

Abstract
Prenatal exposure to elevated testosterone levels induces adult life hypertension associated with selective impairments in endothelium-derived hyperpolarizing factor (EDHF)-mediated relaxation in mesenteric arteries. We tested whether the angiotensin-converting enzyme inhibitor enalapril restores EDHF function through regulating the activities of small (Kcnn3) and intermediate (Kcnn4) conductance calcium-activated potassium channels in mesenteric arteries. Pregnant Sprague-Dawley rats were injected subcutaneously with vehicle or testosterone propionate (0.5 mg/kg/day from Gestation Day 15 to 19), and their 6-mo-old adult male offspring were examined. A subset of rats in these two groups was given enalapril (40 mg/kg/day) for 2 wk through drinking water. Blood pressures were assessed through carotid arterial catheter and endothelium-dependent mesenteric arterial EDHF relaxation, using wire myography. Ace and Kcnn3 and Kcnn4 channel expression levels were also examined. Renal and vascular Ace expression and plasma angiotensin II levels were increased in testosterone offspring. Blood pressure levels were significantly higher in testosterone offspring than in controls, and treatment with enalapril significantly attenuated blood pressure in testosterone offspring. EDHF relaxation in testosterone offspring was reduced compared to that in controls, and it was significantly restored by enalapril treatment. Kcnn4 channel expression and function were similar between control and testosterone rats, but it was not affected by enalapril treatment. Relaxation mediated by Kcnn3 was impaired in testosterone offspring, and it was normalized by enalapril treatment. Furthermore, enalapril treatment restored expression levels of Kcnn3 channels. These findings suggest that enalapril has a positive influence on endothelial function with improvement in EDHF relaxation through normalization of Kcnn3 expression and activity.
Keywords: blood pressure, EDHF, enalapril, endothelium, Kcnn3 channel, mesenteric arteries, pregnancy, testosterone
INTRODUCTION
Endothelium plays a major role in the regulation of vascular tone. It is capable of exerting a profound relaxing influence on underlying smooth muscle through the actions of nitric oxide (NO) and prostacyclin (PGI2) in addition to the more elusive endothelium-derived hyperpolarizing factor (EDHF) [1]. Endothelial hyperpolarization mediated by Ca2+-activated K+ (KCa) channels, such as small (Kcnn3) and intermediate (Kcnn4) conductance KCa channels, has been suggested to play a critical role in initiating EDHF-type relaxation responses in the arteries of many species, including humans [2–5]. The contribution made by EDHF to relaxation is dependent on vessel size. It is suggested that NO is more important in large vessels, whereas EDHF is thought to be more important in small mesenteric arteries, which participate actively in the regulation of systemic peripheral resistance and, thus, blood pressure [6–8]. Also, the expression levels of Kcnn3 and Kcnn4 channels are higher in fourth-order mesenteric arteries than in first-order vessels, stressing the importance of these channels in smaller vessels, where the role of EDHF is more pronounced [8]. Studies using transgenic mice have shown that Kcnn3 and Kcnn4 channels exert a profound hyperpolarizing influence in resistance arteries and that suppression of Kcnn3 [9] and Kcnn4 [10] channel expression or deletion [11] causes pronounced and reversible hypertension. Furthermore, openers of Kcnn3 and Kcnn4 channels reduce blood pressure in mice [12] and dogs [13]. These observations indicate the significance of Kcnn3 and Kcnn4 channels in regulating vasomotor tone and blood pressure. This notion is of particular importance in appreciating the mechanism(s) underlying hypertension and its therapy.
It is well established that hypertension has developmental origins with exposure to adverse insults during the prenatal period, leading to the development of endothelial dysfunction and elevated blood pressure during adult life [14]. It is important to note that endothelial dysfunction has been shown to be a common denominator in all types of hypertension regardless of its pathogenesis [15]. Decreased generation of EDHF, which is particularly critical in resistance arteries, has been shown to contribute to impaired endothelium-dependent vasodilation in genetic as well as developmentally programmed hypertension [16–23]. Recently, reduced expression of Kcnn3 channel mRNA, but not Kcnn4, was observed in mesenteric arteries of rats programmed to develop hypertension following prenatal exposure to testosterone (T) [17]. However, the underlying mechanism that contributes to reduction in expression and function of EDHF components remains largely unclear.
Modulation of the renin-angiotensin system (RAS) by angiotensin-converting enzyme (ACE) inhibitors and angiotensin II (Ang II) receptor blockers has been shown to have vascular protective effects [24–28]. Studies have demonstrated that inhibition of RAS has beneficial endothelial effects because it increases expression and activity of endothelial NO synthase (eNOS) and in turn increases NO bioavailability and function [29]; however, little information is available to indicate whether the RAS blockade might correct EDHF-mediated signaling. Thus, the aims of our study were to assess whether enalapril (an ACE inhibitor) modifies EDHF function and its associated KCa channel expression and functions in mesenteric arteries of prenatally programmed hypertensive rats.
MATERIALS AND METHODS
All experimental procedures were performed in accordance with U.S. National Institutes of Health guidelines (NIH publication no. 85–23, revised 1996) with approval by the Animal Care and Use Committee at the University of Texas Medical Branch. Timed-pregnancy Sprague-Dawley rats (Harlan, Houston, TX) were divided into two groups on Gestational Day 14, and one group received daily subcutaneous injections of T propionate (Sigma, St. Louis, MO) from Gestational Day 15 to 19, 0.5 mg/kg body weight/day (n = 8). The other group received vehicle (sesame oil, n = 8). This dosage and duration of exposure is commonly used to mimic plasma T levels (2-fold increase) observed in pre-eclamptic women [30–33]. Dams in both groups were allowed to deliver at term, and birth weights of pups were recorded. The number of pups in the control and the T litters was adjusted to 10 pups per dam to ensure equal nutrient access for all offspring (pups with weights at each extreme were sacrificed). The male-to-female pup ratios remained equivalent after culling, when possible. Pups were weaned at 3 wk of age, and only the males were used for this study. When pups were 6 mo of age, their mean arterial pressure (MAP) was monitored using a carotid arterial catheter. Following blood pressure measurements, the animals were sacrificed, plasma was separated, and kidney and mesenteric arteries were isolated. A portion of the mesenteric arteries was used for vascular reactivity studies, and the remaining arteries were quickly frozen for RNA isolation. Plasma T and Ang II levels in the samples were measured using a testosterone ELISA kit (Enzo Life Sciences, Farmingdale, NY) and an Ang II enzyme immunoassay kit (Phoenix Pharmaceutical Inc., Burlingame, CA), respectively, as in our previous publications [31,34]. One offspring from each litter was studied, and n refers to the number of litters studied.
Experimental Procedures
Mean arterial pressure.
MAPs were determined in 6-mo-old conscious, freely moving offspring of control and T dams, using indwelling carotid arterial catheters as described in our previous publication [35]. Briefly, rats under anesthesia (isoflurane; Henry Schein Animal Health, Dublin, OH) were surgically instrumented with flexible catheters (PE 50 tubing) in the right carotid artery. Catheters were tunneled to the nape of the neck and exteriorized. After a 24-h recovery period, when the animals were fully conscious and in a free-moving state, the catheter was connected to a pressure transducer, and arterial blood pressure was obtained using a data acquisition system (product DBP001 direct blood pressure system (Kent Scientific, Litchfield, CT) and Workbench for Windows software (Microsoft, Redmond, WA). Following a 30-min stabilization period, the arterial pressure was monitored continuously for 30 min and averaged to determine baseline values.
Enalapril treatment.
A subset of 6-mo-old male offspring was given the ACE inhibitor enalapril (250 mg/L) in their drinking water for 2 wk. All treated groups received a similar dose of enalapril, 40 mg/kg/day, based on average daily water consumption, 65 ml/day, and average daily body weight, 0.43 kg; no significant differences were found between groups. Following enalapril treatment, changes in arterial pressure were recorded using an indwelling arterial catheter.
Ex vivo vascular reactivity studies.
Rats were sacrificed by CO2 inhalation, and the mesenteric arcade was removed. Resistance mesenteric arteries (2-mm segments of the fourth-order branch of the superior mesenteric artery, 150- to 200-μm diameter) were dissected free of fat and connective tissue and mounted in Mulvany-style isometric wire myographs (Danish Myotechnology, Aarhaus, Denmark) for vessel reactivity assessment. Vessels were maintained at 37°C in a physiological Krebs buffer composed of (mM): NaCl, 120; NaHCO3, 25; KCl, 4.8; NaH2PO4, 1.2; MgSO4, 1.2; dextrose, 11.0; CaCl2, 1.8 aerated with 95% O2, and 5% CO2 (pH = 7.4). Rings were bathed in 6- ml Krebs buffer and allowed to equilibrate for 60 min before normalizing to an internal diameter of 0.9 of L13.3kPa by using normalization software (Myodata; Danish Myotechnology). The rings were then assessed for vascular function. Data were captured using a Power Lab data acquisition system (AD Instruments, Colorado Springs, CO). The presence of intact endothelium in vascular preparations was confirmed by observing the relaxation response to acetylcholine (ACh; 10−6 M) in rings precontracted with phenylephrine (PE; 10−6 M), as described previously [36]. In our preliminary experiment, the concentration response curves for PE-induced contractions were not different among the four groups. Once the PE-induced contraction (approximately at 80% of maximally effective dose, i.e., ED80 concentration) had stabilized, relaxation responses to cumulative concentrations of ACh (10−10 to 10−5 M) were elicited. The EDHF-mediated component of ACh vasorelaxation was assessed after inhibiting NO production with NG-nitro-L-arginine methyl ester (L-NAME; 10−4 M) and PGI2 synthesis with indomethacin (10−5 M) for 30 min. In some experiments, the EDHF component of ACh relaxation was generated in the presence of the Kcnn3 inhibitor apamin (10−7 M for 30 min) [4, 37, 38] or the Kcnn4 inhibitor TRAM-34 (10−6 M for 30 min) [4, 37, 38]. Endothelium-independent relaxation responses to levcromakalim (10−10 to 10−5 M) in PE-precontracted rings were also determined.
Quantitative Real-Time qRT-PCR
Total RNA was isolated from mesenteric arties by using TRIzol reagent (Invitrogen, Carlsbad, CA). All RNA isolates were made DNA free by treatment with DNase and further purified with an RNeasy clean-up kit (QIAGEN, Inc., Valencia, CA). Total RNA concentration and purity were determined using an ND-1000 model Nanodrop spectrophotometer (Thermo Fisher Scientific, Newark, DE). Two micrograms of total RNA were reverse transcribed using a modified Maloney murine leukemia virus-derived reverse transcriptase (New England Biolabs Inc., Ipswich, MA) and a blend of oligo(dT) and random hexamer primers (Invitrogen). The reaction was carried out at 28°C for 15 min and then at 42°C for 50 min. It was stopped by heating at 94°C for 5 min, followed by 4°C, and then stored at −20°C until further analysis. One microliter of the resulting cDNA was amplified by real-time RT-PCR, using SYBR Green (Bio-Rad, Hercules, CA) as fluorophore in a CFX96 model real-time thermal cycler (Bio-Rad). Specific pairs of primers from published literature were used for each gene amplification: Kcnn3 forward: 5′-GCATCTCTCTGTGGATCATTGC-3′, and reverse: 5′-AATCTGCTTCTCCAGGTCTTCG-3′; Kcnn4 forward: 5′-CTACTGCACAGCAAAATCTTCACG-3′, and reverse: 5′-CCTGGTATGTTTGTAGATGAGCCAC-3′; Ace forward: 5′-CAGAGGCCAACTGGCATTAT-3′, and reverse: 5′-CTGGAAGTTGCTCACGTCAA-3′; and β-actin forward: 5′-CGTGAAAAGATGACCCAGATC-3′, and reverse: 5′-CACAGCCTAGATGGCTACGT-3.′ PCR conditions were 10 min at 95°C for 1 cycle, 15 sec at 95°C, 30 sec at 60°C, and 15 sec at 72°C for 40 cycles, with a final dissociation step (0.05 sec at 65°C and 0.5 sec at 95°C). Results were calculated using the 2–ΔΔCT method and expressed as fold-level increase or decrease of gene expressions in T offspring versus that in control rats. All reactions were performed in duplicate, and β-actin was used as an internal control.
Statistical Analysis
Differences in tension between PE contraction and basal tension were considered maximal tension (100 %), and relaxation to ACh was expressed as the percentage of relaxation from the maximal response induced by PE. Cumulative concentration-response curves were analyzed by computer fitting to a four-parameter sigmoid curve, using Prism 6 software (GraphPad, San Diego, CA) to evaluate the Emax, the maximum asymptote of the curve. All values are means ± SEM. For comparison, a two-way ANOVA test and a Bonferroni post hoc test were performed. A P value of <0.05 was considered significant.
RESULTS
Plasma T and Ang II Levels and Ace Expression
Plasma T levels were significantly increased in T males (2.4 ± 0.35 ng/ml; n = 7; P ≤ 0.05) compared with that in controls (1.5 ± 0.17 ng/ml; n = 7). Plasma Ang II levels were significantly increased in T males (1.4 ± 0.41 ng/ml; n = 7; P ≤ 0.05) compared with that in controls (0.8 ± 0.04 ng/ml; n = 7). Ace mRNA expression was significantly increased in the kidney and mesenteric artery of T rats compared to that in controls (P < 0.05; n = 5 in each group) (Fig. 1).
FIG. 1.
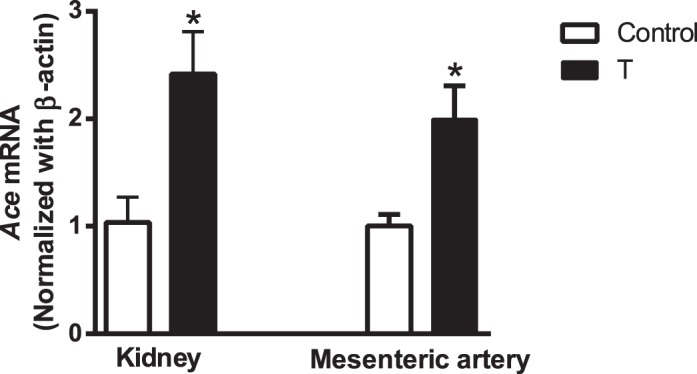
Ace mRNA expression in kidney and mesenteric arteries of control and T rats. Real-time RT-PCR was used to assess vascular Ace mRNA expression. Quantitation of Ace was normalized relative to that of β-actin levels. Values are means ± SEM of 5 rats in each group. *P ≤ 0.05 versus control.
Effects of Enalapril Treatment on Systemic Blood Pressure
MAP was increased significantly in T rats (133 ± 2.5 mm Hg; n = 6) compared with that in control rats (119 ± 2.3 mm Hg; n = 7; P < 0.05) (Fig. 2). Enalapril treatment did not affect MAP in control rats (113 ± 4.2 mm Hg; n = 7), but it significantly lowered MAP in T rats (115 ± 4.5 mm Hg; n = 7; P < 0.05) (Fig. 2).
FIG. 2.
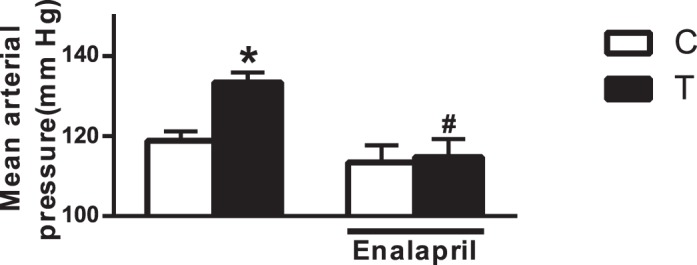
Effect of ACE inhibition on hypertension. ACE inhibitor (enalapril, 0.25 g/L through drinking water) was administered for 2 wk to 6-mo-old offspring, and changes in MAP were measured using a carotid catheter. Values are means ± SEM of 6–7 animals in each group of animals.*P < 0.05 versus control (C); #P < 0.05 versus T counterpart.
Effects of Enalapril Treatment on EDHF-Mediated Relaxation
In order to investigate EDHF-mediated relaxation in the rat mesenteric artery, we examined ACh-induced relaxation in the presence of L-NAME and indomethacin. ACh-induced EDHF-mediated relaxation was significantly lower in rings from T rats (n = 6, P ≤ 0.05) than in those from controls rats (n = 6) (Fig. 3 and Table 1). Enalapril treatment caused no significant alteration in the EDHF relaxation in controls (n = 6) (Fig. 3 and Table 1). In contrast, enalapril treatment of T rats led to a significant enhancement of the EDHF relaxation compared to that in untreated T rats (n = 6) (Fig. 3 and Table 1).
FIG. 3.
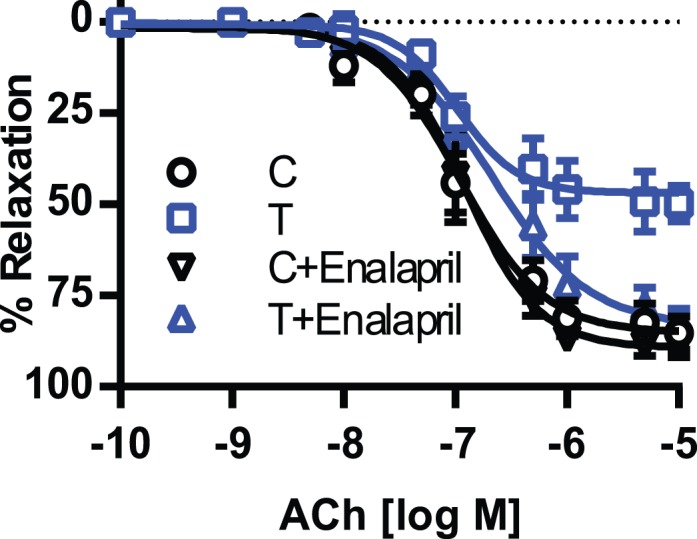
Effect of enalapril treatment on endothelium-dependent EDHF-mediated vascular relaxation in mesenteric arteries. Arterial rings were isolated from control (C) and T rats with and without enalapril treatment, contracted with PE, and examined for relaxation to ACh in the presence of the NOS inhibitor L-NAME (10−4 M) and with the PGI2 blocker indomethacin (10−5 M). Values are means ± SEM (n = 6 rats, 2 vessel segments/rat).
TABLE 1.
Vascular function (Emax values) in offspring of control and T rats with and without enalapril treatment.a

Values are means ± SEM of 10–12 mesenteric arterial rings from 5–7 rats in each group. Emax is a percentage of PE relaxation.
P < 0.05, compared to control group without enalapril.
P < 0.05, compared to T rats without enalapril.
P < 0.05, compared to EDHF-mediated relaxation in their respective control and T groups.
Effects of Enalapril Treatment on Kcnn3 Channel Function
To examine the contribution of Kcnn3 channels in EDHF-mediated relaxation, rings were incubated with apamin, a Kcnn3-channel inhibitor. Preincubation with apamin significantly attenuated ACh-induced EDHF relaxation to a similar magnitude in mesenteric arteries from control rats with and without enalapril treatment (n = 6 in each group) (Fig. 4, A and C, and Table 1). On the other hand, apamin did not affect EDHF relaxation in T rats not treated with enalapril, but it significantly attenuated the EDHF relaxation in T rats treated with enalapril (n = 6–7 in each group) (Fig. 4, B and D, and Table 1).
FIG. 4.

Effect of the Kcnn3 channel inhibitor apamin on EDHF-mediated vascular relaxation in mesenteric arteries from control (A) and T (B) rats. ACE inhibitor enalapril was administered for 2 wk to control (C) and T (D) rats, and the changes in EDHF-mediated vascular relaxation were determined. Arterial rings were contracted with PE and examined for relaxation to ACh in the presence of apamin (10−7 M), the NOS inhibitor L-NAME (10−4M), and the PGI2 blocker indomethacin (10−5M). Values are means ± SEM (n = 6–7 rats, 2 vessel segments/rat). C, control.
Effects of Enalapril Treatment on Kcnn4 Channel Function
To examine the part played by the Kcnn4 channel in the EDHF-mediated relaxation, rings were incubated with TRAM-34, a Kcnn4 channel inhibitor. Presence of TRAM-34 resulted in reductions in EDHF relaxation responses to a magnitude similar to that in the mesenteric arteries of controls and T rats with and without enalapril treatment (n = 6–7 in each group) (Fig. 5 and Table 1).
FIG. 5.

Effect of the Kcnn4 channel inhibitor TRAM-34 on EDHF-mediated vascular relaxation in mesenteric arteries from control (A) and T (B) rats. ACE inhibitor enalapril was administered for 2 wk to control (C) and T (D) rats, and the changes in EDHF-mediated vascular relaxation were determined. Arterial rings were contracted with PE and examined for relaxation to ACh in the presence of TRAM-34 (10−6 M), the NOS inhibitor L-NAME (10−4M), and the PGI2 blocker indomethacin (10−5M). Values are means ± SEM (n = 6–7 rats, 2 vessel segments/rat). C, control.
Effect of Enalapril Treatment on Endothelium-Independent Levcromakalim-Induced Relaxation Responses
Vascular relaxation to levcromakalim, an ATP-sensitive K+ channel opener used to determine the sensitivity of vascular smooth muscle cells to EDHF [39], was not significantly different between the control and T rats with and without enalapril treatment (n = 5 in each group) (Fig. 6 and Table 1).
FIG. 6.
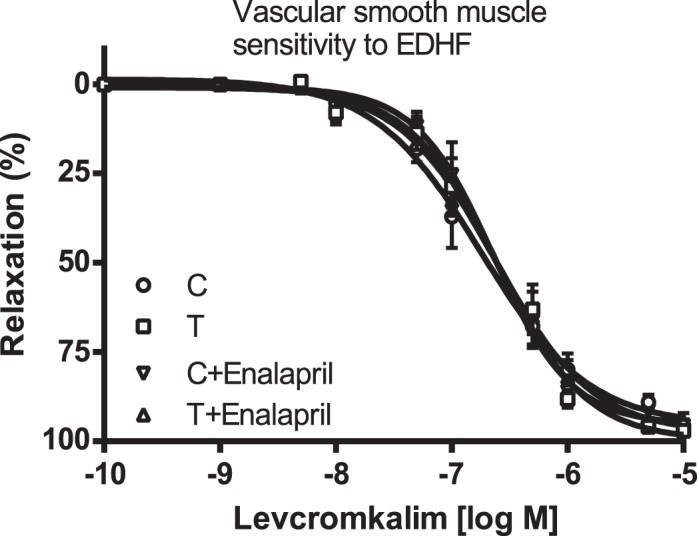
Effect of enalapril treatment on endothelium-independent EDHF relaxation. Arterial rings were isolated from control (C) and T rats with and without enalapril treatment, contracted with PE, and examined for relaxation to cumulative additions of levcromakalim. Values are means ± SEM (n = 5 rats from each group, 2 vessel segments/rat).
Effect of Enalapril Treatment on Expression of Kcnn3 and Kcnn4
Kcnn3 expression was decreased by 50% in T rats (n = 5; P ≤ 0.05) compared with that in controls (n = 5) (Fig 7, left panel). Enalapril treatment increased Kcnn3 expression in T rats but did not have any effect in controls (n = 5 in each group; P ≤ 0.05). Expression of Kcnn4 in control rats was not different from that in T rats (n = 5) (Fig. 7), and enalapril treatment did not have an effect on the expression of Kcnn4 in either controls or T rats (n = 5) (Fig 7). Our attempt to determine Kcnn3 and Kcnn4 protein expression was less conclusive, with many bands at inappropriate molecular weights. Similar difficulties in determining Kcnn3 and Kcnn4 protein expression have been reported [40].
FIG. 7.
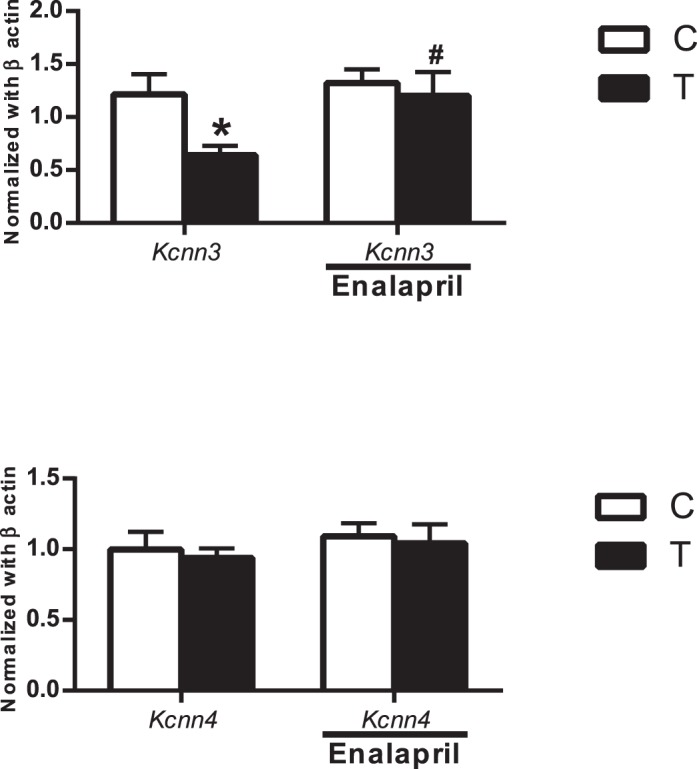
Effect of enalapril treatment on Kcnn3 and Kcnn4 channel mRNA expression in mesenteric arteries of control (C) and T rats with and without enalapril treatment. Real-time RT-PCR was used to assess vascular Kcnn3 and Kcnn4 mRNA expression. Quantitation of vascular Kcnn3 and Kcnn4 components was normalized relative to β-actin levels. Values are means ± SEM of five rats in each group. *P ≤ 0.05 versus control. #P < 0.05 versus T counterpart.
DISCUSSION
We investigated the role of enalapril in modifying EDHF-mediated relaxation responses in mesenteric arteries. The present study confirms our previous finding that prenatal T exposure induces hypertension and impairs the endothelial EDHF component of vasodilation in resistance arteries [17] and expands on it by demonstrating that this impairment is reversed by enalapril treatment. In addition, we demonstrated that enalapril specifically restored the expression and function of the Kcnn3 channel in mesenteric arteries of prenatal T-exposed rats. These findings could explain, at least in part, the importance of RAS signaling in EDHF dysfunction and to the elevated blood pressure observed in prenatal T-exposed rats [17, 36, 41, 42].
It is well established that a variety of insults, when experienced in the prenatal period, can have long-term influences on the health of the individual. Factors, such as maternal undernutrition [43] and placental insufficiency [44], which lead to impaired fetal growth, are known to cause hypertension and other cardiovascular abnormalities in the offspring. We and others have previously reported that elevated maternal T levels are an important stimulus for increased blood pressure in offspring [17, 36, 41, 42]. We not only confirmed the fact that elevated T levels in pregnant rats increase arterial pressure in the offspring, but we also provided a new finding that treatment with the ACE inhibitor enalapril abolished the hypertensive response, which suggests that RAS activation contributes, in part, to increased blood pressure in T rats. The finding of increased ACE expression in the kidney and mesenteric artery along with increased plasma levels of Ang II substantiates the fact that T rats have inappropriate RAS activation.
Because kidneys play an important role in long-term control of blood pressure [45], the decrease in blood pressure in T rats following enalapril treatment could be due to renal ACE inhibition. To determine the effect of enalapril treatment on endothelial function, we examined EDHF function in isolated mesenteric arteries. We previously showed that prenatal T exposure led to endothelial dysfunction with specific impairments in EDHF function [17]. An endothelial dysfunction involving a selective blunting of EDHF-mediated relaxation in the mesenteric artery has also been observed in spontaneously hypertensive rats [46] and in Ang II-treated rats [21]. In the present study, we observed that treatment with the ACE inhibitor enalapril prevented endothelial dysfunction in T rats, as indicated by normal EDHF-mediated relaxations in mesenteric arteries, suggesting that Ang II may be a causal factor contributing to the underlying impairment of the EDHF-mediated response observed in T rats. This observation is consistent with the reported improvement in EDHF function in the conduit internal thoracic artery of hypertensive patients treated with ACE inhibitors [47]. The absence of differences in relaxation responses to levcromakalim between T and control rats with and without enalapril treatment suggests that the enhanced EDHF relaxation in T rats with enalapril is not due to increased vascular smooth muscle sensitivity to EDHF but may be related to improvement in EDHF generation.
Kcnn3 and Kcnn4 channels located in the endothelium are known to play an important role in initiation of hyperpolarization [2–5]. We analyzed the effect of enalapril treatment on the EDHF function with the specific roles of Kcnn3 and Kcnn4 channels. The Kcnn3 channel contribution for EDHF relaxation was similar in controls with and without enalapril treatment, as apamin caused a similar magnitude of inhibition in these groups. The present findings of diminished EDHF relaxation and failure of apamin to alter EDHF relaxation in mesenteric arteries of T rats suggest that prenatal T exposure resulted in a loss of the regulatory role of Kcnn3 channels in initiating hyperpolarization. Our data also suggest that the loss of the regulatory role of Kcnn3 channels in mesenteric arteries of T rats resulted chiefly from reduced channel activities due to suppressed expression of these channels consistent with previous findings [17]. Interestingly, the present study shows that enalapril treatment to T rats restores activities of the Kcnn3 channel as the enhanced EDHF relaxation observed in enalapril-treated T rats was inhibited following Kcnn3 blockade (compared to Kcnn3 blockade, which had no effect in T rats not treated with enalapril). The present finding of enhanced Kcnn3 channel activities in EDHF-mediated relaxation in mesenteric arteries of enalapril-treated T rats correlates well with the restored expression of Kcnn3 channels. The effect of prenatal T exposure and enalapril treatment seems to be specific for Kcnn3 channels, as Kcnn4 channel expression and function were unaffected (TRAM-34 inhibits EDHF relaxation to a similar magnitude in control and T rats with and without enalapril treatment). Taken together, experimental evidence suggests that targeted suppression of Kcnn3 channels is a major mechanism for impaired EDHF relaxation in T rats and that enalapril treatment restores this effect. The present findings are in agreement with previous ones indicating that chronic infusion of Ang II to rats caused a selective downregulation of Kcnn3 levels and attenuation of the Kcnn3 component of EDHF relaxation [22]. It may be possible that the improvement in EDHF function observed in hypertensive patients treated with ACE inhibitor [47] may be due to an improvement in KCNN3 channel function.
Presently, the mechanisms by which enalapril upregulates expression and function of Kcnn3 channels in mesenteric arteries of T rats are not clear. It is conceivable that inhibition of Ang II signaling may contribute to this upregulation. Ang II is known to attenuate vasodilator function via an AT1R/superoxide-dependent pathway [48, 49]. However, reports are contradictory as reactive oxygen species are shown to have no effect [22] or decrease [50, 51] Kcnn3 channel expression and function. Furthermore, whether the improvement in Kcnn3 channel expression and function is the effect of inhibition of Ang II signaling or a consequence of an enalapril-induced decrease in blood pressure is difficult to discern. Bennett et al. [52] showed that long-term antihypertensive treatments with ACE inhibitors, not with hydralazine or amlodipine, increased relaxations in response to ACh and bradykinin in mesenteric resistance arteries from spontaneously hypertensive rats. Thus, the effect of ACE inhibitors on endothelium-dependent relaxations seems to be independent, at least in part, of their antihypertensive actions. Further studies are needed to examine the exact mechanism by which enalapril regulates Kcnn3 channel expression and function.
In conclusion, the present study demonstrates that prenatal exposure to elevated androgen levels results in hypertension and blunting of the ability of endothelial cells to generate normal hyperpolarizing responses due to impairment of Kcnn3 channel function. These alterations were prevented by ACE inhibition with enalapril, indicating a negative influence of RAS on EDHF responses and specifically Kcnn3 function. These findings not only support the beneficial effects of RAS blockers in inhibiting direct vascular smooth muscle contractile effects but also offer an explanation for the beneficial effects on the endothelial EDHF function. We believe our findings should stimulate further interest in RAS blockade as a potential therapeutic target for use against small-vessel vasculopathy.
Footnotes
Supported by U.S. National Institutes of Health (NIH) grants HD069750 and HL119869 to K.S. and HL102866 and HL 58144 to C.Y. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
REFERENCES
- Nilius B, Droogmans G. Ion channels and their functional role in vascular endothelium. Physiol Rev. 2001;81:1415–1459. doi: 10.1152/physrev.2001.81.4.1415. [DOI] [PubMed] [Google Scholar]
- Crane GJ, Gallagher N, Dora KA, Garland CJ. Small- and intermediate-conductance calcium-activated K+ channels provide different facets of endothelium-dependent hyperpolarization in rat mesenteric artery. J Physiol. 2003;553:183–189. doi: 10.1113/jphysiol.2003.051896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eichler I, Wibawa J, Grgic I, Knorr A, Brakemeier S, Pries AR, Hoyer J, Kohler R. Selective blockade of endothelial Ca2+-activated small- and intermediate-conductance K+-channels suppresses EDHF-mediated vasodilation. Br J Pharmacol. 2003;138:594–601. doi: 10.1038/sj.bjp.0705075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feletou M, Vanhoutte PM. EDHF: an update. Clin Sci (Lond) 2009;117:139–155. doi: 10.1042/CS20090096. [DOI] [PubMed] [Google Scholar]
- Hinton JM, Langton PD. Inhibition of EDHF by two new combinations of K+-channel inhibitors in rat isolated mesenteric arteries. Br J Pharmacol. 2003;138:1031–1035. doi: 10.1038/sj.bjp.0705171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stankevicius E, Dalsgaard T, Kroigaard C, Beck L, Boedtkjer E, Misfeldt MW, Nielsen G, Schjorring O, Hughes A, Simonsen U. Opening of small and intermediate calcium-activated potassium channels induces relaxation mainly mediated by nitric-oxide release in large arteries and endothelium-derived hyperpolarizing factor in small arteries from rat. J Pharmacol Exp Ther. 2011;339:842–850. doi: 10.1124/jpet.111.179242. [DOI] [PubMed] [Google Scholar]
- Feletou M, Vanhoutte PM. Endothelial dysfunction: a multifaceted disorder (The Wiggers Award Lecture) Am J Physiol Heart Circ Physiol. 2006;291:H985–1002. doi: 10.1152/ajpheart.00292.2006. [DOI] [PubMed] [Google Scholar]
- Hilgers RH, Todd J, Jr, , Webb RC. Regional heterogeneity in acetylcholine-induced relaxation in rat vascular bed: role of calcium-activated K+ channels. Am J Physiol Heart Circ Physiol. 2006;291:H216–H222. doi: 10.1152/ajpheart.01383.2005. [DOI] [PubMed] [Google Scholar]
- Taylor MS, Bonev AD, Gross TP, Eckman DM, Brayden JE, Bond CT, Adelman JP, Nelson MT. Altered expression of small-conductance Ca2+-activated K+ (SK3) channels modulates arterial tone and blood pressure. Circ Res. 2003;93:124–131. doi: 10.1161/01.RES.0000081980.63146.69. [DOI] [PubMed] [Google Scholar]
- Si H, Heyken WT, Wolfle SE, Tysiac M, Schubert R, Grgic I, Vilianovich L, Giebing G, Maier T, Gross V. Bader M. de WC, Hoyer J, Kohler R. Impaired endothelium-derived hyperpolarizing factor-mediated dilations and increased blood pressure in mice deficient of the intermediate-conductance Ca2+-activated K+ channel. Circ Res. 2006;99:537–544. doi: 10.1161/01.RES.0000238377.08219.0c. [DOI] [PubMed] [Google Scholar]
- Brahler S, Kaistha A, Schmidt VJ, Wolfle SE, Busch C, Kaistha BP, Kacik M, Hasenau AL, Grgic I, Si H, Bond CT, Adelman JP, et al. de WC, Hoyer J, Kohler R. Genetic deficit of SK3 and IK1 channels disrupts the endothelium-derived hyperpolarizing factor vasodilator pathway and causes hypertension. Circulation. 2009;119:2323–2332. doi: 10.1161/CIRCULATIONAHA.108.846634. [DOI] [PubMed] [Google Scholar]
- Sankaranarayanan A, Raman G, Busch C, Schultz T, Zimin PI, Hoyer J, Kohler R, Wulff H. Naphtho1,2-d]thiazol-2-ylamine (SKA-31), a new activator of KCa2 and KCa3.1 potassium channels, potentiates the endothelium-derived hyperpolarizing factor response and lowers blood pressure. Mol Pharmacol. 2009;75:281–295. doi: 10.1124/mol.108.051425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Damkjaer M, Nielsen G, Bodendiek S, Staehr M. Gramsbergen JB de WC, Jensen BL, Simonsen U, Bie P, Wulff H, Kohler R. Pharmacological activation of KCa3.1/KCa2.3 channels produces endothelial hyperpolarization and lowers blood pressure in conscious dogs. Br J Pharmacol. 2012;165:223–234. doi: 10.1111/j.1476-5381.2011.01546.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ligi I, Grandvuillemin I, Andres V, Dignat-George F, Simeoni U. Low birth weight infants and the developmental programming of hypertension: a focus on vascular factors. Semin Perinatol. 2010;34:188–192. doi: 10.1053/j.semperi.2010.02.002. [DOI] [PubMed] [Google Scholar]
- Giles TD, Sander GE, Nossaman BD, Kadowitz PJ. Impaired vasodilation in the pathogenesis of hypertension: focus on nitric oxide, endothelial-derived hyperpolarizing factors, and prostaglandins. J Clin Hypertens (Greenwich) 2012;14:198–205. doi: 10.1111/j.1751-7176.2012.00606.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray C, Li M, Reynolds CM, Vickers MH. Pre-weaning growth hormone treatment reverses hypertension and endothelial dysfunction in adult male offspring of mothers undernourished during pregnancy. PLoS One. 2013;8:e53505. doi: 10.1371/journal.pone.0053505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chinnathambi V, Yallampalli C, Sathishkumar K. Prenatal testosterone induces sex-specific dysfunction in endothelium-dependent relaxation pathways in adult male and female rats. Biol Reprod. 2013;89:1–9. doi: 10.1095/biolreprod.113.111542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weston AH, Porter EL, Harno E, Edwards G. Impairment of endothelial SK(Ca) channels and of downstream hyperpolarizing pathways in mesenteric arteries from spontaneously hypertensive rats. Br J Pharmacol. 2010;160:836–843. doi: 10.1111/j.1476-5381.2010.00657.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goto K, Fujii K, Kansui Y, Iida M. Changes in endothelium-derived hyperpolarizing factor in hypertension and ageing: response to chronic treatment with renin-angiotensin system inhibitors. Clin Exp Pharmacol Physiol. 2004;31:650–655. doi: 10.1111/j.1440-1681.2004.04054.x. [DOI] [PubMed] [Google Scholar]
- Yang Z, Kaye DM. Endothelial dysfunction and impaired L-arginine transport in hypertension and genetically predisposed normotensive subjects. Trends Cardiovasc Med. 2006;16:118–124. doi: 10.1016/j.tcm.2006.02.003. [DOI] [PubMed] [Google Scholar]
- Dal-Ros S, Bronner C, Schott C, Kane MO, Chataigneau M, Schini-Kerth VB, Chataigneau T. Angiotensin II-induced hypertension is associated with a selective inhibition of endothelium-derived hyperpolarizing factor-mediated responses in the rat mesenteric artery. J Pharmacol Exp Ther. 2009;328:478–486. doi: 10.1124/jpet.108.145326. [DOI] [PubMed] [Google Scholar]
- Hilgers RH, Webb RC. Reduced expression of SKCa and IKCa channel proteins in rat small mesenteric arteries during angiotensin II-induced hypertension. Am J Physiol Heart Circ Physiol. 2007;292:H2275–H2284. doi: 10.1152/ajpheart.00949.2006. [DOI] [PubMed] [Google Scholar]
- Torrens C, Brawley L, Anthony FW, Dance CS, Dunn R, Jackson AA, Poston L, Hanson MA. Folate supplementation during pregnancy improves offspring cardiovascular dysfunction induced by protein restriction. Hypertension. 2006;47:982–987. doi: 10.1161/01.HYP.0000215580.43711.d1. [DOI] [PubMed] [Google Scholar]
- Antony I, Lerebours G, Nitenberg A. Angiotensin-converting enzyme inhibition restores flow-dependent and cold pressor test-induced dilations in coronary arteries of hypertensive patients. Circulation. 1996;94:3115–3122. doi: 10.1161/01.cir.94.12.3115. [DOI] [PubMed] [Google Scholar]
- Kishi Y, Ohta S, Kasuya N, Sakita SY, Ashikaga T, Isobe M. Perindopril augments ecto-ATP diphosphohydrolase activity and enhances endothelial anti-platelet function in human umbilical vein endothelial cells. J Hypertens. 2003;21:1347–1353. doi: 10.1097/00004872-200307000-00024. [DOI] [PubMed] [Google Scholar]
- Luscher TF, Wenzel RR, Moreau P, Takase H. Vascular protective effects of ACE inhibitors and calcium antagonists: theoretical basis for a combination therapy in hypertension and other cardiovascular diseases. Cardiovasc Drugs Ther. 1995;9(suppl 3):509–523. doi: 10.1007/BF00877863. [DOI] [PubMed] [Google Scholar]
- Messerli FH. Vascular protection of ACE inhibitors. Arch Intern Med. 2003;163:2791–2792. doi: 10.1001/archinte.163.22.2791-b. [DOI] [PubMed] [Google Scholar]
- Yang D, Zhang M, Huang X, Fang F, Chen B, Wang S, Cai J, Shi X, Qu J, Geng YJ. Protection of retinal vasculature by losartan against apoptosis and vasculopathy in rats with spontaneous hypertension. J Hypertens. 2010;28:510–519. doi: 10.1097/HJH.0b013e328333663f. [DOI] [PubMed] [Google Scholar]
- Morawietz H, Rohrbach S, Rueckschloss U, Schellenberger E, Hakim K, Zerkowski HR, Kojda G, Darmer D, Holtz J. Increased cardiac endothelial nitric oxide synthase expression in patients taking angiotensin-converting enzyme inhibitor therapy. Eur J Clin Invest. 2006;36:705–712. doi: 10.1111/j.1365-2362.2006.01715.x. [DOI] [PubMed] [Google Scholar]
- Chinnathambi V, Balakrishnan M, Ramadoss J, Yallampalli C, Sathishkumar K. Testosterone alters maternal vascular adaptations: role of the endothelial NO system. Hypertension. 2013;61:647–654. doi: 10.1161/HYPERTENSIONAHA.111.00486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chinnathambi V, More AS, Hankins GD, Yallampalli C, Sathishkumar K. Gestational exposure to elevated testosterone levels induces hypertension via heightened vascular angiotensin II type 1 receptor signaling in rats. Biol Reprod. 2014;91:6. doi: 10.1095/biolreprod.114.118968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chinnathambi V, Selvanesan BC, Vincent KL, Saade GR, Hankins GD, Yallampalli C, Sathishkumar K. Elevated testosterone levels during rat pregnancy cause hypersensitivity to angiotensin II and attenuation of endothelium-dependent vasodilation in uterine arteries. Hypertension. 2014;64:405–414. doi: 10.1161/HYPERTENSIONAHA.114.03283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sathishkumar K, Elkins R, Chinnathambi V, Gao H, Hankins GD, Yallampalli C. Prenatal testosterone-induced fetal growth restriction is associated with down-regulation of rat placental amino acid transport. Reprod Biol Endocrinol. 2011;9:1–12. doi: 10.1186/1477-7827-9-110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu LL, Liu JL, Jin CJ, Wang AZ, Guan DX, Wu JB, Yuan FH. [Baseflow separation methods in hydrological process research: a review] Ying Yong Sheng Tai Xue Bao. 2011;22:3073–3080. [PubMed] [Google Scholar]
- Sathishkumar K, Elkins R, Yallampalli U, Yallampalli C. Protein restriction during pregnancy induces hypertension in adult female rat offspring—influence of oestradiol. Br J Nutr. 2011:1–9. doi: 10.1017/S0007114511003448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sathishkumar K, Elkins R, Yallampalli U, Balakrishnan M, Yallampalli C. Fetal programming of adult hypertension in female rat offspring exposed to androgens in utero. Early Hum Dev. 2011;87:407–414. doi: 10.1016/j.earlhumdev.2011.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feletou M. Calcium-activated potassium channels and endothelial dysfunction: therapeutic options? Br J Pharmacol. 2009;156:545–562. doi: 10.1111/j.1476-5381.2009.00052.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grgic I, Kaistha BP, Hoyer J, Kohler R. Endothelial Ca+-activated K+ channels in normal and impaired EDHF-dilator responses–relevance to cardiovascular pathologies and drug discovery. Br J Pharmacol. 2009;157:509–526. doi: 10.1111/j.1476-5381.2009.00132.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matoba T, Shimokawa H, Nakashima M, Hirakawa Y, Mukai Y, Hirano K, Kanaide H, Takeshita A. Hydrogen peroxide is an endothelium-derived hyperpolarizing factor in mice. J Clin Invest. 2000;106:1521–1530. doi: 10.1172/JCI10506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chadha PS, Haddock RE, Howitt L, Morris MJ, Murphy TV, Grayson TH, Sandow SL. Obesity up-regulates intermediate conductance calcium-activated potassium channels and myoendothelial gap junctions to maintain endothelial vasodilator function. J Pharmacol Exp Ther. 2010;335:284–293. doi: 10.1124/jpet.110.167593. [DOI] [PubMed] [Google Scholar]
- Chinnathambi V, Balakrishnan M, Yallampalli C, Sathishkumar K. Prenatal testosterone exposure leads to hypertension that is gonadal hormone-dependent in adult rat male and female offspring. Biol Reprod. 2012;206(507):e1–507. doi: 10.1095/biolreprod.111.097550. e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King AJ, Olivier NB, Mohankumar PS, Lee JS, Padmanabhan V, Fink GD. Hypertension caused by prenatal testosterone excess in female sheep. Am J Physiol Endocrinol Metab. 2007;292:E1837–E1841. doi: 10.1152/ajpendo.00668.2006. [DOI] [PubMed] [Google Scholar]
- McMullen S, Langley-Evans SC. Maternal low-protein diet in rat pregnancy programs blood pressure through sex-specific mechanisms. Am J Physiol Regul Integr Comp Physiol. 2005;288:R85–R90. doi: 10.1152/ajpregu.00435.2004. [DOI] [PubMed] [Google Scholar]
- Alexander BT. Placental insufficiency leads to development of hypertension in growth-restricted offspring. Hypertension. 2003;41:457–462. doi: 10.1161/01.HYP.0000053448.95913.3D. [DOI] [PubMed] [Google Scholar]
- Crowley SD, Coffman TM. In hypertension, the kidney rules. Curr Hypertens Rep. 2007;9:148–153. doi: 10.1007/s11906-007-0026-2. [DOI] [PubMed] [Google Scholar]
- Bussemaker E, Popp R, Fisslthaler B, Larson CM, Fleming I, Busse R, Brandes RP. Aged spontaneously hypertensive rats exhibit a selective loss of EDHF-mediated relaxation in the renal artery. Hypertension. 2003;42:562–568. doi: 10.1161/01.HYP.0000088852.28814.E2. [DOI] [PubMed] [Google Scholar]
- Deja MA, Golba KS, Widenka K, Mrozek R, Biernat J, Kolowca M, Malinowski M, Wos S. Angiotensin-converting enzyme inhibitors reveal non-NO-, non-prostacycline-mediated endothelium-dependent relaxation in internal thoracic artery of hypertensive patients. Int J Cardiol. 2005;102:455–460. doi: 10.1016/j.ijcard.2004.05.050. [DOI] [PubMed] [Google Scholar]
- Rajagopalan S, Kurz S, Munzel T, Tarpey M, Freeman BA, Griendling KK, Harrison DG. Angiotensin II-mediated hypertension in the rat increases vascular superoxide production via membrane NADH/NADPH oxidase activation. Contribution to alterations of vasomotor tone. J Clin Invest. 1996;97:1916–1923. doi: 10.1172/JCI118623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laursen JB, Rajagopalan S, Galis Z, Tarpey M, Freeman BA, Harrison DG. Role of superoxide in angiotensin II-induced but not catecholamine-induced hypertension. Circulation. 1997;95:588–593. doi: 10.1161/01.cir.95.3.588. [DOI] [PubMed] [Google Scholar]
- Zhao L, Wang Y, Ma X, Wang Y, Deng X. Oxidative stress impairs IKCa- and SKCa-mediated vasodilatation in mesenteric arteries from diabetic rats. Nan Fang Yi Ke Da Xue Xue Bao. 2013;33:939–944. [PubMed] [Google Scholar]
- Zhao LM, Wang Y, Ma XZ, Wang NP, Deng XL. Advanced glycation end products impair K(Ca)3.1- and K(Ca)2.3-mediated vasodilatation via oxidative stress in rat mesenteric arteries. Pflugers Arch. 2014;466:307–317. doi: 10.1007/s00424-013-1324-y. [DOI] [PubMed] [Google Scholar]
- Bennett MA, Hillier C, Thurston H. Endothelium-dependent relaxation in resistance arteries from spontaneously hypertensive rats: effect of long-term treatment with perindopril, quinapril, hydralazine or amlodipine. J Hypertens. 1996;14:389–397. doi: 10.1097/00004872-199603000-00017. [DOI] [PubMed] [Google Scholar]