

Abstract
The study of breast cancer dormancy in the bone marrow is an exceptionally difficult undertaking due to the complexity of the interactions of dormant cells with their microenvironment, their rarity and the overwhelming excess of hematopoietic cells. Towards this end, we developed an in vitro 2D clonogenic model of dormancy of estrogen-sensitive breast cancer cells in the bone marrow. The model consists of a few key elements necessary for dormancy. These include 1) the use of estrogen sensitive breast cancer cells, which are the type likely to remain dormant for extended periods, 2) incubation of cells at clonogenic density, where the structural interaction of each cell is primarily with the substratum, 3) fibronectin, a key structural element of the marrow and 4) FGF-2, a growth factor abundantly synthesized by bone marrow stromal cells and heavily deposited in the extracellular matrix. Cells incubated with FGF-2 form dormant clones after 6 days, which consist of 12 or less cells that have a distinct flat appearance, are significantly larger and more spread out than growing cells and have large cytoplasm to nucleus ratios. In contrast, cells incubated without FGF-2 form primarily growing colonies consisting of >30 relatively small cells. Perturbations of the system with antibodies, inhibitors, peptides or nucleic acids on day 3 after incubation can significantly affect various phenotypic and molecular aspects of the dormant cells at 6 days and can be used to assess the roles of membrane-localized or intracellular molecules, factors or signaling pathways on the dormant state or survival of dormant cells. While recognizing the in vitro nature of the assay, it can function as a highly useful tool to glean significant information about the molecular mechanisms necessary for establishment and survival of dormant cells. This data can be used to generate hypotheses to be tested in vivo models.
Keywords: Medicine, Issue 100, Dormancy, Bone marrow stroma, FGF-2, Fibronectin, Breast cancer, Colony assay
Introduction
Breast cancer cells metastasize to the bone marrow before the disease is detectable1, as soon as small tumors develop blood vessels2,3. The metastatic process is rapid but inefficient. Cells enter the new blood vessels rapidly, at millions per day4 but few survive the trip to distant organs5. Nevertheless, some micrometastases survive in the bone and can be found as single cells or small cell clumps in bone marrow aspirates from newly diagnosed patients1. These cells resist adjuvant chemotherapy, which is administered for the very purpose of eliminating them6. This resistance is endowed, substantially, by survival signaling initiated by interactions with the bone marrow microenvironment7,8. Micrometastases can be found in about one third of women with localized breast cancer and represent an independent indicator of survival when analyzed by univariate analysis9. Some micrometastases are growth initiated, but recurrence patterns depend on cell type. Patients with triple negative breast cancer tend to recur between 1 to 4 years, suggesting poor control over the dormant state. Other cell types, including ER/PR+ cells, can remain dormant for up to 20 years, with a steady, continuous rate of recurrence10. While differences in dormancy gene expression signatures between ER+ and ER- breast cell lines and tumors reflect different dormancy potentials11, interactions with bone marrow stroma likely represent a significant contribution to dormancy.
The study of dormancy in vivo is exceptionally difficult because micrometastases are rare and are outnumbered by hematopoietic cells by more than 106-fold. Hence, relevant models must be generated that provide in vitro data that can suggest mechanisms and generate testable hypotheses in vivo. A number of dormancy models, including mathematical models12,13, in vitro models7,8,14,15, in vivo xenograft models16, combinations of in vitro and xenograft models17,18 and spontaneous tumor and metastasis models19, have yielded some insight into cancer cell dormancy20. Each of these models have their own limitations and are of themselves primarily useful for generating hypotheses regarding molecular signaling and interactions that govern dormancy to be tested in more biologically relevant models.
With the overall goal of defining the molecular mechanisms of dormancy, the interactions with the microenvironment that results in cycle arrest, redifferentiation and therapeutic resistance and mechanisms that result in recurrence in ER+ cells, we developed an in vitro model that provides selected relevant elements of the stromal microenvironment7. This model, while relatively sparse in its components, is sufficiently robust to permit investigators to derive specific molecular mechanisms that affect significant functions of dormancy. These experiments generate hypotheses that can be directly tested in vivo. The model relies on a few key elements that we demonstrated to be relevant in dormancy. They include the use of estrogen-dependent breast cancer cells, culture of cells at a clonogenic density where their interaction is primarily with the substratum and soluble components of the medium, a fibronectin substratum and the presence of basic fibroblast growth factor (FGF-2) in the medium.
We characterized mechanisms that govern the system in vitro, including the induction of cell cycle arrest by FGF-221, mediated through TGFβ22, survival signaling through PI3 Kinase7,8 and ERK8 and morphogenic differentiation to an epithelial phenotype, which depended on RhoA inactivation, integrin α5β1 upregulation and ligation of stromal fibronectin for survival7,15 (Figure 1). The in vitro cell cycle effects of FGF-2 on MCF-7 cells begin at concentrations at least one log below 10 ng/ml21,23. The rationale was based on the temporal control of FGF-2 expression governing mammary ductal morphogenesis, cyclic expansion and recession in a number of mammalian systems24-27. We demonstrated that FGF-2 induces differentiation, including ductal morphogenesis in 3D culture28, and that FGF-2 expression are generally lost with malignant transformation of human tumors29. The expression of FGR1 remained intact in breast carcinomas surveyed29 and MCF-7 cells continue to express all 4 FGF receptors30. In the context of dormancy, FGF-2 is exported by and heavily deposited on bone marrow stroma31,32 where it functions in the preservation of hematopoietic stem cells33. We demonstrated that FGF-2 induces a dormant state in ER+ breast cancer cells cultured on fibronectin substrata, also abundant in the marrow, where it induces morphogenic differentiation7. In the model, breast cancer cells are growth inhibited, inactivate Rho A through the RhoGap GRAF, redifferentiate to an epithelial phenotype and re-express integrins α5β1 #lost with malignant progression. They bind fibronectin through integrin α5β1 and activate survival signaling that render them resistant to cytotoxic therapy7,8,15 (Figure 1). Inhibition of Rho class GTPases has been demonstrated previously to induce a dormant phenotype34.
Here we will outline the specific procedures that will permit investigators to establish the model and study specific molecular and cellular mechanisms governing dormancy of ER+ breast cancer cells. In the experiments presented here to illustrate the use of the model, we targeted the PI3K pathway (Figure 1B) with an Akt inhibitor and a PI3K inhibitor and all members of the Rho family (Figure 1B) with a pan-Rho inhibitor and a Rho Kinase (ROCK) inhibitor.
Protocol
1. Clonogenic Assay
- Prepare a single cell suspensions of estrogen-dependent breast cancer cell lines MCF-7 and T47D cells using the steps outlined below
- Aspirate the culture medium (DMEM/10% heat inactivated fetal calf serum/glutamine and pen/strep) from a 10 cm tissue culture dish which is no more than 50% confluent with MCF-7 or T-47D cells. Rinse with PBS. Incubate with trypsin 0.25%/2.21 mM EDTA dissolved in DMEM high glucose at 37 °C for 1-4 min.
- Check cells at 1 min intervals under a phase contrast microscope to ensure a single cell distribution. Resuspend the cells with a 2 ml pipette by pipetting up and down several times to disrupt cell-cell contact to achieve an almost invariable, single cell status.
- Continue to incubate cells in trypsin at 37 °C for up to 4 min if you observe clumps of cells after only 2 min of incubation. Do not use these cells for clonogenic studies if they remain adherent to each other after 4 min of trypsinization because error will be introduced in the colony number yield. NOTE: If cells are clumped, the number of colonies formed will reflect the product of fewer cells than the number incubated. If cells are excessively trypsinized, their clonogenic potential may be diminished.
- Prepare a single cell suspension of 1,500 cells/ml culture medium for 24 well plates, or less, (+ 500 cells/ml, depending on the cell type or passage number), by serial dilutions in one master tube containing the entire volume needed for all of the variables in the experiment. NOTE: The goal is a final cell density of 800 cells/cm2 (range of approximately 500 to 1,100 cells/cm2). The goal is to yield approximately 100 + 50 colonies, which permits relatively easy counting, prevents crowding and permits sufficient colonies to result in significant statistical differences when colonies are increased or decreased by experimental perturbations.
- Incubate Cells at Clonogenic Density Using Steps Outlined Below
- Incubate cells in quadruplicate wells on 24 well fibronectin-coated plates at a clonogenic density of 1,500 cells/well from a master single cell suspension tube of 1,500 cells/ml. Triturate the medium containing cells with a 5 ml pipette by drawing up 3 ml and dispensing 1 ml medium in each of 2 wells. NOTE: Fibronectin-coated plates should be purchased pre-coated from a commercial vendor. Coating plates outside of a quality controlled, automated process results in an uneven surface unsuited for this assay.
- Mix the suspension by pipetting up and down with a 5 ml pipette, draw up 3 ml of cell suspension and fill 2 wells with 1 ml each. Fill only 2 wells at any one time from one pipette. Return the remaining volume in the pipette to the cell suspension in the master tube, resuspend cells again by pipetting up and down and draw up another 3 ml to fill another 2 wells with 1 ml each. NOTE: Continuous mixing of the master tube is necessary because cells will continuously sediment. Drawing up sufficient volume to fill only 2 wells is necessary to add similar cell numbers to each well because cells sediment in the pipette as well.
- Work rapidly to distribute the large volumes of cells because allowing cells to sit in suspension at room temperature and CO2 concentration will modulate their clonogenic potential (unpublished observations).
- Optimize the spatial distribution of cells during the act of pipetting them into wells for colony assays. Do so by slowly pipetting the suspension containing the final cell concentration into the middle of the well. Do not subject the plate to further motion before cells settle to the bottom. Do not swirl the plate because circular mixing will effectively centrifuge the cells to the perimeter of the well creating high cell densities and uncountable confluent colonies at 6 days. NOTE: Do not mix unless necessary since this is less desirable than no mixing at all after introducing the volume with the cells in suspension. If it is necessary to mix cells, do so by moving the plate back and forth in perpendicular directions while it rests on a flat surface.
- Incubate cells at 37 °C 5% CO2 without media change for 6 days. The small number of cells in a well after 6 days will not significantly impact the nutrient or cytokine composition nor the pH of the original medium.
- Design the time course of the assay as follows: Incubate cells on fibronectin coated substrata on day 1. Replace existing medium with 1 ml fresh medium or fresh medium containing FGF-2 10 ng/ml on day 0. Stain cells on day 6 as below in 1.4). Conduct any experimental perturbations on day 3, as below in 1.3).
- Set Up Experimental Perturbations of Molecular Signaling or Adhesion Molecules
- On day 3, add 100 μl of a solution containing 10x of the final intended concentration of the perturbing agent to the 1 ml medium in the wells. Do not mix. Continue to incubate the cells at 37 °C, 5% CO2 for an additional 3 days. NOTE: Perturbing agents can include a variety of inhibitors and blocking agents of adhesion molecules, receptors or other surface proteins, inhibitors of intracellular signaling pathways, molecules, factors, cofactors or structural proteins, that may play roles in supporting the dormant state.
- Stain colonies on day 6, as follows.
- Stain Colonies
- Stain cells after 6 days in culture with a freshly made 0.1% crystal violet in 2% ethanol/10 mM sodium borate (pH 9.0) solution. Aspirate media and add one ml crystal violet solution to each well for 20 min.
- Wash plates by immersing them with the well openings facing down at an acute angle into an ice bucket overflowing with continuously running tap water in the sink. Tilt the plate to a horizontal angle once underwater, well opening down, and then tilt back to an acute angle when removing in one gentle flowing motion.
- Repeat the immersion 2 or 3 times until the water at the bottom of the wells is no longer blue. Vigorous washes may remove cells or colonies that are less adherent due to experimental intervention, adding significantly to the error in counting and the data.
- Dry plates overnight by placing them upside down on towels on the bench top adjacent to their corresponding labeled covers.
- Count Colonies
- Count the number of growing and dormant colonies in each well after 6 days of incubation, staining and drying. Count colonies optimally at 40X magnification in an inverted phase contrast microscope. Count colonies of >30 cells as growing and colonies of 12 or less cells, with the morphological appearance of very large size compared to growing cells, large expanded cytoplasm with large cytoplasm to nucleus ratios, shown in Figure 17,15. NOTE: Clusters of 13-29 cells are not normally counted as they are not very frequent in straightforward dormancy assays. They can be counted if perturbations shift the growth potential of either growing or dormant cells. These results will then need to be correlated with biological significance.
2. Clonogenic Incubation for Immunofluorescence Studies
Adjust cell numbers to approximately 7,500-8,000 cells/well in 6 well plates, to correspond to cell numbers/surface area analogous to 24 well experiments.
Place a round, sterile, fibronectin-coated cover slip into each well base of 6 well plates for imaging studies prior to cell addition. Pipet cells in 3 ml volumes into each well 2 wells at a time at concentrations outlined, as described for the clonogenic experiments above in 1.2).
Incubate cells for 6 days, as above in 1.2.4 and 1.2.5. Add perturbing factors on day 3 at 10x concentrations in 300 μl volumes, as described in the colony assay procedures in 1.3).
- Stain cells on day 6 with antibodies to cell adhesion molecules such as integrins α4, α5, α6, β1, β3, for example, focal adhesion complex molecules FAK, paxillin and vinculin, for example, proteins involved in motility such as α-tubulin, for example, signaling pathway members such as phospho-Akt, phospho-ERK, phospho-p38, phospho-JNK, for example, or any other protein that is the target of investigation for its role in dormancy, using standard techniques for direct or indirect immunofluorescence staining.
- Remove slides with forceps day 6. Fix in acetone/methanol 1:1 at -20°C for 20 min and air dry. An alternative fixative, such as paraformaldehyde, may be used, if necessary. Permeabilize cells with 0.1% Triton X-100, 0.1% sodium citrate for 2 min for detection of intracellular antigens. Wash cells with PBS.
- For indirect immunofluorecence staining, block slides for 1 hr at room temperature with 5% BSA or with 10% preimmune serum from the species in which the secondary antibody was generated.
- Incubate overnight at 4 °C with primary antibodies to cell adhesion molecules, focal complex molecules, signaling pathway members or any other protein that is the target of investigation for its role in dormancy diluted to specific dilutions recommended by the manufacturer in PBS 0.1% TRITON X-100.
- Wash 3 times with PBS. Incubate cover slips with fluorophor-conjugated antibodies at room temperature for 2 hr. As an example, Alexa Fluor 488 Donkey anti-Mouse IgG Antibody can be used to detect murine monoclonal primary antibodies. Mount coverslips cell side down on glass slides using an antifade agent with Dapi. Seal the perimeters with nail polish.
- For direct immunofluorescence, carry out the BSA blocking as above in 2.4.2), incubate with fluorophor-conjugated primary antibody overnight at 4 °C. Wash 3 times with PBS. Incubate the slides with an antifade agent and Dapi and seal with nail polish.
- Cover slide trays with aluminum foil and store at 4°C for imaging and photography anytime up to several weeks later. View and photograph cells using any fluorescence microscopic imaging systems equipped with a camera at 1,000x magnification.
- For fibrillar actin staining, block slides in 1% bovine serum albumin (BSA) for 30 min and incubate in BODIPY FL-Phallacidin (green) or Rhodamine phalloidin (red) at room temperature for 20 min. Add an antifade agent and seal as above.
3. Clonogenic Incubation for Molecular Studies
- Western Blots
- Incubate ER+ breast cancer cells MCF-7 or T47D at clonogenic densities of 20,000 cells/60 mm plate and 50,000 cells/100 mm plate on fibronectin-coated plates at 37 °C in 5% CO2.
- Incubate cells at slightly higher densities of 75,000 cells/100 mm plate for molecular studies requiring mg amounts for protein from lysates. Use up to ten plates per experimental point to collect sufficient protein for molecular studies using Western blots or for RNA isolation for Northern blots.
- Cell number based gel loading is a necessary adjunct for comparing protein expression in vastly different-sized growing and dormant cells. Collect cells by trypsinizing and count an aliquot in a hemocytometer in 0.2% Trypan Blue for preparation of lysates for Western blots instead of scraping off the cells with a single edge blade.
- Centrifuge cells at 10,000 x g for 2 min, remove media by aspiration, add 200 μl lysis buffer, sonicate the cells in lysis buffer and determine the protein concentration.
- Calculate the amount of protein per cell by dividing the protein yield by the number of cells that generated that amount. Load the lysate into each well of separate polyacrylamide gels that represents both a) equivalent amounts of protein and b) protein quantities representing equivalent cell numbers. At least 25 μg protein should be loaded into each well. NOTE: This will permit comparisons between growing and dormant cells, which are significantly different in size and protein content (Figure 1).
- Flow Cytometry
- Collect cells from 100 mm plates by trypsinization, as in 3.1 and analyze by standard fluorescence activated cell sorting (FACS) protocols using primary or secondary immunofluorescence staining for either intracellular or extracellular antigens, as outlined in 2.4. NOTE: Detaching cells from tissue culture plates by trypsinization does not affect the concentration of membrane proteins as determined by antibody labeling.
Representative Results
Experiments were conducted to recapitulate the assay. The time course of the experiment is shown in Figure 2A. Cells are incubated at clonogenic density on day -1, FGF-2 in fresh medium is added on day 0 and cells are cultured until day 6 when they are stained and colonies are counted. Any perturbations to the system are administered on day 3 in 100 μl volumes at 10x final concentrations desired. Figure 2B demonstrates the typical appearance of growing and dormant colonies. Growing colonies contain >30 cells and dormant colonies contain 12 or less cells which are many times larger than growing cells with large cytoplasm/nucleus ratios. Figure 2C demonstrates a typical experimental result conducted in quadruplicate. The predominant distribution of cells without addition of FGF-2 is represented by growing clones while, in the presence of FGF-2, the vast majority of clones are dormant.
Figure 3 is an example of an experiment in which perturbing agents were added on day 3. The figure represents the dose-dependent inhibition of dormant clones by inhibition of the Rho family of GTPases by a pan-Rho inhibitor C3 transferase and a Rho kinase (ROCK) inhibitor Y27632. The assay can assess the ED50 of added inhibitors on dormant clones as well as growing clones. In this experiment, the effect on growing clones was not determined, since it was only presented to illustrate the methodology.
Figures 4 and 5 demonstrate that the assay can be used to assess the combined effects of inhibitors on survival of dormant clones. Our prior studies have demonstrated that the PI3K pathway undergoes a sustained activation in dormant cells in this model and inhibition of both PI3K and Akt partially inhibits the survival of dormant clones7. Here, preliminary observations, demonstrated that nonspecific inhibition of the Rho family of small GTPases also partially inhibits survival of dormant clones. Data presented in Figures 4 and 5 demonstrate that combining inhibition of PI3K and one of its downstream effectors, AKT, with the inhibition of the Rho family with C3 transferase and a ROCK inhibitor can almost completely eliminate the survival of dormant clones in some circumstances. We have previously demonstrated that dormant clones surviving the inhibition of PI3K but not of Akt are comprised of cells that no longer exhibit the dormant phenotype, but rather appear mesenchymal and distressed7. Cells in dormant clones in which the Rho family has been broadly inhibited by C3 transferase and the ROCK inhibitor also lose the typical dormant appearance, assume fibrolastoid appearances and ruffled membranes and also appear distressed (Figure 6). These experiments demonstrate a model that can be queried in a very broad manner to achieve an understanding of the elements that govern dormancy and resistance to therapy.
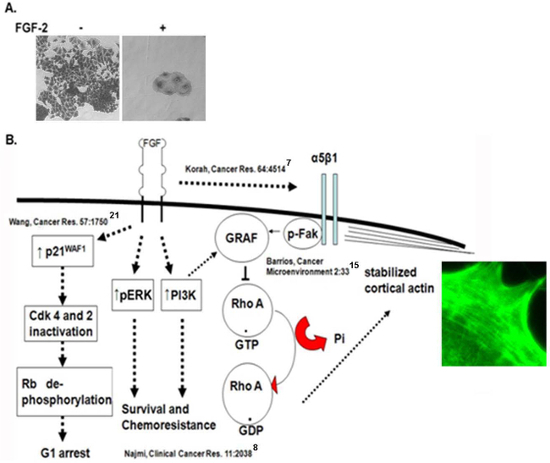 Figure 1: Mechanisms governing our in vitro dormancy model.
(A) Growing and dormant MCF-7 clones after 6 day incubation at
clonogenic density on fibronectin-coated plates with and without FGF2 10 ng/ml7
(100X magnification). (B) Summary schema outlining the data that FGFR and
integrin α5β1 parallel steady state signaling is required to activate and maintain
dormancy7. FGF-2 upregulates cyclin dependent kinase inhibitors resulting in G1
arrest21, activates ERK8 and PI3 kinase7 that
initiatesurvival signaling and upregulate integrin α5β17, which reaches steady
state after several days. Dual signaling through FGFR through PI3K and independently through
ligation of integrin α5β1 by fibronectin are required for activation of FAK and membrane
localization and activation of the RhoA GAP GRAF15. This results in inactivation
of RhoA and a permissive steady state for cortical rearrangement of F-actin
(phalloidin-stained photomicrograph), epithelial re-differentiation and
dormancy15. Please click
here to view a larger version of this figure.
Figure 1: Mechanisms governing our in vitro dormancy model.
(A) Growing and dormant MCF-7 clones after 6 day incubation at
clonogenic density on fibronectin-coated plates with and without FGF2 10 ng/ml7
(100X magnification). (B) Summary schema outlining the data that FGFR and
integrin α5β1 parallel steady state signaling is required to activate and maintain
dormancy7. FGF-2 upregulates cyclin dependent kinase inhibitors resulting in G1
arrest21, activates ERK8 and PI3 kinase7 that
initiatesurvival signaling and upregulate integrin α5β17, which reaches steady
state after several days. Dual signaling through FGFR through PI3K and independently through
ligation of integrin α5β1 by fibronectin are required for activation of FAK and membrane
localization and activation of the RhoA GAP GRAF15. This results in inactivation
of RhoA and a permissive steady state for cortical rearrangement of F-actin
(phalloidin-stained photomicrograph), epithelial re-differentiation and
dormancy15. Please click
here to view a larger version of this figure.
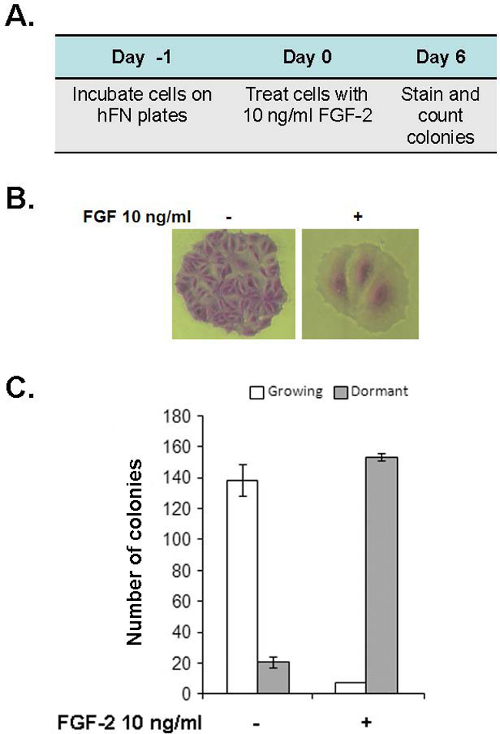 Figure 2: Elements of the in vitro dormancy assay. (A) Schema
of the temporal components of the dormancy assay. Cells are incubated on fibronectin coated
tissue culture plates at clonogenic density, at a maximum density of 1,500 cells/well of a
24 well plate in an incubator at 37 °C and 5% CO2 on day 1. The medium is
replaced on day 0 with fresh medium or fresh medium containing FGF-2 10 ng/ml and the cells
are re-incubated. Cells are stained with crystal violet solution, as described, on day 6.
Any inhibitors or perturbation agents are added on day 3 in 100 μl volumes at 10x desired
concentrations and cells are re-incubated until day 6. (B) The appearance of
stained growing and dormant MCF-7 colonies. Growing colonies contain >30 cells and
dormant colonies contain 12 or less cells. Dormant cells are pancake shaped, many times
larger than growing cells with large cytoplasm/nucleus ratios. (100X magnification).
(C) Graph demonstrating the clonogenic potential of 1,500 MCF-7 human breast
cancer cells incubated with and without FGF-2 10 ng/ml on fibronectin-coated 24 well plates.
Without FGF-2, the predominant distribution of cells is represented by growing clones while
in the presence of FGF-2 the vast majority of clones are dormant. Experiments were done in
quadruplicate. (Error bars are + S.D.)
Figure 2: Elements of the in vitro dormancy assay. (A) Schema
of the temporal components of the dormancy assay. Cells are incubated on fibronectin coated
tissue culture plates at clonogenic density, at a maximum density of 1,500 cells/well of a
24 well plate in an incubator at 37 °C and 5% CO2 on day 1. The medium is
replaced on day 0 with fresh medium or fresh medium containing FGF-2 10 ng/ml and the cells
are re-incubated. Cells are stained with crystal violet solution, as described, on day 6.
Any inhibitors or perturbation agents are added on day 3 in 100 μl volumes at 10x desired
concentrations and cells are re-incubated until day 6. (B) The appearance of
stained growing and dormant MCF-7 colonies. Growing colonies contain >30 cells and
dormant colonies contain 12 or less cells. Dormant cells are pancake shaped, many times
larger than growing cells with large cytoplasm/nucleus ratios. (100X magnification).
(C) Graph demonstrating the clonogenic potential of 1,500 MCF-7 human breast
cancer cells incubated with and without FGF-2 10 ng/ml on fibronectin-coated 24 well plates.
Without FGF-2, the predominant distribution of cells is represented by growing clones while
in the presence of FGF-2 the vast majority of clones are dormant. Experiments were done in
quadruplicate. (Error bars are + S.D.)
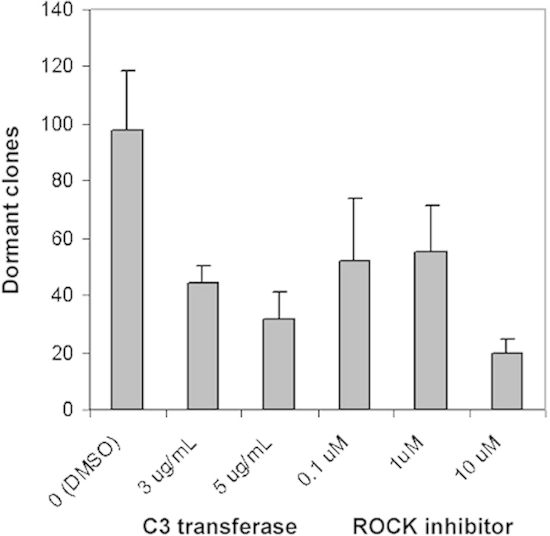 Figure 3: Dose-dependent inhibition of dormant clones by pan-Rho family inhibitors
C3 transferase and the ROCK inhibitor Y27632. T-47D cells were incubated at the
clonogenic density of 1,000 cells per well on 24 well fibronectin-coated plates with FGF-2
10 ng/ml for 6 days. Media, C3 transferase 3 or 5 μg/ml, and the ROCK inhibitor Y27632 0.1,
1 or 10 μg/ml were on day 3 and dormant clones were counted on day 6. The graph demonstrates
a dose-dependent inhibition of stained dormant clones on day 6. The ED50 of C3
was approximately 3 μg/ml while that of the ROCK inhibitor was between 1 and 10 μM in this
assay. Experiments were done in quadruplicate. (Error bars are + S.D.)
Figure 3: Dose-dependent inhibition of dormant clones by pan-Rho family inhibitors
C3 transferase and the ROCK inhibitor Y27632. T-47D cells were incubated at the
clonogenic density of 1,000 cells per well on 24 well fibronectin-coated plates with FGF-2
10 ng/ml for 6 days. Media, C3 transferase 3 or 5 μg/ml, and the ROCK inhibitor Y27632 0.1,
1 or 10 μg/ml were on day 3 and dormant clones were counted on day 6. The graph demonstrates
a dose-dependent inhibition of stained dormant clones on day 6. The ED50 of C3
was approximately 3 μg/ml while that of the ROCK inhibitor was between 1 and 10 μM in this
assay. Experiments were done in quadruplicate. (Error bars are + S.D.)
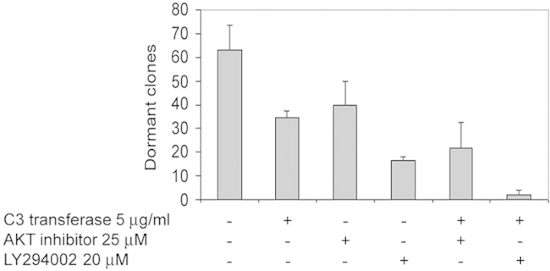 Figure 4: Combined effects of pan-Rho family inhibitor C3 with an Akt inhibitor or
with a PI3 kinase inhibitor on survival of dormant T-47D clones. T-47D cells were
incubated at the clonogenic density of 1,000 cells per well on 24 well fibronectin-coated
plates with FGF-2 10 ng/ml for 6 days. Media, C3 transferase 5 μg/ml, Akt inhibitor 25 μM
and LY294002 20 μM were added individually or in combination on day 3 and dormant clones
were counted on day 6. The combined effects of C3 and the AKT inhibitor appear to be
additive at these concentrations but the combined effects of C3 and the PI3K inhibitor
appear to be synergistic in these experiments on inhibition of dormant clone survival.
Experiments were done in quadruplicate. (Error bars are + S.D.)
Figure 4: Combined effects of pan-Rho family inhibitor C3 with an Akt inhibitor or
with a PI3 kinase inhibitor on survival of dormant T-47D clones. T-47D cells were
incubated at the clonogenic density of 1,000 cells per well on 24 well fibronectin-coated
plates with FGF-2 10 ng/ml for 6 days. Media, C3 transferase 5 μg/ml, Akt inhibitor 25 μM
and LY294002 20 μM were added individually or in combination on day 3 and dormant clones
were counted on day 6. The combined effects of C3 and the AKT inhibitor appear to be
additive at these concentrations but the combined effects of C3 and the PI3K inhibitor
appear to be synergistic in these experiments on inhibition of dormant clone survival.
Experiments were done in quadruplicate. (Error bars are + S.D.)
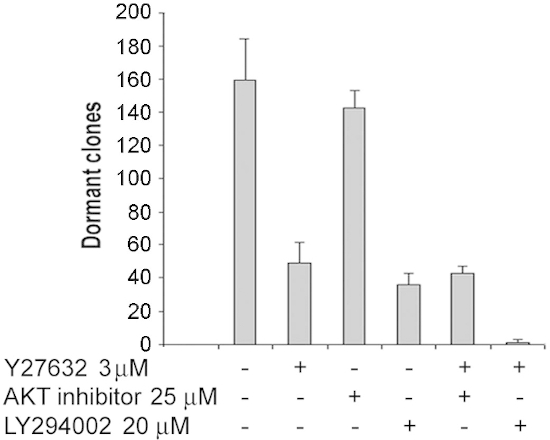 Figure 5: Combined effects of ROCK inhibitor Y27632 with an Akt inhibitor or with
a PI3 kinase inhibitor on survival of dormant T-47D clones. T-47D cells were
incubated at the clonogenic density of 1,000 cells per well on 24 well fibronectin-coated
plates with FGF-2 10 ng/ml for 6 days. Media, ROCK inhibitor Y27632 3 μg/ml, Akt inhibitor
25 μM and LY294002 20 μM were added individually or in combination on day 3 and dormant
clones were counted on day 6. The combined effects of Y27632 and the AKT inhibitor do not
appear to be additive at these concentrations but the combined effects of Y27632 and the
PI3K inhibitor appear to be more than additive in these experiments on inhibition of dormant
clone survival. Experiments were done in quadruplicate. (Error bars are + S.D.)
Figure 5: Combined effects of ROCK inhibitor Y27632 with an Akt inhibitor or with
a PI3 kinase inhibitor on survival of dormant T-47D clones. T-47D cells were
incubated at the clonogenic density of 1,000 cells per well on 24 well fibronectin-coated
plates with FGF-2 10 ng/ml for 6 days. Media, ROCK inhibitor Y27632 3 μg/ml, Akt inhibitor
25 μM and LY294002 20 μM were added individually or in combination on day 3 and dormant
clones were counted on day 6. The combined effects of Y27632 and the AKT inhibitor do not
appear to be additive at these concentrations but the combined effects of Y27632 and the
PI3K inhibitor appear to be more than additive in these experiments on inhibition of dormant
clone survival. Experiments were done in quadruplicate. (Error bars are + S.D.)
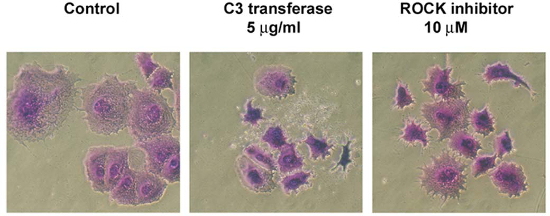 Figure 6: The appearance of surviving dormant T-47D clones after treatment with C3
transferase and ROCK inhibitor Y27632. Colony assays were established in
quadruplicate in 24 well fibronectin coated tissue culture plates at 1,000 cells/well with
FGF-2, as described, inhibitors were added on day 3 at the concentrations shown, cells were
stained and photographed on day 6. Cells treated with pan-Rho family inhibitors lost their
spread appearance, became small, dendritic and appeared distressed. (100X
magnification).
Figure 6: The appearance of surviving dormant T-47D clones after treatment with C3
transferase and ROCK inhibitor Y27632. Colony assays were established in
quadruplicate in 24 well fibronectin coated tissue culture plates at 1,000 cells/well with
FGF-2, as described, inhibitors were added on day 3 at the concentrations shown, cells were
stained and photographed on day 6. Cells treated with pan-Rho family inhibitors lost their
spread appearance, became small, dendritic and appeared distressed. (100X
magnification).
Discussion
Our model is comprised of several key elements of dormancy in the bone marrow. It consists of estrogen sensitive cells, which are the type likely to remain dormant in the marrow for extended periods10, it consists of fibronectin, a key structural element of the marrow, FGF-2, a growth factor abundantly synthesized by the bone marrow stroma and heavily deposited in the extracellular matrix of the bone marrow31,32 and incubation of cells at clonogenic density where their interactions are primarily with the substratum. While the bone marrow microenvironment is far more complex, this simple system can be built up with as much added complexity as needed to ask specific mechanistic questions.
The model can be used to compare dormant with growing cells in a number of ways, including cell biologic techniques, molecular techniques and in vivo techniques. Cellular phenotypes can be assayed by re-cloning, motility and invasion studies35, nonadherent tumor initiating spherule formation36 or by functional studies for interaction with the microenvironment using specific blocking antibodies or peptides7.
The primary output of the clonogenic assay is a numeric count of colonies, either growing or dormant, which translates to the growing or dormant clonogenic potential of the cells. As implied above, a large number of variables have the potential of affecting this outcome, all of which must be controlled as stringently as possible to yield reproducible results. These include the source of cells, overall passage number, cryopreservation technique and the quality of the cell cultures that were cryopreserved, the number of passages since thawing, the confluence of cultures from which the single cell preparations were obtained, the source and quality of the fetal calf serum, the durations of trypsinization and their reproducibility, the duration of cells left at room temperature, the dexterity of pipetting equivalent cell numbers into each well, the dexterity or evenly distributing cells across the entire surface area of a well, the number of times a plate is removed from the incubator for observation, among dozens of others. In addition to representing generally accepted tissue culture quality controls, these variables affect the dormant clonogenic potential from experiment to experiment. Intra-experimental variability is typically less than 8-10%, however, resulting in percentage changes from experiment to experiment that are highly reproducible. Typically, the variance of the data begins to decrease with a direct correlation of increased experience of the experimenter with the system and the time spent conducting this assay.
Gene expression in dormant and growing cells can be assessed by RT PCR, Northern blot, Western blot, immunoprecipitation/Western and functional complex precipitation/Western, immunofluorescence or immunohistochemistry, multiplexing by microfluidics, Raman spectroscopy, flow cytometry and other techniques. These techniques can be used to determine the roles of specific receptors, other membrane proteins, signaling pathways, transcription factors, histone modifiers, organelles and mitochondrial proteins, metabolic pathways and energy utilization, tension and force and interaction with the microenvironment in countless ways on the role of dormancy.
Our prior studies have demonstrated significant roles for the PI3K pathways in survival of dormant clones7 as well as in maintaining the dormant phenotype with the epithelial distribution of cortical actin15. We also demonstrated that RhoA specifically, must be inhibited in order for the dormant phenotype to be activated. This is in contrast to the data presented here, where inactivation of the Rho family with pan-inhibitors diminishes survival of dormant clones. This underscores the danger of using chemical inhibitors to dissect mechanisms and the need for using genetic approaches to inhibit or activate specific pathways. Towards this end, the methodology lends itself to genetic manipulation of the cells, either by permanent transfection or transduction or by transient transfection15, siRNA or nanoparticles. Since the assay is 6 days in duration, transient gene expression or interference with it will result in significant phenotypic effects at 6 days that can be measured.
Recognizing its potential for studying mechanisms of entering as well as exiting dormancy, we are currently investigating mechanisms for these cells to escape dormancy. While entering dormancy is poorly understood, an understanding of the mechanisms governing the exit from the dormant state is even more elusive. We have reported preliminary observations that inflammatory responses by injured stroma can contribute to the reawakening of dormant MCF-7 cells using this model37. The model can be applied to T-47 cells as well, another ER+ breast cancer cell line. While ER- cells do not become dormant in response to FGF-2, it may be possible to adapt the methodology to other cancer cell types with different differentiation agents in future investigations.
Despite being a highly useful technique for identifying key mechanism involved in dormancy, it remains an in vitro model. However, cells that have undergone dormancy or in vitro manipulation can be tested for xenograft tumor forming capacity. The main potential benefit of the system, however, is the identification of mechanisms that will permit the development of a hypothesis which can be tested in highly complex in vivo dormancy models.
Disclosures
Supported by the Department of Defense Grants DAMD17-01-C-0343 and DAMD17-03-1-0524, the New Jersey State Commission on Cancer Research 02-1140-CCR-E0 and the Ruth Estrin Goldberg Memorial for Cancer Research (RW)
Acknowledgments
The authors have nothing to disclose.
References
- Braun S, et al. Cytokeratin-positive cells in the bone marrow and survival of patients with stage I, II, or III breast cancer. N Engl J Med. 2000;342(8):525–533. doi: 10.1056/NEJM200002243420801. [DOI] [PubMed] [Google Scholar]
- Benoy IH, et al. Relative microvessel area of the primary tumour, and not lymph node status, predicts the presence of bone marrow micrometastases detected by reverse transcriptase polymerase chain reaction in patients with clinically non-metastatic breast cancer. Breast Cancer Res. 1186;7(2):R210–R219. doi: 10.1186/bcr980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wapnir IL, Barnard N, Wartenberg D, Greco RS. The inverse relationship between microvessel counts and tumor volume in breast cancer. Breast J. 2001;7(3):184–188. doi: 10.1046/j.1524-4741.2001.007003184.x. [DOI] [PubMed] [Google Scholar]
- Wyckoff JB, et al. A critical step in metastasis: in vivo analysis of intravasation at the primary tumor. Cancer Res. 2000;60(9):2504–2511. [PubMed] [Google Scholar]
- Phadke PA, et al. Kinetics of metastatic breast cancer cell trafficking in bone. Clin Cancer Res. 2006;12(5):1431–1440. doi: 10.1158/1078-0432.CCR-05-1806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braun S, et al. Lack of an effect of adjuvant chemotherapy on the elimination of single dormant tumor cells in bone marrow of high risk breast cancer patients. J Clin Onc. 2000;18(1):80–86. doi: 10.1200/JCO.2000.18.1.80. [DOI] [PubMed] [Google Scholar]
- Korah R, Boots M, Wieder R. Integrin α5β1 promotes survival of growth-arrested breast cancer cells: an in vitro paradigm for breast cancer dormancy in bone marrow. Cancer Res. 2004;64(13):4514–4522. doi: 10.1158/0008-5472.CAN-03-3853. [DOI] [PubMed] [Google Scholar]
- Najmi S, Korah R, Chandra R, Abdellatif M, Wieder R. Flavopiridol blocks integrin-mediated survival in dormant breast cancer cells. Clin Cancer Res. 2005;11(5):2038–2046. doi: 10.1158/1078-0432.CCR-04-1083. [DOI] [PubMed] [Google Scholar]
- Braun S, et al. A pooled analysis of bone marrow micrometastasis in breast cancer. N England J Med. 2005;353(8):793–802. doi: 10.1056/NEJMoa050434. [DOI] [PubMed] [Google Scholar]
- Dent R, et al. Triple-negative breast cancer: clinical features and patterns of recurrence. Clin Cancer Res. 2007;13(15 Part 1):4429–4434. doi: 10.1158/1078-0432.CCR-06-3045. [DOI] [PubMed] [Google Scholar]
- Kim RS, et al. Dormancy signatures and metastasis in estrogen receptor positive and negative breast cancer. PLoS One. 2012;7(4):e35569. doi: 10.1371/journal.pone.0035569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanin L. Seeing the invisible: how mathematical models uncover tumor dormancy, reconstruct the natural history of cancer, and assess the effects of treatment. Adv in Exp Med & Biol. 2013;734:261–282. doi: 10.1007/978-1-4614-1445-2_12. [DOI] [PubMed] [Google Scholar]
- Wilkie KP. A review of mathematical models of cancer-immune interactions in the context of tumor dormancy. Adv in Exp Med & Biol. 2013;734:201–234. doi: 10.1007/978-1-4614-1445-2_10. [DOI] [PubMed] [Google Scholar]
- Barkan D, Green JE. An in vitro system to study tumor dormancy and the switch to metastatic growth. J Vis Exp. 2011. p. e2914. [DOI] [PMC free article] [PubMed]
- Barrios J, Wieder R. Dual FGF-2 and intergrin α5β1 signaling mediate GRAF-induced RhoA inactivation in a model of breast cancer dormancy. Cancer Microenvironment. 2009;2(1):33–47. doi: 10.1007/s12307-009-0019-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganapathy V, et al. Luminal breast cancer metastasis is dependent on estrogen signaling. Clin & Exp Metastasis. 2012;29(5):493–509. doi: 10.1007/s10585-012-9466-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barkan D, et al. Inhibition of metastatic outgrowth from single dormant tumor cells by targeting the cytoskeleton. Cancer Res. 2008;68(15):6241–6250. doi: 10.1158/0008-5472.CAN-07-6849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghajar CM, et al. The perivascular niche regulates breast tumour dormancy. Nat Cell Biology. 2013;15(7):807–817. doi: 10.1038/ncb2767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marshall JC, et al. Effect of inhibition of the lysophosphatidic acid receptor 1 on metastasis and metastatic dormancy in breast cancer. J Nat Cancer Inst. 2012;104(17):1306–1319. doi: 10.1093/jnci/djs319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Almog N. Genes and regulatory pathways involved in persistence of dormant micro-tumors. Adv in Exp Med & Biol. 2013;734:3–17. doi: 10.1007/978-1-4614-1445-2_1. [DOI] [PubMed] [Google Scholar]
- Wang H, et al. Basic FGF causes growth arrest in MCF-7 human breast cancer cells while inducing both mitogenic and inhibitory G1 events. Cancer Res. 1997;57(9):1750–1757. [PubMed] [Google Scholar]
- Fenig E, et al. Role of transforming growth factor beta in the growth inhibition of human breast cancer cells by basic fibroblast growth factor. Breast Cancer Res Treat. 2001;70(1):27–37. doi: 10.1023/a:1012522321762. [DOI] [PubMed] [Google Scholar]
- Fenig E, et al. Basic fibroblast growth factor confers growth inhibition and Mitogen-activated Protein Kinase activation in human breast cancer cells. Clinical Cancer Research. 1997;3(1):135–142. [PubMed] [Google Scholar]
- Coleman-Krnacik S, Rosen JM. Differential temporal and spatial gene expression of fibroblast growth factor family members during mouse mammary gland development. Molecular Endocrinology. 1994;8(2):218–229. doi: 10.1210/mend.8.2.8170478. [DOI] [PubMed] [Google Scholar]
- Plath A, et al. Expression and localization of members of the fibroblast growth factor family in the bovine mammary gland. J Dairy Science. 1998;81(10):2604–2613. doi: 10.3168/jds.S0022-0302(98)75818-7. [DOI] [PubMed] [Google Scholar]
- Lavandero S, Chappuzeau A, Sapag-Hagar M, Oka T. In vivo and in vitro evidence of basic fibroblast growth factor action in mouse mammary gland development. FEBS letters. 1998;439(3):351–356. doi: 10.1016/s0014-5793(98)01370-2. [DOI] [PubMed] [Google Scholar]
- Sinowatz F, Schams D, Plath A, Kolle S. Expression and localization of growth factors during mammary gland development. Adv in Exp Med & Biol. 2000;480:19–27. doi: 10.1007/0-306-46832-8_3. [DOI] [PubMed] [Google Scholar]
- Korah R, Sysounthone V, Scheff E, Wieder R. Intracellular FGF-2 promotes differentiation in T47-D breast cancer cells. Biochem Biophys Res Comm. 2000;277(1):255–260. doi: 10.1006/bbrc.2000.3655. [DOI] [PubMed] [Google Scholar]
- Korah R, Das K, Lindy ME, Hameed M, Wieder R. Co-ordinate loss of FGF-2 and laminin 5 expression during neoplastic progression of mammary duct epithelium. Human Pathology. 2007;38(1):154–160. doi: 10.1016/j.humpath.2006.07.004. [DOI] [PubMed] [Google Scholar]
- Wieder R, et al. Overexpression of basic fibroblast growth factor in MCF-7 human breast cancer cells: lack of correlation between inhibition of cell growth and MAP kinase activation. J. Cellular Physiology. 1998;177:411–425. doi: 10.1002/(SICI)1097-4652(199812)177:3<411::AID-JCP5>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- Brunner G, Gabrilove J, Rifkin DB, Wilson EL. Phospholipase C release of basic fibroblast growth factor from human bone marrow cultures as a biologically active complex with a phosphatidylinositol-anchored heparan sulfate proteoglycan. J Cell Biol. 1991;114(6):1275–1283. doi: 10.1083/jcb.114.6.1275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson EL, Rifkin DB, Kelly F, Hannocks MJ, Gabrilove JL. Basic fibroblast growth factor stimulates myelopoiesis in long-term human bone marrow cultures. Blood. 1991;77(5):954–960. [PubMed] [Google Scholar]
- Gupta P, McCarthy JB, Verfaillie CM. Stromal fibroblast heparan sulfate is required for cytokine-mediated ex vivo maintenance of human long-term culture-initiating cells. Blood. 1996;87(8):3229–3236. [PubMed] [Google Scholar]
- Chatterjee M, van Golen KL. Farnesyl transferase inhibitor treatment of breast cancer cells leads to altered RhoA and RhoC GTPase activity and induces a dormant phenotype. Int J Cancer. 2011;129(1):61–69. doi: 10.1002/ijc.25655. [DOI] [PubMed] [Google Scholar]
- Korah R, Sysounthone V, Golowa Y, Wieder R. Basic fibroblast growth factor confers a more differentiated phenotype in MDA-MB-231 human breast cancer cells. Cancer Res. 2000;60(3):733–740. [PubMed] [Google Scholar]
- Dontu G, et al. In vitro propagation and transcriptional profiling of human mammary stem/progenitor cells. Genes & Dev. 2003;17(10):1253–1270. doi: 10.1101/gad.1061803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tivari S, Lu H, Dasgupta T, Wieder R. Proceedings of the 105th Annual Meeting of the American Association for Cancer Research. San Diego, CA. Philadelphia (PA): AACR; 2014. Reactivation of dormant estrogen-dependent breast cancer micrometastases by bone marrow stromal injury in an in vitro model of dormancy. [Google Scholar]