

Abstract
Maternal stress has been linked to infant birth weight outcomes, which itself may be associated with health later in life. The placenta acts as a master regulator for the fetal environment, mediating intrauterine exposures to stress through the activity of genes regulating glucocorticoids, including the 11beta-hydroxysteroid dehydrogenase (HSD11B) type 1 and 2 genes, and so we hypothesized that variation in these genes will be associated with infant birth weight. We investigated DNA methylation levels at six sites across the two genes, as well as mRNA expression for each, and the relationship to infant birth weight. Logistic regressions correcting for potential confounding factors revealed a significant association between methylation at a single CpG site within HSD11B1 and being born large for gestational age. In addition, our analysis identified correlations between methylation and gene expression, including sex-specific transcriptional regulation of HSD11B2. Our work is one of the first comprehensive views of DNA methylation and expression in the placenta for both HSD11B types 1 and 2, linking epigenetic alterations with the regulation of fetal stress and birth weight outcomes.
Keywords: developmental origins of health and disease, glucocorticoids, glucocorticoid receptor, metabolic syndrome, stress
INTRODUCTION
There is a well-established link between elevated levels of maternal stress during pregnancy and negative infant health outcomes, including size at birth, metabolic disorders, and adult psychiatric disorders [1–4]. The placenta manages the exposure of the fetus to glucocorticoids through the actions of 11β-hydroxysteroid dehydrogenase type 1 (HSD11B1) and type 2 (HSD11B2) [5]. HSD11B1 may both convert inactive cortisone to cortisol, or cortisol back to cortisone, and is the major regulator of circulating excess glucocorticoids [6], while HSD11B2 only inactivates glucocorticoids. Placental activity and expression levels of both HSD11B1 [7, 8] and HSD11B2 [9, 10] have been positively correlated with birth weight, suggesting a link between fetal glucocorticoid metabolism and size at birth.
Epigenetic alterations, specifically DNA methylation, either due to natural variation within a population or as a response to external stimuli, frequently lead to alterations in gene expression of genes with potential physiologic relevance. Recent investigations have demonstrated that there may be epigenetic variation within placental HSD11B2 as a result of differential levels of a maternal stress [11, 12]. These are important findings, as experimental HSD11B2 inactivation during pregnancy has led to alterations in development of the hypothalamic-pituitary-adrenal axis, leading to altered stress and anxiety responses through adulthood in rats [13]. In addition, prenatal exposure to severe stressors has been correlated with placental methylation levels of glucocorticoid receptor (NR3C1) and birth weight [14, 15]. These data suggest that DNA methylation is important in the interplay between cortisol levels managed by HSD11B1 and HSD11B2 and the recognition of stress hormones with NR3C1.
Several studies have recently investigated methylation levels of placental HSD11B2 in relation to factors closely related to birth weight. These include investigations reporting higher methylation in infants following intrauterine growth restriction [16], while another reported lower methylation associated with maternal pre-eclampsia [17]. These studies confirm HSD11B2 as a region of interest in relation to infant birth weight, which, at the extremes, has been shown to predispose infants later in life to both obesity and insulin resistance [18, 19]. HSD11B types 1 and 2 are abundantly expressed in placental tissue, and act as mediators in the transfer of maternal stress to the fetus. Work conducted by our group has previously linked HSD11B2 methylation and expression with predictive neurobehavioral outcomes [20], and glucocorticoid receptor methylation with infant birth weight [21], highlighting the importance of the placental regulation of maternal stress exposure. In addition, placental HSD11B2 expression and activity has been associated with infant birth weight and fetal growth [22], as well as pre-eclampsia [16, 17]. Lowered placental HSD11B1 activity at birth has also been linked to reduced childhood growth of small for gestational age (SGA) infants at 7 yr of age, implicating it in postnatal growth [23]. There are, however, no published data comprehensively investigating DNA methylation, expression, and the interplay between the two HSD11B isoforms on birth weight.
In the present study, we hypothesized that variation exists in placental DNA methylation of HSD11B1 and HSD11B2, and that this variation is associated with mRNA expression of the genes in question, and ultimately is related to offspring birth weight. To examine this hypothesis, placental methylation levels were measured from a birth cohort in which methylation was assessed from 450 and 690 primary placenta samples for HSD11B1 and HSD11B2, respectively, along with gene expression for both isoforms from 72 samples. We then examined how methylation variation within these genes was associated with newborn birth weight grouping.
MATERIALS AND METHODS
Ethics Statement
Study protocols were approved by the Institutional Review Boards for Women and Infants' Hospital of Rhode Island (Providence, RI) and Dartmouth College (Hanover, NH). Mothers provided written informed consent for participation and also for participation of their infants.
Study Population
Subjects involved in the study are part of the Rhode Island Child Health Study (RICHS), which enrolled a total of 899 mother/infant pairs at the Woman and Infant's Hospital from September 1, 2009 through July 31, 2014. Mothers were between the ages of 18 and 40 yr, free of life-threatening conditions, and there were no congenital or chromosomal abnormalities in the infants. Infants were singleton births, with gestation to term (≥37 wk). A structured chart review was conducted to collect maternal in-patient information from the delivery. For this analysis, we included 450 of the RICHS participants with available placental HSD11B1 methylation data, and 690 RICHS participants with HSD11B2 methylation data. Anthropometric and clinical data were collected from the in-patient medical record from delivery, and all participants included had available data on infant birth weight, maternal age, and gestational age. Infants were classified as SGA (<10th percentile), large for gestational age (LGA; >90th percentile), or appropriately sized for gestational age (AGA) from birth weight and gestational age data and calculated based upon the 2013 Fenton growth chart [24]. No participants were administered synthetic glucocorticoids (e.g., prednisone, betamethasone, dexamethasone) during the prenatal period.
Placenta Collection, DNA Extraction, and Bisulfite Modification
For each subject and within 2 h of delivery, 12 samples of placenta parenchyma, 3 samples from each of 4 quadrants (totaling approximately 8–10 g of tissue) were excised. All samples were taken from the fetal side of the placenta, 2 cm from the umbilical cord insertion site, free of maternal decidua. The samples were placed immediately in RNAlater (Life Technologies, Grand Island, NY) and stored at 4°C. At least 72 h later, placenta samples were removed from RNAlater, blotted dry, snap frozen in liquid nitrogen, homogenized by pulverization using a stainless steel cup and piston unit (Cellcrusher, Cork, Ireland), and stored at −80°C until needed for examination. DNA was extracted from the placenta samples using the Qiagen DNAeasy Blood and Tissue Kit (Qiagen, Inc., Valencia, CA). Purified DNA was quantified using an ND-2000 spectrophotometer (ThermoFisher Scientific Inc., Waltham, MA), and DNA samples (500 ng) were bisulfite modified using the EZ DNA Methylation Kit (Zymo Research, Irvine, CA) and stored at −20°C.
Methylation Assay Design and Bisulfite Pyrosequencing
HSD11B1 pyrosequencing was performed on PCR product amplified from bisulfite-modified DNA within the 5′-untranslated region of the gene that includes a site on the Illumina Infinium 450K methylation array to allow for external validation by other investigators using this technology. The genomic locations for the two sites assessed are chromosome 1: 209877942 and 209877970 (Ensembl release 74) for CpG1 and CpG2, respectively. PCR products were amplified from bisulfite-modified DNA using the Pyromark PCR Kit (Qiagen); the following forward and biotinylated reverse primers were used for amplification: HSD11B1-F, 5′-AGAGTGAGATTTGGTTTGAGTT-3′ and HSD11B1-R, 5′-biotin-AACCAAACTCATAACTATACAAAACTAAA-3′ (IDT Inc., Coralville, IA). Cycling conditions were 94°C for 15 min followed by 45 cycles of 94°C for 20 sec, 55°C for 30 sec, and 72°C for 20 sec, with a final extension of 5 min at 72°C. PCR products were sequenced using a PyroMark MD system and the following sequencing primer (IDT): HSD11B1-seq, 5′-AAATTATTTTTGAAAGATTATTGA-3′. For HSD11B2, pyrosequencing was performed on PCR product amplified from bisulfite-modified DNA, as described previously [11, 20] based on the promoter sequence displaying differential methylation in human placenta from Alikhani-Koopaei et al. [25]. In each assay, the percent methylation at each CpG site was quantified using the Pyro Q-CpG software, version 1.0.11 (Qiagen). All samples examined exhibited a bisulfate conversion rate >95%.
Gene Expression Analysis
Total RNA was extracted from homogenized placenta using the RNeasy Mini Kit (Qiagen) following manufacturer's protocols and quantified using a Nanodrop 2000 (ThermoFisher Scientific). RNA samples were aliquoted and stored at −80°C and samples were thawed only once for expression analysis using the Bio-Rad CFX Connect Real-Time PCR Detection System (Bio-Rad, Hercules, CA). First-strand reaction was performed with Bio-Rad iScript cDNA Synthesis Kit and quantitative PCR reaction with Bio-Rad iQ SYBER Green Supermix following the manufacturer's protocols. Expression of HSD11B1 and HSD11B2 mRNA was measured using primers specific to each gene, with SDHA serving as a referent gene. All reactions were run in triplicate and the sample with the lowest expression served as a reference sample to allow normalization using the ΔΔCt method. Expression of HSD11B1, HSD11B2, and SDHA was measured using the following primers: HSD11B1: F-5′-GCTCTGTAGGTTCTCTCTGTG-3′, R-5′-GTCTGAATTCCTGTTTGCAG-3′; HSD11B2: F-5′-CCTGCTTTATTTGTGAGCCA-3′, R-5′-AGCACAAAGGCCTGAGGTGA-3′; SDHA: F-5′-TGCTCAGTATCCAGTAGTGGA, R-5′-TTCTCTTACCTGTGCTGCAA-3′.
Covariates
Maternal age, prepregnancy maternal body mass index (BMI), and infant sex were included in multivariable models based upon their potential to confound birth weight grouping. Maternal BMI was calculated based upon self-reported height and weight. Maternal age and prepregnancy BMI were treated as a continuous variable, while sex was treated as a binomial factor.
Statistical Analysis
Descriptive statistics were calculated for the patients included in the analysis with a Student t-test used to compare demographics of the HSD11B1 and HSD11B2 cohorts. Pearson correlations were calculated to determine the association between methylation levels within each isoform, as well as across the genes. In addition, methylation levels at CpGs 1 and 2 of HSD11B1 were correlated to HSD11B1 gene expression, while the mean methylation level across the four sites of HSD11B2 was correlated to HSD11B2 expression. We chose to use mean methylation for HSD11B2 due to the high correlation between the CpG sites (>0.6 for all comparisons), in order to reduce the number of comparisons within the analysis.
Differences in methylation of HSD11B1 CpGs 1 and 2 and HSD11B2 mean methylation was stratified by birth weight group and analyzed through by one-way ANOVA. Multiple comparisons were calculated using Tukey honest significant difference test. Adjusted analysis was conducted using a logistic regression model in which the response variable consisted of a binary LGA or AGA/SGA binning and methylation quartile. Statistical significance was determined by P values less than 0.05, and all tests were two-sided. All data was analyzed in R version 3.1.0 (The R-Project, Vienna, Austria) and plotted in GraphPad Prism 6.0 (GraphPad Software Inc., La Jolla, CA).
RESULTS
Descriptive Statistics
Statistics profiling characteristics of patients, as well as methylation status at the monitored sites, are listed in Table 1 for HSD11B1 and HSD11B2, respectively. There were no statistical differences (P > 0.05) between the makeup of individuals included in HSD11B1 or HSD11B2 analysis for any characteristic. In total, 450 placental samples were analyzed for HSD11B1 and 690 for HSD11B2. Mothers from the HSD11B1 data set were, on average, 30.3 yr of age with a prepregnancy BMI of 26.2. Infants were nearly evenly split between male and female, with slightly more males analyzed, at 50.2% of the population.
TABLE 1.
Descriptive statistics for HSD11B1 and HSD11B2 methylation analysis.
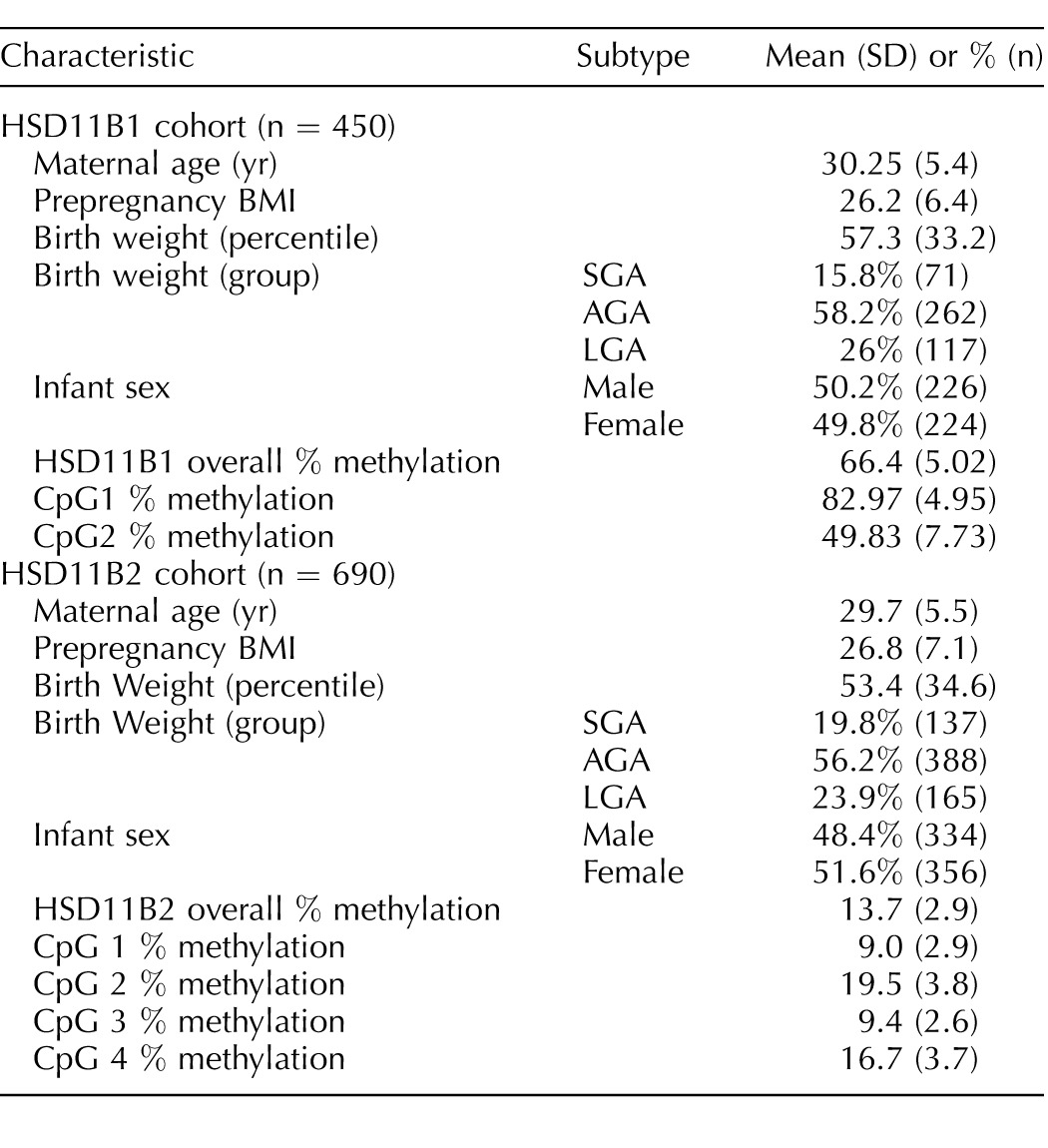
For the HSD11B2 dataset, mothers were, on average, 29.7 yr of age with a prepregnancy BMI of 26.8. Similarly, the numbers of males and females enrolled were nearly identical, with slightly more females (51.6%). AGA infants comprised approximately 56% of those analyzed, with 19.8% SGA infants and 23.9% LGA infants.
HSD11B1 and HSD11B2 Methylation Relationships
Between the two CpG sites interrogated in HSD11B1, we observed a weak correlation (Fig. 1; r = 0.22), while, between the four CpG sites within HSD11B2, there were strong correlations between each of the sites (Fig. 1; r range, 0.66–0.80). Position 1 of HSD11B1 demonstrated weak, negative correlations with each of the CpG sites of HSD11B2 (Fig. 1; r range, −0.16 to −0.21), while no correlations were observed between position 2 of HSD11B1 and methylation of any of the sites within HSD11B2. Due to this correlation structure, we quantified methylation at each of the sites of HSD11B1 individually, but utilized the mean of the sites across HSD11B2 for further analysis.
FIG. 1.
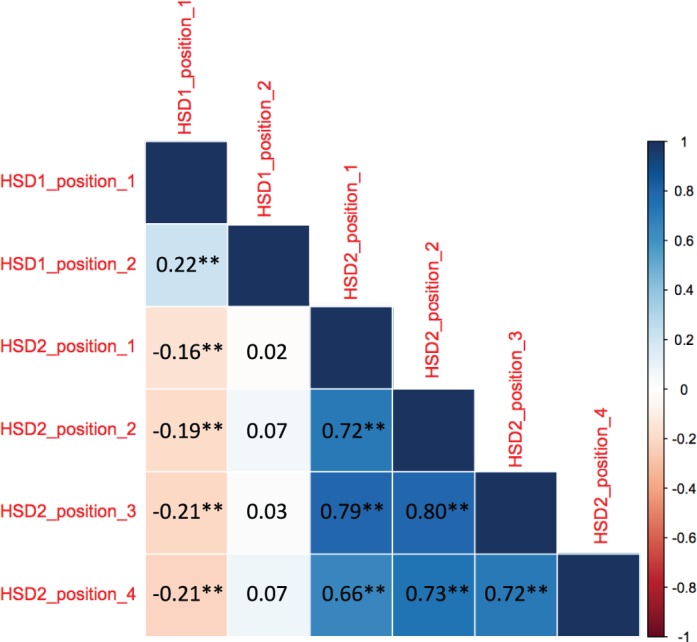
Correlogram of Pearson correlation test for methylation at each individual CpG location measured. Coefficient of correlation (r) value of the relationship is presented for each comparison. **P < 0.01.
HSD11B1 and HSD11B2 Expression
Messenger RNA expression levels of both HSD11B1 and HSD11B2 were examined in a subset of placenta samples (n = 72) in order to determine the extent to which each was controlled by methylation at the measured sites, as well as the association between expression of the two forms of HSD11B. There was no correlation between expression levels of HSD11B1 and HSD11B2 within individuals (r = 0.05, P = 0.62). We observed no correlation between methylation levels at HSD11B1 CpG1 or HSD11B2 and expression of their respective genes. We did, however, see a significant, positive correlation between expression and methylation levels at HSD11B1 CpG2 (r = 0.33, P < 0.01; Fig. 2).
FIG. 2.
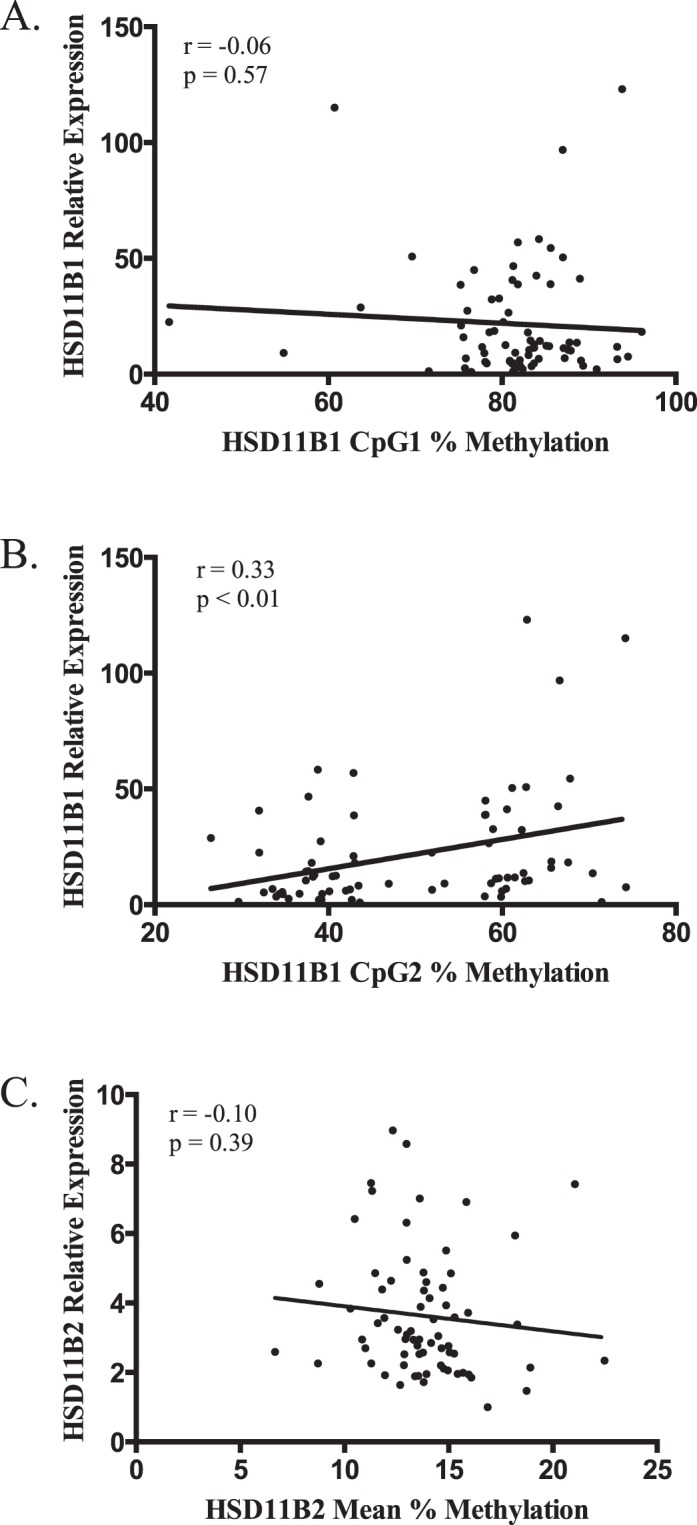
Scatterplots of the correlations between (A) HSD11B1 CpG1, (B) HSD11B1 CpG2, and (C) HSD11B2 mean percent methylation (x axis), and relative expression of the associated gene (y axis). Values of the Pearson correlation coefficient (r) and its P value are presented with each plot.
As we previously noted, infant sex can modify the relationship between DNA methylation and expression of placental leptin [26]; data were stratified by infant sex in order to determine if a similar relationship occurred in HSD11B1 or HSD11B2 (males, n = 38; females, n = 34). Analysis of HSD11B1 methylation yielded similar results when stratified by sex, with a weak negative correlation with expression at CpG1 (males, r = −0.09; females, r = 0.04; Fig. 3, A and B), while CpG2 was positively correlated with expression in both sexes (males, r = 0.44; females, r = 0.24; Fig. 3, C and D). When HSD11B2 data were stratified by sex, however, there was a drastic difference in the role of methylation on gene expression due to sex. Males displayed no correlation between HSD11B2 mean methylation and expression, with an r of 0.002 (P = 0.98). Females, however, demonstrated a moderate negative correlation between expression and mean methylation that approached statistical significance (r = −0.33, P = 0.06; Fig. 3, E and F).
FIG. 3.

Scatterplots of the correlations between (A) HSD11B1 CpG1 males, (B) HSD11B1 CpG1 females, (C) HSD11B1 CpG2 males, (D) HSD11B1 CpG2 females, (E) HSD11B2 Mean males, and (F) HSD11B2 mean female percent methylation (x axis) and relative expression of the associated gene (y axis). Values of the Pearson correlation coefficient (r) and its P value are presented with each plot. For males, n = 38; for females, n = 34.
Relationship Between DNA Methylation and Infant Birth Weight
Methylation levels at CpG1 of HSD11B1 overall were associated with birth weight group (P = 0.017), and, specifically, methylation levels were shown to be significantly lower in LGA infants as compared to AGA infants (P < 0.05; Fig. 4A). We did not observe associations between methylation at HSD11B1 position 2 or the mean of methylation at HSD11B2 and infant birth weight group. Analysis of HSD11B types 1 and 2 methylation and infant birth weight was also stratified by sex to explicitly determine the potential interaction between infant gender and methylation levels. Stratification of the data did not produce significantly different results between male and female infants (data not shown). In a logistic regression model adjusted for maternal age, prepregnancy BMI, and infant sex, individuals with methylation in the lowest quartile for our study population of HSD11B1 CpG1 had odds of being an LGA infant increased by 2.33 times (95% confidence interval [CI] = 1.22–4.44; P < 0.05) as compared to those in the highest quartile. In addition, those in the second-lowest quartile, when compared again to infants with the highest level of methylation, had a risk 1.93 times higher (95% CI = 1.01–3.67; P < 0.05) of being an LGA infant. We also analyzed methylation quartile as a continuous variable to determine whether the resulting changes in infant birth weight were dose dependent, and saw a significant relationship for HSD11B1 CpG1 (P < 0.01), but not CpG2 (Table 2).
FIG. 4.
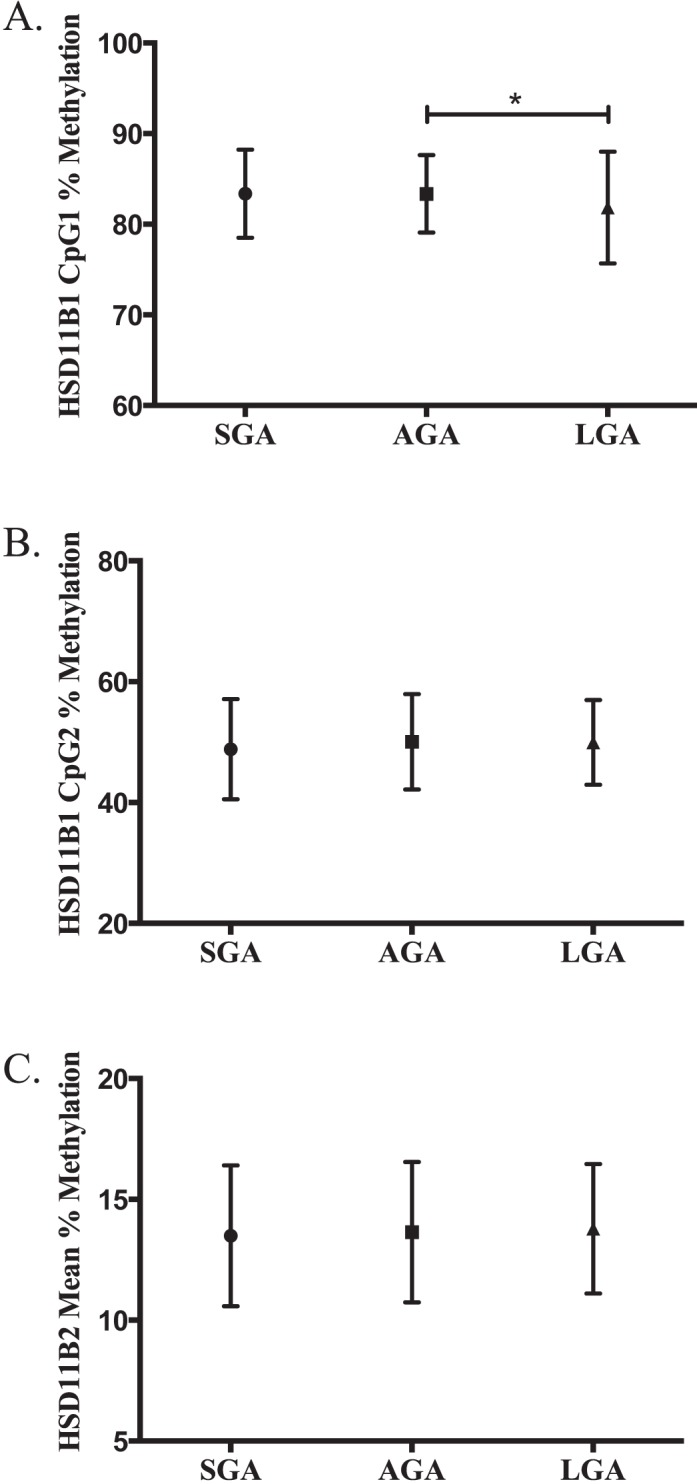
Comparison of the mean extent of methylation of (A) HSD11B1 CpG1, (B) HSD11B1 CpG2, and (C) HSD11B2 mean between birth weight groups. Methylation for HSD11B1 SGA (n = 71), AGA (n = 262), and LGA (n = 117) placentas and HSD11B2 SGA (n = 137), AGA (n = 388), and LGA (n = 165) placentas determined by bisulfite pyrosequencing (HSD11B1, n = 450; HSD11B2, n = 690). Error bars represent SEM. *P < 0.05.
TABLE 2.
Relationship between methylation at HSD11B1 and large-for-gestational-age (LGA) status using logistic regression.*

Adjusted analysis corrected for infant sex, maternal age, and maternal prepregnancy BMI. Each comparison used the fourth quartile of methylation as a referent.
DISCUSSION
In this study, we analyzed placental expression levels of HSD11B1 and HSD11B2, methylation at six sites across the two genes, and the potential association between these genes and infant birth weight. Our results demonstrate that methylation at a single CpG site within HSD11B1 is associated with infant birth weight, and that methylation at several different sites is correlated with gene expression.
Maternal stress, defined to include anxiety and depression, drug use, and tobacco smoke, has been associated with down-regulated expression of HSD11B types 1 and 2 in placental tissue [27–30]. The decrease in expression has been linked with fetal growth restriction [22, 31]. This study is unique because of its novel integration of epigenetic and gene expression data to determine the role of HSD11B1 and HSD11B2 regulation of cortisol in infant birth weight outcomes. The two sites selected for HSD11B1 are both upstream of the gene body, −346 and −318 bp from the transcription start site for CpG1 and CpG2, respectively. This would position them as prime candidates to sit within the promoter region and allow for a potentially large regulatory role in gene expression. This region is sparsely populated by CpG sites, with only eight CpG motifs in total looking 100 bp on either side of the two loci measured. The separation between the sites made it difficult to obtain measurements for all eight sites, which led us to choose two of them within a 28-bp window, for which one is measured on the Illumina 450K chip, allowing for external validation. The sites within HSD11B2 were selected due to having previously been identified as differentially methylated and affecting gene expression within a placenta study [25], classifying this region as a candidate for functional gene control. While we identified no correlation between HSD11B1 CpG1 and expression, there was a strong positive correlation between CpG2 and expression. This is contrary to the common association between DNA promoter methylation and down-regulated gene expression [32]. However, other studies have reported both positive and negative correlations between gene expression and DNA methylation [33, 34], including one that described the relationship between expression and methylation as tissue specific, highlighting the complexity of this relationship [35]. This would suggest that, while the relationship described by our study does not agree with the classical dogma that methylation invariably leads to decreased expression, other instances have introduced exceptions to this rule.
We chose to analyze HSD11B2 methylation levels as the mean of the four sites measured due to the high correlation between the four sites (>0.6 for all comparisons), allowing us to minimize the total number of comparisons within our analysis. Similar to the sites measured for HSD11B1, the four sites measured for HSD11B2 were also within the promoter region of their respective genes, ranging from −419 to −397 bp from the transcription start site. The sites within HSD11B2 were chosen due to previous reports detailing differential methylation within this region [25]. There was a sex-specific correlation between HSD11B2 methylation levels and expression, with females displaying a much stronger correlation between the two factors with no correlation for males. While sex differences in the overall response to prenatal stress has been well described [36], it has not been widely reported in studies focusing on HSD11B2 expression or methylation, although Mericq et al. [7] did describe sex as a mediating factor in gene expression. Many of the previous studies focusing on this gene and birth weight have utilized smaller sample sizes, however [9, 17], likely leaving them underpowered to determine sex-specific differences in comparison to our analysis of DNA methylation and gene expression from 72 infants. It is interesting to note the sexually dimorphic role of methylation on expression in HSD11B2, as there was no sex-specific role of methylation in infant birth weight in either HSD11B type 1 or type 2. This would suggest that the described effect of methylation at HSD11B1 CpG1 on infant birth weight, and the reduced expression of HSD11B2 due to methylation in females, are distinct phenomena with no relative mechanistic association. However, it is also possible that the methylation observed played a greater regulatory role at earlier time points in pregnancy, which we are unable to assess in our term-placenta tissues.
DNA methylation analysis also highlighted HSD11B1 CpG1 as having a significant association with birth weight. We investigated the effect of methylation on birth weight groups (LGA, AGA, or SGA) rather than on infant birth weight to employ the use of binomial logistic regression, which would produce more readily interpretable results than a linear model. An adjusted analysis accounting for potentially confounding factors in which infants were grouped into two categories (LGA vs. SGA/AGA) concluded that infants with methylation in the lowest quartile were 2.33 times more likely to be LGA babies compared to those with methylation in the highest quartile. Those in the second-lowest quartile had a lower risk, though still a significantly higher odds ratio (1.93) of being an LGA infant in comparison to the highest quartile, and a test of trend suggested this was significant. It is important to note that the difference in methylation levels described between LGA and AGA infants in Figure 4, while statistically significant, were very small. There was no correlation between methylation at this site and HSD11B1 gene expression, suggesting that the mechanistic link between methylation and birth weight may not be through a basic relationship between methylation at this site and gene expression. There may be a more complicated relationship beyond what we can observe using this simple correlation, but it could include the activity of various CpGs even distal to the region profiled. In addition, we note that, at least compared to DNA methylation, gene expression as a measure may have more intrinsic variability, and that, although we make extensive efforts to standardize collection, processing, and storage of the samples and resulting RNA, effects beyond our control, including those of labor and delivery, could impact RNA integrity and thus expression measurement. With the current data available, we may only speculate on the mechanism linking birth weight grouping and methylation. It is also difficult to compare these results to other findings, as there have been no publications, to our knowledge, that investigate DNA methylation within the HSD11B1 gene in humans. Several studies have investigated HSD11B1 methylation in animal models; however, a direct comparison to these studies is complicated by the use of different models, as well as the use of different tissues [37, 38].
While this study has many strengths, there were several limitations that warrant addressing. First, samples were obtained from a healthy population, which may have reduced the number of individuals exposed to extreme stressors. This could limit our ability to recruit individuals displaying methylation furthest from the population mean. In addition, our samples were collected from placentas at term, reducing the ability to infer causality within the developing fetus and to understand how these factors play a role throughout development. This also limits our ability to examine cortisol, as the process of labor and delivery would lead to highly increased cortisol levels in mother and infant, thus obfuscating any important variation we would wish to examine. The layout of CpG sites in HSD11B1 allowed us to only measure two CpG loci, and, in conjunction with the complex function of HSD11B1, which has the ability to interconvert cortisol between its active and inactive forms, this makes interpretation of the data and how they relate to birth weight difficult. Even so, our data build upon the previous foundation of knowledge relating to the family of HSD11B genes. While studies exist that have looked at HSD11B1 and HSD11B2, this is the first to comprehensively analyze methylation and gene expression for both at the same time, increasing the overall ability to make associations broadly across the two gene isoforms. Moreover, this investigation utilized a large sample size of healthy infants, increasing the power to identify subtle differences in methylation and gene expression. Our data are consistent with previous publications that have reported a link between HSD11B2 expression and methylation; however, previous studies have not indicated a sex-specific effect, as demonstrated here, potentially due to our large sample size equally distributed across sex.
Our results were able to link DNA methylation levels with birth weight; specifically, that lower methylation levels at HSD11B1 CpG1 led to an increased risk of LGA birth weight classification. In addition, HSD11B1 gene expression was positively correlated with a single CpG site, while HSD11B2 expression was negatively correlated with the mean methylation of four CpG sites within its promoter. These findings, along with previous work conducted by others, suggest that the regulation of fetal stress via HSD11B is a major factor in fetal growth and development, with DNA methylation potentially playing a key role in this process.
Footnotes
Supported by National Institutes of Health (NIH)/National Institute of Mental Health grant R01MH094609, NIH/National Institute of Environmental Health Sciences (NIEHS) grant R01ES022223, NIH/National Cancer Institute grant R25CA134286 to B.B.G., NIH/NIEHS grant P01 ES022832, and by U.S. Environmental Protection Agency (EPA) grant RD83544201. The EPA does not endorse the purchase of any commercial products or services mentioned in this publication.
REFERENCES
- Choe HK, Son GH, Chung S, Kim M, Sun W, Kim H, Geum D, Kim K. Maternal stress retards fetal development in mice with transcriptome-wide impact on gene expression profiles of the limb. Stress. 2011;14:194–204. doi: 10.3109/10253890.2010.529972. [DOI] [PubMed] [Google Scholar]
- Littleton HL, Bye K, Buck K, Amacker A. Psychosocial stress during pregnancy and perinatal outcomes: a meta-analytic review. J Psychosom Obstet Gynaecol. 2010;31:219–228. doi: 10.3109/0167482X.2010.518776. [DOI] [PubMed] [Google Scholar]
- Tamashiro KLK, Moran TH. Perinatal environment and its influences on metabolic programming of offspring. Physiol Behav. 2010;100:560–566. doi: 10.1016/j.physbeh.2010.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Connor T, Heron J, Golding J, Beveridge M, Glover V. Maternal antenatal anxiety and children's behavioural/emotional problems at 4 years: Report from the Avon Longitudinal Study of Parents and Children. Br J Psychiatry. 2002;180:502–508. doi: 10.1192/bjp.180.6.502. [DOI] [PubMed] [Google Scholar]
- Chapman K, Holmes M, Seckl J. 11β-Hydroxysteroid dehydrogenases: intracellular gate-keepers of tissue glucocorticoid action. Physiol Rev. 2013;93:1139–1206. doi: 10.1152/physrev.00020.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan SA, McCabe EL, Gathercole LL, Hassan-Smith ZK, Larner DP, Bujalska IJ, Stewart PM, Tomlinson JW, Lavery GG. 11β-HSD1 is the major regulator of the tissue-specific effects of circulating glucocorticoid excess. Proc Natl Acad Sci U S A. 2014;111:E2482–E2491. doi: 10.1073/pnas.1323681111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mericq V, Medina P, Kakarieka E, Márquez L, Johnson MC, Iñiguez G. Differences in expression and activity of 11β-hydroxysteroid dehydrogenase type 1 and 2 in human placentas of term pregnancies according to birth weight and gender. Eur J Endocrinol. 2009;161:419–425. doi: 10.1530/EJE-09-0308. [DOI] [PubMed] [Google Scholar]
- Muramatsu-Kato K, Itoh H, Kobayashi-Kohmura Y, Murakami H, Uchida T, Suzuki K, Sugihara K, Kanayama N, Tsuchiya KJ, Takei N. Hamamatsu Birth Cohort (HBC) Study Team. Comparison between placental gene expression of 11β-hydroxysteroid dehydrogenases and infantile growth at 10 months of age. J Obstet Gynaecol Res. 2014;40:465–472. doi: 10.1111/jog.12200. [DOI] [PubMed] [Google Scholar]
- Dy J, Guan H, Sampath-Kumar R, Richardson BS, Yang K. Placental 11β-hydroxysteroid dehydrogenase type 2 is reduced in pregnancies complicated with idiopathic intrauterine growth restriction: evidence that this is associated with an attenuated ratio of cortisone to cortisol in the umbilical artery. Placenta. 2008;29:193–200. doi: 10.1016/j.placenta.2007.10.010. [DOI] [PubMed] [Google Scholar]
- McTernan CL, Draper N, Nicholson H, Chalder SM, Driver P, Hewison M, Kilby MD, Stewart PM. Reduced placental 11β-hydroxysteroid dehydrogenase type 2 mRNA levels in human pregnancies complicated by intrauterine growth restriction: an analysis of possible mechanisms. J Clin Endocrinol Metab. 2001;86:4979–4983. doi: 10.1210/jcem.86.10.7893. [DOI] [PubMed] [Google Scholar]
- Appleton A, Armstrong DA, Lesseur C, Lee J, Padbury JF, Lester B, Marsit C. Patterning in placental 11-B hydroxysteroid dehydrogenase methylation according to prenatal socioeconomic adversity. PLoS One. 2013;8:e74691. doi: 10.1371/journal.pone.0074691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen Pena C, Monk C, Champagne FA. Epigenetic effects of prenatal stress on 11β-hydroxysteroid dehydrogenase-2 in the placenta and fetal brain. PLoS One. 2012;7:e39791. doi: 10.1371/journal.pone.0039791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welberg LAM, Seckl JR, Holmes MC. Inhibition of 11β-hydroxysteroid dehydrogenase, the foeto-placental barrier to maternal glucocorticoids, permanently programs amygdala GR mRNA expression and anxiety-like behaviour in the offspring. Eur J Neurosci. 2000;12:1047–1054. doi: 10.1046/j.1460-9568.2000.00958.x. [DOI] [PubMed] [Google Scholar]
- Mulligan CJ, D'Errico NC, Stees J, Hughes DA. Methylation changes at NR3C1 in newborns associate with maternal prenatal stress exposure and newborn birth weight. Epigenetics. 2012;7:853–857. doi: 10.4161/epi.21180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radtke KM, Ruf M, Gunter HM, Dohrmann K, Schauer M, Meyer A, Elbert T. Transgenerational impact of intimate partner violence on methylation in the promoter of the glucocorticoid receptor. Transl Psychiatry. 2011;1:e21. doi: 10.1038/tp.2011.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y, Gong X, Chen L, Li L, Liang Y, Chen S, Zhang Y. Site-specific methylation of placental HSD11B2 gene promoter is related to intrauterine growth restriction. Eur J Hum Genet. 2014;22:734–740. doi: 10.1038/ejhg.2013.226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu W, Weng X, Dong M, Liu Y, Li W, Huang H-F. Alteration in methylation level at 11β-hydroxysteroid dehydrogenase type 2 gene promoter in infants born to preeclamptic women. BMC Genet. 2014;15:96. doi: 10.1186/s12863-014-0096-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okada T, Takahashi S, Nagano N, Yoshikawa K, Usukura Y, Hosono S. Early postnatal alteration of body composition in preterm and small for gestational age infants: implications of catch-up fat. Pediatr Res. 2014 doi: 10.1038/pr.2014.164. [DOI] [PubMed] [Google Scholar]
- Hediger ML, Overpeck MD, McGlynn A, Kuczmarski RJ, Maurer KR, Davis WW. Growth and fatness at three to six years of age of children born small- or large-for-gestational age. Pediatrics. 1999;104:e33. doi: 10.1542/peds.104.3.e33. [DOI] [PubMed] [Google Scholar]
- Marsit C, Maccani MA, Padbury JF, Lester BM. Placental 11-beta hydroxysteroid dehydrogenase methylation is associated with newborn growth and a measure of neurobehavioral outcome. PLoS One. 2014;7:e33794. doi: 10.1371/journal.pone.0033794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filiberto AC, Maccani MA, Koestler DC, Wilhelm-Benartzi C, Avissar-Whiting M, Banister CE, Gagne LA, Marsit CJ. Birthweight is associated with DNA promoter methylation of the glucocorticoid receptor in human placenta. Epigenetics. 2011;6:566–572. doi: 10.4161/epi.6.5.15236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers SL, Hughes BA, Jones CA, Freedman L, Smart K, Taylor N, Stewart PM, Shackleton CHL, Krone NP, Blissett J, Tomlinson JW. Diminished 11β-hydroxysteroid dehydrogenase type 2 activity is associated with decreased weight and weight gain across the first year of life. J Clin Endocrinol Metab. 2014;99:E821–E831. doi: 10.1210/jc.2013-3254. [DOI] [PubMed] [Google Scholar]
- Zuckerman-Levin N, Tsivlin L, Knopf C, Flor O, Shen-Orr Z, Levin M, Hochberg Z. 11B-hydroxysteroid dehydrogenase type 1 activity in short small-for-GA children and in response to GH therapy. Pediatr Res. 2011;70:208–212. doi: 10.1203/PDR.0b013e3182226a0c. [DOI] [PubMed] [Google Scholar]
- Fenton T, Nasser R, Eliasziw M, Kim J, Bilan D, Sauve R. Validating the weight gain of preterm infants between the reference growth curve of the fetus and the term infant. BMC Pediatrics. 2013;13:92. doi: 10.1186/1471-2431-13-92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alikhani-Koopaei R, Fouladkou F, Frey FJ, Frey BM. Epigenetic regulation of 11β-hydroxysteroid dehydrogenase type 2 expression. J Clin Invest. 2004;114:1146–1157. doi: 10.1172/JCI21647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lesseur C, Armstrong DA, Murphy MA, Appleton AA, Koestler DC, Paquette AG, Lester BM, Marsit CJ. Sex-specific associations between placental leptin promoter DNA methylation and infant neurobehavior. Psychoneuroendocrinology. 2014;40:1–9. doi: 10.1016/j.psyneuen.2013.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welberg LAM, Thrivikraman KV, Plotsky PM. Chronic maternal stress inhibits the capacity to up-regulate placental 11β-hydroxysteroid dehydrogenase type 2 activity. J Endocrinol. 2005;186:R7–R12. doi: 10.1677/joe.1.06374. [DOI] [PubMed] [Google Scholar]
- Ponder KL, Salisbury A, McGonnigal B, Laliberte A, Lester B, Padbury JF. Maternal depression and anxiety are associated with altered gene expression in the human placenta without modification by antidepressant use: implications for fetal programming. Dev Psychobiol. 2011;53:711–723. doi: 10.1002/dev.20549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lester BM, Padbury JF. Third pathophysiology of prenatal cocaine exposure. Dev Neurosci. 2009;31:23–35. doi: 10.1159/000207491. [DOI] [PubMed] [Google Scholar]
- Belkacemi L, Jelks A, Chen C-H, Ross M, Desai M. Altered placental development in undernourished rats: role of maternal glucocorticoids. Reprod Biol Endocrinol. 2011;9:105. doi: 10.1186/1477-7827-9-105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy VE, Zakar T, Smith R, Giles WB, Gibson PG, Clifton VL. Reduced 11β-hydroxysteroid dehydrogenase type 2 activity is associated with decreased birth weight centile in pregnancies complicated by asthma. J Clin Endocrinol Metab. 2002;87:1660–1668. doi: 10.1210/jcem.87.4.8377. [DOI] [PubMed] [Google Scholar]
- Smith ZD, Meissner A. DNA methylation: roles in mammalian development. Nat Rev Genet. 2013;14:204–220. doi: 10.1038/nrg3354. [DOI] [PubMed] [Google Scholar]
- van Eijk K, de Jong S, Boks M, Langeveld T, Colas F, Veldink J, de Kovel C, Janson E, Strengman E, Langfelder P, Kahn R, van den Berg L, et al. Genetic analysis of DNA methylation and gene expression levels in whole blood of healthy human subjects. BMC Genomics. 2012;13:636. doi: 10.1186/1471-2164-13-636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner J, Busche S, Ge B, Kwan T, Pastinen T, Blanchette M. The relationship between DNA methylation, genetic and expression inter-individual variation in untransformed human fibroblasts. Genome Biol. 2014;15:R37. doi: 10.1186/gb-2014-15-2-r37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wan J, Oliver VF, Wang G, Zhu H, Zack DJ, Merbs SL, Qian J. Characterization of tissue-specific differential DNA methylation suggests distinct modes of positive and negative gene expression regulation. BMC Genomics. 2015;16:49. doi: 10.1186/s12864-015-1271-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glover V, Hill J. Sex differences in the programming effects of prenatal stress on psychopathology and stress responses: an evolutionary perspective. Physiol Behav. 2012;106:736–740. doi: 10.1016/j.physbeh.2012.02.011. [DOI] [PubMed] [Google Scholar]
- Takaya J, Iharada A, Okihana H, Kaneko K. A calcium-deficient diet in pregnant, nursing rats induces hypomethylation of specific cytosines in the 11β-hydroxysteroid dehydrogenase-1 promoter in pup liver. Nutr Res. 2013;33:961–970. doi: 10.1016/j.nutres.2013.07.015. [DOI] [PubMed] [Google Scholar]
- Takaya J, Iharada A, Okihana H, Kaneko K. Magnesium deficiency in pregnant rats alters methylation of specific cytosines in the hepatic hydroxysteroid dehydrogenase-2 promoter of the offspring. Epigenetics. 2011;6:573–578. doi: 10.4161/epi.6.5.15220. [DOI] [PubMed] [Google Scholar]