

Abstract
Bilirubin is thought to exert anti-inflammatory effects by inhibiting vascular cell adhesion molecule-1 (VCAM-1)-dependent leukocyte migration and by suppressing the expression of inducible nitric oxide synthase (iNOS). As VCAM-1 and iNOS are important mediators of tissue injury in the dextran sodium sulfate (DSS) murine model of inflammatory colitis, we examined whether bilirubin prevents colonic injury in DSS-treated mice. Male C57BL/6 mice were administered 2.5% DSS in the drinking water for 7 days, while simultaneously receiving intraperitoneal injections of bilirubin (30 mg/kg) or potassium phosphate vehicle. Disease activity was monitored, peripheral blood counts and serum nitrate levels were determined, and intestinal specimens were analyzed for histological injury, leukocyte infiltration, and iNOS expression. The effect of bilirubin on IL-5 production by HSB-2 cells and on Jurkat cell transendothelial migration also was determined. DSS-treated mice that simultaneously received bilirubin lost less body weight, had lower serum nitrate levels, and exhibited reduced disease severity than vehicle-treated animals. Concordantly, histopathological analyses revealed that bilirubin-treated mice manifested significantly less colonic injury, including reduced infiltration of eosinophils, lymphocytes, and monocytes, and diminished iNOS expression. Bilirubin administration also was associated with decreased eosinophil and monocyte infiltration into the small intestine, with a corresponding increase in peripheral blood eosinophilia. Bilirubin prevented Jurkat migration but did not alter IL-5 production. In conclusion, bilirubin prevents DSS-induced colitis by inhibiting the migration of leukocytes across the vascular endothelium and by suppressing iNOS expression.
Keywords: bilirubin, VCAM-1, eosinophil, iNOS, DSS colitis
bilirubin is generated during the physiological breakdown of heme, through the sequential activity of heme oxygenase and biliverdin reductase. Elevated serum levels of bilirubin can occur as a consequence of the accelerated release of heme from hemoglobin (e.g., hemolysis) or diminished hepatic conjugating activity (e.g., Gilbert's syndrome). It was first postulated over 75 years ago that bilirubin exerts an anti-inflammatory effect when it was noted that patients with rheumatoid arthritis experienced a remission of symptoms after developing jaundice secondary to superimposed liver disease (28, 61). These observations led to small therapeutic trials in which intravenous bilirubin was found to variably induce sustained analgesia and diminished joint swelling in subjects with chronic arthritis (68), although technical hurdles resulted in the abandonment of this line of investigation (29). More recently, bilirubin has been shown to suppress inflammatory responses in animal models of autoimmune encephalomyelitis (40) and hindpaw inflammation (74). With regard to inflammatory conditions of the intestine, indirect evidence supporting a potential protective effect of bilirubin are derived from the finding that individuals possessing the Gilbert's polymorphism have a decreased risk of Crohn's disease (20).
One mechanism whereby bilirubin has been postulated to exert an anti-inflammatory effect is through inhibition of vascular cell adhesion molecule-1 (VCAM-1)-mediated signaling. VCAM-1 promotes the binding and movement of leukocytes across activated vascular endothelium (43, 54, 72) and is believed to facilitate the immune-mediated tissue injury that occurs in inflammatory bowel disease (18, 27). Specifically, the infiltration of eosinophils into the intestinal mucosa, a VCAM-1-dependent process (13, 50), has been implicated in the pathogenesis of both ulcerative colitis and Crohn's disease (1, 11, 75). In support of this hypothesis, treatment with antibodies (10, 49, 65) or antisense oligonucleotides (56) directed against VCAM-1 suppresses intestinal inflammation in rodent colitis models, and blocking antibodies directed against leukocyte integrins that bind VCAM-1 have been shown to ameliorate Crohn's disease in humans (25, 60). Bilirubin, a potent chain-breaking antioxidant (3, 66), inhibits VCAM-1-dependent migration of lymphocytes across endothelial monolayers by scavenging NADPH oxidase-generated superoxide (33), an early event in VCAM-1 signaling (15). Consistent with these in vitro findings, bilirubin has been demonstrated to attenuate allergen-induced pneumonitis in mice by blocking VCAM-1-mediated eosinophil infiltration into the lungs (33).
Bilirubin also may attenuate intestinal injury by preventing the upregulation of inducible nitric oxide synthase (iNOS). Nitric oxide (NO) generated by the iNOS-catalyzed conversion of l-arginine to l-citrulline is thought to contribute to impaired intestinal mucosal integrity during sepsis (12) and also appears to play a key role in the pathogenesis of intestinal inflammation in animal models of colitis (30, 36, 37, 79). In humans, colonic iNOS expression and NO levels correlate with clinical and endoscopic indices of disease activity in patients with ulcerative colitis (17, 55, 62). Although the influence of bilirubin on intestinal iNOS activity has not been directly examined, bilirubin has been shown to inhibit the upregulation of iNOS in isolated murine macrophages, and in liver, renal, myocardial, and aortic tissues of endotoxin-treated rats (39, 74).
The oral administration of dextran sodium sulfate (DSS) to mice induces a form of colitis that mimics many of the clinical and histological features of human ulcerative colitis (52), including eosinophil infiltration. The fundamental role of eosinophils in inducing colonic injury in this murine model is highlighted by the finding that intestinal inflammation is markedly attenuated in mice lacking eosinophil peroxidase (an enzyme that produces oxidizing compounds implicated in inflammatory responses) or eotaxin (the principal eosinophil chemoattractant) (22). Previous investigations also support a contribution of iNOS activity to DSS-mediated intestinal damage (30, 36). Since bilirubin has been shown to modulate iNOS expression and inhibit VCAM-1-mediated eosinophil migration, we postulated that treatment with this bile pigment would prevent colitis induced by DSS. Hence the aims of the present studies were to examine the influence of bilirubin on colonic inflammation, eosinophil accumulation, and iNOS activity in DSS-treated mice.
MATERIALS AND METHODS
Materials.
Unconjugated bilirubin (bilirubin IXα) was obtained from Porphyrin Products (Logan, UT) and further purified according to the method of McDonagh and Assisi (46) to eliminate potential lipid contaminants. Bilirubin stock solutions were freshly prepared in 0.1 M potassium phosphate (pH 12), as previously described by our group (74). The addition of a small aliquot (≤0.4% vol/vol) of this stock solution had no effect on the pH of the cultured medium or on cell viability. Phorbol myristate acetate (PMA), ionomycin, collagen, fibronectin, tumor necrosis factor-α (TNF-α), Texas red dextran 10,000 mol wt, and CellTrace Far Red were obtained from Life Technologies (Grand Island, NY).
Cell isolation and culture.
Human umbilical vein endothelial cells (HUVEC) were isolated from discarded umbilical cords by use of collagenase, as previously described (19). Cells were cultured in F-12K medium supplemented with 10% fetal calf serum (FCS), endothelial cell growth supplement (Corning; Bedford, MA), 0.1 mg/ml heparin, 100 IU penicillin, and 100 μg/ml streptomycin. All experiments were performed by using passages 3 to 7. The human acute T cell leukemia cell line Jurkat was purchased from ATCC and grown in RPMI 1640 supplemented with 10% FCS, 1 mM l-glutamine, 100 IU penicillin, and 100 μg/ml streptomycin. The following reagent was obtained through the NIH AIDS Reagent Program, Division of AIDS, National Institute of Allergy and Infectious Diseases, National Institutes of Health: HSB-2 from Electro-Nucleonics (59). HSB-2 cells, a human T cell lymphoblastoid line, were cultured in RPMI 1640 medium with 10% FCS.
Induction of colitis by the administration of DSS.
Animals were housed in the Laboratory Animal Medicine Services facility at the University of Cincinnati under controlled conditions (temperature 22 ± 2°C, relative humidity 50 ± 10%, 12 h light-dark cycle) and fed a standard chow diet. Experiments were performed on adult male C57BL6/J mice (Jackson Laboratories, Bar Harbor, ME) because of the previously demonstrated susceptibility of this murine strain to DSS-induced colonic injury (42, 53). Colitis was induced by administering 2.5% DSS (wt/vol) in the drinking water for the entire course of the experiment, with control animals receiving filtered water that did not contain DSS. Twenty-four hours after commencing DSS treatment, animals received intraperitoneal (ip) injections of bilirubin (30 mg/kg), or an equivalent volume of the potassium phosphate vehicle, every 8 h for 7 days. All experiments were approved by the University of Cincinnati Institutional Animal Care and Use Committee.
Assessment of disease activity.
Mice were weighed and examined for signs of colitis on a daily basis, by using a standard clinical disease activity index (DAI) comprised of the following parameters graded on a 4-point scale (16, 69, 71): stool consistency, presence or absence of fecal blood, and weight loss. At the conclusion of the 7-day treatment period, animals were euthanized by CO2 inhalation. Blood was immediately obtained by cardiac puncture. The small and large intestines were resected and colon length was promptly measured.
Histological analyses of tissue specimens.
Longitudinal sections of the entire colon and of the small intestine were fixed in 4% paraformaldehyde and stained with hematoxylin and eosin, by standard histological techniques. Each colon specimen was independently assessed for injury over serial low-power fields by three blinded observers, employing a well-defined quantitative scoring system (38, 71) that grades the severity of inflammation (on a scale of 0–3), the extent of injury (0–3), and crypt damage (0–4). The value assigned to each parameter is multiplied by a factor reflecting the percentage of tissue involvement and then summed, with a maximum achievable severity score of 40. A minimum of 22 independent fields were examined per specimen per observer. To determine whether treatment effects were site specific, a separate experiment was performed in which 2-cm sections of proximal (between the cecum and descending colon) and distal (between the descending colon and anus) colon were isolated, and contiguous low-power fields graded histologically in a blinded fashion by three independent observers.
Eosinophils were identified by staining paraffin-embedded specimens with Sirius red, according to the method of Meyerholz et al. (47). Briefly, slides containing fixed tissue sections were immersed in Harris hematoxylin for 2 min followed by rinses in tap water and 100% ethanol. Slides were then immersed in an alkaline Sirius red solution (pH 8–9) for 2 h and rinsed in tap water. Immunohistochemical staining for inducible nitric oxide synthase was performed by incubating frozen sections of colonic tissue in the presence of primary anti-mouse iNOS antibody (1:250; Transduction Laboratories) or the isotype control antibody (rat anti-mouse Ig; 1:500) at room temperature for 1 h, followed by a horseradish peroxidase-conjugated secondary antibody (67). Immunohistochemical staining of small intestine and colonic tissue for CD3 (prediluted, Ventana Medical Systems), CD68 (1:25; Abcam), and VCAM-1 (1:500; Biorbyt) was performed by the Cincinnati Children's Hospital histology core.
Quantification of leukocytes in blood samples.
Blood samples were collected in heparinized tubes and total white blood cell counts were determined by use of a hemacytometer. Differential leukocyte counts were performed on blood smears treated with Wright's stain, with a total of 200 cells counted per sample. Eosinophils were specifically identified by staining blood samples with Discombe's solution (21, 41).
Serum assay for nitric oxide.
Serum samples were filtered (10,000 mol wt) to exclude erythrocytes, and nitrate levels were measured by using a Nitrate/Nitrite Colorimetric Assay kit (Cayman Chemical, Ann Arbor, MI), as previously described by our group (74).
Quantification of cellular interleukin-5 expression.
HSB-2 cells were seeded in 12-well plates at a density of 2 × 106 per milliliter and grown for 24 h. Monolayers were induced to express interleukin-5 (IL-5) by the addition of 4 ng/ml PMA and 1 μM ionomycin to the culture medium (57). At the end of the incubation period, total RNA was extracted by using a GeneJET PCR Purification Kit (ThermoScientific) and 5 μg of total RNA was reverse transcribed by using a Thermo Scientific cDNA kit, according to the manufacturer's instructions. Quantification of IL-5 and GAPDH mRNA (the latter to control for amplification) was achieved with the ABsolute Blue QPCR SYBR Green detection system (ThermoScientific). Primers pairs were designed by use of Primer-blast NCBI: IL-5 (sense: 5′-gcttctgcatttgagtttgctagct-3′, antisense: 5′-tggccgtcaatgtatttctttattaag-3′) and GAPDH (sense: 5′-agaaggctggggctcatttg-3′, antisense: 5′-aggggccatccacagtcttc-3′).
Transwell migration assay.
HUVEC were seeded in the upper chamber of 24-well Transwells with 8-μm pores (Costar, Cambridge, MA) that were precoated with collagen (0.4 mg/cm2) and fibronectin (2 μg/cm2) at a density of 4 × 105 cells/insert and grown to confluence. Monolayer integrity was validated by overlaying Texas red dextran 10,000 mol wt (20 μg/ml) and measuring fluorescence intensity (ex: 595 nm, em: 625 nm) in the lower chamber. To induce adhesion molecule expression, HUVEC monolayers were stimulated with TNF-α (5 ng/ml) for 16 h prior to performing migration studies (14, 77). Jurkat cells were incubated in the presence of 25 μM CellTrace Far Red for 45 min at 37°C and washed. Migration was initiated by overlaying these fluorescently labeled cells (1 × 105 per insert) onto the HUVEC monolayers in the upper chamber of the Transwell. Studies were performed in the presence of bilirubin (0–20 μM) or vehicle (0.1 M K3PO4), which was added to the assay medium (F-12K plus 0.1% HSA) immediately prior to the start of the experiment. Transendothelial migration was quantified by measuring CellTrace fluorescence intensity (ex: 625 nm, em: 670 nm) in the lower chamber.
Statistical analysis.
Data were analyzed by a computer-based statistical program (SSI SigmaStat, San Jose, CA). Mean values were evaluated by ANOVA with t-test to assess for statistical significance. For data that were not normally distributed, a Kruskal-Wallis analysis of variance on ranks was performed.
RESULTS
Effect of bilirubin on DSS-induced disease activity, colon length, and intestinal histology.
To determine whether bilirubin is able to suppress DSS-induced colitis, C57BL/6 mice were divided into each of four treatment groups comprised of 9 (vehicle treatments) or 10 (bilirubin treatments) animals: no DSS plus ip bilirubin vehicle (vehicle); DSS plus vehicle (DSS + vehicle); no DSS plus bilirubin (bilirubin); and DSS plus bilirubin (DSS + bilirubin). DSS-treated mice that received bilirubin exhibited less weight loss (Fig. 1A), diarrhea, and intestinal bleeding than did animals administered DSS plus vehicle, as reflected in the significantly lower DAI scores (Fig. 1B). Unexpectedly, and despite a normal appearance, mice treated with bilirubin alone failed to gain weight to the same degree as animals receiving vehicle alone. Consistent with the clinical findings, DSS-treated mice that were administered bilirubin demonstrated less colon shortening (Fig. 1C) and reduced mucosal inflammation (Fig. 2, top) compared with animals that received DSS plus vehicle. The colons from DSS-treated mice that were administered vehicle exhibited dense cellular infiltrates, marked architectural destruction, and loss of epithelial lining. Mice receiving DSS plus bilirubin manifest less colonic inflammation and reduced morphological injury, as evidenced by the significantly lower histological injury scores (Fig. 2A). Administration of bilirubin alone was found to have no noticeable effect on colon histology compared with vehicle treatment. Notably, bilirubin was found to exert a less pronounced effect in the distal (Fig. 2B) compared with the proximal (Fig. 2C) colon.
Fig. 1.

Effect of bilirubin on body weight, disease activity, and colonic shortening induced by dextran sodium sulfate (DSS). Mice were administered 2.5% DSS in the drinking water, followed by intraperitoneal injections of bilirubin (shaded squares) or the potassium phosphate vehicle (black squares). Control animals received bilirubin (shaded circles) or vehicle (black circles) without DSS. A: average daily percent change in weight from baseline (±SE) following the initiation of DSS. B: mean disease activity index for each group. C: average length of resected colon specimens. Compared with the other treatment groups, the colons from mice treated with DSS plus vehicle were significantly shorter and lacked formed stool. *P < 0.002 vs. all other groups, **P < 0.03 vs. bilirubin and bilirubin + DSS, ***P < 0.001 vs. vehicle + DSS.
Fig. 2.

Bilirubin treatment reduces colon histological injury following DSS administration. Top: representative low-power (×200) images of hematoxylin and eosin-stained sections of colon from mice treated with vehicle alone, DSS plus vehicle, bilirubin alone, or DSS plus bilirubin. Bottom: histological injury scores based on a blinded analysis of colon specimens using a standard 0–40 scoring system. A: mean histological scores (±SE) assessed over the entire colon. B and C: results of a separate experiment in which histological scoring of sections of distal and proximal colon were performed. *P < 0.002 vs. vehicle plus DSS.
Influence of bilirubin on eosinophil recruitment in response to DSS treatment.
Since eosinophils are believed to play an important role in the pathogenesis of DSS colitis (22), and since bilirubin has previously been shown to inhibit eosinophil influx in a murine model of airway inflammation (33), we assessed whether bilirubin treatment prevents eosinophil recruitment to the colon of DSS-treated mice. Paraffin-embedded sections of distal colon were stained with the eosinophil marker Sirius red (Fig. 3, top), and the average number of eosinophils per high-power field (hpf) was quantified (Fig. 3, bottom). Eosinophil infiltration into the colon of animals that received DSS was significantly lower following bilirubin vs. vehicle treatment, supporting an inhibitory effect of bilirubin on eosinophil infiltration.
Fig. 3.

Bilirubin inhibits eosinophil infiltration and VCAM-1 expression in the colon of DSS-treated mice. Top: representative high-power (×400) images of 2-cm sections of distal colon obtained from DSS-treated mice administered vehicle (left) or bilirubin (right) and stained with Sirius red to highlight eosinophils (arrows). Middle: results of immunohistochemical staining of proximal colon specimens for VCAM-1 (×200). Bottom: graph of mean number of eosinophils (±SE) per high-power field (hpf) averaged over 10 separate fields (n = 9–10 per treatment group). *P < 0.001 vs. vehicle plus DSS.
Since VCAM-1 is upregulated in the colon of mice administered DSS (65) and appears to contribute to tissue injury and eosinophil recruitment (6), we examined the effect of bilirubin on VCAM-1 expression by performing immunohistochemical staining of intestinal specimens. We found that the DSS-induced expression of VCAM-1 in the colon was markedly reduced in mice that were simultaneously administered bilirubin (Fig. 3, middle), approaching levels observed in animals that did not receive DSS (data not shown). These findings provide a potential explanation for the ameliorating effect of bilirubin. However, although the number of colonic eosinophils was not statistically different between the non-DSS control groups, there was a strong trend (P = 0.06) toward lower numbers of eosinophils per hpf in mice that were administered bilirubin vs. vehicle, despite minimal detectable VCAM-1 in both groups. These data raise the possibility that bilirubin modulates eosinophil homing via mechanism(s) that are independent of VCAM-1 expression.
To determine whether the diminished number of eosinophils in the colon of bilirubin-treated animals might be due to fewer circulating cells available for recruitment, we measured total leukocyte and differential counts in the peripheral blood of bilirubin- and vehicle-treated mice, in both the presence and absence of DSS. Although total peripheral blood leukocyte counts did not differ significantly between the various treatment groups (Fig. 4A), mice that received bilirubin exhibited a selective increase in the percentage of circulating eosinophils (Fig. 4B), regardless of whether the animals were administered DSS. An increase in the absolute number of eosinophils in the peripheral circulation of mice treated with bilirubin was confirmed by staining blood smears with Discombe's solution to specifically label eosinophils (Fig. 4C). These data indicate that the observed reduction in the number of colonic eosinophils in mice treated with bilirubin does not result from fewer circulating eosinophils. On the contrary, the increased peripheral blood eosinophilia noted in bilirubin-treated animals suggests that eosinophil access to peripheral tissues is impeded.
Fig. 4.
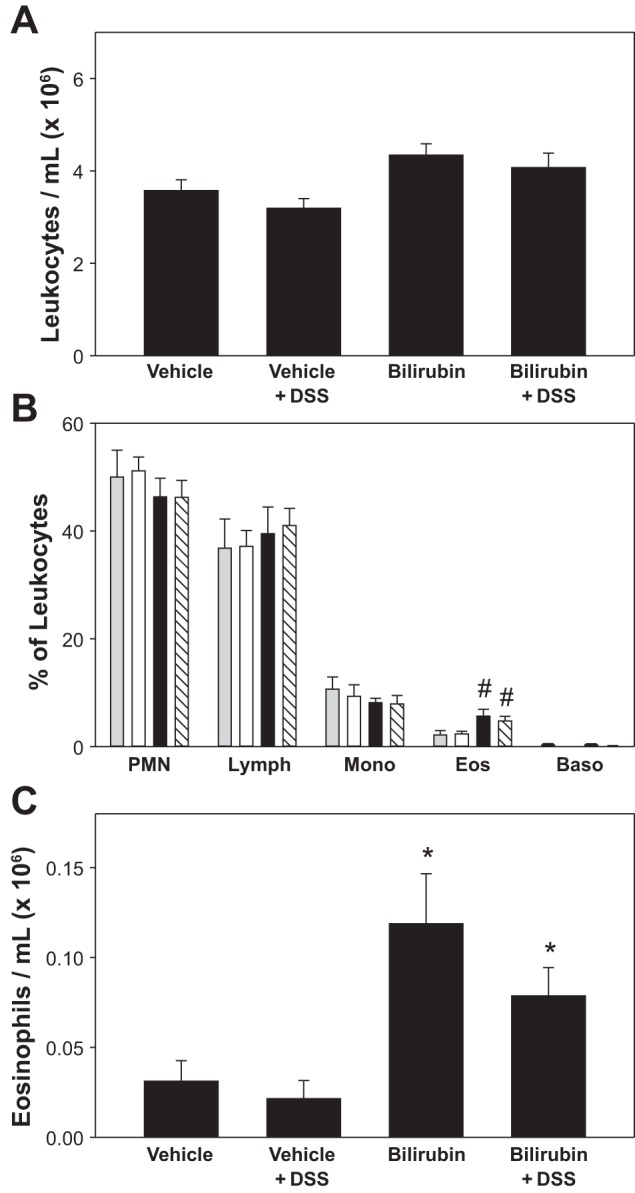
Effect of bilirubin on peripheral blood leukocytes. A: average total leukocyte count (±SE). B: mean percentage of neutrophils (PMN), lymphocytes (Lymph), monocytes (Mono), eosinophils (Eos), and basophils (Baso) in the peripheral blood of mice treated with vehicle alone (shaded bars), DSS plus vehicle (open bars), bilirubin alone (solid bars), or DSS plus bilirubin (hatched bars). C: peripheral blood eosinophil counts determined by Discombe's staining, plotted as the average number of eosinophils/ml (× 106). #P < 0.05 vs. vehicle and vehicle plus DSS; *P < 0.01 vs. vehicle and vehicle plus DSS.
The proliferation, differentiation, and release of eosinophils from the bone marrow into the systemic circulation is mediated primarily by IL-5 (34). To investigate whether the observed increase in peripheral blood eosinophils associated with bilirubin administration might be due to augmented production of IL-5, we examined the effect of bilirubin on cellular IL-5 expression by HSB-2 cells, a human T cell leukemia line that has been shown to produce IL-5 in a manner similar to normal peripheral blood T cells (57, 73). When stimulated with PMA and ionomycin, HSB-2 cells exhibited a marked (1,000-fold) upregulation in IL-5 mRNA that peaked at 6 h and was unaffected by cotreatment with bilirubin (Fig. 5A). When incubated in the presence of bilirubin alone, HSB-2 cells manifested a nonsignificant (<2-fold) increase in IL-5 mRNA out to 24 h (Fig. 5B). In light of previous studies demonstrating no significant effect of bilirubin on IL-5 levels in isolated T cells and lung lavage fluid obtained from mice with allergen-induced pulmonary inflammation (33), these data support that bilirubin does not induce peripheral eosinophilia through modulation of IL-5 production.
Fig. 5.

Influence of bilirubin on cellular IL-5 production, leukocyte transendothelial migration, and serum nitrate concentration. A: effect of 10 μM bilirubin (shaded symbols) on the time course for the expression of IL-5 mRNA in HSB-2 cells, as determined by quantitative PCR. Cells were incubated in the presence (squares) or absence (circles) of 4 ng/ml PMA and 1 μM ionomycin with data reflecting IL-5 expression (±SE) relative to that in untreated cells at time 0 (n = 4). B: HSB-2 cells were incubated with 10 μM bilirubin for the indicated time intervals and IL-5 mRNA levels are plotted relative to expression at time 0. C: time course for Jurkat cell migration across human umbilical vein endothelial cells (HUVEC) monolayers preincubated for 16 h in the presence (squares) or absence (circles) of TNF-α (5 ng/ml). CellTrace Far Red-labeled Jurkat cells were overlaid onto the monolayers in the presence of 20 μM bilirubin (shaded symbols) or vehicle (solid symbols). Displayed is the percentage of overlaid cells that migrated to the lower chamber, as quantified by measuring fluorescence intensity (ex: 625 nm, em: 670 nm). D: mean serum nitrate concentration for each treatment group. *P < 0.05 vs. all other groups.
Bilirubin modulates the physiological homing of eosinophils to the intestinal tract.
We have shown that bilirubin reduces eosinophil infiltration into the colon when inflammation is induced by treatment with DSS (Fig. 3). Since eosinophils primarily reside in intestinal tissue under noninflammatory states (34), we assessed whether bilirubin impairs the normal physiological homing of these cells to the gastrointestinal tract by quantifying the number of eosinophils in Sirius red-stained sections of the midjejunum (Fig. 6, top). Notably, mice that received bilirubin had significantly fewer tissue eosinophils than those administered vehicle, regardless of whether DSS was administered (Fig. 6, bottom). These data, when taken in conjunction with our finding that the number of eosinophils in the peripheral blood increases in bilirubin-treated mice (Fig. 4, B and C), suggest that bilirubin impedes the physiological migration of eosinophils from the circulation to the gut.
Fig. 6.

Bilirubin reduces eosinophil infiltration into the small intestine. Top: representative high-power (×400) images of sections of small intestine stained with Sirius red to identify eosinophils (arrows). Treatment groups are as previously described in Fig. 2. Bottom: mean number of eosinophils per hpf (±SE) for each group. An average of 39 separate hpf was examined per specimen (n = 9–10 per group). *P < 0.001 vs. all other groups.
To further examine this hypothesis, we investigated the effect of bilirubin on the transendothelial migration of leukocytes in vitro. Since VCAM-1 appears to play an essential role in the pathogenesis of DSS colitis (65) and in the recruitment of eosinophils to sites of inflammation (13, 26, 76), we elected to study Jurkat cells, a human acute T cell leukemia cell line that is known to express the VCAM-1-specific integrin VLA-4 (35). HUVEC monolayers were preincubated with TNF-α to induce the expression of VCAM-1 (14, 77), as validated by measuring both cellular mRNA and protein levels (data not shown). Paralleling our in vivo findings and the results of our prior studies (33), bilirubin markedly inhibited the migration of Jurkat cells across TNF-α-stimulated HUVEC monolayers (Fig. 5C) without altering VCAM-1 expression, consistent with a direct modulatory effect on VCAM-1 signaling.
Effect of bilirubin on the intestinal recruitment of T lymphocytes and monocytes/macrophages.
To determine whether the inhibitory effect of bilirubin is specific to eosinophil recruitment, we stained sections of colon and small intestine for the pan-phenotypic T lymphocyte marker CD3 and for CD68, a glycoprotein that is expressed exclusively on monocytes and macrophages. Analogous to our findings for eosinophils (Fig. 3), bilirubin treatment was associated with a significant reduction in the DSS-induced infiltration of CD3- (Fig. 7) and CD68 (Fig. 8)-positive cells into the colon. Although there appears to be no effect of either DSS or bilirubin on the number of CD3-positive cells in the small intestine (Fig. 9), substantially fewer CD68-positive cells were present in small intestinal tissue from bilirubin-treated mice, irrespective of whether they received DSS (Fig. 10). These latter findings parallel our observation for eosinophils (Fig. 6), suggesting that bilirubin also impedes the migration of monocytes/macrophages from the peripheral circulation into the small intestine.
Fig. 7.

Treatment with bilirubin decreases T lymphocyte infiltration into the colon of DSS-treated mice. Shown are representative low-power (×200) images of colon tissue stained for the T lymphocyte marker CD3. Immunohistochemical staining was performed on sections of colon obtained from mice treated with vehicle alone (top left), vehicle plus DSS (top right), bilirubin alone (middle left), or bilirubin plus DSS (middle right). Bottom: mean number of CD3-positive cells per hpf (±SE) for each treatment group. An average of 29 separate hpf was examined per specimen (n = 3–4 per group). *P < 0.02 vs. all other groups.
Fig. 8.

Bilirubin inhibits the recruitment of monocytes/macrophages to the colon in response to DSS. Representative colon specimens stained for CD68, a marker for monocytes and macrophages, are displayed (×200). Conditions are as described under Fig. 7. The marked infiltration of CD68-positive cells in animals receiving DSS plus vehicle precluded accurate quantitation.
Fig. 9.

DSS and bilirubin do not alter T lymphocyte levels in the small intestine. Histological sections of small intestine (×200) were stained for CD3, as detailed in Fig. 7. Quantification of CD3-positive cells is represented as the number (±SE) per intact villus (bottom). An average of 32 villi was examined per specimen (n = 3 per treatment group).
Fig. 10.

Monocyte infiltration into the small intestine is reduced following bilirubin treatment. Representative small intestinal specimens stained for CD68-positive cells (arrows) are shown (×200), as described under Fig. 8. Bottom: mean number of CD68-positive cells per hpf (±SE) for each treatment group. An average of 32 separate hpf was examined per specimen (n = 3 per group). *P < 0.01 vs. both vehicle treatment groups.
Influence of bilirubin on DSS-induced colonic iNOS expression and systemic nitrate production.
Nitric oxide generated by iNOS is thought to contribute to the colonic injury induced by DSS (5, 30, 36). Since bilirubin has been shown to inhibit the upregulation of iNOS in response to inflammatory stimuli (39, 63, 74), we sought to determine whether bilirubin treatment is associated with reduced iNOS expression in the colon of mice treated with DSS. Frozen sections of colon tissue from untreated and DSS-treated mice that were simultaneously administered bilirubin or vehicle were subjected to immunohistochemical staining for murine iNOS, employing isotype antibody as control. Consistent with prior reports (5, 67), the colonic expression of iNOS was markedly augmented by DSS. Additionally, we found that DSS-induced iNOS expression was substantially abrogated by bilirubin treatment (Fig. 11). Consistent with these histological findings, mice treated with vehicle plus DSS manifested elevated serum nitrate levels (a marker of iNOS activity), which were significantly lower in bilirubin-treated animals (Fig. 5D). Taken together, these data support that bilirubin exerts an inhibitory effect on DSS-induced colonic expression and activity of iNOS.
Fig. 11.

Bilirubin inhibits DSS-induced colonic inducible nitric oxide synthase (iNOS) expression. Immunohistochemical staining for iNOS was performed on frozen sections of colon tissue obtained from mice treated with vehicle alone (top left), vehicle plus DSS (top middle), bilirubin alone (bottom left), or bilirubin plus DSS (lower middle). Representative low-power (×200) images are shown. Right: specimens stained with an isotype antibody (iso) as control.
DISCUSSION
The present studies demonstrate that colonic inflammation induced by the oral administration of DSS to C57BL6/J mice is suppressed by bilirubin, as evidenced by reduced disease activity scores and by diminished histological injury. The reason why bilirubin exerts a more pronounced protective effect in the proximal vs. the distal colon is uncertain. We speculate that this could be due to the overall less vigorous inflammatory response in the former region, or possibly because levels of bilirubin in the colonic lumen decline caudally as a result of enterohepatic cycling (70). Our findings are consistent with previous reports that the bilirubin precursor, biliverdin, ameliorates DSS colitis in mice (7). Although the mechanism underlying this protective effect was not examined in this prior study, in light of our present findings, and since biliverdin is known to undergo rapid and quantitative conversion to bilirubin via the action of the ubiquitous biliverdin reductase enzyme (44, 45), we speculate that bilirubin is the principal physiological mediator of the observed cytoprotection. This hypothesis is supported by the results of prior studies in which bilirubin, but not biliverdin, was found to attenuate endothelial cell activation (64).
Our data suggest two potential mechanisms by which bilirubin may exert a modulatory effect on DSS-induced colonic inflammation: 1) inhibition of eosinophil (and other leukocyte) infiltration into intestinal tissues, and 2) suppression of iNOS expression and activity. We previously have demonstrated that bilirubin blocks eosinophil migration into the lungs of mice with allergen-induced pneumonitis through a mechanism that involves the disruption of VCAM-1 signaling within vascular endothelial cells (33). Our present finding that bilirubin administration reduces eosinophil infiltration into the colon, while simultaneously increasing levels of circulating eosinophils, supports a similar mechanism of bilirubin action in the DSS colitis model. Consistent with this proposition, we have directly shown that bilirubin, at physiological concentrations (20 μM ≈ 1.2 mg/dl), effectively prevents the movement of Jurkat cells across TNF-α-activated HUVEC monolayers. Although TNF-α induces HUVEC to express both VCAM-1 and ICAM-1, because Jurkat cells do not bind ICAM-1 (48), our findings imply that bilirubin exerts its effects primarily through disruption of VCAM-1-dependent processes. When taken in conjunction with our previous finding that bilirubin inhibits the movement of lymphocytes across murine endothelial cell monolayers that constitutively express VCAM-1 (33), these data support that the effects of bilirubin are not eosinophil specific but rather that bilirubin interferes with any VCAM-1-mediated migration process. This hypothesis is supported by our demonstration of reduced numbers of CD3- and CD68-positive cells in the colon of DSS-treated mice that also received bilirubin, since both lymphocytes and monocytes/macrophages are known to express VCAM-1-specific integrins (23, 24).
Since VCAM-1 activation (65) and eosinophil infiltration (22) are important contributing factors to the pathogenesis of DSS colitis, the ability of bilirubin to block VCAM-1-dependent leukocyte migration, and thereby inhibit eosinophil recruitment, likely underlies its protective effects. We show that bilirubin-treated animals manifest markedly attenuated colonic VCAM-1 expression in response to DSS, which may, at least in part, explain bilirubin's ameliorating effect on tissue injury. However, since the production of VCAM-1 by endothelial cells is stimulated by proinflammatory cytokines, these data cannot differentiate as to whether this reflects an ability of bilirubin to directly inhibit endothelial VCAM-1 expression or, rather, is a consequence of reduced cytokine production. Our demonstration that bilirubin does not alter basal or TNF-α-stimulated VCAM-1 mRNA or protein levels in cultured HUVEC supports the latter theory, with the caveat that other investigators have reported an inhibitory effect of bilirubin on endothelial cell expression of VCAM-1 (32, 64). Our present findings conflict with a prior study by our group employing a murine model of allergic pneumonitis in which we observed that bilirubin did not alter pulmonary VCAM-1 expression. Notably, in this same study, bilirubin also did not influence the production of IL-2, IL-4, IL-6, IL-10, IL-12, TNF-α, or the main eosinophil chemoattractants IL-5 and eotaxin (33). Although an important limitation of the present experiments is our inability to quantitate IL-5 or eotaxin levels in intestinal tissue, the disparate findings could possibly reflect the fact that allergic pneumonitis is a Th2 cytokine-dependent model (9) whereas DSS-induced colitis is Th1 mediated (2).
Because eosinophils are tissue-resident leukocytes that localize primarily to the gastrointestinal tract (58), our observation that eosinophil levels in the jejunum are significantly reduced in bilirubin-treated animals (irrespective of whether they received DSS) supports that bilirubin also inhibits the normal physiological migration of eosinophils into the gut. This theory is bolstered by the concomitant increase in circulating eosinophils associated with bilirubin administration, a finding that corroborates previous observations by our group (33). Our finding that bilirubin does not alter IL-5 message in unstimulated or activated HSB-2 T cell leukemia cells, which are a generally accepted in vitro model of IL-5 regulation (57, 73), indicates that the peripheral eosinophilia associated with bilirubin administration is unlikely to be the result of increased eosinophil production and/or release from the bone marrow. Notably, whereas the number of T lymphocytes in the small intestine was unaffected by bilirubin treatment, the level of monocytes/macrophages was reduced, paralleling our eosinophil results. These data suggest that eosinophils and monocytes may share a similar trafficking mechanism to the small bowel. Since there was no detectable VCAM-1 expression in the small intestine in any of the treatment groups, it seems unlikely that bilirubin reduces leukocyte infiltration by modulating the expression of VCAM-1 in noninflamed intestinal tissue. In previous studies, we have shown that physiological concentrations of bilirubin, a potent chain-breaking antioxidant (66), inhibits the transmigration of VLA-4-expressing leukocytes across murine endothelial monolayers by scavenging VCAM-1-dependent reactive oxygen species signaling intermediaries (33). We speculate that this same mechanism may underlie the inhibitory effect of bilirubin on the infiltration of eosinophils and monocytes into the small intestine.
We are perplexed by the observed difference in growth curves between vehicle- and bilirubin-treated control animals, since we have not previously observed this phenomenon in mice receiving similar dosing of ip (33) or oral (63) bilirubin, which we have shown produces very modest three- to fourfold elevations in serum bilirubin levels (≤0.4 mg/dl) (74). Notably, Gunn rats, which are homozygous (j/j) for a mutation in the UGT1A gene locus leading to persistent, marked hyperbilirubinemia (∼5 mg/dl) (63), do not manifest abnormal growth. Since bilirubin is generated as part of the physiological degradation of heme, it is normally present in the circulation. In newborns, markedly elevated serum bilirubin concentrations (generally over 20 mg/dl ≈ 340 μM) can cause neurological injury (kernicterus); however, toxicity in adults is negligible. Indeed, bilirubin has been administered intravenously to patients without overt sequelae (68), reaching serum levels as high as 22 mg/dl (normal ≤ 1.2 mg/dl ≈ 20 μM). More modest (<3-fold) chronic elevations in serum bilirubin are commonly encountered in individuals with Gilbert's syndrome, a benign condition resulting from polymorphisms in the gene encoding UGT1A1 (8), the principal bilirubin-conjugating enzyme. It is notable that our in vitro studies utilizing human cells support that bilirubin exerts substantial inhibitory effects on leukocyte migration at concentrations that are at the upper end of the normal physiological range (20 μM).
The administration of DSS to mice has been shown to induce iNOS expression in the colon (31, 37) and to increase nitrate concentrations in the serum (5, 37). Although conflicting data exist (4, 78), it is generally held that iNOS-derived NO contributes to the pathogenesis of DSS-induced colitis, as evidenced by the findings that iNOS-deficient mice (30, 36, 37) and that animals administered iNOS inhibitors (37, 51) manifest reduced colonic injury. It is notable that bilirubin has previously been shown to suppress the endotoxin stimulated upregulation of iNOS (39, 74). Consistent with these prior investigations, our experiments demonstrate that bilirubin treatment reduces colonic iNOS expression and serum nitrate levels in DSS-treated mice, suggesting that bilirubin may act, in part, by preventing peroxynitrite-induced tissue injury (79). Both iNOS (55, 62) and VCAM-1 (18, 27) are believed to contribute to the pathogenesis of inflammatory bowel disease in humans and, notably, natalizumab (an antibody directed against the α4 integrin subunit that mediates leukocyte binding to VCAM-1) has shown efficacy in inducing and maintaining clinical remission in Crohn's disease (25, 60). Hence our demonstration that bilirubin is able to disrupt both of these processes raises the specter of a potential therapeutic application.
GRANTS
This study was supported by National Institutes of Health Research Grant DK063954 (S. D. Zucker).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
S.D.Z., M.E.V., T.L.K., G.I., and U.A. conception and design of research; S.D.Z., M.E.V., T.L.K., D.L.S., G.I., U.A., G.K., and M.E.M. performed experiments; S.D.Z., M.E.V., T.L.K., D.L.S., G.I., U.A., G.K., and M.E.M. analyzed data; S.D.Z., M.E.V., T.L.K., D.L.S., G.I., and U.A. interpreted results of experiments; S.D.Z., M.E.V., and G.I. prepared figures; S.D.Z., M.E.V., and G.I. drafted manuscript; S.D.Z., M.E.V., T.L.K., D.L.S., G.I., U.A., and G.K. edited and revised manuscript; S.D.Z., M.E.V., T.L.K., D.L.S., G.I., U.A., G.K., and M.E.M. approved final version of manuscript.
ACKNOWLEDGMENTS
Present address for T. Kindel: Division of General Surgery, University of Nebraska Medical Center, Omaha, NE 68198.
Present address for U. Avissar: Section of Gastroenterology, Boston University School of Medicine, Boston, MA 02118.
REFERENCES
- 1.Ahrens R, Waddell A, Seidu L, Blanchard C, Carey R, Forbes E, Lampinen M, Wilson T, Cohen E, Stringer K, Ballard E, Munitz A, Xu H, Lee N, Lee JJ, Rothenberg ME, Denson L, Hogan SP. Intestinal macrophage/epithelial cell-derived CCL11/eotaxin-1 mediates eosinophil recruitment and function in pediatric ulcerative colitis. J Immunol 181: 7390–7399, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Alex P, Zachos NC, Nguyen T, Gonzales L, Chen TE, Conklin LS. Distinct cytokine patterns identified from multiplex profiles of murine DSS and TNBS-induced colitis. Inflamm Bowel Dis 15: 341–352, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Barañano DE, Rao M, Ferris CD, Snyder SH. Biliverdin reductase: a major physiologic cytoprotectant. Proc Natl Acad Sci USA 99: 16093–16098, 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Beck PL, Li Y, Wong J, Chen CW, Keenan CM, Sharkey KA, McCafferty DM. Inducible nitric oxide synthase from bone marrow-derived cells plays a critical role in regulating colonic inflammation. Gastroenterology 132: 1778–1790, 2007. [DOI] [PubMed] [Google Scholar]
- 5.Beck PL, Xavier R, Wong J, Ezedi I, Mashimo H, Mizoguchi A, Mizoguchi E, Bhan AK, Podolsky DK. Paradoxical roles of different nitric oxide synthase isoforms in colonic injury. Am J Physiol Gastrointest Liver Physiol 286: G137–G147, 2004. [DOI] [PubMed] [Google Scholar]
- 6.Bell LV, Else KJ. Mechanisms of leucocyte recruitment to the inflamed large intestine: redundancy in integrin and addressin usage. Parasite Immunol 30: 163–170, 2008. [DOI] [PubMed] [Google Scholar]
- 7.Berberat PO, A-Rahim YI, Yamashita K, Warny MM, Csizmadia E, Robson SC, Bach FH. Heme oxygenase-1-generated biliverdin ameliorates experimental murine colitis. Inflamm Bowel Dis 11: 350–359, 2005. [DOI] [PubMed] [Google Scholar]
- 8.Bosma PJ, Roy-Chowdhury J, Bakker C, Gantla S, de Boer A, Oostra BA, Lindhout D, Tytgat GNJ, Jansen PLM, Oude Elferink RPJ, Roy-Chowdhury N. The genetic basis of the reduced expression of bilirubin UDP-glucuronosyltransferase 1 in Gilbert's syndrome. N Engl J Med 333: 1171–1175, 1995. [DOI] [PubMed] [Google Scholar]
- 9.Brusselle G, Kips J, Joos G, Bluethmann H, Pauwels R. Allergen-induced airway inflammation and bronchial responsiveness in wild-type and interleukin-4-deficient mice. Am J Respir Cell Mol Biol 12: 254–259, 1995. [DOI] [PubMed] [Google Scholar]
- 10.Burns RC, Rivera-Nieves J, Moskaluk CA, Matsumoto S, Cominelli F, Ley K. Antibody blockade of ICAM-1 and VCAM-1 ameliorates inflammation in the SAMP-1/Yit adoptive transfer model of Crohn's disease in mice. Gastroenterology 121: 1428–1436, 2001. [DOI] [PubMed] [Google Scholar]
- 11.Carvalho AT, Elia CC, de Souza HS, Elias PR, Pontes EL, Lukashok HP, de Freitas FC, Lapa e Silva JR. Immunohistochemical study of intestinal eosinophils in inflammatory bowel disease. J Clin Gastroenterol 36: 120–125, 2003. [DOI] [PubMed] [Google Scholar]
- 12.Chen K, Inoue M, Okada A. Expression of inducible nitric oxide synthase mRNA in rat digestive tissues after endotoxin and its role in intestinal mucosal injury. Biochem Biophys Res Commun 224: 703–708, 1996. [DOI] [PubMed] [Google Scholar]
- 13.Chin JE, Hatfield CA, Winterrowd GE, Brashler JR, Vonderfecht SL, Fidler SF, Griffin RL, Kolbasa KP, Krzesicki RF, Sly LM, Staite ND, Richards IM. Airway recruitment of leukocytes in mice is dependent on α4-integrins and vascular cell adhesion molecule-1. Am J Physiol Lung Cell Mol Physiol 272: L219–L229, 1997. [DOI] [PubMed] [Google Scholar]
- 14.Cid MC, Kleinman HK, Grant DS, Schnaper HW, Fauci AS, Hoffman GS. Estradiol enhances leukocyte binding to tumor necrosis factor (TNF)-stimulated endothelial cells via an increase in TNF-induced adhesion molecules E-selectin, intercellular adhesion molecule type 1, and vascular cell adhesion molecule type 1. J Clin Invest 93: 17–25, 1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cook-Mills JM, Johnson JD, Deem TL, Ochi A, Wang L, Zheng Y. Calcium mobilization and Rac1 activation are required for VCAM-1 (vascular cell adhesion molecule-1) stimulation of NADPH oxidase activity. Biochem J 378: 539–547, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cooper HS, Murthy SN, Shah RS, Sedergran DJ. Clinicopathologic study of dextran sulfate sodium experimental murine colitis. Lab Invest 69: 238–249, 1993. [PubMed] [Google Scholar]
- 17.Cross RK, Wilson KT. Nitric oxide in inflammatory bowel disease. Inflamm Bowel Dis 9: 179–189, 2003. [DOI] [PubMed] [Google Scholar]
- 18.Danese S, Smeraro S, Marini M, Roberto I, Armuzzi A, Papa A, Gasbarrini A. Adhesion molecules in inflammatory bowel disease: therapeutic implications for gut inflammation. Dig Liver Dis 37: 811–818, 2005. [DOI] [PubMed] [Google Scholar]
- 19.Davis J, Crampton SP, Hughes CCW. Isolation of human umbilical vein endothelial cells (HUVEC). J Vis Exp 3: 183, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.de Vries HS, te Morsche RHM, Jenniskens K, Peters WHM, de Jong DJ. A functional polymorphism in UGT1A1 related to hyperbilirubinemia is associated with a decreased risk for Crohn's disease. J Crohns Colitis 6: 597–602, 2011. [DOI] [PubMed] [Google Scholar]
- 21.Discombe G. Criteria of eosinophilia. Lipids 247: 195–196, 1946. [DOI] [PubMed] [Google Scholar]
- 22.Forbes E, Murase T, Yang M, Matthaei KI, Lee JJ, Lee NA, Foster PS, Hogan SP. Immunopathogenesis of experimental ulcerative colitis is mediated by eosinophil peroxidase. J Immunol 172: 5664–5675, 2004. [DOI] [PubMed] [Google Scholar]
- 23.Fu H, Wang A, Mauro C, Marelli-Berg F. T lymphocyte trafficking: molecules and mechanisms. Front Biosci 18: 422–440, 2013. [DOI] [PubMed] [Google Scholar]
- 24.Gerhardt T, Ley K. Monocyte trafficking across the vessel wall. Cardiovasc Res 107: 321–330, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ghosh S, Goldin E, Gordon FH, Malchow HA, Rask-Madsen J, Rutgeerts P, Vyhnálek P, Zádorová Z, Palmer T, Donoghue S. Natalizumab for active Crohn's disease. N Engl J Med 348: 24–32, 2003. [DOI] [PubMed] [Google Scholar]
- 26.Gonzalo JA, Lloyd CM, Kremer L, Finger E, Martinez AC, Siegelman MH, Cybulsky M, Gutierrez-Ramos J. Eosinophil recruitment to the lung in a murine model of allergic inflammation: the role of T cells, chemokines, and adhesion receptors. J Clin Invest 98: 2332–2345, 1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gulubova MV, Manolova IM, Vlaykova TI, Prodanova M, Jovchev JP. Adhesion molecules in chronic ulcerative colitis. Int J Colorectal Dis 22: 581–589, 2007. [DOI] [PubMed] [Google Scholar]
- 28.Hench PS. The analgesic effect of hepatitis and jaundice in chronic arthritis, fibrositis and sciatic pain. Ann Intern Med 7: 1278–1294, 1934. [Google Scholar]
- 29.Hench PS. Effect of jaundice on chronic infectious arthritis and on primary fibrositis: further observations, attempts to reproduce the phenomenon. Arch Intern Med 61: 451–500, 1938. [Google Scholar]
- 30.Hokari R, Kato S, Matsuzaki K, Kuroki M, Iwai A, Kawaguchi A, Nagao S, Miyahara T, Itoh K, Sekizuka E, Nagata H, Ishii H, Miura S. Reduced sensitivity of inducible nitric oxide synthase-deficient mice to chronic colitis. Free Radic Biol Med 31: 153–163, 2001. [DOI] [PubMed] [Google Scholar]
- 31.Jin Y, Kotakadi VS, Ying L, Hofseth AB, Cui X, Wood PA, Windust A, Matesic LE, Pena EA, Chiuzan C, Singh NP, Nagarkatti M, Nagarkatti PS, Wargovich MJ, Hofseth LJ. American ginseng suppresses inflammation and DNA damage associated with mouse colitis. Carcinogenesis 29: 2351–2359, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kawamura K, Ishikawa K, Wada Y, Kimura S, Matsumoto H, Kohro T, Itabe H, Kodama T, Maruyama Y. Bilirubin from heme oxygenase-1 attenuates vascular endothelial activation and dysfunction. Arterioscler Thromb Vasc Biol 25: 155–160, 2005. [DOI] [PubMed] [Google Scholar]
- 33.Keshavan P, Deem TL, Schwemberger SJ, Babcock GF, Cook-Mills JM, Zucker SD. Unconjugated bilirubin inhibits VCAM-1-mediated transendothelial leukocyte migration. J Immunol 174: 3709–3718, 2005. [DOI] [PubMed] [Google Scholar]
- 34.Kita H. Eosinophils: multifaceted biologic properties and roles in health and disease. Immunol Res 242: 161–177, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kitani A, Nakashima N, Izumihara T, Inagaki M, Baoui X, Yu S, Matsuda T, Matsuyama T. Soluble VCAM-1 induces chemotaxis of Jurkat and synovial fluid T cells bearing high affinity very late antigen-4. J Immunol 161: 4931–4938, 1998. [PubMed] [Google Scholar]
- 36.Krieglstein C, Anthoni C, Cerwinka WH, Stokes KY, Russell J, Grisham MB, Granger DN. Role of blood- and tissue-associated inducible nitric-oxide synthase in colonic inflammation. Am J Pathol 170: 490–496, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Krieglstein CF, Cerwinka WH, Laroux FS, Salter JW, Russel JM, Schuermann G, Grisham MB, Ross CR, Granger DN. Regulation of murine intestinal inflammation by reactive metabolites of oxygen and nitrogen: divergent roles of superoxide and nitric oxide. J Exp Med 194: 1207–1218, 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Krieglstein CF, Cerwinka WH, Sprague AG, Laroux FS, Grisham MB, Koteliansky VE, Senninger N, Granger DN, de Fougerolles AR. Collagen-binding integrin α1β1 regulates intestinal inflammation in experimental colitis. J Clin Invest 110: 1773–1782, 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lanone S, Bloc S, Foresti R, Almolki A, Taille C, Callebert J, Conti M, Goven D, Aubier M, Dureuil B, El-Benna J, Motterlini R, Boczkowski J. Bilirubin decreases NOS2 expression via inhibition of NAD(P)H oxidase: implications for protection against endotoxic shock in rats. FASEB J 19: 1890–1892, 2005. [DOI] [PubMed] [Google Scholar]
- 40.Liu Y, Zhu B, Wang X, Luo L, Li P, Paty DW, Cynader MS. Bilirubin as a potent antioxidant suppresses experimental autoimmune encephalomyelitis: implications for the role of oxidative stress in the development of multiple sclerosis. J Neuroimmunol 139: 27–35, 2003. [DOI] [PubMed] [Google Scholar]
- 41.Ma W, Bryce PJ, Humbles AA, Laouini D, Yalcindag A, Alenius H, Friend DS, Oettgen HC, Gerard C, Geha RS. CCR3 is essential for skin eosinopilia and airway hyperresponsiveness in a murine model of allergic skin inflammation. J Clin Invest 109: 621–628, 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mahler M, Bristol IJ, Leiter EH, Workman AE, Birkenmeier EH, Elson CO, Sundberg JP. Differential susceptibility of inbred mouse strains to dextran sulfate sodium-induced colitis. Am J Physiol Gastrointest Liver Physiol 274: G544–G551, 1998. [DOI] [PubMed] [Google Scholar]
- 43.Matheny HE, Deem TL, Cook-Mills JM. Lymphocyte migration through monolayers of endothelial cell lines involves VCAM-1 signaling via endothelial cell NADPH oxidase. J Immunol 164: 6550–6559, 2000. [DOI] [PubMed] [Google Scholar]
- 44.McCoubrey WK, Cooklis MA, Maines MD. The structure, organization and differential expression of the rat gene encoding biliverdin reductase. Gene 160: 235–240, 1995. [DOI] [PubMed] [Google Scholar]
- 45.McDonagh AF. Turning green to gold. Nat Struct Biol 8: 198–200, 2001. [DOI] [PubMed] [Google Scholar]
- 46.McDonagh AF, Assisi F. The ready isomerization of bilirubin IXα in aqueous solution. Biochem J 129: 797–800, 1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Meyerholz DK, Griffin MA, Castilow EM, Varga SM. Comparison of histochemical methods for murine eosinophil detection in an RSV vaccine-enhanced inflammation model. Toxicol Pathol 37: 249–255, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mobley JL, Ennis E, Shimizu Y. Differential activation-dependent regulation of integrin function in cultured human T-leukemic cell lines. Blood 83: 1039–1050, 1994. [PubMed] [Google Scholar]
- 49.Molla M, Gironella M, Miquel R, Tovar V, Engel P, Biete A, Pique JM, Panes J. Relative roles of ICAM-1 and VCAM-1 in the pathogenesis of experimental radiation-induced intestinal inflammation. Int J Radiat Oncol Biol Phys 57: 264–273, 2003. [DOI] [PubMed] [Google Scholar]
- 50.Nakajima H, Sano H, Nishimura T, Yoshida S, Iwamoto I. Role of vascular cell adhesion molecule 1/very late activated antigen 4 and intercellular adhesion molecule 1/lymphocyte function-associated antigen 1 interactions in antigen-induced eosinophil and T cell recruitment into the tissue. J Exp Med 179: 1145–1154, 1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Obermeier F, Kojouharoff G, Hans W, Schölmerich J, Gross V, Falk W. Interferon-gamma (IFN-γ)- and tumour necrosis factor (TNF)-induced nitric oxide as atoxic effector molecule in chronic dextran sulphate sodium (DSS)-induced colitis in mice. Clin Exp Immunol 116: 238–245, 1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Okayasu I, Hatakeyama S, Yamada M, Ohkusa T, Inagaki Y, Nakaya R. A novel method in the induction of reliable experimental acute and chronic ulcerative colitis in mice. Gastroenterology 98: 694–702, 1990. [DOI] [PubMed] [Google Scholar]
- 53.Qualls JE, Kaplan AM, van Rooijen N, Cohen DA. Suppression of experimental colitis by intestinal mononuclear phagocytes. J Leukoc Biol 80: 802–815, 2006. [DOI] [PubMed] [Google Scholar]
- 54.Quarmby S, Kumar P, Kumar S. Radiation-induced normal tissue injury: role of adhesion molecules in leukocyte-endothelial cell interactions. Int J Cancer 82: 385–395, 1999. [DOI] [PubMed] [Google Scholar]
- 55.Rachmilewitz D, Eliakim R, Ackerman Z, Karmeli F. Direct determination of colonic nitric oxide level: a sensitive marker of disease activity in ulcerative colitis. Am J Gastroenterol 93: 409–412, 1998. [DOI] [PubMed] [Google Scholar]
- 56.Rijcken E, Krieglstein C, Anthoni C, Laukoetter MG, Mennigen R, Spiegel HU, Senninger N, Bennett CF, Schuermann G. ICAM-1 and VCAM-1 antisense oligonucleotides attenuate in vivo leucocyte adherence and inflammation in rat inflammatory bowel disease. Gut 51: 529–535, 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Rolfe FG, Valentine JE, Sewell WA. Cyclosporin A and FK506 reduce interleukin-5 mRNA abundance by inhibiting gene transcription. Am J Respir Cell Mol Biol 17: 243–250, 1997. [DOI] [PubMed] [Google Scholar]
- 58.Rothenberg ME, Mishra A, Brandt EB, Hogan SP. Gastrointestinal eosinophils. Immunol Rev 179: 139–155, 2001. [DOI] [PubMed] [Google Scholar]
- 59.Salahuddin SZ, Ablashi DV, Markham PD, Josephs SF, Sturzenegger S, Kaplan M, Halligan G, Biberfeld P, Wong-Staal F, Kramarsky B, Gallo RC. Isolation of a new virus, HBLV, in patients with lymphoproliferative disorders. Science 234: 596–601, 1986. [DOI] [PubMed] [Google Scholar]
- 60.Sanborn WJ, Colombel JF, Enns R, Feagan BG, Hanauer SB, Lawrance IC, Panaccione R, Sanders M, Schreiber S, Targan S, van Deventer S, Goldblum R, Despain D, Hogge GS, Rutgeerts P. Natalizumab induction and maintenance therapy for Crohn's disease. N Engl J Med 353: 1912–1925, 2005. [DOI] [PubMed] [Google Scholar]
- 61.Sidel N, Abrams MI. Jaundice in arthritis: its analgesic action. N Engl J Med 210: 181–182, 1934. [Google Scholar]
- 62.Singer II, Kawaka DW, Scott S, Weidner JR, Mumford RA, Riehl TE, Stenson WF. Expression of inducible nitric oxide synthase and nitrotyrosine in colonic epithelium in inflammatory bowel disease. Gastroenterology 111: 871–885, 1996. [DOI] [PubMed] [Google Scholar]
- 63.Smith DLH, Keshavan P, Avissar U, Ahmed K, Zucker SD. Sodium taurocholate inhibits intestinal adenoma formation in APCMin/+ mice, potentially through activation of the farnesoid X receptor. Carcinogenesis 31: 1100–1109, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Soares MP, Seldon MP, Gregoire IP, Vassilevskaia T, Berberat PO, Yu J, Tsui TY, Bach FH. Heme oxygenase-1 modulates the expression of adhesion molecules associated with endothelial cell activation. J Immunol 172: 3553–3563, 2004. [DOI] [PubMed] [Google Scholar]
- 65.Soriano A, Salas A, Salas A, Sans M, Gironella M, Elena M, Anderson DC, Pique JM, Panes J. VCAM-1, but not ICAM-1 or MAdCAM-1, immunoblockade ameliorates DSS-induced colitis in mice. Lab Invest 80: 1541–1551, 2000. [DOI] [PubMed] [Google Scholar]
- 66.Stocker R, Yamamoto Y, McDonagh AF, Glazer AN, Ames BN. Bilirubin is an antioxidant of possible physiological importance. Science 235: 1043–1046, 1987. [DOI] [PubMed] [Google Scholar]
- 67.Tanaka T, Suzuki R, Kohno H, Sugie S, Takahashi M, Wakabayashi K. Colonic adenocarcinomas rapidly induced by the combined treatment with 2-amino-1-methyl-6-phenlyimidazo[4,5-b]pyridine and dextran sodium sulfate in male ICR mice possess β-catenin gene mutations and increases immunoreactivity for β-catenin, cyclooxygenase-2 and inducible nitric oxide synthase. Carcinogenesis 26: 229–238, 2005. [DOI] [PubMed] [Google Scholar]
- 68.Thompson HE, Wyatt BL. Experimentally induced jaundice (hyperbilirubinemia): report of animal experimentation and of the physiologic effect of jaundice in patients with atrophic arthritis. Arch Intern Med 61: 481–500, 1938. [Google Scholar]
- 69.Tokuyama H, Ueha S, Kurachi M, Matsushima K, Moriyasu F, Blumberg RS, Kakimi K. The simultaneous blockade of chemokine receptors CCR2, CCR5 and CXCR3 by a non-peptide chemokine receptor antagonist protects mice from dextran sodium sulfate-mediated colitis. Int Immunol 17: 1023–1034, 2005. [DOI] [PubMed] [Google Scholar]
- 70.Vitek L, Carey MC. Enterohepatic cycling of bilirubin as a cause of ‘black’ pigment gallstones in adult life. Eur J Clin Invest 33: 799–810, 2003. [DOI] [PubMed] [Google Scholar]
- 71.Vowinkel T, Anthoni C, Wood KC, Stokes KY, Russell J, Gray L, Bharwani S, Senninger N, Alexander JS, Krieglstein CF, Grisham MB, Granger DN. CD40-CD40 ligand mediates the recruitment of leukocytes and platelets in the inflamed murine colon. Gastroenterology 132: 955–965, 2007. [DOI] [PubMed] [Google Scholar]
- 72.Wang J, Springer TA. Structural specializations of immunoglobulin superfamily members for adhesion to integrins and viruses. Immunol Rev 163: 197–215, 1998. [DOI] [PubMed] [Google Scholar]
- 73.Wang J, Young IG. Eosinophilic inflammation: mechanisms regulating IL-5 transcription in human T lymphocytes. Allergy 62: 1131–1138, 2007. [DOI] [PubMed] [Google Scholar]
- 74.Wang WW, Smith DLH, Zucker SD. Bilirubin inhibits iNOS expression and nitric oxide production in response to endotoxin. Hepatology 40: 424–433, 2004. [DOI] [PubMed] [Google Scholar]
- 75.Wedemeyer J, Vosskuhl K. Role of gastrointestinal eosinophils in inflammatory bowel disease and intestinal tumours. Best Pract Res Clin Gastroenterol 22: 537–549, 2008. [DOI] [PubMed] [Google Scholar]
- 76.Weg VB, Williams TJ, Lobb RR, Nourshargh S. A monoclonal antibody recognizing very late activation antigen-4 inhibits eosinophil accumulation in vivo. J Exp Med 177: 561–566, 1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.William C, Schindler R, Frei U, Eckardt KU. Increases in oxygen tension stimulate expression of ICAM-1 and VCAM-1 on human endothelial cells. Am J Physiol Heart Circ Physiol 276: H2044–H2052, 1999. [DOI] [PubMed] [Google Scholar]
- 78.Yoshida Y, Iwai A, Itoh K, Tanaka M, Kato S, Hokari R, Miyahara T, Koyama H, Miura S, Kobayashi M. Role of inducible nitric oxide synthase in dextran sulphate sodium-induced colitis. Aliment Pharmacol Ther 14: 26–32, 2000. [DOI] [PubMed] [Google Scholar]
- 79.Yue G, Lai PS, Yin K, Sun FF, Nagele RG, Liu X, Linask KK, Wang C, Lin KT, Wong PYK. Colon epithelial cell death in 2,4,6-trinitrobenzenesulfonic acid-induced colitis is associated with increased inducible nitric-oxide synthase expression and peroxynitrite production. J Pharmacol Exp Ther 297: 915–925, 2001. [PubMed] [Google Scholar]