

Abstract
Thyroid gland involvement by Langerhans cell histiocytosis is extremely rare. A 35-year-old woman with a history of a suprasellar mass previously diagnosed as a ganglioglioma and complicated by diabetes insipidus, hypogonadotropic hypogonadism, and central hypothyroidism presented with acute onset of neck enlargement. On ultrasound examination, almost the entire thyroid appeared replaced by abnormal lobulated hypoechoic tissue with increased vascularity. Fine needle aspiration (FNA) of the thyroid was performed and revealed singly scattered and loosely cohesive large cells with abundant cytoplasm, including some with irregular nuclear contours and nuclear grooves. No thyroid follicular cells were noted. Based on the cytomorphologic findings and ancillary studies (immunohistochemistry and flow cytometry analysis) a cytological diagnosis of “positive for neoplastic cells” with features suggestive of monocytic/histiocytic origin, possibly Langerhans cell histiocytosis (LCH) was rendered. Following FNA, the patient underwent an incisional thyroid biopsy that confirmed the cytological impression of LCH. In light of the new diagnosis of LCH, the prior suprasellar mass biopsy slides were re-reviewed and rare cells suspicious for LCH were observed. Appropriate treatment for systemic LCH was initiated successfully. This case demonstrates that the presence of enlarged and loosely cohesive cells, especially those with irregular nuclear contours, in thyroid FNA specimens should raise suspicion for LCH. The diagnosis of LCH in FNA specimens is challenging. Additional material should be allocated for ancillary studies to confirm the morphological impression. In our case, not only was the thyroid FNA crucial in diagnosing LCH, but instrumental in initiating a thorough diagnostic work-up for multisystem involvement and thus unmasking the true etiology of the patient’s suprasellar mass and associated endocrinopathies.
Keywords: Thyroid, Langerhans cell histiocytosis, Fine needle aspiration, Cytology, Cytopathology
Clinical Presentation
A 35-year-old woman with a history of a suprasellar mass, previously diagnosed as a ganglioglioma and complicated by diabetes insipidus (DI), hypogonadotropic hypogonadism, and central hypothyroidism, presented with acute onset of neck enlargement. On physical exam, she appeared euthyroid, and a bilaterally enlarged soft, non-tender thyroid of about 60 g was palpated.
Radiographic Features
On thyroid ultrasound, the right lobe measured 7.3 × 2.9 × 3.4 cm, and the left lobe measured 5.8 × 2.3 × 2.5 cm; replacement of almost the entire thyroid with abnormal lobulated hypoechoic tissue with increased vascularity was identified (Fig. 1a, b). The rate of growth and the ultrasound findings prompted fine needle aspiration (FNA) of the left and right thyroid lobes.
Fig. 1.

Representative view of the heterogenous parenchyma seen on ultrasound of the right thyroid lobe in transverse (a) and sagittal (b) views
Diagnosis
Fine Needle Aspiration and Biopsy
At the time of on-site assessment, the smears showed large atypical mononuclear cells (Fig. 2a). Due to the dyscohesion of the cells, the samples were submitted for flow cytometry analysis and additional aspirates were requested for cell block preparation. The Diff-Quik and Papanicolaou-stained smears and hematoxylin and eosin (H&E)-stained cell block sections showed mononuclear cells with moderate-to-abundant cytoplasm. The nuclei were enlarged, and some cells had convoluted nuclei, with occasional cells exhibiting nuclear grooves. Only rare eosinophils were evident in the background, and no significant follicular cell component was seen (Figs. 2b–d, 3). On immunohistochemical staining, the lesional cells were positive for S100, CD1a, Langerin, and negative for pancytokeratin, thyroid transcription factor-1 (TTF-1), synaptophysin, chromogranin, CEA, CD138, and PAX-8 (Fig. 4a, b). A few cells expressed CD68 and CD163. The corresponding flow cytometry of the lesional cells showed the following profile: CD4+, CD13±, CD33+, HLA-DR+, and CD11c+. Based on the morphologic and immunophenotypic analysis, a diagnosis of “positive for neoplastic cells” with features suggestive of monocytic/histiocytic origin, possibly Langerhans cell histiocytosis (LCH), was rendered.
Fig. 2.

FNA smears of the thyroid mass. a Singly scattered atypical mononuclear cells seen during on-site evaluation of the aspirate (Diff-Quik stain, ×600). b, c Alcohol-fixed slides showing loosely cohesive clusters of Langerhans cells with moderate cytoplasm and convoluted nuclei (PAP stain, ×600). d Rare eosinophils were noted (Diff Quik stain, ×600, cropped and enlarged)
Fig. 3.
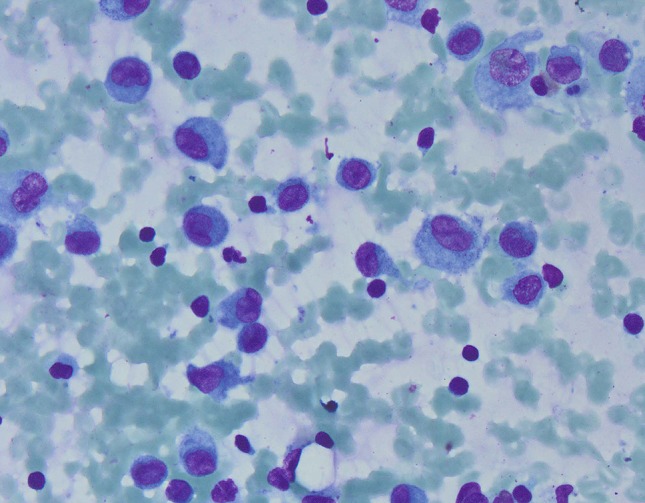
Air-dried smear with scattered atypical mononuclear cells with convoluted nuclei (Diff-Quik stain, ×400)
Fig. 4.

Cell block preparations showing immunoreactivity of the neoplastic cells for a S-100 protein and b CD1a (Immunohistochemistry, 600)
Following FNA, the patient underwent an incisional thyroid biopsy that confirmed the cytological impression of LCH. Flow cytometry using a comprehensive panel of markers detected 20 % events (cells) in the CD45 dim-bright, intermediate-high side scatter gate, which displayed the following phenotype: CD34−, CD117−, TdT−, CD1a+, CD13±, CD33+, CD64±, CD4+, CD11c+, CD14−, HLA-DR+, CD2±, surface/cytoplasmic CD3−, CD5−, CD7−, CD8−, CD19−, CD20−, cCD79a−, CD16/56− and cMPO−. On immunohistochemical staining, the neoplastic cells showed variable CD45 expression and they were immunoreactive for CD43, CD33, vimentin, S100, CD1a and Langerin (Fig. 5a–d). A subset expressed monocytic/histiocytic antigens (CD14+, CD68/PGM1+, CD163+). Absence of CD34, CD117, CD15 or myeloperoxidase expression was confirmed. The Ki-67 labeling index was 30 %. Stains for CD3, CD20, CD21 and CD23 showed scattered reactive lymphoid aggregates. Stains for pan-cytokeratin and TTF-1 demonstrated preserved, as well as, disrupted thyroid follicles.
Fig. 5.

Incisional biopsy specimen from the thyroid mass. a Sheets of the neoplastic Langerhans cells and entrapped residual thyroid follicles (H&E, ×200). b Same microscopic field with higher magnification demonstrating nuclear pleomorphism of the tumor cells (H&E, ×600). c, d The diagnosis is supported by diffuse strong immunoreactivity of the tumors cells to c Langerin (Immunohistochemistry, ×400) and d CD1a (Immunohistochemistry, ×600)
Real-time PCR analysis using fluorescently labeled hydrolysis probes to detect the BRAF V600E mutation was performed on the cytology and incisional biopsy samples; however, both specimens lacked the mutation. Conventional cytogenetic (G-band) analysis showed a normal female karyotype and no chromosome abnormalities were detected. On performing SNP array analysis, using the CytoScan HD array (Affymetrix), no distinct somatic copy number alteration (chromosome gain or loss) or loss of heterozygosity was observed, supporting the G-band karyotype results.
In light of the new diagnosis of LCH, the prior suprasellar mass biopsy histopathology was re-reviewed at another institution, where rare cells suspicious for LCH were noted. Immunohistochemistry performed on the suprasellar mass showed S-100 stain to be positive in glial cells and CD68 in macrophages. Although CD1a was reportedly negative, Langerin immunoreactivity in rare cells was observed.
Oncologic evaluation was negative for skeletal, bone marrow, and gingival involvement. A PET/CT scan showed a markedly enlarged, FDG-avid thyroid (SUV 14.7), increased FDG activity in the superior mediastinum likely due to thyroid extension, and FDG-avid level IIB lymph nodes. The patient’s clinical course was complicated by growth (fungation) of the tumor into the skin of the neck. She underwent two 6-weeks courses of induction chemotherapy with vinblastine and prednisone, followed by three cycles of vinblastine, methotrexate, 6-mercaptopurine and steroids due to persistence of thyroid FDG-avidity. Follow-up PET/CT showed improvement of FDG-avid disease and decrease in thyroid size. There has been no recurrence of disease till present. A brain MRI showed interval improvement in the size of the suprasellar lesion. She remains on stable doses of desmopressin and levothyroxine.
Discussion
LCH is a rare monoclonal disease of a subtype of antigen-presenting dendritic cells. First described in 1893 by Hand in association with polyuria and tuberculosis, it was subsequently recognized to comprise a triad of symptoms including DI, exophtalmosis, and lytic bone lesions and became known as Hand-Schuller-Christian disease [1]. Since then it has been determined that LCH can be unifocal, mutifocal unisystem, or a multifocal multisystem disorder, and depending upon the location and distribution of the abnormal cells, the disorder has been referred to by other names, including eosinophilic granuloma, histiocytosis X and Letterer-Siwe disease. More recently, with the application of electron microscopy and immunohistochemistry, the various disease presentations have been unified under a single entity—LCH [2, 3].
The morphologic features of LCH are characteristic. The lesions are composed of a mixture of proliferating Langerhans cells (antigen-presenting dendritic cells) that exhibit prominent nuclear indentations and grooves, multinucleated giant cells, eosinophils and lymphocytes, at times forming lymphoid follicles, diffusely infiltrating the affected organ. Electron microscopy demonstrates cytoplasmic rod-shaped organelles (Birbeck granules) within the dendritic cells. Langerhans cells express MHC Class 1 and 2-related surface glycoproteins, a variety of myeloid antigens, S-100, CD1a and Langerin (CD207). It has been suggested that LCH is a disorder of a Langerin-expressing subset of tissue dendritic cells that are derived from blood-borne precursors and are present in most tissue types affected by LCH. It is the co-expression of Langerin (CD207) that defines CD1a positive dendritic cells as Langerhans cells [4]. This fact is important as recent studies have revealed the existence of subsets of Langerin (+), non-Langerhans dendritic cells, especially in the dermis [4, 5]. Langerhans cell histiocytes less often express maturation associated antigens such as CD83 compared to other types of myeloid derived dendritic cells [6, 7]. Despite recent progress, the etiology and pathogenesis of LCH remains unclear. Willman et al. demonstrated that LCH is a clonal proliferative disorder, confirming its neoplastic nature [8]. Establishing clonality is essential in the diagnosis of LCH, especially in cases of unifocal disease, as LCH-like proliferative lesions may be seen in different reactive conditions and also in association with a variety of hematologic and non-hematologic neoplasms [9].
Recent studies have identified recurrent BRAF V600E point mutations in 50–60 % of LCH cases. Moreover, Berres et al. demonstrated that BRAF V600E expression in precursor versus differentiated dendritic cells defines clinically distinct LCH risk groups [10]. The authors hypothesized that high-risk LCH arises from somatic mutations in a hematopoietic progenitor with potential multisystem involvement, while low-risk disease arises from somatic mutations in tissue-restricted dendritic cell precursors. Moreover in a recent study Chakraborty et al. stated that the high frequency of BRAF mutations in LCH, the presence of this mutation in myelomonocyic precursors in patients with high risk LCH, and the ability of enforced expression of BRAF V600E in mice that recapitulates an LCH-like phenotype, supports the concept of LCH being a myeloid neoplasm that arises as the result of MAPK pathway activation at critical stages in myeloid differentiation [11]. It should be noted, however, that detection of the BRAF V600E mutation is not specific for LCH even among histiocytic disorders, as this mutation has been detected in 54 % cases of Erdheim-Chester disease [12] Whether the presence of BRAF V600E mutations reflects biologic relatedness of these types of histiocytoses remains to be determined [13]. More recent studies have uncovered a high frequency of MAP2K1 mutations (33–50 %) in LCH cases that have wild-type BRAF as well as occasional somatic mutations in other mitogen-activated protein kinase (MAPK) pathway genes, ARAF and ERBB3 [11, 14, 15].
Thyroid gland involvement by LCH is extremely rare. A recent review by Patten et al. summarized the clinico-pathological features of 75 reported cases of thyroid gland involvement by LCH, with 65 having been documented in the English literature [6]. In most of the cases, thyroid involvement was part of multisystem disease, and adults were affected more often than children, with a slight female predominance (female to male ratio 1.4:1) [6]. In 59 % of cases, the patients presented with diffuse enlargement of the thyroid gland, as noted in our case. Posterior pituitary involvement was a more frequent endocrine presentation of LCH, particularly in children. Of all thyroid LCH cases reviewed by Patten et al. eleven (15 %) patients had associated central DI (four males and seven females, age range 1.5–61 years), and only one 28-year-old patient presented with amenorrhea [6]. Association of LCH with chronic lymphocytic thyroiditis has been described [16]. Thompson et al. showed that patients with localized disease have a better prognosis compared to those with thyroid involvement as part of systemic disease [17].
Moreover, rare association of LCH with Graves’ disease has been reported. In the first reported case with this association, isolated LCH was diagnosed in a 26-year-old woman who was suffering from Graves’ disease with goiter and bilateral exophthalmoses for 24 months [18]. In another case, a 27-year-old woman was diagnosed with LCH in childhood, and presented to the endocrinology clinic with goiter and thyrotoxicosis. Her diagnostic work-up was consistent with Graves’ thyrotoxicosis [19]. Co-occurrence of LCH and papillary thyroid carcinoma (PTC) has also been reported. The first reported case of thyroidmegaly secondary to simultaneous PTC and LCH was in a patient with longstanding LCH [20]. It has been suggested that both processes could have common pathways of pathogenesis, and the underlying inflammatory process and BRAF mutation could also trigger PTC development [21].
FNA has been proven as a reliable tool in the initial diagnostic work up of thyroid lesions. However, FNA diagnosis of LCH involving the thyroid gland may be challenging, especially if there is no known history of systemic LCH. In fact, to the best of our knowledge, there are only four reported cases where a tentative cytologic diagnosis of LCH was proposed and subsequently confirmed by biopsy and subtotal thyroidectomy [20, 22–24]. Table 1 summarizes the clinical and cytologic findings of LCH cases diagnosed in thyroid FNA specimens. In one of these cases, a diagnosis of PTC in a background suggestive of LCH was rendered [15]. In the other reported cases of thyroid LCH where FNA was performed, the cytologic findings were misinterpreted as “atypical follicular epithelial cells”, “epithelial neoplasm” and papillary or medullary carcinoma, or were non-diagnostic [6, 25]. Therefore, a high index of suspicion is required to correctly diagnose LCH in thyroid FNA specimens. Other differential diagnostic considerations in thyroid cytology specimens include anaplastic carcinoma, lymphocytic thyroiditis, chronic granulomatous thyroiditis, cystic degeneration in goiter, tuberculosis, non-Hodgkin or Hodgkin lymphoma, and leukemic infiltration of the thyroid among others.
Table 1.
Clinical presentation and cytologic features in cases with tentative diagnosis of LCH by FNA
| Patient age/sex | Clinical presentation | Other organ involvement | Cytologic findings | Cytologic diagnosis | Authors |
|---|---|---|---|---|---|
| 31-year-old female | Swelling of the left thyroid lobe | History of systemic LCH involving external genitalia, respiratory tract and oral cavity | Numerous papillary clusters with nuclear grooves and pseudoinclusions. Occasional LC and rare eosinophils | Papillary thyroid carcinoma. Background suggestive of LCH | Goldstein and Layfield [20] |
| 16- year-old male | Diffuse enlargement of the thyroid | Panhypopituitarism, supracellar hemangioblastoma | Large number of LC, eosinophils follicular cells | LCH | Kirchgraber et al. [22] |
| 23-year-old female | Thyroid swelling | None | LC with abundant pale cytoplasm with centrally-placed, large nuclei with frequent prominent grooves. Large number of reactive lymphoid cells and eosinophils. Frequent mitoses | LCH | Dey et al. [23] |
| 13-year-old male | Swelling of the left thyroid lobe and isthmus | None | Mono- and multinucleated LC in loosely cohesive clusters and singly scattered, follicular cells, eosinophils, and lymphocytes. LC with abundant pale cytoplasm, indistinct cell borders, nuclei 10–20 micron, prominent nuclear grooves | LCH | Sahoo et al. [24] |
LCH Langerhans cell histiocytosis, FNA fine needle aspiration, LC Langerhans cells
A smear with dyshesive cells serves as a diagnostic clue. Typically, these are common in cases containing a lymphoid component. Unlike lymphocytes, which range in size from small to large cells, LCH cells are relatively large. Presence of histiocytes, reactive lymphocytes and eosinophils should raise the suspicion of LCH [24]. Interestingly, only rare eosinophils were noted in our case. Akhtar et al. have reported that the degree of eosinophilic infiltrate in cytology specimens varies from scant to abundant in different areas of LCH lesions and also in different organs. However, presence of eosinophils or Charcot–Leyden crystals is a subtle feature that should alert the pathologist to include LCH in the differential diagnosis [26, 27]. On-site adequacy assessment with procurement of extra material for ancillary studies is helpful in clinching the diagnosis of this rare entity in thyroid FNA specimens.
In our case, not only was thyroid FNA crucial in diagnosing LCH, it was also instrumental in initiating a thorough multimodality diagnostic work-up for multisystem involvement, thus unmasking the true etiology of the patient’s suprasellar mass and associated endocrinopathies. Our case lacked the BRAF V600E mutation, and genomic aberrations were not detected by SNP array analysis. We did not analyze the recently described mutations in genes encoding constituents of the MAP kinase pathway, which occur in approximately 50 % of BRAF wild-type LCH, and such analyses might be helpful in establishing clonality in cases similar to ours in the future.
Acknowledgments
Presented in part at the 84th Annual American Thyroid Association Meeting, October 29–November 2, 2014, Coronado, CA and received Trainee’s Poster Contest Award.
References
- 1.Hand A. Polyuria and tuberculosis. Proc Pathol Soc Phila. 1893;16:282–284. [Google Scholar]
- 2.Willman CL, McClain KL. An update on clonality, cytokines, and viral etiology in Langerhans cell histiocytosis. Hematol Oncol Clin North Am. 1998;12(2):408–416. doi: 10.1016/S0889-8588(05)70519-0. [DOI] [PubMed] [Google Scholar]
- 3.Egeler RM, D’Angio GJ. Langerhans cell histiocytosis. J Pediatr. 1995;127(1):1–11. doi: 10.1016/S0022-3476(95)70248-2. [DOI] [PubMed] [Google Scholar]
- 4.Merad M, Ginhoux F, Collin M. Origin, homeostasis and function of Langerhans cells and other Langerin-expressing dendritic cells. Nat Rev Immunol. 2008;8(12):935–947. doi: 10.1038/nri2455. [DOI] [PubMed] [Google Scholar]
- 5.Romani N, Clausen BE, Stoitzner P. Langerhans cells and more: langerin-expressing dendritic cell subsets in the skin. Immunol Rev. 2010;234(1):120–141. doi: 10.1111/j.0105-2896.2009.00886.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Patten DK, Wani A, Tolley N. Solitary Langerhans histiocytosis of the thyroid gland: a case report and literature review. Head Neck Pathol. 2012;6(2):279–289. doi: 10.1007/s12105-011-0321-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Merad M, Manz MG, Karsunky H, Wagers A, Peters W, Charo I, Weissman IL, Cyster JG, Engleman EG. Langerhans cells renew in the skin throughout life under steady-state conditions. Nat Immunol. 2002;3(12):1135–1141. doi: 10.1038/ni852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Willman CL, Busque L, Griffith BB, Favara BE, McClain KL, Duncan MH, Gilliland DG. Langerhans’-cell histiocytosis (histiocytosis X)—a clonal proliferative disease. N Engl J Med. 1994;331(3):154–160. doi: 10.1056/NEJM199407213310303. [DOI] [PubMed] [Google Scholar]
- 9.Mete Ö, Doğan Ö, Kapran Y, Tihan D, Erbil Y, Ozarmağan S. Interstitial Langerhans cell histiocytosis-like lesion in an adult presented with diverticulitis: a reactive or neoplastic condition? Pathol Oncol Res. 2011;17(2):403–407. doi: 10.1007/s12253-010-9313-3. [DOI] [PubMed] [Google Scholar]
- 10.Berres ML, Lim KP, Peters T, et al. BRAF-V600E expression in precursor versus differentiated dendritic cells defines clinically distinct LCH risk groups. J Exp Med. 2014;211(4):669–683. doi: 10.1084/jem.20130977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chakraborty R, Hampton OA, Shen X, et al. Mutually exclusive recurrent somatic mutations in MAP2K1 and BRAF support a central role for ERK activation in LCH pathogenesis. Blood. 2014;123(19):3007–15. doi: 10.1182/blood-2014-05-577825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Haroche J, Charlotte F, Arnaud L, et al. High prevalence of BRAF V600E mutations in Erdheim–Chester disease but not in other non-Langerhans cell histiocytoses. Blood. 2012;120(13):2700–2703. doi: 10.1182/blood-2012-05-430140. [DOI] [PubMed] [Google Scholar]
- 13.Hervier B, Harcoche J, Arnaud L. Association of both Langerhans cell histiocytosis and Erheim–Chester disease linked to the BTAFV600E mutation. Blood. 2014;124(7):1119–1126. doi: 10.1182/blood-2013-12-543793. [DOI] [PubMed] [Google Scholar]
- 14.Brown NA, Furtado LV, Betz BL, et al. High prevalence of somatic MAP2K1 mutations in BRAF V600E-negative Langerhans cell histiocytosis. Blood. 2014;124(10):1655–1658. doi: 10.1182/blood-2014-05-577361. [DOI] [PubMed] [Google Scholar]
- 15.Nelson DS, Quispel W, Badaliam-Very G, et al. Somatic activating ARAF mutations in Langerhans cell histiocytosis. Blood. 2014;123(20):3152–3155. doi: 10.1182/blood-2013-06-511139. [DOI] [PubMed] [Google Scholar]
- 16.Thompson LD. Langerhans cell histiocytosis isolated to the thyroid gland. Eur Arch Otorhinolaryngol. 1996;253:62–65. doi: 10.1007/BF00176706. [DOI] [PubMed] [Google Scholar]
- 17.Thompson LD, Wenig BM, Adair CF, Smith BC, Heffess CS. Langerhans cell histiocytosis of the thyroid: a series of seven cases and a review of the literature. Mod Pathol. 1996;9:145–149. [PubMed] [Google Scholar]
- 18.Lassalle A, Hofman V, Santini J, Sadoul JL, Hofman P. Isolated Langerhans cell histiocytosis of the thyroid and Graves’ diseases: an unreported association. Pathology. 2008;40:525–527. doi: 10.1080/00313020802198002. [DOI] [PubMed] [Google Scholar]
- 19.Abarra A, Chhina N, Joharatnam J, Tharakkan G, Todd J. Langerhans cell histiocytosis and Graves’ disease. Presented at society for endocrinology BES. Harrogate; 2012. Endocrine Abstracts, 28, p. 372.
- 20.Goldstein N, Layfield LJ. Thyromegaly secondary to simultaneous papillary thyroid carcinoma and histiocytosis X. Report of a case and review of the literature. Acta Cytol. 1991;35(4):422–426. [PubMed] [Google Scholar]
- 21.Diego E, Biagetti B, Iglesias C, Gonzales O, Mesa J. Langerhans cell histiocytosis and papillary thyroid carcinoma. Rev Clin Esp. 2014;214(2):e19–e21. doi: 10.1016/j.rce.2013.11.005. [DOI] [PubMed] [Google Scholar]
- 22.Kirchgraber PR, Weaver MG, Arafah BM, Abdul-Karim FW. Fine needle aspiration cytology of Langerhans cell histiocytosis involving the thyroid. Acta Cytol. 1994;38:101–106. [PubMed] [Google Scholar]
- 23.Dey P, Luthra UK, Sheikh ZA. Fine needle aspiration cytology of Langerhans cell histiocytosis of the thyroid. Acta Cytol. 1999;43:429–431. doi: 10.1159/000331093. [DOI] [PubMed] [Google Scholar]
- 24.Sahoo M, Karak AS, Bhatnagar D, Bal CS. Fine-needle aspiration cytology in a case of isolated involvement of thyroid with Langerhans cell histiocytosis. Diagn Cytopathol. 1998;19:33–37. doi: 10.1002/(SICI)1097-0339(199807)19:1<33::AID-DC7>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- 25.Ma JT, Ho FC, Wang C, Lam KS, Yeung RT. Primary hypothyroidism and essential hypernatremia in a patient with histiocytosis X. Aust NZ J Med. 1985;15:72–74. doi: 10.1111/j.1445-5994.1985.tb02742.x. [DOI] [PubMed] [Google Scholar]
- 26.Akhtar M, Ali MA, Bakry M, Sackey K, Sabbah R. Fine-needle aspiration biopsy of Langerhans histiocytosis (histiocytosis-X) Diagn Cytopathol. 1993;9(5):527–533. doi: 10.1002/dc.2840090511. [DOI] [PubMed] [Google Scholar]
- 27.Kumar N, Sayed S, Vinayak S. Diagnosis of Langerhans cell histiocytosis on fine needle aspiration cytology: a case report and review of the cytology literature. Pathol Res Int. 2011;20(2011):439518. doi: 10.4061/2011/439518. [DOI] [PMC free article] [PubMed] [Google Scholar]