

Abstract
AIM
To explore the effects and mechanism of vascular endothelial cadherin (VE-cadherin) on experimental corneal neovascularization (CRNV).
METHODS
Mouse corneas were burned with sodium hydroxide to build a CRNV model. The burned corneas were locally administrated with anti-mouse VE-cadherin neutralizing antibody. Annexin V and cluster of differentiation 31 (CD31) double staining was used to measure vascular endothelial cell apoptosis with the use of flow cytometry (FCM). The protein expression of NADPH oxidase 2 (Nox2), caspase-3, and protein kinase C (PKC) in the burned corneas were examined by Western blot. Human retinal endothelial cell (HREC) proliferation was detected using a Cell Counting Kit 8 (CCK-8) assay in vitro.
RESULTS
The amount of CRNV peaked two weeks after the alkali burn. FCM confirmed that VE-cadherin neutralizing antibody treatment increased CD31 positive cell apoptosis. Western blot revealed that the intracorneal protein expression of Nox2 and caspase-3 were up-regulated, while PKC was down-regulated in the VE-cadherin neutralizing antibody administrated group. CCK-8 assay showed that VE-cadherin neutralizing antibody markedly inhibited HREC proliferation.
CONCLUSION
VE-cadherin exhibited an anti-apoptosis effect through enhanced PKC signaling and an enhanced cell proliferation pathway.
Keywords: vascular endothelial cadherin, neovascularization, cornea, chemokine
INTRODUCTION
The cornea is a transparent tissue that lacks vessels under normal physiological conditions. If infection, physical injury, or chemical or other pathological damages occur, corneal neovascularization (CRNV) will take place. CRNV can reduce the transparency of the corneas and cause serious visual impairment [1],[2], eventually leading to blindness[3]. Therefore, effective prevention of the occurrence of CRNV is important to reduce corneal injuries and restore vision[4].
CRNV is one of a multitude of angiogenesis-dependent diseases[5]. Many studies have been carried out using anti-angiogenic drugs because medications such as bevacizumab have been proven to be of benefit for the treatment of corneal disease[6]; however, clinical data on the safety and efficacy of such drugs are currently limited to non-randomized, largely non-comparative case series, and antiangiogenic agents developed and approved specifically for CRNV are not yet available. Hence, although we have a thorough understanding of the molecules relevant to vascular development, we are still on the cusp of translating that knowledge into specific therapeutic agents, which will be useful in the ophthalmic clinic to treat and prevent CRNV[7].
Vascular endothelial cadherin (VE-cadherin) is a member of the adhesion molecule family[8]. It is mainly expressed in vascular endothelial cells[9]. As a component of endothelial cell-to-cell adhesion junctions, it plays a key role in the maintenance of vascular integrity[10]. During embryo development, VE-cadherin is required for the organization of a stable vascular system, while in the adult it controls vascular permeability and inhibits unrestrained vascular growth[11],[12]. The mechanisms of action of VE-cadherin are complex and include reshaping and organization of the endothelial cell cytoskeleton and modulation of gene transcription[13].
The roles of VE-cadherin in mediating CRNV subsequent to severe injury remain unclear. To further address the roles of VE-cadherin in CRNV, alkali-induced CRNV was induced and anti-VE-cadherin neutralizing antibody was topically administrated into the injured eyes. The anti-VE-cadherin neutralizing antibody is a kind of reagent which is used for inhibiting VE-cadherin function and thereby blocking the VE-cadherin signal pathway. Here, we provide the definitive evidence to indicate the important role and mechanism of VE-cadherin in CRNV.
MATERIALS AND METHODS
Reagents and Antibodies
Rat anti-mouse VE-cadherin monoclonal antibodies (mAbs) (Clone: BV13, Cat No. 138003) were acquired from BioLegend (San Diego, CA, USA). Goat anti-mouse cluster of differentiation 31 (CD31) (ABIN958222) mAbs were purchased from antibodies-online.com (Atlanta, GA, USA). 3H-Indocyanines (Cy3)-labeled donkey anti-goat IgG antibody was obtained from Pierce (Rockford, IL, USA). Rabbit anti-NADPH oxidase 2 (Nox2) polyclonal antibody (Cat No. NBP1-02160), rabbit anti-caspase-3 polyclonal antibody (Cat No. NBP1-51398), and rabbit anti-protein kinase C (PKC) polyclonal antibody (Cat No. NBP1-61529) were supplied by Novus Biologicals (Littleton, CO, USA). We purchased horseradish peroxidase (HRP)-goat anti-rabbit IgG antibody from Huamei Biological Engineering Co., Ltd. (Shanghai, China). The Cell Counting Kit-8 (CCK-8) was sold by Biyuntian Biological Engineering Co., Ltd. (Shanghai, China), and the Human Retinal Endothelial cell line (HREC) was purchased from Yaji Biological Engineering Co., Ltd. (Shanghai, China).
Animals
All animal experiments were approved by the Guidelines for the Care and Use of Laboratory Animals of the Chinese Medical Academy and the Soochow University Animal Care Committee and were performed in accordance with the Association for Research in Vision and Ophthalmology (ARVO) Statement for the Use of Animals in Ophthalmic and Vision Research. Specific pathogen-free 7- to 8-week-old male BALB/c mice weighing 20-25 g were obtained from Shanghai Laboratory Animal Co., Ltd (SLAC), and were kept in our animal facility under specific pathogen-free conditions. Forty-eight mice were equally assigned into either the control group or the VE-cadherin antibodies group according to a randomized number table. A 12-hour day/12-hour night cycle was maintained during the entire course of the study. Animals were kept in groups of five and fed regular lab chow and water ad libitum.
Alkali-induced Corneal Injury Model
Mice were anesthetized with an intraperitoneal injection of 1.8% (v/v) avertin at a dosage of 0.20 mL/10 g body weight. A 2-mm disc of filter paper saturated with 1 mol/L sodium hydroxide (NaOH) was placed onto the left cornea of each mouse for 40s and then followed by rinsing extensively with 10 mL of phosphate-buffered saline (PBS). The corneal epithelia were removed using a corneal knife in a rotary motion parallel to the limbus by gently scraping over the corneal surface without injuring the underlying corneal stroma[14]. Erythromycin ophthalmic ointment was instilled immediately following epithelial denudation. Anti-mouse VE-cadherin antibody was dissolved in 0.2% sodium hyaluronate (Sigma-Aldrich) at 10 µg/mL. Next, 5 mL of VE-cadherin antibody preparation or vehicle was applied topically to the alkali-treated eye three times a day for 7d. At the indicated time on day 14, the mice were sacrificed and the corneas were removed from experimented eyes.
Flow Cytometrical Analysis
Mononuclear cells were isolated from corneas according to a previously described procedure with some modifications[15]. Briefly, at day 14 after the alkali injury, the corneas were removed, teased away with scissors, and incubated at 37°C for 30min with constant shaking in the presence of 0.5 mg/mL collagenase type D (Roche Diagnostics, Mannheim, Germany). Cell suspensions were then passed over a nylon filter with a pore size of 100 µm. The resultant cells were stained with propidium iodide (PI), Annexin V, and goat anti-mouse CD31 following by staining with Cy3-labeled donkey anti-goat IgG antibody. The samples stained with non-immunized goat IgG were used as an isotype control. In apoptotic cells, the membrane phospholipid phosphatidylserine (PS) is translocated from the inner to the outer leaflet of the plasma membrane, thereby exposing the PS to the external cellular environment. Annexin V is a 35-36 kDa Ca2+-dependent phospholipid-binding protein that has a high affinity for PS and binds to cells with exposed PS. These characteristics allow it to retain its high affinity for PS and thus to serve as a sensitive probe for the flow cytometric analysis of cells that are undergoing apoptosis.
Western Blotting
Caspase-3, Nox2, and PKC protein expression levels in vehicle- and VE-cadherin antibody-treated corneas were evaluated by Western blotting. The eyes were enucleated, and the corneal samples were placed in a 150-mL lysis buffer [20 mmol/L imidazole hydrochloric acid (HCl), 10 mmol/L potassium chloride (KCl), 1 mmol/L magnesium chloride (MgCl2), 10 mmol/L ethylene glycol tetra acetic acid (EGTA), 1% Triton, 10 mmol/L sodium fluoride (NaF), 1 mmol/L sodium molybdate, 1 mmol/L ethylenediaminetetraacetic acid (EDTA)], pH 6.8 supplemented with a protease inhibitor cocktail (Boehringer Mannheim, Indianapolis, IN, USA) and were sonicated. The lysate was centrifuged at 12 000 rpm for 15min at 4°C. Samples (20 µg/each lane) were boiled for 5min and then separated by sodium dodecyl sulfate (SDS)-polyacrylamide gel electrophoresis under denaturing conditions and electroblotted to a polyvinylidene difluoride membrane (Bio-Rad, Hercules, CA, USA). The membranes were blocked with PBS/5% nonfat dry milk for nonspecific binding and incubated with antibodies as follows: anti-caspase-3, anti-Nox2, and anti-PKC (Novus Biologicals, Littleton, CO, USA). Immunoblot assays were then washed and incubated with a HRP-labeled secondary antibody (Amersham Pharmacia, Piscataway, NJ, USA). The blot was visualized with enhanced chemiluminescence (ECL Plus; Amersham Pharmacia) according to the manufacturer's instructions[16].
CCK-8 Assay
Cell proliferation was analyzed using CCK-8 (Biyuntian Biological Engineering Co. Ltd, Shanghai, China). This kit measures the metabolic activity of dehydrogenases by employing a tetrazolium salt. In the presence of dehydrogenases, tetrazolium salt produces a yellow-colored, water-soluble Formosan. The amount of the Formosan is directly proportional to the number of living cells and can be measured by a thermo multi-scan EX plate reader (Thermo Multiskan EX plate reader, VWR, CA, USA). After the HRECs were attached to the plate and stimulated with anti-VE-cadherin neutralizing antibody, the absorbance was measured at 6, 12, and 24h. The inhibition rate (IR) for the proliferation of cells in different groups was compared to the control groups.
Statistical Analysis
The means and standard error of the mean (SEM) were calculated for all parameters determined in the study. Data were analyzed statistically using one-way analysis of variance (ANOVA) or two-tailed Student's t-test. A value of P<0.05 was considered statistically significant.
RESULTS
Exacerbated Cell Death in Vascular Endothelial Cadherin Antibody-treated Animals
Firstly, we compared the expression of apoptosis-related protein of caspase-3 in control versus VE-cadherin antibody-treated mice. Western blot demonstrated that the caspase-3 relative expression level was increased to 2.14±0.45 in VE-cadherin antibody-treated corneas in comparison to controls (1.32±0.39) (Figure 1A). Statistical significance was shown between these two groups (P<0.05) (Figure 1B).
Figure 1. Protein of caspase-3 expression in the injured corneas of vehicle and anti-VE-cadherin mAbs-treated BALB/c mice.

A: Protein extracts were obtained and subjected to Western blotting analysis. Representative results from three independent experiments are shown here. B: Ratios of caspase-3 to glyceraldehyde-3-phosphate dehydrogenase (GAPDH) protein bands of vehicle- (open bars) and anti-mouse VE-cadherin antibody (black bars) were determined as described in Methods. All values represent mean±SEM (n=6-8 animals). aP <0.05 compared with vehicle-treated mice.
Dependence of Vascular Endothelial Viability upon Vascular Endothelial Cadherin
To determine whether VE-cadherin's protective effects in CRNV were targeted at vascular endothelial cells, PI and Annexin V-positive cells co-localized with CD31 were examined using FCM and the apoptotic cells were quantified. The results showed that the apoptotic cells were vascular endothelial cells. Apoptotic cells co-localized with CD31 were significantly increased to 11.27%±1.66% in VE-cadherin antibody-treated mice compared to vehicle-treated mice (6.81%±1.14%) (P<0.05) (Figure 2). These data suggested that the protective effect of VE-cadherin on alkali-induced CRNV was directly targeted at vascular endothelial cells by suppressing vascular endothelial cell apoptosis.
Figure 2. The proportion of PI/Annexin V-positive cells co-localized with CD31 in corneas after alkali injury.

Corneal tissues were obtained 14d after injury from vehicle- or anti-VE-cadherin mAbs-treated mice, and the tissues from 7-8 mice were combined and subjected to analysis using a flow cytometer after being immunostained with anti-mouse PI/Annexin V and CD31 antibody. Isotype IgG derived from the same species of the test antibody was used as a negative control. Representative results from three to four tests of intracorneal PI/Annexin V and CD31-positive cells from either vehicle- or anti-VE-cadherin mAbs-treated BABL/c are shown.
Vascular Endothelial Cadherin Mediated Repression of Nox2 Activation in Corneal Neovascularization
To determine the mechanism by which VE-cadherin regulates cell apoptosis, we next measured Nox2 levels in the burned corneas. Nox2 levels increased to 0.42±0.08 in the VE-cadherin antibody-treated mice, which was much more than the control mice (0.20±0.05) (P<0.05) (Figure 3A, 3B), suggesting that Nox2's effect was VE-cadherin-dependent. Nox2 is a very important cytokine that promotes cell apoptosis. Our results suggested that VE-cadherin plays an important role in suppressing cell apoptosis at least in part by inhibiting Nox2 expression in hypoxia after alkali injury. Furthermore, we also detected PKC expression in the burned corneas. PKC is one of the most important transcription factors for cell survival and proliferation. We found that PKC decreased to 0.26±0.07 in VE-cadherin antibody-treated mice, which was a greater decrease than that of the control mice (0.53±0.09) (P<0.05) (Figure 3A, 3C), suggesting that VE-cadherin antibody suppresses PKC expression.
Figure 3. Protein of Nox2 and PKC expression in the injured corneas of vehicle- and anti-VE-cadherin mAbs-treated BALB/c mice.

A: Protein extracts were obtained and subjected to Western blotting analysis. Representative results from three independent experiments are shown here. B, C: Ratios of Nox2 and PKC to GAPDH protein bands of vehicle- (open bars) and anti-mouse VE-cadherin antibody (black bars) were determined as described in Methods. All values represent mean±SEM (n=6-8 animals). aP <0.05 compared with vehicle-treated mice.
Reduced Cell Proliferation with Vascular Endothelial Cadherin Antibody Stimulation
To delineate the effects of VE-cadherin on vascular endothelial cell function, we also examined the role of VE-cadherin in cell proliferation of HRECs in vitro. After incubation and stimulation with anti-VE-cadherin antibody for 24h, cell viability was examined; HRECs incubated with anti-VE-cadherin antibody showed a significant reduction in cell proliferation compared to the vehicles (Figure 4). Optical density (OD) value quantification and statistical analysis on inhibition rate (IR) demonstrated that the anti-VE-cadherin antibody was able to reduce cell proliferation (Table 1). These data indicated that a decrease in proliferation of HRECs after anti-VE-cadherin antibody stimulation was responsible for the protective effect of VE-cadherin in vivo.
Figure 4. CCK-8 assays to detect the effects of VE-cadherin antibody on cell proliferation.
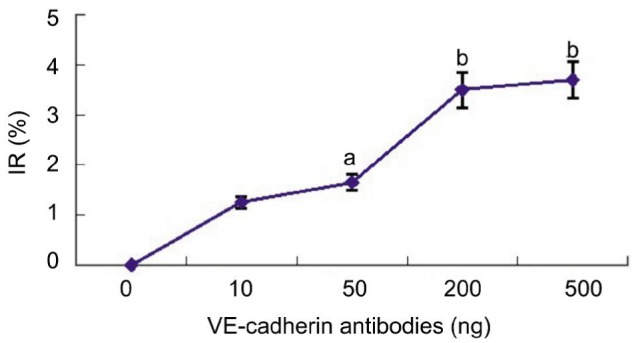
The inhibition rate (IR) on HREC cell proliferation was determined. Each value represents the mean and SEM (n=3). aP <0.05 and bP <0.01 compared with untreated animals.
Table 1. The effects of VE-cadherin antibody on cell proliferation.
| Vaule | The concetration of VE-cadherin antibodies intervention |
||||
| 0 ng | 10 ng | 50 ng | 200 ng | 500 ng | |
| OD | 1.61±0.25 | 1.59±0.19 | 1.56±0.16 | 1.55±0.08 | 1.54±0.06 |
| IR | 1.4±0.27 | 1.8±0.35 | 3.5±0.76 | 3.7±0.82 | |
| P | 0.510 | 0.040 | 0.003 | 0.002 | |
DISCUSSION
We have previously found that VE-cadherin was intracorneally expressed and had a protective effect on CRNV[17]. In the present study, we explored the specific mechanism and latent efficacy of VE-cadherin. Here, we have identified for the first time that VE-cadherin has a potent protective effect in CRNV. Our results demonstrated that blocking of VE-cadherin increased intracorneal vascular endothelial cell apoptosis, while PKC was down-regulated in the group that received VE-cadherin neutralizing antibody. Further, we also identified that VE-cadherin neutralizing antibody was able to markedly inhibit HREC proliferation, establishing a molecular mechanism of action.
The injured cornea consists of fibroblasts, corneal epithelial cells, and vascular endothelial cells[18]. To assess the possibility that VE-cadherin neutralizing antibody increases endothelial cells (EC) death, we labeled corneal cells with Annexin V, PI, and CD31 (an EC marker). Our results demonstrated that in CRNV animals, Annexin V/PI-positive cells also co-expressed CD31[19]. In addition, we found that the percentage of Annexin V, PI, and CD31 co-expression cells in the VE-cadherin neutralizing antibody administrated group was higher than the control group. We also examined the intraocorneal caspase-3 protein expression. Based upon our results, the caspase-3 protein expression was higher in the VE-cadherin neutralizing antibody administrated group than in the control group. Caspase-3 is one of the most important proteins expressed by apoptotic cells[20],[21]. The quality of caspase-3 is closely related with the number of apoptotic cells. More specifically, the caspase-3 protein directly reflects the pathologic degree of cell apoptosis. Thus, our results indicated that VE-cadherin had a protective effect on cell viability by suppressing cell apoptosis. Taken together, these results suggest that apoptotic cells at this experiment model are vascular endothelial cells, and VE-cadherin exerts a protective effect on newly developed vascular endothelial corneal cells from cell apoptosis, as this protective effect was blocked by VE-cadherin neutralizing antibody in our current experiment. Therefore, VE-cadherin improved vascular endothelial survival and integrity under alkali injury. Furthermore, we used VE-cadherin neutralizing antibody to stimulate HRECs in vitro. In CCK-8 assay, we found that HREC proliferation was suppressed with the stimulation of VE-cadherin neutralizing antibody. These results are consistent with our in vivo experiments, suggesting that VE-cadherin was the most important protective factor for vascular endothelial cell survival and proliferation.
Our prior studies have demonstrated that the VE-cadherin gene is expressed in the mouse cornea, and we have also shown that VE-cadherin exerted a direct protective effect on CRNV, as evidenced by corneal whole-mount immunofluorescence where the alkali injury markedly reduced the vascular areas in corneas to a larger extent in VE-cadherin neutralizing antibody-treated mice than in controls. Hence, in the present study, we concluded that VE-cadherin's vascular protective effects may result from a direct effect of inhibiting cell apoptosis in vascular endothelial cells.
VE-cadherin is an important signaling molecule. Signaling via VE-cadherin influences endothelial cell behavior by modulating the activity of growth factor receptors, intracellular messengers, and proteins that regulate gene transcription[22]. Experimental studies have revealed that VE-cadherin protects the stabilization of the vasculature[23]; however, they have provided little insight into which pathophysiological parameters VE-cadherin was able to improve in cornea neovascularization or how it could do so on a molecular level[24]. Therefore, in order to delineate VE-cadherin's protective effects in CRNV, we also examined intracorneal Nox2 and PKC protein expression. We found that VE-cadherin suppressed Nox2 expression. Moran et al[25] reported that the transcription of Nox2 and Nox4 expression was enhanced under pathologic conditions and Nox2 was one of the promoters of reactive oxygen species (ROS), which was the crucial factor in cell apoptosis. In turn, VE-cadherin suppression of Nox2 expression may be one of the imperative pathways that protect endothelial cells from apoptosis. Our results in this experiment of CRNV are consistent with previous findings, suggesting that VE-cadherin is an important anti-apoptosis factor.
In summary, our studies identified novel anti-apoptotic activities of VE-cadherin in CRNV, and established that it negatively regulates the Nox2 pathway, which is likely responsible in part for its anti-apoptosis effect. Therefore, VE-cadherin has therapeutic potential as target for CRNV. Investigation of the physiologic regulation of VE-cadherin may thus be useful for the treatment in clinical settings characterized by persistent neovascularization.
Acknowledgments
Foundations: Supported by the National Natural Science Foundation of China (No. 81200727; No. 30972712); Jiangsu Province's Key Provincial Talents Program (No. RC2011104); Suzhou Municipal Natural Science Foundation (No. SYS201448); the Soochow Scholar Project of Soochow University.
Conflicts of Interest: Liu GQ, None; Wu HY, None; Xu J, None; Wang MJ, None; Lu PR, None; Zhang XG, None.
REFERENCES
- 1.Zhang SX, Ma JX. Ocular neovascularization: Implication of endogenous angiogenic inhibitors and potential therapy. Prog Retin Eye Res. 2007;26(1):1–37. doi: 10.1016/j.preteyeres.2006.09.002. [DOI] [PubMed] [Google Scholar]
- 2.Usui T. Mechanisms and regulation of corneal neovascularization. Nihon Ganka Gakkai Zasshi. 2009;113(11):1041–1049. [PubMed] [Google Scholar]
- 3.Mazhdrakova I, Demerdjieva Z. Neovascularisation of the cornea. Klin Monbl Augenheilkd. 2005;222(8):623–629. doi: 10.1055/s-2005-858449. [DOI] [PubMed] [Google Scholar]
- 4.Koenig Y, Bock F, Kruse FE, Stock K, Cursiefen C. Angioregressive pretreatment of mature corneal blood vessels before keratoplasty: fine-needle vessel coagulation combined with anti-VEGFs. Cornea. 2012;31(8):887–892. doi: 10.1097/ICO.0b013e31823f8f7a. [DOI] [PubMed] [Google Scholar]
- 5.Folkman J. Angiogenesis: an organizing principle for drug discovery? Nat Rev Drug Discov. 2007;6(4):273–286. doi: 10.1038/nrd2115. [DOI] [PubMed] [Google Scholar]
- 6.Bock F, Onderka J, Dietrich T, Bachmann B, Kruse FE, Paschke M, Zahn G, Cursiefen C. Bevacizumab as a potent inhibitor of inflammatory corneal angiogenesis and lymphangiogenesis. Invest Ophthalmol Vis Sci. 2007;48(6):2545–2552. doi: 10.1167/iovs.06-0570. [DOI] [PubMed] [Google Scholar]
- 7.Menzel-Severing J. Emerging techniques to treat corneal neovascularisation. Eye (Lond) 2012;26(1):2–12. doi: 10.1038/eye.2011.246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wallez Y, Vilgrain I, Huber P. Angiogenesis: the VE-cadherin switch. Trends Cardiovasc Med. 2006;16(2):55–59. doi: 10.1016/j.tcm.2005.11.008. [DOI] [PubMed] [Google Scholar]
- 9.Vestweber D, Winderlich M, Cagna G, Nottebaum AF. Cell adhesion dynamics at endothelial junctions: VE-cadherin as a major player. Trends Cell Biol. 2009;19(1):8–15. doi: 10.1016/j.tcb.2008.10.001. [DOI] [PubMed] [Google Scholar]
- 10.Giannotta M, Trani M, Dejana E. VE-cadherin and endothelial adherens junctions: active guardians of vascular integrity. Dev Cell. 2013;26(5):441–454. doi: 10.1016/j.devcel.2013.08.020. [DOI] [PubMed] [Google Scholar]
- 11.Takeichi M. The cadherins: cell-cell adhesion molecules controlling animal morphogenesis. Development. 1988;102(4):639–655. doi: 10.1242/dev.102.4.639. [DOI] [PubMed] [Google Scholar]
- 12.Noda K, Zhang J, Fukuhara S, Kunimoto S, Yoshimura M, Mochizuki N. Vascular endothelial-cadherin stabilizes at cell-cell junctions by anchoring to circumferential actin bundles through alpha- and beta-catenins in cyclic AMP-Epac-Rap1 signal-activated endothelial cells. Mol Biol Cell. 2010;21(4):584–596. doi: 10.1091/mbc.E09-07-0580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schulte D, Küppers V, Dartsch N, Broermann A, Li H, Zarbock A, Kamenyeva O, Kiefer F, Khandoga A, Massberg S, Vestweber D. Stabilizing the VE-cadherin-catenin complex blocks leukocyte extravasation and vascular permeability. EMBO J. 2011;30(20):4157–4170. doi: 10.1038/emboj.2011.304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Liu GQ, Lu PR, Li LB, Zhang XG. Inhibited experimental corneal neovascularization by neutralizing anti-SDF-1α antibody. Int J Ophthalmol. 2012;5(1):7–12. doi: 10.3980/j.issn.2222-3959.2012.01.02. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Liu G, Zhang W, Xiao Y, Lu P. Critical Role of IP-10 on Reducing Experimental Corneal Neovascularization. Curr Eye Res. 2014;13:1–11. doi: 10.3109/02713683.2014.968934. [DOI] [PubMed] [Google Scholar]
- 16.Liu G, Lu P, Li L, Jin H, He X, Mukaida N, Zhang X. Critical role of SDF-1α-induced progenitor cell recruitment and macrophage VEGF production in the experimental corneal neovascularization. Mol Vis. 2011;17:2129–138. [PMC free article] [PubMed] [Google Scholar]
- 17.Liu GQ, Chen L, Xiao YH, Chen ZG, Lu PR. The effect and mechanism of VE-cadherin on the development of experimental corneal neovascularization. Chin J Optom Ophthalmol Vis Sci. 2013;15(3):174–180. [Google Scholar]
- 18.Hassell JR, Birk DE. The molecular basis of corneal transparency. Exp Eye Res. 2010;91(3):326–335. doi: 10.1016/j.exer.2010.06.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kaur M, Velmurugan B, Rajamanickam S, Agarwal R, Agarwal C. Gallic acid, an active constituent of grape seed extract, exhibits anti-proliferative, pro-apoptotic and anti-tumorigenic effects against prostate carcinoma xenograft growth in nude mice. Pharm Res. 2009;26(9):2133–2140. doi: 10.1007/s11095-009-9926-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Abu-Qare AW, Abou-Donia MB. Biomarkers of apoptosis: release of cytochrome c, activation of caspase-3, induction of 8-hydroxy-2′-deoxyguanosine, increased 3-nitrotyrosine, and alteration of p53 gene. J Toxicol Environ Health B Crit Rev. 2001;4(3):313–332. doi: 10.1080/109374001301419737. [DOI] [PubMed] [Google Scholar]
- 21.Shimizu S, Tsujimoto Y. Molecular mechanism of apoptosis. Seikagaku. 1998;70(1):14–21. [PubMed] [Google Scholar]
- 22.Mukherjee S, Tessema M, Wandinger-Ness A. Vesicular trafficking of tyrosine kinase receptors and associated proteins in the regulation of signaling and vascular function. Circ Res. 2006;98(6):743–756. doi: 10.1161/01.RES.0000214545.99387.e3. [DOI] [PubMed] [Google Scholar]
- 23.Wang Y, Kaiser MS, Larson JD, Nasevicius A, Clark KJ, Wadman SA, Roberg-Perez SE, Ekker SC, Hackett PB, McGrail M, Essner JJ. Moesin1 and Ve-cadherin are required in endothelial cells during in vivo tubulogenesis. Development. 2010;137(18):3119–3128. doi: 10.1242/dev.048785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liao F, Doody JF, Overholser J, Finnerty B, Bassi R, Wu Y, Dejana E, Kussie P, Bohlen P, Hicklin DJ. Selective targeting of angiogenic tumor vasculature by vascular endothelial-cadherin antibody inhibits tumor growth without affecting vascular permeability. Cancer Res. 2002;62(9):2567–2575. [PubMed] [Google Scholar]
- 25.Moran E, Ding L, Wang Z, Cheng R, Chen Q, Moore R, Takahashi Y, Ma JX. Protective and antioxidant effects of PPARα in the ischemic retina. Invest Ophthalmol Vis Sci. 2014;55(7):4568–4576. doi: 10.1167/iovs.13-13127. [DOI] [PMC free article] [PubMed] [Google Scholar]