vegf promoter have marked reduction of ischemia-induced retinal NV and choroidal NV at Bruch’s membrane rupture sites (Vinores et al., 2006). Intravitreous injection of an adenoviral vector containing an expression cassette for a stabilized form of HIF1α causes increased expression of all of the factors listed above and results in retinal NV (Kelly et al., 2003). Subretinal injection of the same vector causes choroidal NV.
High throughput screening of a drug library with a reporter cell line for HIF-1 transcriptional activity has determined that digoxin and doxorubicin are strong inhibitors of HIF-1 activity (Lee et al., 2009; Zhang et al., 2008). This has provided additional tools to probe the role of HIF-1 in retinal and choroidal NV. Subcutaneous injections or intraocular injections of digoxin reduce levels of VEGF, PDGF-B, SDF-1 and their receptors in ischemic retina and cause a dose-dependent reduction in retinal NV with optimal doses eliminating the NV (Yoshida et al., 2010). Systemic or intraocular digoxin also dramatically suppresses choroidal NV. Similar results were obtained with doxyrubicin and sustained intraocular delivery of microparticle-incorporated doxyrubicin resulted in prolonged suppression of choroidal NV (Iwase et al., 2013). Therefore HIF-1 plays an important role in both retinal and choroidal NV and reduction of HIF-1-activity provides a means to reduce multiple angiogenic stimulators and their receptors.
3.1.7 Signals from extracellular matrix
In addition to secreted factors that are primarily regulated by HIF-1, signals derived from extracellular matrix (ECM) and surrounding cells participate in CNV. As new vessels become established and more mature their endothelial cells communicate with surrounding cells and ECM which provide survival and stabilizing signals that make the endothelial cells less dependent upon soluble signals such as VEGF. CNV grows into dense ECM and rapidly recruits surrounding cells, so that blockage of VEGF even relatively early in the process suppresses growth but does not cause regression, at least over the short-term. Retinal NV grows on the surface of the retina into the vitreous and it appears that it takes longer to establish interactions with surrounding cells and ECM and remains dependent upon soluble signals so that when VEGF is suppressed, retinal NV regresses. Over time, retinal NV lays down matrix and recruits cells forming a fibrovascular membrane and new vessels in such membranes do not regress when VEGF is blocked.
3.1.7.1 Integrins
Much of the communication between vascular cells and the ECM occurs through integrins on the vascular cells which bind components of the ECM which stimulates intracellular signaling. In normal quiescent vessels, endothelial cells bind to collagen IV, collagen XVIII, laminin, and other component of their basement membrane which leads to stimulation of focal adhesion kinase and activation of the Akt pathway which provides survival signals. When there are sufficient soluble signals discussed above to stimulate NV, this is accompanied by production of proteases that degrade the extracellular matrix and also stimulate endothelial cells to begin expressing some integrins that they don’t normally express α5β1, α5β1, and α5β1, which allow them to bind to fibronectin and vitronectin in provisional matrix, which enhances signaling through VEGF receptor 2 and some other tyrosine kinase receptors. The signaling promoted by the newly upregulated integrins is important in ocular NV, because a small molecule antagonist of α5β1 causes apoptosis of endothelial cells participating in CNV, resulting in regression of CNV (Umeda et al., 2006).
3.1.7.2 Endogenous inhibitors of angiogenesis
While the shift in endothelial cell binding from ECM in basement membrane to components of provisional ECM facilitates the actions of soluble stimulators like VEGF and fuels angiogenesis, the proteolytic degradation of basement membrane components and other proteins results in fragments that participate in a negative feedback system that puts on the brakes to stop angiogenesis when normal wound repair is approaching completion. Angiostatin, a fragment of plasminogen, endostatin, a fragment of collagen XVIII, and multiple fragments of collagen IV have antiangiogenic activity.(O’Reilly et al., 1997; O’Reilly et al., 1994; Petitclerc et al., 2000) There are also full-length proteins including pigment epithelium-derived factor (PEDF)(Dawson et al., 1999) and thrombospondins that participate in the suppression of angiogenesis. A third component of the anti-angiogensis defense system is soluble receptor formed from alternative splicing of full-length receptor such as soluble VEGF receptor 1 also known as sFlt1. Endogenous antiangiogenic proteins are appealing candidates for treatment of ocular NV because they are likely to have a good safety profile and also because they can be delivered by gene transfer to provide prolonged sustained delivery.
3.1.7.2.1 PEDF
Intravitreous or subretinal injection of an adenoviral or AAV2 vectors expressing PEDF suppressed the development of retinal or choroidal NV (Auricchio et al., 2002; Mori et al., 2001b; Mori et al., 2002b; Raisler et al., 2002). When given after NV was established, the adenoviral vector expressing PEDF caused apoptosis of endothelial cells in new vessels, but not in normal, quiescent blood vessels, resulting in regression of the NV (Mori et al., 2001c). A phase I clinical trial was done to test the effect of intravitreous injection of an adenoviral vector expressing PEDF in patients with advanced neovascular AMD (Campochiaro et al., 2006). There were no serious adverse events and no dose-limiting toxicities, but mild, transient intraocular inflammation occurred in 25% of patients. Six patients experienced increased intraocular pressure that was easily controlled by topical medication. At 3 and 6 months after vector injection, 55% and 50%, respectively, of patients treated with106–107.5 pu and 94% and 71% of patients treated with 108–109.5 pu had either no change or had an improvement in lesion size from baseline. The median increase in lesion size at 6 and 12 months was 0.5 and 1.0 disc areas in the low dose group compared to 0 and 0 disc areas in the high dose group. Thus a single intravitreous injection of a dose greater than 108 pu of AdPEDF.11 resulted in anti-angiogenic activity that lasted for several months demonstrating that the activity identified in mouse models was predicitive of effects in patients and providing proof-of-concept for the strategy of gene transfer of anti-angiogenic proteins to treat neovascular AMD.
3.1.7.2.2 Endostatin and angiostatin
Mice deficient in collagen XVIII, the source of endostatin, show delayed regression of the hyaloid vasculature and poor development of the retinal vasculature (Fukai et al., 2002). After tail vein injection of an adenoviral vector expressin endostatin, there was suppression of CNV with strong correlation between endostatin serum level and amount of CNV suppression (Mori et al., 2001a). Subretinal injection of a bovine immunodeficiency lentiviral (BIV) vector coding for endostatin (BIVendostatin) resulted in high expression of endostatin in the RPE that diffused into the overlying retina and outlined retinal vessels suggesting endostatin binding to a component of vessel walls (Takahashi et al., 2003). Gene transfer of endostatin in Tet/opsin/VEGF mice with induced expression of VEGF significantly reduced vascular leakage and exudative retinal detachments. These data suggest that endostatin may be an endogenous inhibitor of vasopermeability as well as a strong inhibitor of NV. Similarly, intraocular injection of angiostatin packaged in AAV or lentiviral vectors suppressed retinal and choroidal NV (Igarashi et al., 2003; Lai et al., 2001; Raisler et al., 2002). Subretinal injection of an equine infectious anemia lentiviral vector (EIAV) expression of endostatin and angiostatin suppressed leakage and growth of choroidal NV in mice (Balaggan et al., 2006; Kachi et al., 2008). This same vector is being tested in a phase I trial in patients with advanced neovascular AMD.
3.1.7.2.3 sFlt1
Intraocular injection of adenoviral or AAV2 vector expressing sFlt1 strongly suppressed retinal and choroidal NV and in mice and injection of AAV2.sFlt-1 suppressed retinal NV in mice (Bainbridge et al., 2002; Honda et al., 2000; Rota et al., 2004) and choroidal NV in monkeys (Lai et al., 2005; Lai et al., 2002). Intravitreous injection of an AAV2 vector expressing a modified version of sFlt1 consisting of domain 2 of Flt1 linked to a human IgG Fc fragment (sFLT01), choroidal NV in primates (Lukason et al., 2011; Pechan et al., 2009). This vector is being tested in a phase 1 trial in patients with neovascular AMD. Another phase 1 trial is testing subretinal injection of an AAV2 vector expressing native sFlt1 in patients with advanced neovascular AMD.
4.1 Difference between retinal and choroidal NV
There is considerable overlap regarding vasoactive proteins involved in retinal and choroidal NV, because HIF-1 is involved in both leading to up-regulation of the same complement of HIF-1-regulated gene products (Figures 8 and 9). However, retinal NV is highly dependent upon VEGF for a prolonged period of time, because even a single intravitreous injection of a VEGF antagonist causes regression of retinal NV. Choroidal NV in patients with neovascular AMD is usually sensitive to VEGF in that VEGF antagonists reduce leakage and prevent further growth, but rarely cause elimination of the choroidal NV over the short-term (Rosenfeld et al., 2006). In patients with choroidal NV due to ocular histoplasmosis or pathologic myopia, regression of choroidal NV often occurs after one or a small series of anti-VEGF injections resulting in a disease free period that can last many months or years until there is recurrence of choroidal NV requiring additional injections. As discussed above, a possible explanation is differences in ECM in the different disease settings. Retinal NV grows into the vitreous, a loose matrix with few surrounding cells. In ocular histoplasmosis there is little excess/abnormal ECM, but in AMD, there is a great deal of abnormal ECM that may promote survival signaling that makes the endothelial cells in new vessels less dependent upon VEGF for survival. More experiments are needed to test this hypothesis, but the practical outcome of the clearly observable difference in VEGF dependence is that despite the contribution of other vasoactive proteins to retinal NV and choroidal NV in diseases other than AMD as described above, VEGF antagonists are sufficient for those disease processes and development of other pharmacologic treatments is not needed. In contrast, while VEGF antagonists have revolutionized the treatment of NVAMD, there is still substantial unmet need for additional pharmacologic treatments. It should be noted however, that while VEGF antagonists do not cause regression of CNV in the majority of patients with AMD over the initial stages of treatment, prolonged treatment can result in regression of CNV that leaves macular atrophy in its wake (Channa et al., 2015). While subfoveal CNV seems to be less susceptible to regression and therefore atrophy often spares the fovea, there are some patients who lose vision from foveal atrophy. This raises the question as to whether the goal of treatment in patients with neovascular AMD should be to reduce leakage, stabilize and promote survival of CNV rather than cause regression.
Figure 8. Molecular pathogenesis of retinal neovascularization (NV).

A simplified version of the molecular pathogenesis of retinal NV illustrates several important molecular signals. It highlights soluble mediators and omits cell-cell and cell-matrix signaling. Retinal NV occurs in diabetic retinopathy and other ischemic retinopathies. The underlying disease process (e.g. high glucose in diabetic retinopathy) damages retinal vessels causing vessel closure and retinal ischemia, which results in elevated HIF-1 levels. HIF-1 upregulates several vasoactive gene products including angiopoietin 2 (Angpt2), vascular endothelial-protein tyrosine phosphatase (VE-PTP), vascular endothelial growth factor (VEGF), platelet-derived growth factor (PDGF-B), stromal-derived growth factor (SDF-1), placental growth factor (PLGF), and several of their receptors. VEGF causes vascular leakage and in combination with Angpt2 and VE-PTP causes sprouting of new vessels. VEGF, SDF-1, and PLGF recruit bone marrow-derived cells which provide paracrine stimulation. PDGF-B recruits pericytes which also provide paracrine stimulation.
Figure 9. Molecular pathogenesis of subretinal neovascularization (NV).

This schematic highlights several important molecular signals involved in subretinal NV. Oxidative stress in the retinal pigmented epithelium (RPE) and photoreceptors causes increased levels of HIF-1, which upregulates vasoactive gene products as described above. Retinal angiomatous proliferation (RAP) occurs if VEGF levels in photoreceptors are sufficiently high to cause a gradient that reaches the deep capillary bed of the retina where there is constitutive expression of Angpt2. Choroidal NV occurs if there is elevation of VEGF and Angpt2 combined with perturbation of Bruch’s membrane and the RPE. The other HIF-1-responsive gene products fuel the process similar to the situation in retinal NV.
5.1 Retinal vascular leakage
5.1.1 Diabetic macular edema
While retinal NV is one complication of retinal vascular diseases, it is not the most prevalent nor the most difficult to treat; that distinction is held by excessive vascular leakage resulting in macular edema. A major challenge in understanding pathogenesis and developing treatments for DME or macular edema due to RVO is the lack of animal models. An important observation was that sustained release of VEGF in the vitreous cavity of monkeys caused severe leakage and macular edema indicating that high levels of VEGF in the vitreous cavity had the ability to cause macular edema (Ozaki et al., 1997). When there is a lack of animal models, an alternative is the use of small human trials for proof-of-concept. One important question was, does retinal hypoxia contribute to macular edema? To address this question, patients with DME were given supplemental inspired oxygen for 3 months. This resulted in significant improvement in macular edema and visual acuity which was lost when patients returned to room air demonstrating that retinal hypoxia contributes to DME (Nguyen et al., 2003). Oral administration of an inhibitor of VEGF receptor tyrosine kinases that had been demonstrated to cause dramatic benefits in models of choroidal NV and retinal NV (Kwak et al., 2000; Ozaki et al., 2000) caused a dose-dependent reduction in DME (Campochiaro and Group, 2004) This study was notable because it was the first DME trial to use OCT as an outcome measure in a subgroup of patients to show drug-induced reduction of macular edema, but more importantly was the first demonstration of a beneficial effect of a nonspecific VEGF antagonist in DME. The next step was to use a completely specific VEGF antagonist. In patients with DME, 3 injections of 0.5mg ranibizumab were given at baseline and months 1, 2, 4, and 6 with primary endpoint at month 7 when there was mean improvement from baseline best-corrected visual acuity (BCVA) of 12.1 ETDRS letters, mean improvement from baseline foveal thickness of 257 μm, and elimination of 85% of excess foveal thickness (Nguyen et al., 2006). In a follow-up study, READ2, injections of ranibizumab were found to cause significantly greater improvement in BCVA compared to focal/grid laser therapy (Nguyen et al., 2009). These results were confirmed in large multicenter clinical trials (Network et al., 2010; Nguyen et al., 2012). Treatment consisting of intraocular injections of a VEGF-neutralizing protein is now first line therapy for DME.
5.1.2 Macular edema after retinal vein occlusion
Retinal vein occlusions (RVOs) consist of central RVO (CRVO) in which the major outflow vessel becomes occluded and branch RVO (BRVO) in which a second or third order vein is occluded. In CRVO, there are hemorrhages, cotton wool patches, and variable amounts of retinal nonperfusion throughout the entire retina and in BRVO similar findings occur throughout the retina drained by the occluded branch vein which is usually about half of the retina. The major cause of reduced vision in patients with RVO is macular edema which was felt to be due to high intravenous pressure behind the obstruction resulting in transudation of fluid into the retina. However, a small pilot trial demonstrated that high levels of VEGF are the major cause macula edema and reduced vision, because after an injection of ranibizumab every month for 3 months, 90% of excess foveal thickness was eliminated and visual acuity was dramatically improved (Campochiaro et al., 2013). These results were confirmed in large multicenter clinical trials (Brown et al., 2011; Brown et al., 2010; Campochiaro et al., 2011a; Campochiaro et al., 2010) and treatment with injections of VEGF-neutralizing proteins is now first line therapy in RVO.
6.1 Retinal nonperfusion and background diabetic retinopathy
Observations in clinical trials for macular edema have shown that VEGF has additional activities in addition to stimulation of ocular NV and vascular leakage. Suppression of VEGF slows the progression of retinal nonperfusion and even causes reopening of recently closed retinal vessels in patients with retinal vein occlusion or diabetic retinopathy (Campochiaro et al., 2013; Campochiaro et al., 2014). VEGF promotes leukostasis (Tolentino et al., 1996) and this is likely the cause of VEGF-induced capillary closure in the retina. Suppression of VEGF also improves background diabetic retinopathy (Nguyen et al., 2012) indicating that VEGF participates earlier in the pathogenesis of diabetic retinopathy than was previously thought. Leukostasis may also underlie this effect of high VEGF levels, because sustained delivery of a steroid in the eye also improves background diabetic retinopathy (Campochiaro et al., 2012; Campochiaro et al., 2011b).
7.1 Future directions
7.1.1 Conclusions
Studies in animal models have elucidated many aspects of the molecular pathogenesis of retinal and subretinal NV. There is considerable overlap between retinal and choroidal NV in vasoactive molecules involved, because both types of NV are driven by stabilization of HIF-1 resulting in upregulation of several vasoactive proteins and their receptors (Figures 7 and 8). Despite the participation of multiple factors, suppression of VEGF is sufficient to cause regression of retinal NV until late in the disease process when the NV is encased in fibrous tissue containing numerous accessory cells and extensive ECM which make the endothelial cells less dependent upon VEGF for survival. For this reason, VEGF antagonists are the only pharmacologic treatment needed for retinal NV and are a useful adjunct to scatter photocoagulation which provides long-term stabilization by reducing VEGF production by the ischemic retina. In contrast, choroidal and subretinal NV are located in the sub-RPE and/or subretinal spaces where they are surrounded by RPE cells which interact with the endothelial cells and lay down extensive ECM; this provides survival factors that make the endothelial cells less dependent upon VEGF for survival. None-the-less VEGF antagonists are the primary treatment because VEGF is required for continued growth and leakage of the NV and VEGF antagonists stop growth and leakage. The persistence of choroidal NV despite VEGF blockade and the resumption of NV growth and leakage during treatment lapses, leave considerable unmet need which can be addressed by targeting other participating vasoactive proteins, such as PDGF-B, VE-PTP, Ang2, SDF-1, or the prime mover, HIF-1.
Due to lack of good animal models for macular edema, small pilot clinical trials have provided critical information regarding molecular pathogenesis. Despite small subject numbers, studies using the specific VEGF antagonist ranibizumab clearly showed that VEGF plays a very important role in the retinal vascular leakage that leads to DME (Nguyen et al., 2006) or macular edema due to RVO (Campochiaro et al., 2008). These results were confirmed by large multicenter trials and suppression of VEGF is now first line therapy for treatment of macular edema in ischemic retinopathies.
7.1.2 Where next?
Inability to eliminate macular edema with VEGF antagonists in some patients and the need for prolonged treatment to control edema in many, leaves considerable unmet need. Activators of Tie2 may fill much of that need, and there is substantial ongoing effort to identify other contributors to macular edema that can be targeted. Finally, the contribution of VEGF to retinal nonperfusion and progression of background diabetic retinopathy, as well as the need for prolonged intravitreous injections in patients with choroidal NV due to AMD, diabetic macular edema, or macular edema due to retinal vein occlusions makes sustained delivery of VEGF antagonists a major priority.
Acknowledgments
Supported by EY012609 from the National Eye Institute
Footnotes
PAC is a consultant for Genentech, Regeneron, Allergan, and Aerpio for which his employer, the Johns Hopkins University, receives remuneration. PAC is a consultant for Kala, AsclepiX, Advanced Cell Technologies, Eleven, Applied Genetic Technologies Corporation, Alimera, Allegro, Novaritis, and Graybug for which he receives remuneration. PAC receives research funding for clinical trials from Genentech, Regeneron, Allergan, Aerpio, Genzyme, and Oxford BioMedica.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Age-Related Eye Disease Study Research Group. A randomized, placebo-controlled, clinical trial of high-dose supplementation with vitamins C and E, beta carotene, and zinc for age-related macular degeneration and vision loss. Arch Ophthalmol. 2001;119 (10):1417–1436. doi: 10.1001/archopht.119.10.1417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aiello LP, Pierce EA, Foley ED, Takagi H, Chen H, Riddle L, Ferrara N, King GL, Smith LEH. Suppression of retinal neovascularization in vivo by inhibition of vascular endothelial growth factor (VEGF) using soluble VEGF-receptor chimeric proteins. Proc Natl Acad Sci USA. 1995;92 (23):10457–10461. doi: 10.1073/pnas.92.23.10457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alon T, Hemo I, Itin A, Pe’er J, Stone J, Keshet E. Vascular endothelial growth factor acts as a survival factor for newly formed retinal vessels and has implications for retinopathy of prematurity. Nature Med. 1995;1 (10):1024–1028. doi: 10.1038/nm1095-1024. [DOI] [PubMed] [Google Scholar]
- Auricchio A, Behling KC, Maguire AM, O’Conner EM, Bennett J, Wilson JM, Tolentino MJ. Inhibition of retinal neovascularization by intraocular viral-mediated delivery of anti-angiogenic agents. Mol Ther. 2002;6 (4):490–494. doi: 10.1006/mthe.2002.0702. [DOI] [PubMed] [Google Scholar]
- Avery RL, Castellarin AA, Steinle NC, Dhoot DS, Pieramici DJ, See R, Couvillion S, Nasir MA, Rabena MD, Le K, Maia M, Visich JE. Systemic pharmacokinenetics following intravitreal injections of ranibizumab, bevacizumab or aflibercept in patients with neovascular AMD. Br J Ophthalmol. 2014;98 (12):1543–1546. doi: 10.1136/bjophthalmol-2014-305252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bainbridge J, Mistry A, Alwis MD, Paleolog E, Baker A, Thrasher AJ, Ali RR. Inhibition of retinal neovascularization by gene transfer of soluble VEGF receptor sFlt-1. Gene Ther. 2002;9 (5):320–326. doi: 10.1038/sj.gt.3301680. [DOI] [PubMed] [Google Scholar]
- Balaggan KS, Binley K, Esapa M, MacLaren RE, Iqball S, Duran Y, Pearson RA, Kan O, Barker SE, Smith AJ, Bainbridge JWB, Naylor S, Ali RR. EIAV vector-mediated delivery of endostatin or angiostatin inhibits angiogenesis and vascular hyperpermeability in experimental CNV. Gene Ther. 2006;13 (15):1153–1165. doi: 10.1038/sj.gt.3302769. [DOI] [PubMed] [Google Scholar]
- Barleon B, Sozzani S, Zhou D, Weich HA, Mantovani A, Marme D. Migration of human monocytes in response to vascular endothelial growth factor (VEGF) is mediated via the VEGF receptor flt-1. Blood. 1996;87 (8):3336–3343. [PubMed] [Google Scholar]
- Betsholtz C. Insight into the physiological functions of PDGF through genetic studies in mice. Cytokine Growth Factor Rev. 2004;15 (4):215–228. doi: 10.1016/j.cytogfr.2004.03.005. [DOI] [PubMed] [Google Scholar]
- Boyer DS the Ophthotech Anti-PDGF in AMD Study Group. Combined inhibition of platelet-derived (PDGF) and vascular endothelial (VEGF) growth factors for the treatment of neovascular age-related macular degeneration (NV-AMD). Results of a phase 1 study. Invest Ophthalmol Vis Sci. 2009 Online ARVO abstract 1260. [Google Scholar]
- Brown DM, Campochiaro PA, Bhisitkul RB, Ho AC, Gray S, Saroj N, Adamis AP, Rubio RG, Murahashi WY. Sustained benefits from ranibizumab for macular edema following branch retinal vein occlusion: 12-month outcomes of a phase III study. Ophthalmology. 2011;118 (8):1594–1602. doi: 10.1016/j.ophtha.2011.02.022. [DOI] [PubMed] [Google Scholar]
- Brown DM, Campochiaro PA, Singh RP, Gray S, Rundle AC, Li Z, Rubio RG, Murahashi WY the CRUISE Study Group. Efficacy and safety of ranibizumab in the treatment of macular edema secondary to central retinal vein occlusion:6-month results of the phase III CRUISE study. Ophthalmology. 2010;117 (6):1124–1133. doi: 10.1016/j.ophtha.2010.02.022. [DOI] [PubMed] [Google Scholar]
- Brown DM, Kaiser PK, Michels M, Soubrane G, Heier JS, Kim RY, Sy JP, Schneider S the Anchor Study Group. Ranibizumab versus verteporfin for neovascular age-related macular degeneration. N Eng J Med. 2006;355 (14):1432–1444. doi: 10.1056/NEJMoa062655. [DOI] [PubMed] [Google Scholar]
- Campochiaro PA. Ocular versus extraocular neovascularization: mirror images or vague resemblances. Invest Ophthalmol Vis Sci. 2006;47 (2):462–474. doi: 10.1167/iovs.05-1494. [DOI] [PubMed] [Google Scholar]
- Campochiaro PA, Bhistikul RB, Shapiro H, Rubio RG. Vascular endothelial growth factor promotes progressive retinal nonperfusion in patients with retinal vein occlusion. Ophthalmology. 2013;120 (4):795–802. doi: 10.1016/j.ophtha.2012.09.032. [DOI] [PubMed] [Google Scholar]
- Campochiaro PA, Brown DM, Awh CC, Lee Y, Gray S, Saroj N, Murahashi WY, Rubio RG. Sustained benefits from ranibizumab for macular edema following central retinal vein occlusion: 12-month outcomes of a phase III study. Ophthalmology. 2011a;118 (10):2041–2049. doi: 10.1016/j.ophtha.2011.02.038. [DOI] [PubMed] [Google Scholar]
- Campochiaro PA, Brown DM, Pearson A, Chen S, Boyer D, Ruiz-Moreno J, Garretson B, Gupta A, Seenu M, Hariprasad SM, Bailey C, Reichel E, Soubrane G, Kapik B, Billman K, Kane FE, Green K the FAME Study Group. Sustained delivery fluocinolone acetonide vitreous inserts provide benefit for at least 3 years in patients with diabetic macular edema. Ophthalmology. 2012;119 (10):2125–2132. doi: 10.1016/j.ophtha.2012.04.030. [DOI] [PubMed] [Google Scholar]
- Campochiaro PA, Brown DM, Pearson A, Ciulla T, Boyer D, Holz FG, Tolentino M, Gupta A, Duarte L, Madreperla S, Gonder J, Kapik B, Billman K, Kane FE the FAME Study Group. Long-term benefit of sustained-delivery fluocinolone acetonide vitreous inserts for diabetic macular edema. Ophthalmology. 2011b;118 (4):626–635. doi: 10.1016/j.ophtha.2010.12.028. [DOI] [PubMed] [Google Scholar]
- Campochiaro PA the C99-PKC412-003 Study Group. Reduction of diabetic macular edema by oral administration of the kinase inhibitor PKC412. Invest Ophthalmol Vis Sci. 2004;45 (3):922–931. doi: 10.1167/iovs.03-0955. [DOI] [PubMed] [Google Scholar]
- Campochiaro PA, Hackett SF. Corneal endothelial cell matrix promotes expression of differentiated features of retinal pigmented epithelial cells: implication of laminin and basic fibroblast growth factor as active components. Exp Eye Res. 1993;57 (5):539–537. doi: 10.1006/exer.1993.1158. [DOI] [PubMed] [Google Scholar]
- Campochiaro PA, Hafiz G, Shah SM, Nguyen QD, Ying H, Do DV, Quinlan E, Zimmer-Galler I, Haller JA, Solomon S, Sung JU, Hadi Y, Janjua KA, Choy DF, Arron JR. Ranibizumab for macular edema due to retinal vein occlusions; implication of VEGF as a critical stimulator. Molec Ther. 2008;16 (4):791–799. doi: 10.1038/mt.2008.10. [DOI] [PubMed] [Google Scholar]
- Campochiaro PA, Heier JS, Feiner L, Gray S, Saroj N, Rundle AC, Murahashi WY, Rubio RG the BRAVO Study Group. Ranibizumab for macular edema following branch retinal vein occlusion: 6-month primary endpoint results of a phase III study. Ophthalmology. 2010;117 (6):1102–1112. doi: 10.1016/j.ophtha.2010.02.021. [DOI] [PubMed] [Google Scholar]
- Campochiaro PA, Nguyen QD, Shah SM, Klein ML, Holz E, Frank RN, Saperstein DA, Gupta A, Stout JT, Macko J, DiBartolomeo R, Wei LL. Adenoviral vector-delivered pigment epithelium-derived factor for neovascular age-related macular degeneration: results of a phase I clinical trial. Hum Gene Ther. 2006;17 (2):167–176. doi: 10.1089/hum.2006.17.167. [DOI] [PubMed] [Google Scholar]
- Campochiaro PA, Sophie R, Tolentino M, Miller DM, Browning D, Boyer DS, Heier JS, Gambino L, Withers B, Brigell M, Peters K. Treatment of diabetic macular edema with an inhibitor of vascular endothelial-protein tyrosine phosphatase that activates Tie2. Ophthalmology. 2015;122 (3):545–554. doi: 10.1016/j.ophtha.2014.09.023. [DOI] [PubMed] [Google Scholar]
- Campochiaro PA, Wykoff CC, Shapiro H, Rubio RG, Ehrlich JS. Neutalization of vascular endothelial growth factor slows progression of retinal nonperfusion in patients iwth diabetic macular edema. Ophthalmology. 2014b;121 (9):1783–1789. doi: 10.1016/j.ophtha.2014.03.021. [DOI] [PubMed] [Google Scholar]
- Channa R, Sophie R, Bagheri S, Shah SM, Wang J, Adeyemo O, Sophie A, Wenick A, Ying HS, Campochiaro PA. Regression of choroidal neovascularization results in macular atrophy in anti-vascular endothelial growth factor-treated eyes. Am J Ophthalmol. 2015;159 (1):9–19. doi: 10.1016/j.ajo.2014.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis AA, Berstein PS, Bok D, Turner J, Nachtigal M, Hunt RC. A human retinal pigment epithelial cell line that retains epithelial characteristics after prolonged culture. Invest Ophthalmol Vis Sci. 1995;36 (5):955–964. [PubMed] [Google Scholar]
- Davis S, Aldrich TH, Jones P, Acheson A, Ryan TE, Bruno J, Radziejewski C, Maisonpierre PC, Yancopoulos GD. Isolation of angiopoietin-1, a ligand for the TIE2 receptor, by secretion-trap expression cloning. Cell. 1996;87 (7):1161–1169. doi: 10.1016/s0092-8674(00)81812-7. [DOI] [PubMed] [Google Scholar]
- Dawson DW, Volpert OV, Gillis P, Crawford SE, Xu HJ, Benedict W, Bouck NP. Pigment epithelium-derived factor: a potent inhibitor of angiogenesis. Science. 1999;285 (5425):245–248. doi: 10.1126/science.285.5425.245. [DOI] [PubMed] [Google Scholar]
- Dobi ET, Puliafito CA, Destro M. A new model of choroidal neovascularization in the rat. Arch Ophthalmol. 1989;107 (2):264–269. doi: 10.1001/archopht.1989.01070010270035. [DOI] [PubMed] [Google Scholar]
- Dong A, Seidel C, Snell D, Ekawardhani S, Ahlskog JK, Baumann M, Shen J, Iwase T, Tian J, Stevens R, Hackett SF, Stumpp MT, Campochiaro PA. Antagonism of PDGF-BB suppresses subretinal neovascularization and enhances the effects of blocking VEGF-A. Angiogenesis. 2014;17 (3):553–562. doi: 10.1007/s10456-013-9402-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elice F, Rodeghiero F, Falanga A, Rickels F. Thrombosis associated with angiogenesis inhibitors. Best Pract Res Clin Haematol. 2009;22 (1):115–128. doi: 10.1016/j.beha.2009.01.001. [DOI] [PubMed] [Google Scholar]
- Eremina V, Jefferson JA, Kowalewska J, Hochster H, Haas M, Weisstuch J, Richardson CD, Kopp JB, Gerber HP, Ferrara N, Barisoni L, Alpers CE, Quaggin SE. VEGF inhibition and renal thrombotic microangiopathy. New Engl J Med. 2008;358 (11):1129–1136. doi: 10.1056/NEJMoa0707330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fachinger G, Deutsch U, Risau W. Functional interaction of vascular endothelial-protein-tyrosine phosphatase with the angiopoietin receptor Tie-2. Oncogene. 1999;18 (43):5948–5943. doi: 10.1038/sj.onc.1202992. [DOI] [PubMed] [Google Scholar]
- Gale NW, Thurston G, Hackett SF, Renard R, Wang Q, McClain J, Martin C, Witte C, Witte M, Jackson D, Suri C, Campochiaro PA, Wiegand SJ, Yancopoulos GD. Angiopoietin-2 is required for postnatal angiogenesis and lymphatic patterning, and only the latter role is rescued by angiopoietin-1. Devel Cell. 2002;3 (3):411–423. doi: 10.1016/s1534-5807(02)00217-4. [DOI] [PubMed] [Google Scholar]
- Grunwald JE, Daniel E, Huang J, Ying G, Maguire MG, Toth CA, Jaffe GJ, Fine SL, Martin DF the CATT Research Group. Risk of geographic atrophy in the comparison of age-related macular degeneration treatments trials. Ophthalmology. 2012;121(1):150–161. doi: 10.1016/j.ophtha.2013.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hackett SF, Ozaki H, Strauss RW, Wahlin K, Suri C, Maisonpierre P, Yancopoulos G, Campochiaro PA. Angiopoietin 2 expression in the retina: upregulation during physiologic and pathologic neovascularization. J Cell Physiol. 2000;184 (3):275–284. doi: 10.1002/1097-4652(200009)184:3<275::AID-JCP1>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- Hackett SF, Wiegand SJ, Yancopoulos G, Campochiaro P. Angiopoietin-2 plays an important role in retinal angiogenesis. J Cell Physiol. 2002;192 (2):182–187. doi: 10.1002/jcp.10128. [DOI] [PubMed] [Google Scholar]
- Heier JS, Brown DM, Chong V, Korobelnik JF, Kaiser PK, Nguyen QD, Kirchhof B, Ho A, Ogura Y, Yancopoulos GD, Stahl N, Vitti R, Berliner AJ, Soo Y, Anderesi M, Groetzbach G, Sommerauer B, Sandbrink R, Simader C, Schmidt-Erfurth U V.a.V.S Groups. Intravitreal Aflibercept (VEGF Trap-Eye) in wet age-related macular degeneration. Ophthalmology. 2012;119 (12):2537–2548. doi: 10.1016/j.ophtha.2012.09.006. [DOI] [PubMed] [Google Scholar]
- Honda M, Sakamoto T, Ishibashi T, Inomata H, Ueno H. Experimental subretinal neovascularization is inhibited by adenovirus-mediated soluble VEGF. flt-1 receptor gene transfection: a role of VEGF and possible treatment for SRN in age-related macular degeneration. Gene Ther. 2000;7 (11):978–985. doi: 10.1038/sj.gt.3301203. [DOI] [PubMed] [Google Scholar]
- Igarashi T, Miyake K, Kato K, Watanabe A, Ishizaki M, Ohara K, Shimada T. Lentivirus-mediated expression of angiostatin efficiently inhibits neovascularization in a murine proliferative retinopathy model. Gene Ther. 2003;10 (3):219–226. doi: 10.1038/sj.gt.3301878. [DOI] [PubMed] [Google Scholar]
- Inai T, Mancuso M, Hashizume H, Baffert F, Haskell A, Baluk P, HuLowe DD, Shalinsky DR, Thurston G, Yancopoulos GD, McDonald DM. Inhibition of vascular endothelial growth factor (VEGF) signaling in cancer causes loss of endothelial fenestrations, regression of tumor vessels, and appearance of basement membrane ghosts. Am J Pathol. 2004;165 (1):35–52. doi: 10.1016/S0002-9440(10)63273-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwase T, Fu J, Yoshida T, Muramatusu D, Miki A, Hashida N, Lu L, Oveson B, Lime e Silva R, Seidel C, Yang M, Connelly S, Shen J, Han B, Wu M, Semenza GL, Hanes J, Campochiaro PA. Sustained delivery of a HIF-1 antagonist for ocular neovascularization. J Control Release. 2013;172 (3):625–633. doi: 10.1016/j.jconrel.2013.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamba T, Tam BYY, Hashizume H, Haskell A, Sennino B, Mancuso MR, Norberg SM, O’Brien SM, Davis RB, Gowen LC, Anderson KD, Thurston G, Joho S, Springer ML, Kuo CJ, McDonald DM. VEGF-dependent plasticity of fenestrated capillaries in the normal adult microvasculature. Am J Physiol Heart Circ Physiol. 2005;290 (2):H560–H576. doi: 10.1152/ajpheart.00133.2005. [DOI] [PubMed] [Google Scholar]
- Kelly BD, Hackett SF, Hirota K, Oshima Y, Cai Z, Berg-Dixon S, Rowan A, Yan Z, Campochiaro PA, Semenza GL. Cell type-specific regulation of angiogenic growth factor gene expression and induction of angiogenesis in nonischemic tissue by a constitutively active form of hypoxia-inducible factor 1. Circ Res. 2003;93 (11):1074–1081. doi: 10.1161/01.RES.0000102937.50486.1B. [DOI] [PubMed] [Google Scholar]
- Kim I, Kim HG, So JN, Kim JH, Kwak HJ, Koh GY. Angiopoietin-1 regulates endothelial cell survival through the phosphatidylinositol 3′-kinase/Akt signal transduction pathway. Circ Res. 2000a;86 (1):24–29. doi: 10.1161/01.res.86.1.24. [DOI] [PubMed] [Google Scholar]
- Kim I, Kim JH, Moon SO, Kwak HJ, Kim NG, Koh GY. Angiopoietin-2 at high concentration can enhance endothelial cell survival through the phosphatidylinositol 3′-kinase/Akt signal transduction pathway. Oncogene. 2000b;19 (39):4549–4552. doi: 10.1038/sj.onc.1203800. [DOI] [PubMed] [Google Scholar]
- Klein ML, Ferris FLI, Armstrong J, Hwang TS, Chew EY, Bressler SB, Chandra SR AREDS Reseach Group. Retinal precursors and the development of geographic atrophy in age-related macular degeneration. Ophthalmology. 2008;115 (6):1026–1031. doi: 10.1016/j.ophtha.2007.08.030. [DOI] [PubMed] [Google Scholar]
- Krueger NX, Streuli M, Saito H. Structural diversity and evolution of human receptor-like protein tyrosine phosphatases. EMBO J. 1990;9 (10):3241–3252. doi: 10.1002/j.1460-2075.1990.tb07523.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kryzstolik MG, Afshari MA, Adamis AP, Gaudreault J, Gragoudas ES, Michaud NM, Li W, Connolly E, O’Neill CA, Miller JW. Prevention of experimental choroidal neovascularization with intravitreal anti-vascular endothelial growth factor antibody fragment. Arch Ophthalmol. 2002;120 (3):338–346. doi: 10.1001/archopht.120.3.338. [DOI] [PubMed] [Google Scholar]
- Kurihara T, Westenskow PD, Bravo S, Afuilar E, Friedlander M. Targeted deletion of Vegfa in adult mice induces vision loss. J Clin Invest. 2012;122 (11):4213–4117. doi: 10.1172/JCI65157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwak N, Okamoto N, Wood JM, Campochiaro PA. VEGF is an important stimulator in a model of choroidal neovascularization. Invest Ophthalmol Vis Sci. 2000;41 (10):3158–3164. [PubMed] [Google Scholar]
- Lai CC, Wu WC, Chen SL, Xiao X, Tsai TC, Huan SJ, Chen TL, Tsai RJF, Tsao YP. Suppression of choroidal neovascularization by adeno-associated virus vector expressing angiostatin. Invest Ophthalmol Vis Sci. 2001;42 (10):2401–2407. [PubMed] [Google Scholar]
- Lai CM, Shen WY, Brankov M, Lai YK, Barnett NL, Lee SY, Yeo IY, Mathur R, Ho JE, Pineda P, Barathi A, Ang CL, Constable IJ, Rakoczy EP. Long-term evaluation of AAV-mediated sFlt-1 gene therapy for ocular neovascularization in mice and monkeys. Mol Ther. 2005;12 (4):659–668. doi: 10.1016/j.ymthe.2005.04.022. [DOI] [PubMed] [Google Scholar]
- Lai YK, Shen WY, Brankov M, Lai CM, Constable IJ, Rakoczy PE. Potential long-term inhibition of ocular neovascularization by recombinant adeno-associated virus-mediated secretion gene therapy. Gene Ther. 2002;9 (12):804–813. doi: 10.1038/sj.gt.3301695. [DOI] [PubMed] [Google Scholar]
- Lee K, Qian DZ, Rey S, Wei H, Liu JO, Semenza GL. Antracycline chemotherpy inhibits HIF-1 transcriptional activity and tumor induced mobilizatio of circulating angiogenic cells. Proc Natl Acad Sci USA. 2009;106 (7):2353–2358. doi: 10.1073/pnas.0812801106. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Lima e Silva R, Shen J, Hackett SF, Kachi S, Akiyama H, Kiuchi K, Yokoi K, Hatara C, McLauer T, Aslam S, Gong YY, Xiao WH, Khu NH, Thut C, Campochiaro P. The SDF-1/CXCR4 ligand/receptor pair is an important contributor to several types of ocular neovascularization. FASEB J. 2007;21 (12):3219–3230. doi: 10.1096/fj.06-7359com. [DOI] [PubMed] [Google Scholar]
- Lindahl P, Johansson BR, Leveen P, Betsholtz C. Pericyte loss and microaneurysm formation in PDGF-B-deficient mice. Science. 1997;277 (5323):242–245. doi: 10.1126/science.277.5323.242. [DOI] [PubMed] [Google Scholar]
- Lindblom P, Gerhardt H, Liebner S, Abramsson A, Enge M, Hellstrom M, Backstrom G, Fredriksson S, Landergren U, Nystrom HC, Bergstrom G, Dejana E, Ostman A, Lindahl P, Betsholtz C. Endothelial PDGF-B retention is required for proper investment of pericytes in the microvessel wall. Genes & Dev. 2003;17 (15):1835–1840. doi: 10.1101/gad.266803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukason M, Dufresne E, Hillard R, Pechan P, Li Q, Kim I, Kiss S, Flaxel C, Collins M, Miller J, Hauswirth W, MacLachlan T, Wadsworth S, Scaria A. Inhibition of choroidal neovascularization in a nonhuman primate model by intravitreal administration of an AAV2 vector expressing a novel anti-VEGF molecule. 2011;19(2):260–265. doi: 10.1038/mt.2010.230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maisonpierre PC, Suri C, Jones PF, Bartunkova S, Wiegand SJ, Radziejewski C, Compton D, McClain J, Aldrich TH, Papadopoulos N, Daly TJ, Davis S, Sato TN, Yancopoulos GD. Angiopoietin-2, a natural antagonist for Tie2 that disrupts in vivo angiogenesis. Science. 1997;277 (5322):55–60. doi: 10.1126/science.277.5322.55. [DOI] [PubMed] [Google Scholar]
- Mameros AG, Fan J, Yokoyama Y, Gerber HP, Ferrara N, Crouch RK, Olsen BR. Vascular endothelial growth factor expression in the retinal pigment epithelium is essential for choriocapillaris development and visual function. Am J Pathol. 2005;167 (5):1451–1459. doi: 10.1016/S0002-9440(10)61231-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mori K, Ando A, Gehlbach P, Nesbitt D, Takahashi K, Goldsteen D, Penn M, Chen CT, Melia M, Phipps S, Moffat D, Brazzell K, Liau G, Dixon KH, Campochiaro PA. Inhibition of choroidal neovascularization by intravenous injection of adenoviral vectors expressing secretable endostatin. Am J Pathol. 2001a;159 (1):313–320. doi: 10.1016/S0002-9440(10)61697-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mori K, Duh E, Gehlbach P, Ando A, Takahashi K, Pearlman J, Mori K, Yang HS, Zack DJ, Ettyreddy D, Brough DE, Wei LL, Campochiaro PA. Pigment epithelium-derived factor inhibits retinal and choroidal neovascularization. J Cell Physiol. 2001b;188 (2):253–263. doi: 10.1002/jcp.1114. [DOI] [PubMed] [Google Scholar]
- Mori K, Gehlbach P, Ando A, Dyer G, Lipinsky E, Chaudhry AG, Hackett SF, Campochiaro PA. Retina-specific expression of PDGF-B versus PDGF-A: vascular versus nonvascular proliferative retinopathy. Invest Ophthalmol Vis Sci. 2002;43 (6):2001–2006. [PubMed] [Google Scholar]
- Mori K, Gehlbach P, Ando A, McVey D, Wei L, Campochiaro PA. Regression of ocular neovascularization by increased expression of pigment epithelium-derived factor. Invest Ophthalmol Vis Sci. 2001c;43 (7):2428–2434. [PubMed] [Google Scholar]
- Mori K, Gehlbach P, Yamamoto S, Duh E, Zack DJ, Li Q, Berns KI, Raisler BJ, Hauswirth WW, Campochiaro PA. AAV-mediated gene transfer of pigment epithelium-derived factor inhibits choroidal neovascularization. Invest Ophthalmol Vis Sci. 2002b;43 (6):1994–2000. [PubMed] [Google Scholar]
- Mousa SA, Lorelli W, Campochiaro PA. Extracellular matrix-integrin binding modulates secretion of angiogenic growth factors by retinal pigmented epithelial cells. J Cell Biochem. 1999;74 (1):135–143. [PubMed] [Google Scholar]
- Nambu H, Nambu R, Oshima Y, Hackett SF, Wiegand SJ, Yancopoulos G, Zack DJ, Campochiaro PA. Angiopoietin 1 inhibits ocular neovascularization and breakdown of the blood-retinal barrier. Gene Ther. 2004;11 (10):865–873. doi: 10.1038/sj.gt.3302230. [DOI] [PubMed] [Google Scholar]
- Nambu H, Umeda N, Kachi S, Oshima Y, Nambu R, Campochiaro PA. Angiopoietin 1 prevents retinal detachment in an aggressive model of proliferative retinopathy, but has no effect on established neovascularization. J Cell Physiol. 2005;204 (1):227–235. doi: 10.1002/jcp.20292. [DOI] [PubMed] [Google Scholar]
- The Diabetic Retinopathy Clinical Research Network. Elman MJ, Aiello LP, Beck RW, Bressler NM, Bressler SB, Edwards AR, Ferris FL, 3rd, Friedman SM, Glassman AR, Miller KM, Scott IU, Stockdale CR, Sun JK. Randomized trial evaluating ranibzumab plus prompt or deferred laser or triamcinolone plus prompt laser for diabetic macular edema. Ophthalmolgy. 2010;117 (6):1064–1077. doi: 10.1016/j.ophtha.2010.02.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen QD, Brown DM, Marcus DM, Boyer DS, Patel S, Feiner L, Gibson A, Sy J, Rundle AC, Hopkins JJ, Rubio RG, Ehrlich JS the RISE and RIDE Research Group. Ranibizumab for Diabetic Macular Edema. Results from 2 Phase III Randomized Trials: RISE and RIDE. Ophthalmology. 2012;119 (4):789–801. doi: 10.1016/j.ophtha.2011.12.039. [DOI] [PubMed] [Google Scholar]
- Nguyen QD, Shah SM, Heier JS, Do DV, Lim J, Boyer D, Abraham P, Campochiaro PA READ-2 Study Group. Primary End Point (Six Months) Results of the Ranibizumab for Edema of the mAcula in Diabetes (READ-2) Study. Ophthalmology. 2009;116 (11):2175–2181. doi: 10.1016/j.ophtha.2009.04.023. [DOI] [PubMed] [Google Scholar]
- Nguyen QD, Shah SM, Van Anden E, Sung JU, Vitale S, Campochiaro PA. Supplemental inspired oxygen improves diabetic macular edema; a pilot study. Invest Ophthalmol Vis Sci. 2003;45 (2):617–624. doi: 10.1167/iovs.03-0557. [DOI] [PubMed] [Google Scholar]
- Nguyen QD, Tatlipinar S, Shah SM, Haller JA, Quinlan E, Sung J, Zimmer-Galler I, Do DV, Campochiaro PA. Vascular endothelial growth factor is a critical stimulus for diabetic macular edema. Am J Ophthalmol. 2006;142 (6):961–969. doi: 10.1016/j.ajo.2006.06.068. [DOI] [PubMed] [Google Scholar]
- O’Reilly MS, Boehm T, Shing Y, Fukai N, Vasios G, Lane WS, Flynn E, Birknead JR, Olsen BR, Folkman J. Endostatin: an endogenous inhibitor of angiogenesis and tumor growth. Cell. 1997;88 (2):277–285. doi: 10.1016/s0092-8674(00)81848-6. [DOI] [PubMed] [Google Scholar]
- O’Reilly MS, Holmgren S, Shing Y, Chen C, Rosenthal RA, Moses M, Lane WS, Cao Y, Sage HE, Folkman J. Angiostatin: a novel angiogenesis inhibitor that mediates the suppression of metastases by a Lewis lung carcinoma. Cell. 1994;79 (2):315–328. doi: 10.1016/0092-8674(94)90200-3. [DOI] [PubMed] [Google Scholar]
- Ohno-Matsui K, Hirose A, Yamamoto S, Saikia J, Okamoto N, Gehlbach P, Duh EJ, Hackett SF, Chang M, Bok D, Zack DJ, Campochiaro PA. Inducible expression of vascular endothelial growth factor in photoreceptors of adult mice causes severe proliferative retinopathy and retinal detachment. Am J Pathol. 2002;160 (2):711–719. doi: 10.1016/S0002-9440(10)64891-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okamoto N, Tobe T, Hackett SF, Ozaki H, Vinores MA, LaRochelle W, Zack DJ, Campochiaro PA. Transgenic mice with increased expression of vascular endothelial growth factor in the retina: a new model of intraretinal and subretinal neovascularization. Am J Pathol. 1997;151 (1):281–291. [PMC free article] [PubMed] [Google Scholar]
- Oshima Y, Deering T, Oshima S, Nambu H, Reddy PS, Kaleko M, Connelly S, Hackett SF, Campochiaro PA. Angiopoietin-2 enhances retinal vessel sensitivity to vascular endothelial growth factor. J Cell Physiol. 2004a;199 (3):412–417. doi: 10.1002/jcp.10442. [DOI] [PubMed] [Google Scholar]
- Oshima Y, Oshima S, Nambu H, Kachi S, Hackett SF, Melia M, Kaleko M, Connelly S, Esumi N, Zack DJ, Campochiaro PA. Increased expression of VEGF in retinal pigmented epithelial cells is not sufficient to cause choroidal neovascularization. J Cell Physiol. 2004b;201 (3):393–400. doi: 10.1002/jcp.20110. [DOI] [PubMed] [Google Scholar]
- Oshima Y, Oshima S, Nambu H, Kachi S, Takahashi K, Umeda N, Shen J, Dong A, Apte RS, Duh E, Hackett SF, Okoye G, Ishibashi K, Handa J, Melia M, Wiegand S, Yancopoulos G, Zack DJ, Campochiaro PA. Different effects of angiopoietin 2 in different vascular beds in the eye; new vessels are most sensitive. FASEB J. 2005;19 (8):963–965. doi: 10.1096/fj.04-2209fje. [DOI] [PubMed] [Google Scholar]
- Oshima Y, Takahashi K, Oshima S, Saishin Y, Saishin Y, Silva RL, Liang X, Reddy PS, Ganesh S, Brann R, Liau G, Kaleko M, Connelly S, Campochiaro PA. Intraocular gutless adenoviral vectored VEGF stimulates anterior segment but not retinal neovascularization. J Cell Physiol. 2004c;199 (3):399–411. doi: 10.1002/jcp.10441. [DOI] [PubMed] [Google Scholar]
- Ozaki H, Hayashi H, Vinores SA, Moromizato Y, Campochiaro PA, Oshima K. Intravitreal sustained release of VEGF causes retinal neovascularization in rabbits and breakdown of the blood-retinal barrier in rabbits and primates. Exp Eye Res. 1997;64 (4):505–517. doi: 10.1006/exer.1996.0239. [DOI] [PubMed] [Google Scholar]
- Ozaki H, Seo MS, Ozaki K, Yamada H, Yamada E, Hofmann F, Wood J, Campochiaro PA. Blockade of vascular endothelial cell growth factor receptor signaling is sufficient to completely prevent retinal neovascularization. Am J Pathol. 2000;156 (2):697–707. doi: 10.1016/S0002-9440(10)64773-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozaki H, Yu A, Della N, Ozaki K, Luna JD, Yamada H, Hackett SF, Okamoto N, Zack DJ, Semenza GL, Campochiaro PA. Hypoxia inducible factor-1a is increased in ischemic retina: temporal and spatial correlation with VEGF expression. Invest Ophthalmol Vis Sci. 1999;40 (1):182–189. [PubMed] [Google Scholar]
- Park JE, Chen HH, Winer J, Houck KA, Ferrara N. Placenta Growth Factor: potentiation of vascular endothelial growth factor bioactivity, in vitro and in vivo, and high affinity binding to Flt-1 but not to Flk-1/KDR. J Biol Chem. 1994;269 (41):25646–25654. [PubMed] [Google Scholar]
- Patz A, Eastham A, Higgenbotham DH, Kleh T. Oxygen studies in retrolental figroplasia: Production of the microscopic changes of retrolental fibroplasia in experimental animals. Am J Ophthalmol. 1953;36 (11):1511–1522. [PubMed] [Google Scholar]
- Pechan P, Rubin H, Lukason M, Ardinger J, DuFresne E, Hauswirth WW, Wadsworth SC, Scaria A. Novel anit-VEGF chimeric molecules delivered by AAV vectors for inhibition of retinal neovascularization. Gene Ther. 2009;16 (1):10–16. doi: 10.1038/gt.2008.115. [DOI] [PubMed] [Google Scholar]
- Petitclerc C, Boutaud A, Prestayko A, Xu J, Sado Y, Ninomiya Y, Sarras MP, Jr, Hudson BG, Brooks PC. New functions for non-collagenous domains of human collagen type IV. Novel integrin ligands inhibiting angiogenesis and tumor growth in vivo. J Biol Chem. 2000;275 (11):8051–8061. doi: 10.1074/jbc.275.11.8051. [DOI] [PubMed] [Google Scholar]
- Pierce EA, Foley ED, Smith LE. Regulation of vascular endothelial growth factor by oxygen in a model of retinopathy of prematurity. Arch Ophthalmol. 1996;114 (10):1219–1228. doi: 10.1001/archopht.1996.01100140419009. [DOI] [PubMed] [Google Scholar]
- Raisler BJ, Berns KI, Grant MB, Beliaev D, Hauswirth WW. Adeno-associated virus type 2 expression of pigmented epithelium-derived factor or Kringles 1–3 of angiostatin reduce retinal neovascularization. Proc Natil Acad Sci USA. 2002;99 (13):8909–8914. doi: 10.1073/pnas.122247299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenfeld PJ, Brown DM, Heier JS, Boyer DS, Kaiser PK, Chung CY, Kim RY Marina Study Group. Ranibizumab for neovascular age-related macular degeneration. N Eng J Med. 2006;355 (14):1419–1431. doi: 10.1056/NEJMoa054481. [DOI] [PubMed] [Google Scholar]
- Rota R, Riccioni T, Zaccarini M, Lamartina S, Gallo AD, Fusco A, Kovesdi I, Balestrazzi E, Abeni DC, Ali RR, Capogrossi MC. Marked inhibition of retinal neovascularization in rats following soluble-flt-1 gene transfer. J Gene Med. 2004;6 (9):992–1002. doi: 10.1002/jgm.586. [DOI] [PubMed] [Google Scholar]
- Ryan SJ. Subretinal neovascularization: natural history of an experimental model. Arch Ophthalmol. 1982;100 (11):1804–1809. doi: 10.1001/archopht.1982.01030040784015. [DOI] [PubMed] [Google Scholar]
- Saint-Geniez M, Kurihara T, Sekiyama E, Maldonado AE, D’Amore PA. An essential role for RPE-derived soluble VEGF in the maintenance of the choriocapillaris. Proc Natl Acad Sci USA. 2009;106 (44):18751–18756. doi: 10.1073/pnas.0905010106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saishin Y, Saishin Y, Takahashi K, Lima Silva R, Hylton D, Rudge JS, Wiegand SJ, Campochiaro PA. VEGF-TRAPR1R2 suppresses choroidal neovascularization and VEGF-induced breakdown of the blood-retinal barrier. J Cell Physiol. 2003;195 (2):241–248. doi: 10.1002/jcp.10246. [DOI] [PubMed] [Google Scholar]
- Sato TN, Tozawa Y, Deutsch U, Wolburg-Buchholz K, Fujiwara Y, Gendron-Maguire M, Gridley T, Wolburg H, Risau W, Qin Y. Distinct roles of the receptor tyrosine kinases Tie-1 and Tie-2 in blood vessel formation. Nature. 1995;376 (6535):70–74. doi: 10.1038/376070a0. [DOI] [PubMed] [Google Scholar]
- Semenza GL. HIF-1: mediator of physiological and pathophysiological responses to hypoxia. J Appl Physiol. 2000;88 (4):1474–1480. doi: 10.1152/jappl.2000.88.4.1474. [DOI] [PubMed] [Google Scholar]
- Seo MS, Okamoto N, Vinores MA, Vinores SA, Hackett SF, Yamada H, Yamada E, Derevjanik NL, LaRochelle W, Zack DJ, Campochiaro PA. Photoreceptor-specific expression of PDGF-B results in traction retinal detachment. Am J Pathol. 2000;157 (3):995–1005. doi: 10.1016/S0002-9440(10)64612-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen J, Frye M, Lee BL, Reinardy JL, McClung JM, Ding K, Kojima M, Xia H, Seidel C, Lime e Silva R, Dong A, Hackett SF, Wang J, Howard BW, Vestweber D, Kontos CD, Peters KG, Campochiaro PA. Targeting VE-PTP activates TIE2 and stabilizes the ocular vasculature. J Clin Invest. 2014;124 (10):4564–4576. doi: 10.1172/JCI74527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen J, Xie B, Dong A, Swaim M, Hackett SF, Campochiaro PA. In vivo immunostaining demonstrates macrophages associate with growing and regressing vessels. Invest Ophthalmol Vis Sci. 2007;48 (9):4335–4341. doi: 10.1167/iovs.07-0113. [DOI] [PubMed] [Google Scholar]
- Smith LEH, Wesolowski E, McLellan A, Kostyk SK, D’Amato R, Sullivan R, D’Amore PA. Oxygen-induced retinopathy in the mouse. Invest Ophthalmol Vis Sci. 1994;35 (1):101–111. [PubMed] [Google Scholar]
- Stone J, Itin A, Alon T, Pe’er J, Gnessin H, Chan-Ling T, Keshet E. Development of retinal vasculature is mediated by hypoxia-induced vascular endothelial growth factor (VEGF) expression by neuroglia. J Neurosci. 1995;15 (7 pt 1):4738–4747. doi: 10.1523/JNEUROSCI.15-07-04738.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tobe T, Okamoto N, Vinores MA, Derevjanik NL, Vinores SA, Zack DJ, Campochiaro PA. Evolution of neovascularization in mice with overexpression of vascular endothelial growth factor in photoreceptors. Invest Ophthalmol Vis Sci. 1998a;39 (1):180–188. [PubMed] [Google Scholar]
- Tobe T, Ortega S, Luna JD, Ozaki H, Okamoto N, Derevjanik NL, Vinores SA, Basilico C, Campochiaro PA. Targeted disruption of the FGF2 gene does not prevent choroidal neovascularization in a murine model. Am J Pathol. 1998b;153 (5):1641–1646. doi: 10.1016/S0002-9440(10)65753-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tolentino MJ, Miller JW, Gragoudas ES, Chatzistefanou K, Ferrara N, Adamis AP. Vascular endothelial growth factor is sufficient to produce iris neovascularization and neovascular glaucoma in a nonhuman primate. Arch Ophthalmol. 1996;114 (8):964–970. doi: 10.1001/archopht.1996.01100140172010. [DOI] [PubMed] [Google Scholar]
- Ueno S, Pease ME, Wersinger DMB, Masuda T, Vinores SA, Licht T, Zack DJ, Quigley H, Keshet E, Campochiaro PA. Prolonged blockade of VEGF family members does not cause identifiable damage to retinal neurons or vessels. J Cell Physiol. 2008;217 (1):13–22. doi: 10.1002/jcp.21445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Umeda N, Kachi S, Akiyama H, Zahn G, Vossmeyer D, Stragies R, Campochiaro PA. Suppression and regression of choroidal neovascularization by systemic administration of an Alpha5Beta1 integrin antagonist. Mol Pharmacol. 2006;69 (6):1820–1828. doi: 10.1124/mol.105.020941. [DOI] [PubMed] [Google Scholar]
- Vinores SA, Xiao WH, Aslam S, Shen J, Oshima Y, Nambu H, Liu H, Carmeliet P, Campochiaro PA. Implication of the hypoxia response element of the VEGF promoter in mouse models of retinal and choroidal neovascularization, but not retinal vascular development. J Cell Physiol. 2006;206 (3):749–758. doi: 10.1002/jcp.20525. [DOI] [PubMed] [Google Scholar]
- Weber BHF, Vogt G, Pruett RC, Stohr H, Felbor U. Mutations in the tissue inhibitor of metalloproteinases-3 (TIMP3) in patients with Sorsby’s fundus dystrophy. Nat Genet. 1994;8 (4):352–356. doi: 10.1038/ng1294-352. [DOI] [PubMed] [Google Scholar]
- Yacyshyn OK, Lai PFH, Forse K, Teichert-Kuliszewska K, Jurasz P, Stewart DJ. Tyrosine phosphatase beta regulates angiopoietin-Tie2 signaling in human endothelial cells. Angiogenesis. 2009;12 (1):25–33. doi: 10.1007/s10456-008-9126-0. [DOI] [PubMed] [Google Scholar]
- Yamada H, Yamada E, Hackett SF, Ozaki H, Okamoto N, Campochiaro PA. Hyperoxia causes decreased expression of vascular endothelial growth factor and endothelial cell apoptosis in adult retina. J Cell Physiol. 1999;179 (2):149–156. doi: 10.1002/(SICI)1097-4652(199905)179:2<149::AID-JCP5>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- Yannuzzi LA, Negrao S, Iida T, Carvalho C, Rodriguez-Coleman H, Slakter JS, Freund KB, Sorenson J, Orlock D, Borodoker N. Retinal angiomatous proliferation in age-related macular degeneration. Retina. 2001;21 (5):416–434. doi: 10.1097/00006982-200110000-00003. [DOI] [PubMed] [Google Scholar]
- Yoshida T, Zhang H, Iwase T, Shen J, Semenza G, Campochiaro PA. Digoxin inhibits retinal ischemia-induced HIF-1alpha expression and ocular neovascularization. FASEB J. 2010;24 (6):1759–1767. doi: 10.1096/fj.09-145664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu DY, Cringle SJ, Su EN, Yu PK. Intraretinal oxygen levels before and after photoreceptor loss in the RCS rat. Invest Ophthalmol Vis Sci. 2000;41 (12):3999–4006. [PubMed] [Google Scholar]
- Yuan HT, Khankin EV, Karumanchi SA, Parikh SM. Angiopoietin 2 is a partial agonist/antagonist of Tie2 signaling in the endothelium. Molec Cell Biol. 2009;29 (8):2011–2022. doi: 10.1128/MCB.01472-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Qian DZ, Tan YS, Lee K, Gao P, Ren YR, Rey S, Hammers H, Chang D, Pili R, Dang CV, Liu JO, Semenza GL. Digoxin and other cardiac glycosides inhibit HIF-1alpha sythesis and block tumor growth. Proc Natl Acad Sci USA. 2008;105 (50):19579–19586. doi: 10.1073/pnas.0809763105. [DOI] [PMC free article] [PubMed] [Google Scholar]