

Abstract
AIM
To identify the disease-causing gene mutation in a Chinese pedigree with autosomal dominant cone-rod dystrophy (adCORD).
METHODS
A southern Chinese adCORD pedigree including 9 affected individuals was studied. Whole-exome sequencing (WES), coupling the Agilent whole-exome capture system to the Illumina HiSeq 2000 DNA sequencing platform was used to search the specific gene mutation in 3 affected family members and 1 unaffected member. After a suggested variant was found through the data analysis, the putative mutation was validated by Sanger DNA sequencing of samples from all available family members.
RESULTS
The results of both WES and Sanger sequencing revealed a novel nonsense mutation c.C766T (p.Q256X) within exon 5 of CRX gene which was pathogenic for adCORD in this family. The mutation could affect photoreceptor-specific gene expression with a dominant-negative effect and resulted in loss of the OTX tail, thus the mutant protein occupies the CRX-binding site in target promoters without establishing an interaction and, consequently, may block transactivation.
CONCLUSION
All modes of Mendelian inheritance in CORD have been observed, and genetic heterogeneity is a hallmark of CORD. Therefore, conventional genetic diagnosis of CORD would be time-consuming and labor-intensive. Our study indicated the robustness and cost-effectiveness of WES in the genetic diagnosis of CORD.
Keywords: cone-rod dystrophy, autosomal dominant cone-rod dystrophy, whole-exome sequencing, Sanger sequencing, CRX gene, mutation
INTRODUCTION
Cone-rod dystrophies (CORDs; prevalence, 1/40 000) are progressive inherited retinal disorders characterized predominantly by cone dysfunction in the early stage and subsequent rod degeneration[1]. The clinical manifestations of CORDs include photophobia, reduced visual acuity, color vision defects, and central scotoma. Nystagmus may present in some cases. Absent or severely impaired cone function on electroretinography (ERG) is the typical sign of CORDs[2]. Impairment of rod function is frequently observed soon after significant cone dysfunction[1]. In extreme cases, these progressive symptoms are accompanied by widespread, advancing retinal pigmentation along with central and peripheral chorioretinal atrophy. At the advanced stage, CORDs can be difficult to differentiate from retinitis pigmentosa based on the clinical signs alone[3],[4].
CORDs are exceptionally heterogeneous, both genetically and phenotypically[3]–[5]. Not only is the diagnosis of some of these diseases difficult because of overlapping phenotypes, but mutations within a single gene can cause very different phenotypes[3]–[5]. CORD may be transmitted as an autosomal dominant (adCORD), autosomal recessive (arCORD), or X-linked trait (xlCORD). To date, mutations in at least 26 genes have been reported to be associated with different forms of CORDs. Of these, 10 genes with mutations responsible for adCORD are: AIPL1[6], CRX[7], GUCA1A[8], GUCY2D[9], PITPNM3[10], PROM1[11], PRPH2[12], RIMS1[13], SEMA4A[14], and UNC119[15]. The next 13 for arCORD are: ABCA4, ADAM9, C8ORF37, CACNA2D4, CDHR1, CERKL, CNGB3, CNNM4, KCNV2, PDE6C, RAX2, RDH5, and RPGRIP1[15]. Other mapped loci or chromosomal regions for arCORD are: NF1, 18q21.1-q21.3, 1q12-q24, and 10q26[16]. xlCORD is caused by mutation in an alternative terminal exon 15 (ORF15) of the RPGR gene, which maps to chromosome Xp21.1[17]. Additional forms of xlCORD are Xq27.2-q28 and the CACNA1F gene on chromosome Xp11.23[18].
Whole-exome sequencing (WES) is a direct, reproducible, and robust method for the confirmation of novel pathogenic genes and the genetic diagnosis of both Mendelian and complex diseases[19]. Protein-coding genes constitute only about 1% of the human genome but harbor nearly 85% of the disease-causing mutations at individual Mendelian loci[20]. Therefore, selectively sequencing complete coding regions can serve as a genome-wide scan for pathogenic genes[20]. Moreover, WES may be a better choice for some diseases with overlapping symptoms that might be ambiguously associated with many pathogenic genes, in which event the detection of mutation by Sanger sequencing could be time-consuming and labor-intensive[19],[21].
Here, we used WES, coupling the Agilent whole-exome capture system to the Illumina HiSeq 2000 DNA sequencing platform, to identify a Chinese pedigree with adCORD.
SUBJECTS AND METHODS
Participants and Examinations
A five-generation Chinese family including 9 affected individuals was investigated in this study(Figure 1). Ophthalmic examination and diagnostic testing was based on color vision testing of three color axes using the Hardy-Rand-Rittler tests(HRR), full-field ERG, Goldmann perimetry, optical coherence tomography (OCT) scans and retinal thickness measurements. One hundred unrelated healthy matched controls were included. This study was conducted in conformity with the Declaration of Helsinki and was approved by the Ethics Committee of Zhejiang University. Written informed consent was given by all participants.
Figure 1. Pedigree of the family investigated Arrow indicates the proband (III-12).

Exome Sequencing and the Data Filtering and Analysis Pipeline
Peripheral blood genomic DNA samples from each of 4 members of the family, III-4, III-10, III-12 and IV-7, were prepared for WES (Figure 1). The whole-exome capture array design, library construction, next-generation sequencing, data filtering, and analysis pipeline followed protocols described previously[21].
Linkage Analysis
MERLIN(Multipoint Engine for Rapid Likelihood Inference) software was used to analyze the compiled pedigree structures of the WES dataset[22]. The filter threshold as a minimum LOD score of 6.0 was set for further analysis.
Prediction of Functional Impact
In an effort to assess the functional significance of the gene variations identified in this study, we used Sorting Intolerant from Tolerant(SIFT; http://sift.bii.a-star.edu.sg/) to predict the functional effect of non-synonymous single-nucleotide polymorphisms (SNPs)[23].
Sanger Sequencing
The putative mutations were validated by Sanger DNA sequencing of samples from available family members (Figure 1).
RESULTS
Clinical Examination
The proband (Figure 1, III-12) was a 50-year-old male, who had been diagnosed with progressively reduced visual acuity and mild color vision abnormalities when he was 40 years old. Fundus photographs indicated a 2PD gold-foil-like reflection of the macula, diffuse hypopigmentation. The peripheral retina exhibited bone spicule-like hyperpigmentation with attenuation of the retinal arteries (Figure 2A). OCT revealed retinal thinning in the macular region (Figure 2B). Fundus autofluorescence showed hyperfluorescence in the macula without peripheral choroidal atrophy (Figure 2C). ERG revealed mildly reduced cone responses and normal rod responses. The other affected members exhibited a similar clinical phenotype, and showed a clear autosomal dominant pattern of inheritance. Therefore, a diagnosis of CORD was made based on the published criteria[2],[3],[24],[25].
Figure 2. Ophthalmic examination results of the proband (III-12).
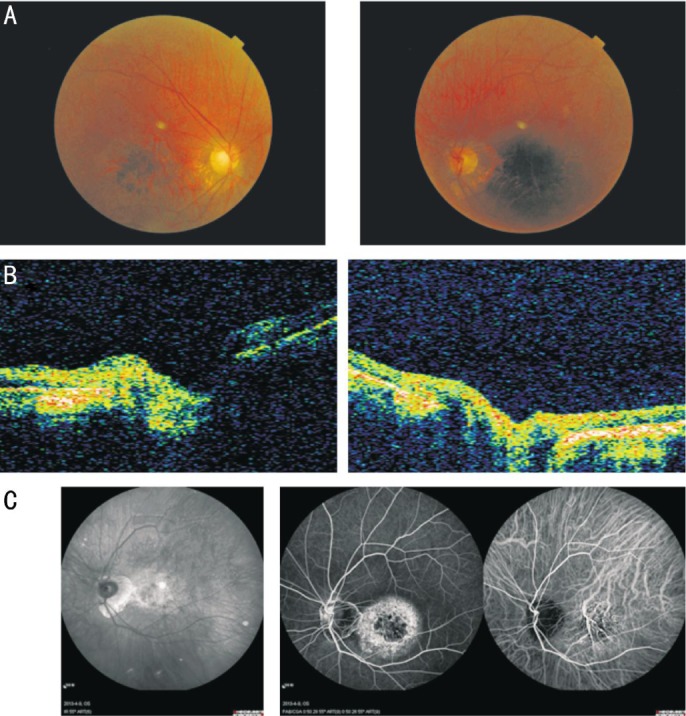
A: Fundus photographs of the proband showed diffuse hypopigmentation and reduced sensitivity in a central scotoma accompanied by maculopathy. The peripheral retina exhibited bone spicule-like hyperpigmentation with attenuation of retinal arteries. B: OCT of the proband revealed retinal thinning in the macular region. C: Fundus autofluorescence of the proband showed hyperfluorescence in the macula without peripheral choroidal atrophy.
Exome Sequencing Identified 4 Candidate Pathogenic Genes
We generated an average of about 4.5 billion bases of sequence per affected individual as paired-end, 90bp reads, and the fraction of effective bases on-target was about 95% with a minimum 20-fold average sequencing depth on-target. At this depth of coverage, >99% of the targeted bases were sufficiently covered to pass our thresholds for variant-calling (Tables 1, 2). We focused on non-synonymous (NS) variants, splice acceptor, and donor site mutations (SS), anticipating that synonymous variants would be far less likely to be pathogenic. Filtering against public SNP databases, the 1000 Genome Project, eight HapMap exomes, the in-house exome database provided by BGI and one normal individual from the family, 26 genes harboring 26 different NS/SS were shared by the three patients (Table 1).
Table 1. Overview of whole-exome sequencing data production.
| Exome capture statistics | III-12 (Proband) | III-10 (Unaffected) | III-4 (Affected) | IV-7 (Affected) |
| 1Target region (bp) | 51339787 | 51391525 | 51339787 | 51391525 |
| Raw reads | 133565546 | 147779980 | 136823886 | 140113624 |
| Raw data yield (Mb) | 12021 | 13300 | 12314 | 12610 |
| Reads mapped to genome | 118841720 | 129869013 | 119724061 | 122370094 |
| 2Reads mapped to target region | 57175597 | 66226757 | 63991407 | 62336567 |
| Data mapped to target region (Mb) | 4440.09 | 5159.21 | 4981.38 | 4850.57 |
| Mean depth of target region (×) | 86.48 | 100.39 | 97.03 | 94.38 |
| Coverage of target region (%) | 99.73 | 99.72 | 99.72 | 99.69 |
| Average read length (bp) | 89.95 | 89.97 | 89.94 | 89.93 |
| Rate of nucleotide mismatch (%) | 0.22 | 0.18 | 0.22 | 0.23 |
| Fraction of target covered ≥4× (%) | 99.30 | 99.35 | 99.32 | 99.26 |
| Fraction of target covered ≥10× (%) | 98.27 | 98.50 | 98.45 | 98.31 |
| Fraction of target covered ≥20× (%) | 95.45 | 96.24 | 96.10 | 95.75 |
| 3Capture specificity (%) | 48.93 | 52.08 | 54.32 | 51.92 |
| 4Reads mapped to flanking region | 9072073 | 9020405 | 9210197 | 8861715 |
| Mean depth of flanking region (×) | 20.97 | 22.39 | 22.24 | 21.54 |
| Coverage of flanking region (%) | 98.92 | 98.67 | 98.75 | 98.59 |
| Fraction of flanking covered ≥4× (%) | 91.19 | 89.02 | 90.10 | 89.20 |
| Fraction of flanking covered ≥10× (%) | 66.13 | 63.98 | 65.45 | 64.00 |
| Fraction of flanking covered ≥20× (%) | 39.15 | 40.16 | 40.49 | 39.22 |
| Fraction of unique mapped bases on or near target (%) | 56.14 | 58.73 | 61.64 | 58.83 |
| 5Duplication rate (%) | 6.80 | 9.41 | 8.49 | 8.39 |
| Mean depth of chrX (×) | 96.40 | 58.22 | 107.72 | 54.62 |
| Mean depth of chrY (×) | - | 55.05 | - | 53.52 |
| GC rate (%) | 45.46 | 45.92 | 46.14 | 45.50 |
| Gender test result | F | M | F | M |
1Target regions refer to the regions that are actually covered by the designed probes; 2Reads mapped to target regions are reads that are within or overlap with the target region; 3Capture specificity is defined as the percentage of uniquely mapped reads aligning to the target region; 4Flanking region refers to regions ±200 bp on both sides of each target region; 5PCR duplicates would have the same start and end for both mates, which rarely occurs by chance. Duplication rate is the fraction of duplicated reads in the raw data.
Table 2. Summary of SNPs for exome capture sample.
| Categories | III-12 (proband) | III-10 (unaffected) | III-4 (affected) | IV-7 (affected) |
| 1Number of genomic positions for calling SNPs | 134975362 | 135126064 | 134975362 | 135126064 |
| 2Number of high-confidence genotypes | 128995083 | 128400486 | 128538834 | 128338404 |
| Number of high-confidence genotypes in TR | 50782259 | 50828750 | 50782665 | 50812238 |
| Total number of SNPs | 110292 | 108907 | 109560 | 108350 |
| Nonsense | 114 | 115 | 122 | 120 |
| Readthrough | 57 | 50 | 53 | 55 |
| Missense | 11030 | 11033 | 11010 | 10849 |
| 3Splice site | 2704 | 2767 | 2729 | 2673 |
| 5-UTR | 3813 | 3777 | 3836 | 3767 |
| 3-UTR | 7426 | 7276 | 7327 | 7187 |
| NR_exon | 10219 | 10113 | 10086 | 9933 |
| Synonymous-coding | 5856 | 5939 | 5929 | 5908 |
| Intron | 65755 | 64506 | 65128 | 64450 |
| Intergenic | 3318 | 3331 | 3340 | 3408 |
| Homozygous | 45692 | 45421 | 45114 | 45639 |
| Heterozygous | 64600 | 63486 | 64446 | 62711 |
1Genomic positions for calling SNPs include capture target regions and their 200-bp flanking regions; 2Consensus genotype with quality score of at least 20; 3Intronic SNPs within 10 bp of an exon/intron boundary.
Functional Significance of Gene Variations
c.C766T(p.Q256X) in CRX was considered to be a definite pathogenic mutation due to the creation of a novel upstream stop-site which truncates the normal protein. The pathogenicity of the other 25 variants was excluded by SIFT(Table 1) and linkage analysis.
Validation of the CRX Germline Mutation
The Sanger DNA sequencing scan of the whole CRX gene was completely consistent with WES. c.C766T (p.Q256X) within exon 5 of CRX was found in the affected family members but absent from the unaffected members and 100 normal controls (Figure 3).
Figure 3. Results of nucleotide sequencing analysis.
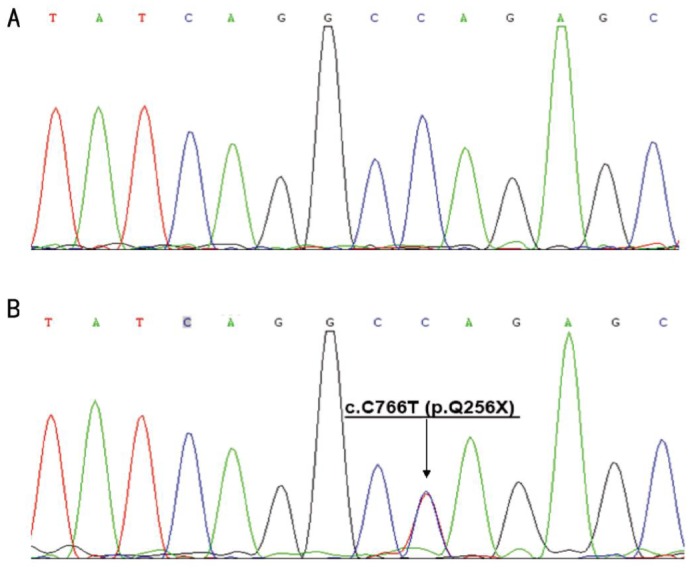
A: Sequence analysis of the CRX coding region in an unrelated normal individual was reference homozygote. B: The proband(III-12) showed a heterozygous c.C766T(p.Q256X) mutation. Arrow indicates the position of the mutation.
DISCUSSION
In this study, we used WES to identify a novel nonsense mutation c.C766T (p.Q256X) within exon 5 of CRX in a Chinese adCRD family. WES analyzes the exons and coding regions of thousands of genes simultaneously using next-generation sequencing technique[20]. By sequencing the exome of a patient and comparing it with a normal reference sequence, variations in an individual's DNA sequence can be identified and related to the individual's medical concerns in an effort to discover the cause of the disorder[20]. The overall molecular diagnostic rate is higher than several comparable genetic tests, including chromosome studies(5%-10%) and chromosomal microarray analysis (15%-20%)[26]–[28]. Recent increases in accuracy have enabled the development of clinical exome sequencing for mutation identification in patients with suspected genetic diseases[29]–[31]. We anticipate applications for WES that include the discovery of genes and alleles contributing to Mendelian and complex traits, especially for disease like CORDs, which are exceptionally heterogeneous in genotype and phenotype[5],[19],[29],[30].
The CRX gene located on 19q13 contains 5 exons and encodes a protein with 299 amino acid residues; it is expressed in the inner nuclear layer, where it plays a significant role in the differentiation and maintenance of photoreceptor cells by synergistic interaction with other transcription factors such as NRL and RX[7],[31],[32]. CRX molecule possesses a paired-like homeodomain followed by a basic region, a WSP domain, and a C-terminal OTX tail. The OTX tail is important for its transcriptional activation[32],[33]. CRX exerts its activity on dimerization such as with the neural retina leucine zipper via the OTX tail[34],[35]. Therefore, mutations in CRX abolishing the OTX tail (e.g. c.504delA, c.587delCCCC, c.816delCACinsAA, and c.C766T in this study) affect photoreceptor-specific gene expression with a dominant-negative effect because the OTX tail establishes interactions with other transcription factors[3],[7],[36],[37]. Upon loss of the OTX tail, the mutant protein occupies the CRX-binding site in target promoters without establishing an interaction and, consequently, may block transactivation.
To date, more than 31 CRX mutations have been reported [http://www.retina-international.org/files/sci-news/crxmut.htm (update from June 16, 2005)] to underlie 3 different phenotypes: CORD, autosomal dominant retinitis pigmentosa (RP), and autosomal dominant Leber congenital amaurosis (LCA)[3],[5]. The first mutation in CRX was detected in a Greek family with adCORD[7]. Thereafter, a number of mutations in CRX have been identified as being responsible for CORD as well as LCA or RP[24]. However, the severity and progression vary considerably both within and across families[38]. All CRX mutations appear to be completely penetrant and cause disease in heterozygotes. Missense mutations preferentially affect the conserved homeobox(codons 39-98), and all frameshift mutations leave the homeodomain intact but alter the OTX motif encoded by codons 284-295 at the carboxy terminus[3],[5]. So far, there does not appear to be an obvious relationship between the type of dominant mutation (missense vs frameshift) in CRX and the severity of the resulting retinal disease[3],[5].
In conclusion, we have used WES to identify a novel nonsense mutation c.C766T(p.Q256X) of CRX in a Chinese pedigree with adCORD. Although the assessment of disease-associated variation in CORD is still a difficult task for ophthalmologists and geneticists, WES offers a direct and robust diagnostic tool to substantially advance our knowledge, specifically in CORD-associated diseases and in ophthalmic genetics in general.
Acknowledgments
Foundations: Supported by the Zhejiang Provincial Natural Science Foundation of China (No.LY12H12001); the Ningbo Key Foundation of Society Development (No.2014C50091); the Ningbo Natural Science Foundation (No. 2012A610192); the Ningbo Yinzhou District S&T Foundation (No.YK2013-90); the Shenzhen Municipal Government of China (No.GJHZ20130417140916986).
Conflicts of Interest: Lu QK, None; Zhao N, None; Lv YS, None; Gong WK, None; Wang HY, None; Tong QH, None; Lai XM, None; Liu RR, None; Fang MY, None; Zhang JG, None; Du ZF, None; Zhang XN, None.
References
- 1.Hamel CP. Cone rod dystrophies. Orphanet J Rare Dis. 2007;2:7. doi: 10.1186/1750-1172-2-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Itabashi T, Wada Y, Sato H, Kawamura M, Shiono T, Tamai M. Novel 615delC mutation in the CRX gene in a Japanese family with cone-rod dystrophy. Am J Ophthalmol. 2004;138(5):876–877. doi: 10.1016/j.ajo.2004.05.067. [DOI] [PubMed] [Google Scholar]
- 3.Hull S, Arno G, Plagnol V, Chamney S, Russell-Eggitt I, Thompson D, Ramsden SC, Black GC, Robson A, Holder GE, Moore AT, Webster AR. The phenotypic variability of retinal dystrophies associated with mutations in CRX, with report of a novel macular dystrophy phenotype. Invest Ophthalmol Vis Sci. 2014;55(10):6934–6944. doi: 10.1167/iovs.14-14715. [DOI] [PubMed] [Google Scholar]
- 4.Huang L, Zhang Q, Li S, Guan L, Xiao X, Zhang J, Jia X, Sun W, Zhu Z, Gao Y, Yin Y, Wang P, Guo X, Wang J, Zhang Q. Exome sequencing of 47 Chinese families with cone-rod dystrophy: mutations in 25 known causative genes. PLoS One. 2013;8(6):e65546. doi: 10.1371/journal.pone.0065546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Roosing S, Thiadens AA, Hoyng CB, Klaver CC, den Hollander AI, Cremers FP. Causes and consequences of inherited cone disorders. Prog Retin Eye Res. 2014;42:1–26. doi: 10.1016/j.preteyeres.2014.05.001. [DOI] [PubMed] [Google Scholar]
- 6.Sohocki MM, Perrault I, Leroy BP, Payne AM, Dharmaraj S, Bhattacharya SS, Kaplan J, Maumenee IH, Koenekoop R, Meire FM, Birch DG, Heckenlively JR, Daiger SP. Prevalence of AIPL1 mutations in inherited retinal degenerative disease. Mol Genet Metab. 2000;70(2):142–150. doi: 10.1006/mgme.2000.3001. [DOI] [PubMed] [Google Scholar]
- 7.Freund CL, Gregory-Evans CY, Furukawa T, Papaioannou M, Looser J, Ploder L, Bellingham J, Ng D, Herbrick JA, Duncan A, Scherer SW, Tsui LC, Loutradis-Anagnostou A, Jacobson SG, Cepko CL, Bhattacharya SS, McInnes RR. Cone-rod dystrophy due to mutations in a novel photoreceptor-specific homeobox gene (CRX) essential for maintenance of the photoreceptor. Cell. 1997;91(4):543–553. doi: 10.1016/s0092-8674(00)80440-7. [DOI] [PubMed] [Google Scholar]
- 8.Payne AM, Downes SM, Bessant DA, Taylor R, Holder GE, Warren MJ, Bird AC, Bhattacharya SS. A mutation in guanylate cyclase activator 1A (GUCA1A) in an autosomal dominant cone dystrophy pedigree mapping to a new locus on chromosome 6p21.1. Hum Mol Genet. 1998;7(2):273–277. doi: 10.1093/hmg/7.2.273. [DOI] [PubMed] [Google Scholar]
- 9.Kelsell RE, Gregory-Evans K, Payne AM, Perrault I, Kaplan J, Yang RB, Garbers DL, Bird AC, Moore AT, Hunt DM. Mutations in the retinal guanylate cyclase (RETGC-1) gene in dominant cone-rod dystrophy. Hum Mol Genet. 1998;7(7):1179–1184. doi: 10.1093/hmg/7.7.1179. [DOI] [PubMed] [Google Scholar]
- 10.Kohn L, Kadzhaev K, Burstedt MS, Haraldsson S, Hallberg B, Sandgren O, Golovleva I. Mutation in the PYK2-binding domain of PITPNM3 causes autosomal dominant cone dystrophy (CORD5) in two Swedish families. Eur J Hum Genet. 2007;15(6):664–671. doi: 10.1038/sj.ejhg.5201817. [DOI] [PubMed] [Google Scholar]
- 11.Yang Z, Chen Y, Lillo C, et al. Mutant prominin 1 found in patients with macular degeneration disrupts photoreceptor disk morphogenesis in mice. J Clin Invest. 2008;118(8):2908–2916. doi: 10.1172/JCI35891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fishman GA, Stone EM, Alexander KR, Gilbert LD, Derlacki DJ, Butler NS. Serine-27-phenylalanine mutation within the peripherin/RDS gene in a family with cone dystrophy. Ophthalmology. 1997;104(2):299–306. doi: 10.1016/s0161-6420(97)30320-0. [DOI] [PubMed] [Google Scholar]
- 13.Kelsell RE, Gregory-Evans K, Gregory-Evans CY, Holder GE, Jay MR, Weber BH, Moore AT, Bird AC, Hunt DM. Localization of a gene (CORD7) for a dominant cone-rod dystrophy to chromosome 6q. Am J Hum Genet. 1998;63(1):274–279. doi: 10.1086/301905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Abid A, Ismail M, Mehdi SQ, Khaliq S. Identification of novel mutations in the SEMA4A gene associated with retinal degenerative diseases. J Med Genet. 2006;43(4):378–381. doi: 10.1136/jmg.2005.035055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kobayashi A, Higashide T, Hamasaki D, Kubota S, Sakuma H, An W, Fujimaki T, McLaren MJ, Weleber RG, Inana G. HRG4 (UNC119) mutation found in cone-rod dystrophy causes retinal degeneration in a transgenic model. Invest Ophthalmol Vis Sci. 2000;41(11):3268–3277. [PubMed] [Google Scholar]
- 16.Kamenarova K, Cherninkova S, Romero Duran M, Prescott D, Valdes Sanchez ML, Mitev V, Kremensky I, Kaneva R, Bhattacharya SS, Tournev I, Chakarova C. A novel locus for autosomal dominant cone-rod dystrophy maps to chromosome 10q. Eur J Hum Genet. 2013;21(3):338–342. doi: 10.1038/ejhg.2012.158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Demirci FY, Rigatti BW, Wen G, Radak AL, Mah TS, Baic CL, Traboulsi EI, Alitalo T, Ramser J, Gorin MB. X-linked cone-rod dystrophy (locus COD1): identification of mutations in RPGR exon ORF15. Am J Hum Genet. 2002;70(4):1049–1053. doi: 10.1086/339620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jalkanen R, Mantyjarvi M, Tobias R, Isosomppi J, Sankila EM, Alitalo T, Bech-Hansen NT. X linked cone-rod dystrophy, CORDX3, is caused by a mutation in the CACNA1F gene. J Med Genet. 2006;43(8):699–704. doi: 10.1136/jmg.2006.040741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Choi M, Scholl UI, Ji W, Liu T, Tikhonova IR, Zumbo P, Nayir A, Bakkaloğlu A, Ozen S, Sanjad S, Nelson-Williams C, Farhi A, Mane S, Lifton RP. Genetic diagnosis by whole exome capture and massively parallel DNA sequencing. Proc Natl Acad Sci U S A. 2009;106(45):19096–19101. doi: 10.1073/pnas.0910672106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ng SB, Turner EH, Robertson PD, Flygare SD, Bigham AW, Lee C, Shaffer T, Wong M, Bhattacharjee A, Eichler EE, Bamshad M, Nickerson DA, Shendure J. Targeted capture and massively parallel sequencing of 12 human exomes. Nature. 2009;461(7261):272–276. doi: 10.1038/nature08250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Qi XP, Du ZF, Ma JM, Chen XL, Zhang Q, Fei J, Wei XM, Chen D, Ke HP, Liu XZ, Li F, Chen ZG, Su Z, Jin HY, Liu WT, Zhao Y, Jiang HL, Lan ZZ, Li PF, Fang MY, Dong W, Zhang XN. Genetic diagnosis of autosomal dominant polycystic kidney disease by targeted capture and next-generation sequencing: utility and limitations. Gene. 2013;516(1):93–100. doi: 10.1016/j.gene.2012.12.060. [DOI] [PubMed] [Google Scholar]
- 22.Abecasis GR, Cherny SS, Cookson WO, Cardon LR. Merlin--rapid analysis of dense genetic maps using sparse gene flow trees. Nat Genet. 2002;30(1):97–101. doi: 10.1038/ng786. [DOI] [PubMed] [Google Scholar]
- 23.Ng PC, Henikoff S. Accounting for human polymorphisms predicted to affect protein function. Genome Res. 2002;12(3):436–446. doi: 10.1101/gr.212802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Berger W, Kloeckener-Gruissem B, Neidhardt J. The molecular basis of human retinal and vitreoretinal diseases. Prog Retin Eye Res. 2010;29(5):335–375. doi: 10.1016/j.preteyeres.2010.03.004. [DOI] [PubMed] [Google Scholar]
- 25.Michaelides M, Hunt DM, Moore AT. The cone dysfunction syndromes. Br J Ophthalmol. 2004;88(2):291–297. doi: 10.1136/bjo.2003.027102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shevell M, Ashwal S, Donley D, Flint J, Gingold M, Hirtz D, Majnemer A, Noetzel M, Sheth RD, Quality Standards Subcommittee of the American Academy of Neurology; Practice Committee of the Child Neurology Society Practice parameter: evaluation of the child with global developmental delay: report of the Quality Standards Subcommittee of the American Academy of Neurology and The Practice Committee of the Child Neurology Society. Neurology. 2003;60(3):367–380. doi: 10.1212/01.wnl.0000031431.81555.16. [DOI] [PubMed] [Google Scholar]
- 27.Shaffer LG, American College of Medical Genetics Professional Practice and Guidelines Committee American College of Medical Genetics guideline on the cytogenetic evaluation of the individual with developmental delay or mental retardation. Genet Med. 2005;7(9):650–654. doi: 10.1097/01.gim.0000186545.83160.1e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Miller DT, Adam MP, Aradhya S, et al. Consensus statement: chromosomal microarray is a first-tier clinical diagnostic test for individuals with developmental disabilities or congenital anomalies. Am J Hum Genet. 2010;86(5):749–764. doi: 10.1016/j.ajhg.2010.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lee H, Deignan JL, Dorrani N, Strom SP, Kantarci S, Quintero-Rivera F, Das K, Toy T, Harry B, Yourshaw M, Fox M, Fogel BL, Martinez-Agosto JA, Wong DA, Chang VY, Shieh PB, Palmer CG, Dipple KM, Grody WW, Vilain E, Nelson SF. Clinical exome sequencing for genetic identification of rare Mendelian disorders. JAMA. 2014;312(18):1880–1887. doi: 10.1001/jama.2014.14604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yang Y, Muzny DM, Xia F, et al. Molecular findings among patients referred for clinical whole-exome sequencing. JAMA. 2014;312(18):1870–1879. doi: 10.1001/jama.2014.14601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gomez CM, Das S. Clinical exome sequencing: the new standard in genetic diagnosis. JAMA Neurol. 2014;71(10):1215–1216. doi: 10.1001/jamaneurol.2014.2015. [DOI] [PubMed] [Google Scholar]
- 32.Furukawa T, Morrow EM, Cepko CL. Crx, a novel otx-like homeobox gene, shows photoreceptor-specific expression and regulates photoreceptor differentiation. Cell. 1997;91(4):531–541. doi: 10.1016/s0092-8674(00)80439-0. [DOI] [PubMed] [Google Scholar]
- 33.Chau KY, Chen S, Zack DJ, Ono SJ. Functional domains of the cone-rod homeobox (CRX) transcription factor. J Biol Chem. 2000;275(47):37264–37270. doi: 10.1074/jbc.M002763200. [DOI] [PubMed] [Google Scholar]
- 34.Chen S, Wang QL, Nie Z, Sun H, Lennon G, Copeland NG, Gilbert DJ, Jenkins NA, Zack DJ. Crx, a novel Otx-like paired-homeodomain protein, binds to and transactivates photoreceptor cell-specific genes. Neuron. 1997;19(5):1017–1030. doi: 10.1016/s0896-6273(00)80394-3. [DOI] [PubMed] [Google Scholar]
- 35.Mitton KP, Swain PK, Chen S, Xu S, Zack DJ, Swaroop A. The leucine zipper of NRL interacts with the CRX homeodomain. A possible mechanism of transcriptional synergy in rhodopsin regulation. J Biol Chem. 2000;275(38):29794–29799. doi: 10.1074/jbc.M003658200. [DOI] [PubMed] [Google Scholar]
- 36.Swain PK, Chen S, Wang QL, Affatigato LM, Coats CL, Brady KD, Fishman GA, Jacobson SG, Swaroop A, Stone E, Sieving PA, Zack DJ. Mutations in the cone-rod homeobox gene are associated with the cone-rod dystrophy photoreceptor degeneration. Neuron. 1997;19(6):1329–1336. doi: 10.1016/s0896-6273(00)80423-7. [DOI] [PubMed] [Google Scholar]
- 37.Paunescu K, Preising MN, Janke B, Wissinger B, Lorenz B. Genotype-phenotype correlation in a German family with a novel complex CRX mutation extending the open reading frame. Ophthalmology. 2007;114(7):1348–1357. doi: 10.1016/j.ophtha.2006.10.034. [DOI] [PubMed] [Google Scholar]
- 38.Kitiratschky VB, Nagy D, Zabel T, Zrenner E, Wissinger B, Kohl S, Jagle H. Cone and cone-rod dystrophy segregating in the same pedigree due to the same novel CRX gene mutation. Br J Ophthalmol. 2008;92(8):1086–1091. doi: 10.1136/bjo.2007.133231. [DOI] [PubMed] [Google Scholar]