

Abstract
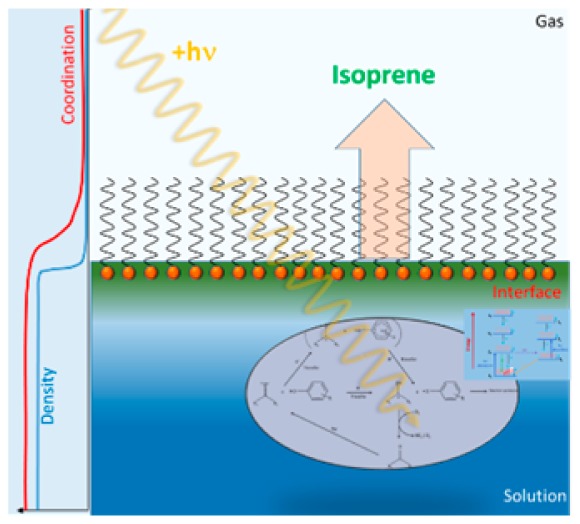
Isoprene is an important reactive gas that is produced mainly in terrestrial ecosystems but is also produced in marine ecosystems. In the marine environment, isoprene is produced in the seawater by various biological processes. Here, we show that photosensitized reactions involving the sea-surface microlayer lead to the production of significant amounts of isoprene. It is suggested that H-abstraction processes are initiated by photochemically excited dissolved organic matter which will the degrade fatty acids acting as surfactants. This chemical interfacial processing may represent a significant abiotic source of isoprene in the marine boundary layer.
Introduction
Aerosols and low-altitude reflective clouds in the marine boundary layer have been shown to impact the climate system as a whole,1,2 triggering substantial research on the origin and chemical composition of marine aerosols. While primary sources originate from bubble bursting (producing sea-salt), secondary sources are believed to derive from the gas-to-particle conversion of phytoplankton-emitted trace gases such as dimethylsulfide (DMS) and various organic precursors such as isoprene and other terpenes.3−10 While global modeling tends to identify a possible missing source of organic aerosol in the troposphere,11−14 the nature of the chemical compounds and their source fluxes remain poorly characterized.
Since the initial identification of marine emissions of isoprene,15,16 its production rates have been determined for algaes,11,17−21 macroalgae,22 and various microbial communities,23 with light-sensitivity of isoprene production from phytoplankton observed resulting in a midday maximum.24 Field observations have shown isoprene levels up to several hundreds of pptv,25 which could be high enough to affect marine pristine regions. Vaattovaara et al.26 found that under coastal nucleation events, 11–47% of the mass fraction could be attributed to an organic fraction produced by secondary organic aerosols (SOA) formation processes. These authors suggested that isoprene was a probable precursor to SOA. Despite this potential importance, large discrepancies still exists between emission flux measurements, so-called “bottom-up” methods11,27 and the “top-down” approach which tries to combine in situ observations and simulations14,27,28 with fluxes ranging from ∼0.1 to 1.9 Tg C yr–1 and ∼11.6 Tg C yr–1, respectively.11,18,29,30 There is therefore a clear need of identifying possible sources of isoprene, including abiotic processes.
It has been shown that various alkenes can be photochemically produced, possibly via the degradation of the dissolved organic matter, in the upper region of the oceanic mixed layer.31 To date, only little or even no attention has been devoted to atmospherically relevant photochemistry taking place at the air–water interface.32 The sea-surface microlayer (SML), defined as the uppermost tens to hundreds of micrometers of the surface of the ocean, covers more than 70% of the Earth’s surface.33 The SML is known to concentrate organic matter (i.e., dissolved organic matter including UV absorbing humic substances, amino acids, proteins, lipids, phenolic compounds) and surfactants (i.e., fatty acids).33 This interface, having different chemical, physical and biological properties compared to subsurface waters, plays a significant role in biogeochemical processes on a global scale (i.e., air–sea gas exchange, trace gas deposition to the ocean, and secondary organic aerosol formation).33,34
It is only very recently, that the interfacial photoproduction of a series of unsaturated volatile organic compounds (VOCs) has been reported as arising from the photosensitized degradation of organic surfactants.35,36 The main produced VOCs were corresponding to the oxidation products of the surfactant, but in addition a series of functionalized and unsaturated compounds was also observed. A mechanism initiated by the H-abstraction of the surfactant (nonanoic acid) in the presence of exited photosensitizer (humic acid) was proposed. We present here new experimental evidence that such photosensitized reactions at the air–sea interface can also led to the abiotic production of isoprene.
Experimental Section
Photochemical Reactor
Gas phase products produced upon illumination of various SML samples were investigated in a quartz cell (2 cm diameter and 5 cm length) filled with 7 mL of solutions (see below), creating a 5 cm2 air–water interface irradiated by means of a 150W, water filtered, xenon lamp positioned 10 cm away so that the photon flux entering the cell was almost mimicking solar irradiation (for more details, see Ciuraru et al.35). This cell was continuously purged with 200 sccm of purified air, entraining the gas phase products to various analytical tools described below. This simple approach35 enabled the reproduction of the air–sea exchange on quiescent water covered by an (synthetic or authentic) organic film, mimicking low wind conditions.
Blank experiments were routinely performed on 18 MΩ deionized water and solutions containing solely the surfactant or the humic acids to assess background signals when irradiating. The resulting data are presented in the Supporting Information.
Analytical Methods
The gas-phase products were analyzed using a commercial SRI-PTR-ToF-MS 8000 (Selective Reagent Ionization Proton Transfer Reaction Time of Flight Mass Spectrometer) instrument from Ionicon Analytik GmbH (Innsbruck, Austria) in its standard configuration (V mode)37 with H3O+ and NO+ as reagent ion. The PTR-MS sampled continuously 50 sccm from the emissions stream through 1.5 m of 6 mm i.d. peek tubing. No particle filter was placed at the inlet to avoid artifact from possible organics deposition on the filter. Internal calibration of the ToF data and peak extraction were performed according to the procedure described in detail by Lindinger et al.37 PTR-ToF-MS spectra were collected at a time resolution of 8s. Measurements using H3O+ ionization were performed using a drift voltage of 600 V, drift temperature of 60 °C and a drift pressure of 2.25 mbar resulting in an E/N of about 130 Td (1 Td = 10–17 cm2 V–1). The ionization conditions in the NO+ mode were maintained at a drift voltage of 600 V and a drift pressure of 2.25 mbar. The instrument was operated at an E/N value of 132 Td. The VOCs concentrations are expressed in ppbv and they have been calculated according to the formula described in Cappellin et al.38 We have used the same value of k = 2 × 10–9 cm3/s for all masses excepting isoprene, which has a value of k = 1.94 × 10–9 cm3/s.39 Uncertainties in our data may arise from systematic errors in the concentration determination because the accuracy for compounds concentration has been estimated using calculated values for the collision rate constant which should equal the reaction rate constant within ±30%.
One of the main weaknesses of PTR-ToF-MS is that the VOC identification relies solely on mass spectrometry; therefore, comparison of PTR-ToF-MS measurements to those by means of a GC-MS technique was used for accurate identification of individual VOCs. This comparison was only used to validate the chemical speciation made with the PTR-ToF-MS. The GC-MS data were not used to extract yield or fluxes information. An advantage of the PTR-ToF-MS used in this study relies on the possibility of the simultaneous use of different ionization modes (H3O+ and NO+) in order to eliminate or reduce possible interferences and therefore obtain a more accurate compound identification.
Analysis of VOCs were also performed by Automatic Thermal Desorption (ATD Markes Thermal Desorber Unity with an Markes Autosampler Ultra 2) coupled with GC/MS, using an Agilent 6890 GC (DB-VRX column of 60 m × 0.25 mm × 1.4 μm, J&W, 122–1564) and an Agilent MSD 5973 N MS with electron impact ionization mode. A Tenax sorption tube of 200 mg (L = 3 1/2 in., o.d. = 1/4 in.) has been used.
The surface tension was measured using a Krüss tensiometer K6 (measuring range between 0 and 90 mN/m). For the measured surface tension, the errors are estimated at 10%.
Chemicals
The proxy organic layer for the experiments within the present study was either nonanoic acid (a simple proxy for fatty acids) as an organic surfactant or authentic SML samples from Bergen (Norway). The bulk solutions contained dissolved organic matter in the form of humic acids as environmental photosensitizer present in dissolved organic matter (DOM).40
To our knowledge, there is no direct measurements of nonanoic acid concentration in SML. This acid is a product of the oxidation of oleic acid; elevated concentrations of C9 acid being attributed to the ozonolysis of the latter which is enriched in the SML.41 King et al.42 reported a high yield of nonanoic acid (87%), which remained at the surface of the water after being produced by the reaction of ozone with oleic acid. Some other measurements showed 4.9 ng m–3 of nonanoic acid in fine particulate organic acids in marine areas.41
For the experiments with artificial SML, a salt solution composed of 1 mol L–1 sodium chloride (99+%, Sigma-Aldrich), 1 mmol L–1 sodium bromide (99+%, Sigma-Aldrich), and 10–3 mmol L–1 sodium iodide (99+%, Sigma-Aldrich), with additional concentrations of humic acids (Fluka), was prepared by dissolving known amounts of the products in 18 MΩ deionized water. The surfactant monolayer was prepared by adding a known concentration of nonanoic acid in these solutions. The pH of the solution was raised to approximately 8 using a sodium hydroxide solution; experiments were performed using solutions to which base was not added, these having a pH in the range of 4–5. The nonanoic acid was purchased at Alfa Aesar (97%).
In some experiments, nonanoic acid was previously purified by bubbling ozone before irradiation. In this case, the PTR-ToF-MS spectra showed no differences compared to those acquired without purification. All the experiments presented here represent the results without purification.
The authentic SML samples were collected by means of the glass plate technique, out of the coast in the Raune fjord and are therefore “coastal samples”, which can be naturally more continentally influenced compared to samples collected in the open ocean. Sampling was performed on a sunny day under calm conditions (low wind speed of 4 m s–1) at 1:00 p.m. UTC (Coordinated Universal Time), and the collected SML film was around 200 μm thick. Salinity and dissolved organic carbon (DOC) and particulate organic carbon (POC) content of the SML sample are reported elsewhere.35 Nonfiltered samples were used in this study. We did not add any reagents such as Ag or Hg to terminate biological activity.
Quantification of Fluxes
Fluxes (molec mW–1 s–1) were calculated according to
| 1 |
where C is the isoprene concentration (molecules cm–3), Q is the flow rate of air into the cell (cm3 s–1), S is the surface area of the solution (cm2), and L is the light intensity (mW cm–2). Assuming, that only the UV-A fraction of the Xe lamp output triggers the observed photochemistry, the calculated flux was 8 mW cm–2 for the wavelength below 400 nm.
Results
Figure 1 shows that when nonanoic acid is used as a synthetic proxy of the SML, isoprene is readily produced after switching on the light. That is the outcome of a typical experiment. The time scale required to produce it is very short as the residence time of the carrier gas in the glass reactor is in the order of a few seconds depending on the gas flow. It can be seen that a steady-state concentration is reached very rapidly and maintained through the irradiation period. In contrast, no isoprene formation was observed using only water, humic acid, or the surfactant (Figures S1 and S2). In addition to the formation of isoprene, methacrolein, and methyl vinyl ketone are also detected and separated by PTR-ToF-MS in NO+ ionization mode.43 These compounds are isoprene first-generation oxidation products.
Figure 1.

Typical irradiation experiment of a solution containing salt water, humic acid (30 mg L–1) and nonanoic acid (1 mM) showing the formation of isoprene (red line) and sum of methyl vinyl ketone and methacrolein (MVK+MACR, blue line) measured by PTR-ToF-MS with H3O+ as reagent ion.
It is well described in the literature that the chromophores contained within the humic acid substances of dissolved organic matter will be excited.44 Subsequently, they will initiate the degradation of an organic surfactant at the air–water interface. Such processes are well documented44 for bulk water and are known to initiate a series of potential oxidative pathways such H-abstraction, charge transfer reactions, hydrated electrons, HOx or singlet oxygen production.45,46 All these processes have the potential to chemically degrade aqueous organic compounds leading to formation of different classes of molecules (i.e., unsaturated compound, alcohols, aldehydes, or ketones), their structure depending on their parent molecule.35
In addition to these classes of products, in the experiments performed here, we also observed the photoinduced formation of dienes (Figure 1), including isoprene at m/z 69.069 (detected in H3O+ ionization mode) and 68.062 (in NO+ mode).43 These chemical attributions were verified by two techniques (i.e., ATD-GC-MS measurements and PTR-ToF-MS in different ionization gases (H3O+ and NO+)).
In H3O+ mode, the proton transfer reaction of MACR and MVK with H3O+ giving rise dominantly to a single ion, thus only the sum of their mixing ratios can be reported.47 Gas chemistry in our experiments is expected to be unimportant due to the absence of added oxidant and to the short residence time preventing, for instance, the building-up of ozone. Therefore, these compounds may not only arise for the air–water interface chemistry but also, depending on the ionization mode (especially for H3O+), from secondary processes in the ion cell of the PTR-MS. Table 1 also lists the emission fluxes of these compounds assuming that their photochemical production do not interfere with ionization processes in the mass spectrometer.
Table 1. Marine Isoprene Seawater Flux Measurements and Estimates Taken from Shaw et al.24 Compared to our Estimated Values (Laboratory Maximal and Minimal Fluxes and from the Authentic Sample)a.
| location | dates | measured isoprene flux (24) (molecules cm–2 s–1) × 109 |
|---|---|---|
| North Sea | July 1993–July 1994 | 0.017 |
| Northeast Pacific | July 2002 | 0.02 |
| Raunefjord, Southern Norway | May–June 2005 | 0.1 |
| Mace Head, Ireland | Sep–Oct 1998 | 0.68 |
| Coastal Crete | Feb–Oct 2004 | 6 |
| this study (molecules cm–2 s–1) × 109 | ||||||
|---|---|---|---|---|---|---|
| isoprene | MACR | MVK | ||||
| lab conditions | EF 2 | EF 1.5 | EF 1 | |||
| max | 230 | 23 | 17.3 | 11.5 | 23 | 37 |
| min | 7 | 0.14 | 0.1 | 0.07 | 12 | 4.7 |
| SML | 17 | 1.7 | 1.3 | 0.8 | 2.1 | 1.1 |
The values from this study have been multiplied by the mean solar flux (21 mW cm–2)56 estimated for latitude and time of the year. The laboratory conditions refer to the measured flux with a saturated surface and high enrichment factors (EF), while the column at different EFs present the data scaled to observed marine conditions (see text).
Similar observations were also made with authentic SML samples taken in Bergen. In these samples, 30 mg L–1 humic acid was added to mimic DOM. When irradiating the samples, the results reveal a close matching with the artificial samples (Figure S4).
Using the authentic SML samples, a formation of 17 × 109 molecules cm–2 s–1 of isoprene is observed during irradiation with λ > 300 nm, which represents the same order of magnitude as for isoprene production by phytoplankton.24 A production of methacrolein and methyl vinyl ketone was also observed during irradiation.
To obtain further insights into the underlying processes, we measured the yield of isoprene on synthetic samples as a function of the nonanoic and humic acids concentrations. The actual isoprene yield was observed to linearly depend on the humic acid concentration (Figure S3) but nonlinearly with the nonanoic acid concentration. The surface concentration of the later is related to the surface tension of the corresponding acid/water mix.35 A strong correlation between the measured surface tension and the isoprene concentration was clearly evidenced (Figure 2). In fact, both the yield of isoprene and the surface tension are leveling-off at surface tension lower than 39 mN m–1, corresponding to surfactant concentrations larger than ca. 1.5 mM, and clearly the isoprene yield is not proportional to the nonanoic acid bulk concentration. Furthermore, in the absence of a surfactant film, isoprene production was never observed. These results represent direct evidence of photochemical production of isoprene by interfacial chemistry (i.e., the air–aqueous interface tends to concentrate the organic molecules at the surface, enhancing chemical pathways otherwise too slow to occur).
Figure 2.

Evolution of gaseous isoprene concentration as a function of the nonanoic acid surface tension.
Some experiments have been performed with longer irradiation times (about 12 h). In these experiments, the isoprene signal was stable during the entire irradiation time. This means that the surfactant replenishes itself, just by diffusion from the bulk water to the surface, during the irradiation time. At this point, a loss of surfactant over time cannot be calculated because the nonanoic acid concentration is constant over time and we are only measuring the emitted VOCs.
We varied the carbon chain length of the surfactant to help understand if the length will contribute to the isoprene formation. The experimental results, performed with pentanoic and hexanoic acid as surfactants, showed no photochemical isoprene formation, indicating that gaseous isoprene is emitted from organic monolayers containing at least seven atoms of carbon. Because pentanoic and hexanoic acids are more water-soluble than nonanoic acid, the equilibrium will be then displaced toward the bulk, leading to a lower concentration of the acid at the surface than in the bulk. It has been shown that if the carbon number of the hydrocarbon tail of an acid is greater than eight, it will be immiscible in water and act as a surfactant, as it is the case of nonanoic acid.48 Pentanoic acid is then unable to form a monolayer on the water surface under our experimental conditions. These results highlight again the importance of the surfactant for this specific chemistry at the air–water interface.
Experiments as a function of pH have also been performed using nonanoic acid as surfactant (pKa = 4.9) within a pH range of 4 to 12. The pH of the solution was raised to approximately 8 using a sodium hydroxide solution; experiments were performed using solutions to which base was not added, these having a pH in the range of 4–5. When the pH of the solution was increased to 12, no isoprene was identified in the PTR MS spectra. This can be explained by the ability of an alkaline solution to dissolve completely nonanoic acid and depleting the organic surfactant (i.e., corresponding to a reduced surface concentration which directly reduces the emitted VOCs). Figure 3 illustrates the flux of isoprene as a function of the pH of the solution.
Figure 3.

Evolution of gaseous isoprene flux as a function of the pH of the solution.
A key feature of interfacial reactions is that they favor protonation and self-reactions (due to higher concentrations), which will lead to the formation of dimers as already suggested by Griffith et al.49 (Scheme 1).
Scheme 1. Suggested Reaction Mechanisms Leading to Nonanoic Acid Dimer.

The isoprene production may be explained by the formation of these dimers followed by subsequent oxidation via H-transfer and unimolecular decay of the peroxy intermediates. Further hydrogen abstraction reactions result in shorter alcohols, acids, aldehydes, or ketones (due to the variety of highly oxygenated and substituted products obtained in the various oxidations steps) which can in turn lead to the formation of unsaturated products as identified in the PTR-ToF-MS spectra in our previous study.35
Due to the high concentration of nonanoic acid at the interface, we expect a direct abstraction of an H atom at the alkyl chain by OH radicals to form an α-carboxyalkyl radical. The H-abstraction followed by oxygen addition will lead to a hydroxy acid. Primary photochemical reactions of an excited carbonyl group (as hydrogen-atom abstraction) will produce radical-pair or biradical intermediates.50 The reaction of these excited triplet ketones with the hydroxy acid can lead, after decarboxylation, to the formation of a ramified unsaturated diol. The hydoxyalkyl-hydroxy-elimination from the diol formed will lead to isoprene and smaller ketones. The reactions of diols in which at least one OH group is tertiary, can be cleaved in an acid-catalyzed environment.51 This reaction is then privileged by the surfactant excess at the interface and its favorable H-abstraction and decarboxylation51 (Scheme 2,a). Another plausible pathway for the isoprene formation is represented by the reaction of the α-carboxyalkyl radical (Scheme 2) with the excited triplet ketone. After decarboxylation and H-abstraction of this radical, the O2 addition will form a peroxyl radical. The recombination of two RO2 radicals will lead to an alcohol and a carbonyl formation. The alcohol can be afterward converted into isoprene by water elimination and fragmentation (Scheme 2, b).
Scheme 2. Suggested Reaction Mechanisms Leading to the Photosensitized Production of Isoprene at the Air–Sea Interface.

Discussion
Hydrophobic surfactant films are typically believed to play the role of a physical barrier to air–sea exchanges, especially at low wind speed.52 This inhibiting effect targets only gases present the underlying bulk water. However, and as shown here, if chemistry is taking place in the SML, then the associated chemical products may still be released into the gas phase. In other words, while surfactant films may isolate the bulk seawater from the gas phase, the composition of the latter will be mostly affected by products arising from interfacial processes, corresponding to the degradation products of the surface layer.
In fact, our results suggest that when H-abstraction reactions are predominately occurring in an organic monolayer at the air–water interface (i.e., on a fully covered surface), unsaturated gaseous compounds, such as isoprene, can be emitted. Wurl53 et al. studied the surfactants concentration in the SML in different regions of the ocean (subtropical, temperate, polar). They suggested that the ocean surface microlayer is enriched with surfactants to a much larger extent than previously recognized and can be assumed as fully covered by an organic layer, mostly during fall. It has been also shown that the surfactant enrichments persisted at wind speeds of up to 10 ms–1, without any observed depletion above 5 ms–1.53Table 1 compares favorably the isoprene flux derived from our studies, at 1 and 5 mM surfactant concentrations, to those of Shaw et al.24 (real marine fluxes), so far assigned to phytoplankton and macroalgae,24 for a solar flux of 21 mW cm–2, estimated for a solar zenith angle of 40° corresponding to fall conditions54 (as those encountered during the sampling of the SML samples). Considering such typical actinic fluxes, the chemical (photosensitized) isoprene production under the present conditions is ranging from 7 × 109 to 23 × 1010 molecules cm–2 s–1, which are at the upper limit or much larger than reported in situ measurements (Table 1). It must be emphasized that these data arise from a laboratory study that certainly tends to maximize such fluxes by isolating these processes or by using slightly broader wavelength regions as compared to the actinic fluxes, or by environmentally too large enrichment factors (EF). In fact, the maximum achievable surface concentration by a C9 carboxylic acid is ca. 0.01 mol m–2;55 assuming that this evenly distributed throughout a 100 μm thick water layer, matching operationally defined SML thickness, one can calculate enrichment factors of 20 or even 100 for 5 and 1 mM nonanoic acid solutions respectively, which is much larger than typically encountered values. While the laboratory based values are providing unrealistic high fluxes, scaling to marine conditions (i.e., EF = 1, 1.5 or 2, matching for instance the values reported by Wurl et al.53), realistic fluxes ranging from 0.01 to 23 × 109 molecules cm–2 s–1 are derived (see Table 1), which are in the order of the observed emission fluxes.
However, as pointed out by Wurl et al.,53 surfactant production by nonchlorophyll-containing organisms (i.e., bacteria and zooplankton) may become more important in oligotrophic conditions, leading to a much larger extent of surfactant as compared to chlorophyll-based estimates. In turn, this would mean that photochemical production of isoprene discussed above could take place on a wider extent than those estimates based solely on biological activity, estimated again on chlorophyll levels.29 This may help reducing the gap between “top-down” and “bottom-up” estimations of the isoprene fluxes. Finally, while this study is focusing on photochemistry at the air–sea interface, one could also expect that particles produced by bubble bursting would carry some features similar to the air–sea interface allowing such photochemical process to take place also on the surface of aerosols and therefore at various altitudes, which could be a source not captured by bottom-up approaches. If we roughly estimate that around 30% of the ocean surface (assumed to be 3.8 × 108 km2) is enriched by surfactant, depending on season,53 and that 50% of the time the surface of the ocean is illuminated, then we can estimate that the photochemical flux of isoprene ranges from 0.2 to 35 Tg yr–1, adding to reported biological sources. Arnold et al.,27 combining satellite maps of the global distribution of phytoplankton functional type and new measurements of phytoplankton-specific isoprene productivities, found a mean “bottom-up” oceanic isoprene emission of 0.31 ± 0.08 (1σ)Tgyr–1, while modeling produced a “top-down” source estimate of 1.9 Tg yr–1. Therefore, such differences could be partially explain by the suggested abiotic source of isoprene.
This study clearly points toward a global impact of the interfacial chemistry at the air–sea interface. The existence of organic films on the ocean surface due to biological activities influences therefore air–sea exchanges in an unexpected significant manner, as interfacial photosensitized chemistry may represent a significant source of isoprene in the absence of any biological sources in the marine boundary layer. This interfacial chemistry involves only fatty acids as surfactants and dissolved organic matter as photosentitizers, and both are ubiquitous in the marine environment.
Acknowledgments
The research leading to these results has received funding from the European Research Council under the European Union’s Seventh Framework Programme (FP/2007-2013)/ERC Grant Agreement 290852 - AIRSEA. Helpful discussions with O. Piva and M. Medebielle are gratefully acknowledged. The authors thank C. Ferronato for the access to the ATD-GC/MS instrumentation. We also thank the anonymous reviewers for their thorough and helpful comments. R.C. thanks S. Feil for support and helpful discussions.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.est.5b02388.
Information about the blank experiments routinely performed during this study. (PDF)
Author Present Address
∥ University of Bordeaux, EPOC UMR 5805, F-33405 Talence cedex, France; CNRS, EPOC UMR 5805, F-33405 Talence cedex, France.
Author Contributions
C.G. conceived the idea, designed the experiments, and led the analysis. R.C. performed the experiments and analyzed the data. L.F. performed the ATD-GCMS measurements. M.P. and H.H. sampled and analyzed the authentic sea surface microlayer. B.D. provided expertise. C.G. and R.C. wrote the paper. All authors contributed comments on the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Randall D. A.; Coakley J. A.; Lenschow D. H.; Fairall C. W.; Kropfli R. A. Outlook for Research on Subtropical Marine Stratification Clouds. Bull. Am. Meteorol. Soc. 1984, 65121290–1301 10.1175/1520-0477(1984)065<1290:OFROSM>2.0.CO;2. [DOI] [Google Scholar]
- Stevens B.; Vali G.; Comstock K.; Wood R.; Van Zanten M. C.; Austin P. H.; Bretherton C. S.; Lenschow D. H. Pockets of open cells and drizzle in marine stratocumulus. Bull. Am. Meteorol. Soc. 2005, 86151–57 10.1175/BAMS-86-1-51. [DOI] [Google Scholar]
- Blanchard D. C. Sea-to-Air Transport of Surface Active Material. Science 1964, 1463642396–397 10.1126/science.146.3642.396. [DOI] [PubMed] [Google Scholar]
- Hoffman E. J.; Duce R. A. Factors influencing the organic carbon content of marine aerosols: A laboratory study. J. Geophys. Res. 1976, 81213667–3670 10.1029/JC081i021p03667. [DOI] [Google Scholar]
- Charlson R. J.; Lovelock J. E.; Andreae M. O.; Warren S. G. Oceanic phytoplankton, atmospheric sulphur, cloud albedo and climate. Nature 1987, 3266114655–661 10.1038/326655a0. [DOI] [Google Scholar]
- O’Dowd C. D.; de Leeuw G. Marine aerosol production: a review of the current knowledge. Philos. Trans. R. Soc., A 2007, 36518561753–1774 10.1098/rsta.2007.2043. [DOI] [PubMed] [Google Scholar]
- de Leeuw G.; Andreas E. L.; Anguelova M. D.; Fairall C. W.; Lewis E. R.; O’Dowd C.; Schulz M.; Schwartz S. E.. Production flux of sea spray aerosol. Rev. Geophys. 2011, 49 (2). 10.1029/2010RG000349 [DOI] [Google Scholar]
- Carpenter L. J.; Archer S. D.; Beale R. Ocean-atmosphere trace gas exchange. Chem. Soc. Rev. 2012, 41196473–6506 10.1039/c2cs35121h. [DOI] [PubMed] [Google Scholar]
- Meskhidze N.; Nenes A. Phytoplankton and Cloudiness in the Southern Ocean. Science 2006, 31458041419–1423 10.1126/science.1131779. [DOI] [PubMed] [Google Scholar]
- Meskhidze N.; Petters M. D.; Tsigaridis K.; Bates T.; O’Dowd C.; Reid J.; Lewis E. R.; Gantt B.; Anguelova M. D.; Bhave P. V.; et al. Production mechanisms, number concentration, size distribution, chemical composition, and optical properties of sea spray aerosols. Atmos. Sci. Lett. 2013, 144207–213 10.1002/asl2.441. [DOI] [Google Scholar]
- Gantt B.; Meskhidze N.; Kamykowski D. A new physically-based quantification of marine isoprene and primary organic aerosol emissions. Atmos. Chem. Phys. 2009, 9144915–4927 10.5194/acp-9-4915-2009. [DOI] [Google Scholar]
- Heald C. L.; Jacob D. J.; Park R. J.; Russell L. M.; Huebert B. J.; Seinfeld J. H.; Liao H.; Weber R. J. A large organic aerosol source in the free troposphere missing from current models. Geophys. Res. Lett. 2005, 3218L18809. 10.1029/2005GL023831. [DOI] [Google Scholar]
- Roelofs G. J. A GCM study of organic matter in marine aerosol and its potential contribution to cloud drop activation. Atmos. Chem. Phys. 2008, 83709–719 10.5194/acp-8-709-2008. [DOI] [Google Scholar]
- Spracklen D. V.; Arnold S. R.; Sciare J.; Carslaw K. S.; Pio C.. Globally significant oceanic source of organic carbon aerosol. Geophys. Res. Lett. 2008, 35 (12). 10.1029/2008GL033359 [DOI] [Google Scholar]
- Bonsang B.; Polle C.; Lambert G. Evidence for marine production of isoprene. Geophys. Res. Lett. 1992, 19111129–1132 10.1029/92GL00083. [DOI] [Google Scholar]
- Broadgate W. J.; Liss P. S.; Penkett S. A. Seasonal emissions of isoprene and other reactive hydrocarbon gases from the ocean. Geophys. Res. Lett. 1997, 24212675–2678 10.1029/97GL02736. [DOI] [Google Scholar]
- Moore R. M.; Oram D. E.; Penkett S. A. Production of isoprene by marine phytoplankton cultures. Geophys. Res. Lett. 1994, 21232507–2510 10.1029/94GL02363. [DOI] [Google Scholar]
- Milne P. J.; Riemer D. D.; Zika R. G.; Brand L. E. Measurement of vertical distribution of isoprene in surface seawater, its chemical fate, and its emission from several phytoplankton monocultures. Mar. Chem. 1995, 483–4237–244 10.1016/0304-4203(94)00059-M. [DOI] [Google Scholar]
- McKay W. A.; Turner M. F.; Jones B. M. R.; Halliwell C. M. Emissions of hydrocarbons from marine phytoplankton—Some results from controlled laboratory experiments. Atmos. Environ. 1996, 30142583–2593 10.1016/1352-2310(95)00433-5. [DOI] [Google Scholar]
- Bonsang B.; Gros V.; Peeken I.; Yassaa N.; Bluhm K.; Zoellner E.; Sarda-Esteve R.; Williams J. Isoprene emission from phytoplankton monocultures: the relationship with chlorophyll-a, cell volume and carbon content. Envir. Chem. 2010, 76554. 10.1071/EN09156. [DOI] [Google Scholar]
- Exton D. A.; Suggett D. J.; McGenity T. J.; Steinke M. Chlorophyll-normalized isoprene production in laboratory cultures of marine microalgae and implications for global models. Limnol. Oceanogr. 2013, 5841301–1311 10.4319/lo.2013.58.4.1301. [DOI] [Google Scholar]
- Broadgate W. J.; Malin G.; Küpper F. C.; Thompson A.; Liss P. S. Isoprene and other non-methane hydrocarbons from seaweeds: a source of reactive hydrocarbons to the atmosphere. Mar. Chem. 2004, 881–261–73 10.1016/j.marchem.2004.03.002. [DOI] [Google Scholar]
- Alvarez L. A.; Exton D. A.; Timmis K. N.; Suggett D. J.; McGenity T. J. Characterization of marine isoprene-degrading communities. Environ. Microbiol. 2009, 11123280–3291 10.1111/j.1462-2920.2009.02069.x. [DOI] [PubMed] [Google Scholar]
- Shaw S. L.; Gantt B.; Meskhidze N. Production and Emissions of Marine Isoprene and Monoterpenes: A Review. Adv. Meteorol. 2010, 2010, 1–24 10.1155/2010/408696. [DOI] [Google Scholar]
- Yassaa N.; Peeken I.; Zöllner E.; Bluhm K.; Arnold S.; Spracklen D.; Williams J. Evidence for marine production of monoterpenes. Environ. Chem. 2008, 56391–401 10.1071/EN08047. [DOI] [Google Scholar]
- Vaattovaara P.; Huttunen P. E.; Yoon Y. J.; Joutsensaari J.; Lehtinen K. E. J.; O’Dowd C. D.; Laaksonen A. The composition of nucleation and Aitken modes particles during coastal nucleation events: evidence for marine secondary organic contribution. Atmos. Chem. Phys. 2006, 6124601–4616 10.5194/acp-6-4601-2006. [DOI] [Google Scholar]
- Arnold S. R.; Spracklen D. V.; Williams J.; Yassaa N.; Sciare J.; Bonsang B.; Gros V.; Peeken I.; Lewis A. C.; Alvain S.; et al. Evaluation of the global oceanic isoprene source and its impacts on marine organic carbon aerosol. Atmos. Chem. Phys. 2009, 941253–1262 10.5194/acp-9-1253-2009. [DOI] [Google Scholar]
- Heald C. L.; Jacob D. J.; Park R. J.; Alexander B.; Fairlie T. D.; Yantosca R. M.; Chu D. A.. Transpacific transport of Asian anthropogenic aerosols and its impact on surface air quality in the United States. J. Geophys. Res. 2006, 111 (D14). 10.1029/2005JD006847 [DOI] [Google Scholar]
- Palmer P. I.; Shaw S. L. Quantifying global marine isoprene fluxes using MODIS chlorophyll observations. Geophys. Res. Lett. 2005, 329L09805. 10.1029/2005GL022592. [DOI] [Google Scholar]
- Luo G.; Yu F. A numerical evaluation of global oceanic emissions of α-isoprene. Atmos. Chem. Phys. 2010, 1042007–2015 10.5194/acp-10-2007-2010. [DOI] [Google Scholar]
- Riemer D. D.; Milne P. J.; Zika R. G.; Pos W. H. Photoproduction of nonmethane hydrocarbons (NMHCs) in seawater. Mar. Chem. 2000, 713–4177–198 10.1016/S0304-4203(00)00048-7. [DOI] [Google Scholar]
- Liss P. S.; Liss P. S.; Duce R. A.. The Sea Surface and Global Change; Cambridge University Press: Cambridge, 2005. [Google Scholar]
- Liss P. S.; Duce R. A.. The Sea Surface and Global Change; Cambridge University Press: Cambridge, 1997. [Google Scholar]
- Donaldson D. J.; George C. Sea-Surface Chemistry and Its Impact on the Marine Boundary Layer. Environ. Sci. Technol. 2012, 461910385–10389 10.1021/es301651m. [DOI] [PubMed] [Google Scholar]
- Ciuraru R.; Fine L.; van Pinxteren M.; D’Anna B.; Herrmann H.; George C. Photosensitized production of functionalized and unsaturated organic compounds at the air–sea interface. Sci. Rep. 2015, 5, 12741. 10.1038/srep12741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu H.; Ciuraru R.; Dupart Y.; Passananti M.; Tinel L.; Rossignol S.; Perrier S.; Donaldson D. J.; Chen J.; George C. Photosensitized Production of Atmospherically Reactive Organic Compounds at the Air/Aqueous Interface. J. Am. Chem. Soc. 2015, 137268348–8351 10.1021/jacs.5b04051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindinger W.; Hansel A.; Jordan A. On-line monitoring of volatile organic compounds at pptv levels by means of proton-transfer-reaction mass spectrometry (PTR-MS) medical applications, food control and environmental research. Int. J. Mass Spectrom. Ion Processes 1998, 1733191–241 10.1016/S0168-1176(97)00281-4. [DOI] [Google Scholar]
- Cappellin L.; Karl T.; Probst M.; Ismailova O.; Winkler P. M.; Soukoulis C.; Aprea E.; Märk T. D.; Gasperi F.; Biasioli F. On Quantitative Determination of Volatile Organic Compound Concentrations Using Proton Transfer Reaction Time-of-Flight Mass Spectrometry. Environ. Sci. Technol. 2012, 4642283–2290 10.1021/es203985t. [DOI] [PubMed] [Google Scholar]
- Zhao J.; Zhang R. Proton transfer reaction rate constants between hydronium ion (H3O+) and volatile organic compounds. Atmos. Environ. 2004, 38142177–2185 10.1016/j.atmosenv.2004.01.019. [DOI] [Google Scholar]
- Clark C. D.; Zika R. G.. Marine Organic Photochemistry: From the Sea Surface to Marine Aerosols. In Marine Chemistry; Wangersky P. J., Ed.; The Handbook of Environmental Chemistry; Springer: Berlin, Heidelberg, 2000; pp 1–33. [Google Scholar]
- Fraser M. P.; Cass G. R.; Simoneit B. R. T. Air quality model evaluation data for organics. 6. C3-C24 organic acids. Environ. Sci. Technol. 2003, 373446–453 10.1021/es0209262. [DOI] [PubMed] [Google Scholar]
- King M. D.; Rennie A. R.; Thompson K. C.; Fisher F. N.; Dong C. C.; Thomas R. K.; Pfrang C.; Hughes A. V. Oxidation of oleic acid at the air–water interface and its potential effects on cloud critical supersaturations. Phys. Chem. Chem. Phys. 2009, 11357699–7707 10.1039/b906517b. [DOI] [PubMed] [Google Scholar]
- Karl T.; Hansel A.; Cappellin L.; Kaser L.; Herdlinger-Blatt I.; Jud W. Selective measurements of isoprene and 2-methyl-3-buten-2-ol based on NO+ ionization mass spectrometry. Atmos. Chem. Phys. 2012, 122411877–11884 10.5194/acp-12-11877-2012. [DOI] [Google Scholar]
- D’Anna B.; Jammoul A.; George C.; Stemmler K.; Fahrni S.; Ammann M.; Wisthaler A. Light-induced ozone depletion by humic acid films and submicron aerosol particles. J. Geophys. Res. 2009, 114D12D12301. 10.1029/2008JD011237. [DOI] [Google Scholar]
- Zhang Y.; Del Vecchio R.; Blough N. V. Investigating the mechanism of hydrogen peroxide photoproduction by humic substances. Environ. Sci. Technol. 2012, 462111836–11843 10.1021/es3029582. [DOI] [PubMed] [Google Scholar]
- Frimmel F. H. Photochemical aspects related to humic substances. Environ. Int. 1994, 203373–385 10.1016/0160-4120(94)90123-6. [DOI] [Google Scholar]
- Liu Y. J.; Herdlinger-Blatt I.; McKinney K. A.; Martin S. T. Production of methyl vinyl ketone and methacrolein via the hydroperoxyl pathway of isoprene oxidation. Atmos. Chem. Phys. 2013, 13115715–5730 10.5194/acp-13-5715-2013. [DOI] [Google Scholar]
- Gilman J. B.; Eliason T. L.; Fast A.; Vaida V. Selectivity and stability of organic films at the air–aqueous interface. J. Colloid Interface Sci. 2004, 2801234–243 10.1016/j.jcis.2004.07.019. [DOI] [PubMed] [Google Scholar]
- Griffith E. C.; Rapf R. J.; Shoemaker R. K.; Carpenter B. K.; Vaida V. Photoinitiated Synthesis of Self-Assembled Vesicles. J. Am. Chem. Soc. 2014, 136103784–3787 10.1021/ja5006256. [DOI] [PubMed] [Google Scholar]
- Canonica S.; Jans U.; Stemmler K.; Hoigne J. Transformation Kinetics of Phenols in Water: Photosensitization by Dissolved Natural Organic Material and Aromatic Ketones. Environ. Sci. Technol. 1995, 2971822–1831 10.1021/es00007a020. [DOI] [PubMed] [Google Scholar]
- March J.Advanced Organic Chemistry: Reactions, Mechanisms, and Structure, 4th ed.; Wiley-Interscience: New York, 1992. [Google Scholar]
- Carpenter L. J.; Nightingale P. D. Chemistry and Release of Gases from the Surface Ocean. Chem. Rev. 2015, 115104015–4034 10.1021/cr5007123. [DOI] [PubMed] [Google Scholar]
- Wurl O.; Wurl E.; Miller L.; Johnson K.; Vagle S. Formation and global distribution of sea-surface microlayers. Biogeosciences 2011, 81121–135 10.5194/bg-8-121-2011. [DOI] [Google Scholar]
- Finlayson-Pitts B. J.; Pitts J. N. Jr.. Chemistry of the Upper and Lower Atmosphere: Theory, Experiments, and Applications; Academic Press, 1999. [Google Scholar]
- Washburn E. W.; National Academy of Sciences (U.S.); National Research Council (U.S.). International critical tables of numerical data, physics, chemistry and technology; McGraw-Hill: New York, 1928. [Google Scholar]
- Seinfeld J. H.; Pandis S. N.. Atmospheric chemistry and physics: From air pollution to climate change; Wiley-Interscience, 2006. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.