

Abstract
Nucleophosmin (NPM1) is a multifunctional phosphoprotein localized predominantly within the nucleoli of eukaryotic cells. Mutations within its C-terminal domain are frequently observed in patients with acute myeloid leukemia (AML), are thought to play a key role in the initiation of the disease, and result in aberrant, cytoplasmic localization of the mutant protein. We have previously shown that the electrophilic antiproliferative natural product (+)-avrainvillamide (1) binds to proteins, including nucleophosmin, by S-alkylation of cysteine residues. Here, we report that avrainvillamide restores nucleolar localization of certain AML-associated mutant forms of NPM1 and provide evidence that this relocalization is mediated by interactions of avrainvillamide with mutant NPM1 and exportin-1 (Crm1). Immunofluorescence and mass spectrometric experiments employing a series of different NPM1 constructs suggest that a specific interaction between avrainvillamide and Cys275 of certain NPM1 mutants mediates the relocalization of these proteins to the nucleolus. Avrainvillamide treatment is also shown to inhibit nuclear export of Crm1 cargo proteins, including AML-associated NPM1 mutants. We also observe that avrainvillamide treatment displaces Thr199-phosphorylated NPM1 from duplicated centrosomes, leads to an accumulation of supernumerary centrosomes, and inhibits dephosphorylation of Thr199-phosphorylated NPM1 by protein phosphatase 1. Avrainvillamide is the first small molecule reported to relocalize specific cytoplasmic AML-associated NPM1 mutants to the nucleolus, providing an important demonstration of principle that small molecule induction of a wild-type NPM1 localization phenotype is feasible in certain human cancer cells.
Nucleophosmin (NPM1, also known as B23, NO38, or numatrin) is a multifunctional nucleolar phosphoprotein with roles in cell-cycle progression,1,2 ribosome biogenesis,3 and histone assembly.4 NPM1 readily forms oligomers through its N-terminal domain,5 is one of the most abundant proteins in the nucleolus,6 and plays a role in the maintenance of nucleolar and nuclear structure.7 It also functions as a molecular chaperone8 and has been associated with both tumor suppresion9,10 and oncogenesis.11–14 NPM1 is frequently overexpressed in human cancers, and this has been associated with c-Myc-mediated transformation and increased cellular proliferation.15–17 Additionally, mutant forms of NPM1 characterized by distinct, aberrant localization patterns are frequently observed in blasts from patients with acute myeloid leukemia (AML).18–20
The subcellular localization of NPM1 is controlled by several dynamic processes. Although NPM1 is predominantly localized within nucleoli, it shuttles continuously between the nucleolus, nucleoplasm, and cytoplasm in its role as a chaperone protein. Additionally, a small fraction of NPM1 localizes to the centrosomes, where it plays an important role in cell-cycle regulation. The specific structural elements of NPM1 that determine its subcellular localization have been studied extensively.21,22 Wild-type NPM1 contains a bipartite nuclear localization sequence (NLS), two leucine-rich nuclear export sequences (NES), and a nucleolar localization sequence (NoLS), tryptophan residues 288 and 290.23 The eGFP-NPM1-W288,290A fusion protein, which lacks the NoLS, is observed to localize throughout the nucleus, but not within the nucleolus.24 Although the NoLS is required to ensure nucleolar localization of NPM1, several other features of NPM1 have been demonstrated to play critical roles in its proper localization as well (for a further discussion of factors influencing the subcellular localization of NPM1, please refer to the Supporting Information).25
Mutations in the NPM1 gene are among the most common genetic aberrations in normal karyotype AML, with a prevalence of between 33 and 60% in adult patients.18–20 Despite molecular heterogeneity, the majority of these mutations induce a reading-frame shift that leads to replacement of one or both residues Trp288 and Trp290, and thereby full or partial loss of the NoLS, as well as the introduction of a C-terminal NES at positions 289–296. The NES motifs in NPM1 mediate its transport out of the nucleus through interactions with the nuclear export receptor protein exportin-1 (Xpo1, also known as chromosome region maintenance 1 protein homologue, Crm1).25 The addition of a third NES in AML-associated NPM1 mutants results in increased efficiency of Crm1-mediated nuclear export. The combination of the loss of the NoLS and the gain of an additional NES leads to an accumulation of NPM1 in the cytoplasm of cells, a phenotype referred to as NPMc+.19 In NPM1Mutant A, the most common NPMc+ variant in AML patients,18–20 both tryptophan residues comprising the NoLS are mutated, whereas the less common NPM1Mutant E contains a partial NoLS due to retention of Trp288, but not Trp290 (Figure 1A). Attempts thus far to correct the aberrant localization of NPMc+ proteins in AML cells with small molecules have focused on the development of selective inhibitors of Crm1, which induce nuclear retention of NPMc+ proteins but do not restore these variants to the nucleolus.26–28
Figure 1.
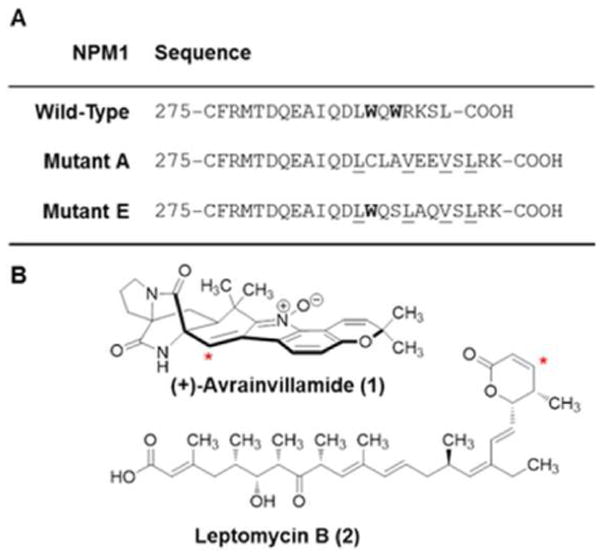
Primary structures of three NPM1 proteins and chemical structures of avrainvillamide and leptomycin B. (A) C-terminal sequences of wild-type NPM1 and two AML-NPMc+ variants. The NoLS is shown in bold; the C-terminal NES introduced in the mutants is underlined. (B) Structures of (+)-avrainvillamide and leptomycin B. Red asterisks indicate sites of cysteine addition.
In addition to the essential role of Crm1 in the nuclear export of NPM1 and its leukemic NPMc+ variants, interactions between Crm1 and wild-type NPM1 have also been established to play a key role in cell-cycle regulation. Interactions between RanGTP and Crm1 recruit a fraction of Crm1 to the centrosomes;29 Crm1 in turn interacts with the two NESs of NPM1, thereby recruiting NPM1 to centrosomes of nonmitotic cells.30 NPM1 is phosphorylated on Thr199 by Cdk2/cyclin E late in G1 phase,31 which causes dissociation of the majority of NPM1 from the centrosomes, a step required for centrosome duplication to occur.2,32 Following centrosome duplication, Thr199-phosphorylated NPM1 reassociates with the duplicated centrosomes, ensuring that reduplication does not occur. It has been demonstrated that inhibiting NPM1 from associating with centrosomes leads to premature centrosome duplication and accumulation of supernumerary centrosomes.1 Thus, both spatial control of Thr199-phosphorylated NPM1 during mitosis and precise control of the NPM1 Thr199 phosphorylation state throughout the cell cycle are required to ensure proper cell-cycle regulation.
We reported the first evidence of a small molecule-NPM1 interaction with our discovery that activity-based probes based on the electrophilic antiproliferative natural product (+)-avrainvillamide (1, AVA, Figure 1B) bind to NPM1 in cultured human cancer cells and in cell lysates.33 Avrainvillamide binds reversibly to thiols, including cysteine residues of certain proteins, by conjugate addition of sulfhydryl groups to the α,β-unsaturated nitrone function of avrainvillamide.33 Affinity isolation experiments employing a biotinylated avrainvillamide conjugate demonstrated that avrainvillamide binds both NPM1 and Crm1. Subsequent competition assays using a panel of fully synthetic avrainvillamide analogs revealed a correlation between their antiproliferative activities and NPM1 binding affinities. A similar correlation was not observed between antiproliferative activities and Crm1 binding affinities; for example, avrainvillamide and its non-natural enantiomer were essentially equally effective at inhibiting affinity isolation of Crm1 by the biotinylated avrainvillamide conjugate despite an approximately 3-fold difference in their antiproliferative activities in whole cell assays. To identify the cysteine residue (or residues) mediating the interaction between NPM1 and avrainvillamide, we generated three NPM1 constructs with single cysteine to alanine mutations of each of the cysteine residues present in wild-type NPM1. We found that that the C275A mutation, but not the C21A nor the C104A mutation, was uniquely able to reduce affinity isolation of NPM1 by the biotinylated avrainvillamide conjugate, indicating that avrainvillamide binds NPM1 specifically at Cys275. Following our initial report, other NPM1-interacting compounds have been identified. These include NSC348884, a synthetic polybenzimidazole alkaloid,34 and a synthetic 76-nucleotide RNA aptamer,35 both of which disrupt NPM1 oligomer formation and induce apoptosis in cancer cells. Recently, novel synthetic indoloquinolizines were reported that bind both NPM1 and Crm1 and are potent modulators of centrosome integrity.36 Small molecules that specifically bind to NPMc+ proteins have not previously been described, so far as we are aware.
Here, we report that avrainvillamide binds to both wild-type and AML-associated mutant forms of NPM1, has profound effects on the localization of cytoplasmic mutants of NPM1, and provides experimental evidence that this activity arises from the interaction of avrainvillamide with both NPM1 and Crm1. We also show that avrainvillamide treatment influences the localization of Thr199-phosphorylated NPM1 during mitosis, causes deregulation of centrosome duplication, and leads to an accumulation of Thr199-phosphorylated NPM1 by inhibiting its dephosphorylation by the beta isoform of protein phosphatase 1 (PP1β). Avrainvillamide is the first small molecule demonstrated to interact with the C-terminal domain of wild-type NPM1 and NPMc+ variants and is the first small molecule shown to restore an apparent wild-type NPM1 localization phenotype in cells expressing NPMc+ proteins. Taken together, our results may begin to explain the antiproliferative effects of avrainvillamide and suggest new avenues for the study of NPM1 as a potential therapeutic target in human cancer.
Results and Discussion
Avrainvillamide Binds Wild-Type NPM1 and AML-Associated NPMc+ Mutants
To assess binding of avrainvillamide to mutant forms of NPM1, we conducted time-resolved mass spectrometric analyses of peptide constructs corresponding to the C-terminal domain of NPMc+ proteins (Ser243-Lys298) admixed with an excess of the natural product. In order to establish the validity of this approach, we first examined the interaction of avrainvillamide with a peptide corresponding to the C-terminal 52 residues of wild-type NPM1 (Ser243-Leu294). When an excess of avrainvillamide (150 μM, 3 equiv. relative to NPM1 peptide) was added to a solution of wild-type NPM1 peptide at 23 °C, we observed the formation of an avrainvillamide–NPM1 peptide complex by ESI-TOF mass spectrometry (Figure 2A), in accord with our previous affinity isolation assays.33 The avrainvillamide–NPM1 peptide complex was observable within 1 min, and the fraction of NPM1 peptide bound by avrainvillamide appeared to reach a maximum value in approximately 2 h. When we repeated the same experiment with peptides corresponding to NPM1Mutant A and NPM1Mutant E, we observed the formation of the corresponding avrainvillamide–NPMc+ peptide adducts, suggesting that avrainvillamide binds to NPMc+ variants as well as to wild-type NPM1. The NPM1Mutant A and NPM1Mutant E constructs appeared to bind avrainvillamide more rapidly than the wild-type construct; the fraction of these NPMc+ constructs bound by avrainvillamide appeared to reach a maximum value within approximately 45 min (Figure 2B). Mass spectrometry also revealed that Cys288 of NPM1Mutant A is capable of binding avrainvillamide, as an avrainvillamide– NPM1 complex was observed for the NPM1Mutant A–C275A peptide. Furthermore, a complex between NPM1Mutant A (containing both Cys275 and Cys288) and two molecules of avrainvillamide was observed when a greater excess of avrainvillamide was added to the peptide solution (500 μM, 10 equiv. relative to NPM1 peptide). On the other hand, avrainvillamide-NPM1 adducts were not detected for the NPM1Mutant A-C275,288A and NPM1Mutant E-C275A peptides, consistent with our hypothesis that avrainvillamide interacts with proteins exclusively through cysteine residues.
Figure 2.
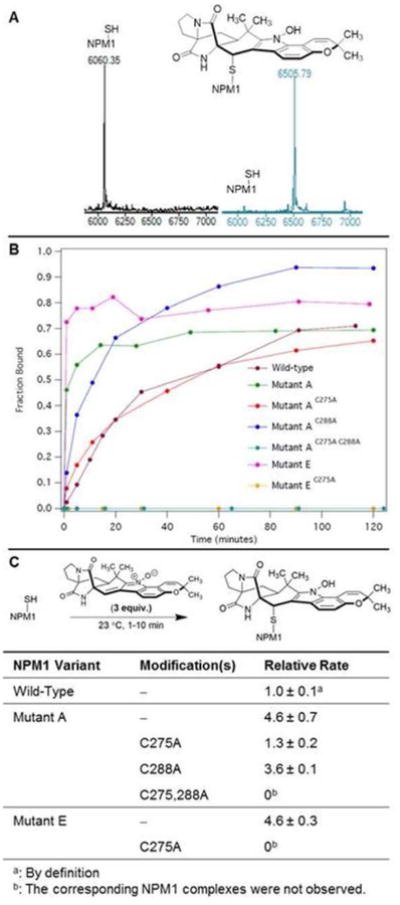
Avrainvillamide binds the C-terminal domain of NPM1 and AML-associated NPM1 variants. (A) ESI-TOF mass spectrum of wild-type NPM1 C-terminal peptide prior to (black trace) and 2 h after the addition of 3 mol equiv of avrainvillamide (blue trace). (B) Fraction of NPM1 C-terminal peptides bound by avrainvillamide vs time. (C) Relative rates of formation of the NPM1 C-terminal peptide– avrainvillamide complexes. Results are the mean ± standard deviation of three independent experiments.
The apparent kinetic selectivity for NPMc+ variants was further explored by assessing the relative rates of formation of the avrainvillamide–NPM1 complexes. To accomplish this, we studied the reaction between avrainvillamide and the NPM1 peptides at early time points (ca. 1–10 min), where the rates of formation of the adducts are essentially linear. In agreement with the above observations, avrainvillamide was found to alkylate the NPM1Mutant A and NPM1Mutant E peptides more rapidly than the wild-type NPM1 peptide (Figure 2C). We hypothesize that the faster rates of reaction of the NPMc+ peptides arise from the fact that these peptides are unfolded24 and presumably possess more accessible cysteine residues than wild-type NPM1. Of the two C-terminal cysteines in NPM1Mutant A, the C288A mutation resulted in a 1.3-fold reduction in reaction rate, whereas the C275A mutation exhibited a corresponding 3.5-fold reduction, supporting our hypothesis that Cys275 is the primary avrainvillamide binding site within the C-terminal domain of NPM1Mutant A. We also found that avrainvillamide preferentially alkylated Cys275 of NPM1Mutant A in direct competition experiments. Thus, avrainvillamide bound a greater fraction of NPM1Mutant A than wild-type NPM1 in a solution containing equimolar amounts of the two peptides and a substoichiometric amount of avrainvillamide (spectra available within the Supporting Information). Similarly, avrainvillamide displayed a pronounced selectivity for the NPM1Mutant A–C288A peptide in solutions containing both the NPM1Mutant A–C275A and NPM1Mutant A–C288A peptides. Taken together, our data indicate that Cys275 of NPM1Mutant A is the primary binding site of avrainvillamide, and that alkylation of this residue has functional consequences in cells expressing both NPM1 and NPM1Mutant A (vide infra).
Avrainvillamide Influences the Localization of NPM1 Proteins in Cancer Cells
We next sought to determine whether avrainvillamide influences the subcellular localization of NPM1 and NPMc+ proteins in cultured cancer cells. To this end, we elected to study the localization of NPM1 in the OCI-AML3 cell line, which is heterozygous for NPM1/NPM1Mutant A37 and, as a control, the OCI-AML2 cell line, which is homozygous for wild-type NPM1. Avrainvillamide was found to have moderate antiproliferative activity in both cell lines, with GI50 values at 72 h of 0.35 ± 0.09 μM and 0.52 ± 0.15 μM for OCI-AML2 and OCI-AML3 cells, respectively. As expected, NPM1 was observed predominantly within the nucleoli of untreated OCI-AML2 cells; this localization was unchanged following treatment with sublethal doses of avrainvillamide (250 nM, 48 h; Figure 3A). In contrast, NPM1 was observed in both the cytoplasm and nucleoli of control OCI-AML3 cells (Figure 3B). When these cells were treated with sublethal doses of avrainvillamide (250 nM, 48 h), NPM1 was observed predominantly in the nucleolus, although NPM1 was also observed diffusely throughout the nucleoplasm. At the highest concentration of avrainvillamide studied (4.0 μM, 48 h), which induces an apoptotic phenotype in both cell lines, we observed condensed or fragmented nuclei and a loss of nucleolar structure. However, the subcellular localization of NPM1 in apoptotic cells was markedly different between the two cell lines—NPM1 was displaced from the nuclei of apoptotic OCI-AML2 cells, whereas the protein appeared to colocalize with the highly condensed nuclei of apoptotic OCI-AML3 cells. We also observed an apparent reduction in nucleolin expression levels in OCI-AML3 (but not OCI-AML2) cells, suggesting that the effects of avrainvillamide on the biology of leukemic cells may be cell line dependent.
Figure 3.
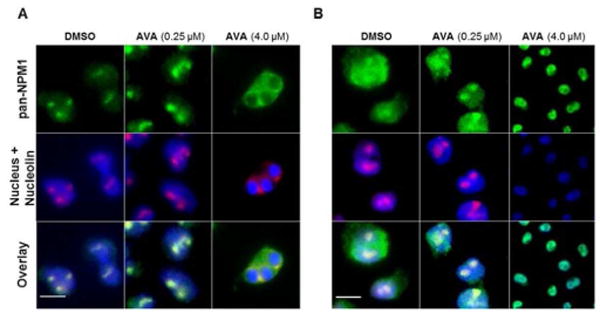
Avrainvillamide influences the localization of NPM1 and an NPMc+ variant in AML cells. (A) OCI-AML2 and (B) OCI-AML3 cells treated with DMSO (vehicle control) or avrainvillamide as indicated for 48 h. Colors: green, pan-NPM1; red, nucleolin (nucleolar marker); blue, DNA (nuclear marker). Scale bars = 10 μm.
To better understand the relocalization of NPM1 by avrainvillamide, we generated a series of eGFP-NPM1 constructs (Supporting Information Figure 1). For these experiments, we selected the HCT-116 human colon cancer cell line, which is homozygous wt-NPM1, on the basis of its moderate sensitivity toward avrainvillamide (GI50 = 1.10 ± 0.04 μM) and the facility of plasmid DNA transfection within this cell line. Since the nuclear export of NPMc+ proteins is mediated by Crm1, we included leptomycin B (LMB, 2, Figure 1B), a selective Crm1 inhibitor, as a control in these experiments. When HCT-116 cells were transfected with an eGFP-NPM1 wild-type plasmid, the eGFP fluorescence emission signal was detected exclusively in the nucleolus, as expected and in agreement with previous reports (Figure 4).24,25 This phenotype was not altered upon treatment with avrainvillamide. When cells were instead transfected with an eGFP-NPM1Mutant A plasmid, a cytoplasmic eGFP signal was observed, as expected.24,25 Following avrainvillamide treatment (1.0 μM, 24 h), the eGFP-NPM1Mutant A fusion protein was observed predominantly in the nucleolus, mimicking the distribution of the eGFP-NPM1 wild-type fusion protein. In contrast, and in agreement with previous reports,25 LMB treatment (100 nM, 4 h) resulted in nuclear, but not nucleolar, retention of the eGFP-NPM1 Mutant A protein (Supporting Information Figure 2), suggesting that a specific interaction between avrainvillamide and NPM1Mutant A was responsible for the observed relocalization of this protein to the nucleolus in HCT-116 cells.
Figure 4.
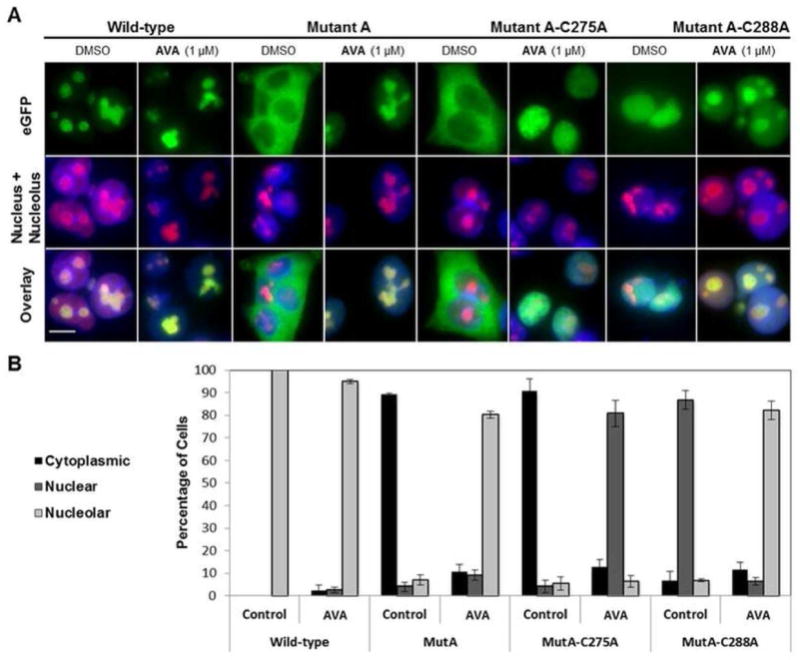
Cys275 of NPM1 Mutant A is required for the nucleolar relocalization of NPM1 by avrainvillamide. (A) HCT-116 cells transfected with eGFP-NPM plasmids and treated with DMSO (vehicle control) or avrainvillamide (1 μM, 24 h). Colors: green, eGFP; red, nucleolin; blue, DNA. Scale bar = 10 μm. (B) Subcellular localization of the eGFP-NPM Mutant A-derived constructs following treatment with DMSO or avrainvillamide. For each dosing condition, a minimum of 150 cells was counted in each of three separate experiments. Error bars indicate mean ± standard deviation.
To help elucidate the specific molecular interactions responsible for the relocalization of NPMc+ variants by avrainvillamide, we synthesized a series of N-terminal eGFP-tagged NPM1 constructs containing mutations of one or both residues Cys275 and Cys288. The eGFP–NPM1Mutant A– C275A protein was observed in the cytoplasm of untreated cells, indicating that the C275A mutation does not alter the localization of NPM1Mutant A. When cells expressing this construct were treated with avrainvillamide, the protein was observed to localize within nuclei (but not specifically within nucleoli). In contrast, the eGFP–NPM1Mutant A–C288A mutant was observed in the nuclei of untreated cells38 and was found to relocalize to nucleoli upon treatment with avrainvillamide. Since our mass spectrometric data indicated that avrainvillamide binds to the C-terminal domains of both NPM1Mutant A–C275A and NPM1Mutant A–C288A, our findings suggested that there is a functional difference between Cys275– and Cys288–avrainvillamide bound forms of NPM1Mutant A. Specifically, the interaction between avrainvillamide and Cys275 of NPM1Mutant A appears to uniquely restore nucleolar localization of the protein. We found that LMB induced nuclear retention of the cytoplasmic eGFP– NPM1Mutant A–C275A protein but had no effect on the localization of the nuclear eGFP–NPM1Mutant A–C288A protein, consistent with the documented ability of LMB to inhibit nuclear export through its binding to Crm1.24,25 Finally, neither avrainvillamide nor LMB treatment affected the localization of the eGFP–NPM1Mutant A–C275,288A protein (Supporting Information Figure 3); this was expected since this protein does not bind avrainvillamide and was observed to localize within nuclei (and not within the cytoplasm) of untreated cells.
We conducted an identical series of experiments using the less common NPM1Mutant E variant, which possesses a W290S mutation, retains Trp288, and localizes within nucleoli and the cytoplasm, but not within the nucleoplasm (Supporting Information Figure 4). We found that both avrainvillamide and LMB treatment restored both eGFP-NPM1Mutant E and eGFP-NPM1Mutant E-C275A proteins to nucleoli. Since the eGFP–NPM1Mutant E–C275A protein does not possess a C-terminal cysteine residue and does not bind avrainvillamide (Figure 2B), this result implied that avrainvillamide binds to a cellular target or targets other than NPM1 in human cancer cells. Indeed, LMB treatment has previously been demonstrated to restore the eGFP–NPM1Mutant E protein to nucleoli,25 suggesting that avrainvillamide may influence the localization of NPMc+ proteins both directly, by binding to NPM1, and indirectly, by inhibiting Crm1-mediated nuclear export, a hypothesis supported by experiments described below. The localization of all eGFP–NPM1 constructs following avrainvillamide and LMB treatment was also studied in HeLa S3 cells with identical results (Supporting Information Figure 5), indicating that the observed effects of avrainvillamide (and LMB) on NPMc+ localization are conserved across certain human cancer cell lines.
Avrainvillamide Does Not Appear to Alter the C-terminal Tertiary Structure of NPM1 Proteins
Our next goal was to develop a mechanistic understanding of the ability of avrainvillamide to relocalize NPMc+ proteins. Since the nucleolar localization of NPM1 is known to be highly dependent on the tertiary structure of its C-terminal domain,24 we chose to study the effects of avrainvillamide on the tertiary structure of NPM1 C-terminal constructs by circular dichroism (CD) spectroscopy. These experiments, which are discussed fully in the Supporting Information, revealed that the tertiary structure of isolated C-terminal domains of NPM1 and NPMc+ proteins appears to be unaltered in the presence of avrainvillamide.
Avrainvillamide Inhibits the Nuclear Export of Crm1 Cargo Proteins
The cytoplasmic localization of NPMc+ proteins is believed to involve two factors—loss of the NoLS and the addition of a C-terminal NES, which enhances nuclear export by Crm1.25 It has previously been demonstrated that LMB causes nuclear retention of NPMc+ proteins;24,25 LMB is proposed to inhibit the interaction between Crm1 and its cargo proteins by conjugate addition of the sulfhydryl group of Cys528 of Crm1 to the α,β-unsaturated lactone function of LMB.39 As noted above, we also identified Crm1 as a potential binding partner of avrainvillamide in initial affinity-isolation assays employing a biotinylated derivative of avrainvillamide.33 Based on our prior identification of Crm1 as a potential binding partner of avrainvillamide and the observation that avrainvillamide treatment influenced the localization of an NPMc+ variant which it did not bind (the eGFP-NPM1Mutant E-C275A protein), we reasoned that avrainvillamide may inhibit nuclear export by directly binding to Crm1.
To test the hypothesis that avrainvillamide inhibits the export function of Crm1 independent of its NPM1-binding activity, we studied the effects of avrainvillamide on subcellular localization of another Crm1 cargo protein, Ran Binding Protein 1 (RanBP1) by immunofluorescence microscopy. RanBP1 is exported from the nucleus by Crm1,40 has no known interactions with NPM1, and was observed to localize predominantly in the cytoplasm of untreated HCT-116 cells (Figure 5). When HCT-116 cells were treated with avrainvillamide (1.0 μM, 24 h) or leptomycin B (100 nM, 4h), RanBP1 was observed to localize primarily within the nucleus (Figure 5), suggesting that avrainvillamide influences Crm1-mediated nuclear export on a global level. We also studied the subcellular localization of an additional Crm1 export substrate, a cytoplasmic GFP-Rev fusion protein (Supporting Information Figure 6).41 Both avrainvillamide and LMB treatment caused nuclear retention of this protein, further supporting the hypothesis that Crm1 is an additional cellular target of avrainvillamide. In order to further confirm these results we sought to observe the formation of a Crm1– avrainvillamide complex by mass spectroscopy. Although we found that avrainvillamide did bind to an 18-amino acid peptide corresponding to residues 513–530 of human Crm1 (it has previously been reported that this peptide is also alkylated by leptomycin B;39 spectra are available within the Supporting Information), our attempts to observe longer Crm1 constructs (including a Trp491–Ala557 peptide construct and a His6-ZZ-Crm1 fusion protein) or their avrainvillamide adducts by mass spectrometry were unsuccessful.
Figure 5.
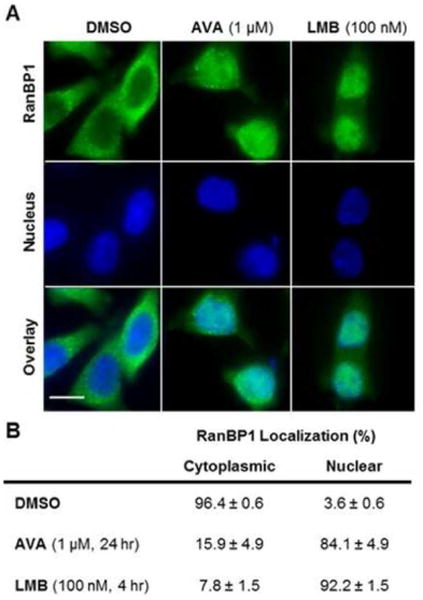
Avrainvillamide inhibits Crm1-mediated nuclear export. (A) HCT-116 cells treated with DMSO (vehicle control), avrainvillamide (1 μM, 4 h), or leptomycin B (100 nM, 4 h). Colors: green, RanBP1; blue DNA (nuclear marker). Scale bar = 10 μm. (B) Percentage of cells with RanBP1 observed in the cytoplasm or nucleus. Results are the mean ± standard deviation of three separate experiments; a minimum of 150 cells was counted per experiment.
Avrainvillamide Alters the Mitotic Localization of Thr199-Phosphorylated NPM1
As we have previously discussed, the interaction between Crm1 and wild-type NPM1 also plays a key role in cell-cycle regulation. Specifically, the reassociation of NPM1pThr199 with duplicated centrosomes during metaphase is thought to be a critical step to ensure that centrosome duplication occurs only once per cell cycle.30 If NPM1 is displaced from the centrosomes, such as by siRNA knock-down of NPM1 or treatment with LMB, mitotic defects, including supernumerary centrosomes, are observed.1 Since our data suggested that avrainvillamide inhibits the interaction between NPMc+ variants and Crm1, we sought to determine whether the interaction between wild-type NPM1 and Crm1 was similarly affected by avrainvillamide treatment.
When HCT-116 cells blocked in the S phase were released into medium containing avrainvillamide and examined during their first mitotic phase, we observed that Thr199-phosphory-lated NPM1 was displaced from the centrosomes, although no other obvious morphological aberrations were apparent (Figure 6A). However, in subsequent mitotic cycles, supernumerary centrosomes were observed in the majority of cells (Figure 6A,B), in some cases resulting in tripolar mitotic morphologies. These observations are consistent with the hypothesis that displacement of Thr199-phosphorylated NPM1 from duplicated centrosomes enables centrosome reduplication, and suggest that avrainvillamide alters essential cellular processes by preventing NPM1 and Crm1 from interacting with their native binding partners.
Figure 6.
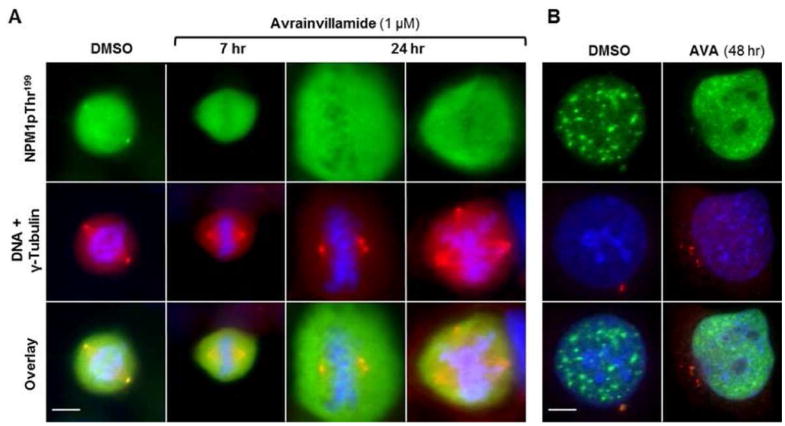
Avrainvillamide interferes with proper centrosome duplication. (A) Mitotic and (B) interphase HCT-116 cells treated with DMSO (vehicle control) or avrainvillamide (1 μM, time points as indicated). Twenty-four-hour images taken at 1.6× magnification relative to the 7 h images. Colors: green, NPM1pThr199; red, γ-tubulin; blue, DNA. Scale bar = 5 μm.
Avrainvillamide Increases Cellular Levels of Thr199-Phosphorylated NPM1
NPM1 is phosphorylated on Thr199 by several kinases, including Cdk1,42 Cdk2,43 Cdk4,44 and Cdk6,45 and is dephosphorylated by PP1β.46 Increases in NPM1 thr199 phosphorylation are known to repress pre-mRNA splicing,47 reduce NPM1-mediated DNA repair activity,46 and target NPM1 to nuclear speckles.47 Over the course of our studies, we observed an apparent increase in the amount of NPM1pThr199 in avrainvillamide-treated cells (Figure 6B); this finding was confirmed by subsequent immunoblotting experiments (Supporting Information Figure 7). To identify potential mechanisms behind this observation, we studied the effects of avrainvillamide on PP1β activity. Avrainvillamide inhibited in vitro dephosphorylation of NPM1pThr199 by PP1β (Figure 7A). On the other hand, avrainvillamide did not affect the phosphatase activity of PP1β in a standard assay employing 4-nitrophenyl phosphate as a substrate (Figure 7B). Our results indicate that avrainvillamide increases the relative amount of NPM1pThr199 by preventing the action of PP1β on NPM1, rather than by direct inhibition of PP1β itself. Leptomycin B did not inhibit NPM1 dephosphorylation in these assays, consistent with the hypothesis that the NPM1-binding activity of avrainvillamide is responsible for the observed increase of Thr199-phosphorylated NPM1. Taken together, our results suggest that avrainvillamide may influence the cellular functions of NPM1 by causing an accumulation of Thr199-phosphorylated NPM1. Since Thr199 phosphorylation of NPM1 displaces it from the nucleolus,47 we hypothesize that elevated levels of NPM1pThr199 may cause significant disruption of the normal structure and function of nucleoli. Indeed, cells treated with toxic concentrations of avrainvillamide for ≥2 days routinely displayed abnormal nuclear and nucleolar morphologies (Figures 3, 6B).
Figure 7.
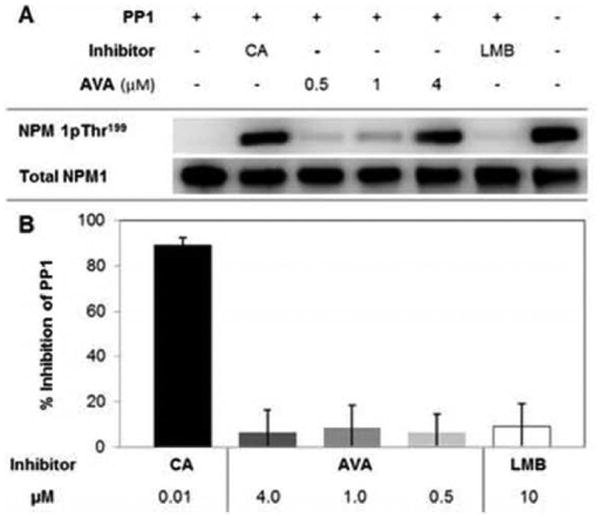
Avrainvillamide inhibits the dephosphorylation of NPM1 by PP1β. (A) Inhibition of NPM1pTh199 dephosphorylation in an in vitro PP1β activity assay. CA = calyculin A, 10 nM, LMB = leptomycin B, 10 μM. Total NPM1 shown as a loading control. (B) Inhibition of PP1β activity in a standard activity assay with 4-nitrophenyl phosphate as the substrate.
Conclusions
We have found that the natural product avrainvillamide has remarkable effects upon the biology of cancer cells, likely due to its interactions with NPM1 and Crm1. Avrainvillamide relocalizes certain cytoplasmic NPM1 mutants to the nucleoli of HCT-116 and HeLa S3 cells and inhibits the nuclear export of NPM1 in NPMc+ AML cells. Although avrainvillamide was equally effective in inhibiting the growth of OCI-AML2 and OCI-AML3 cells, lethal doses of avrainvillamide induced different NPM1 localization phenotypes in the two cell lines, suggesting that the effects of avrainvillamide on leukemic cell biology are cell-line- (and potentially NPM1 mutational status-) dependent. We hypothesize that avrainvillamide relocalizes NPMc+ proteins by altering both key factors responsible for its cytoplasmic localization—loss of the C-terminal NoLS of NPM1 and enhanced nuclear export by Crm1. Avrainvillamide, when bound to Cys275 (but not Cys288) of NPM1Mutant A appears to serve as a surrogate NoLS in HCT-116 and HeLa S3 cells. Additionally, avrainvillamide inhibits Crm1-mediated export of NPMc+ proteins, presumably by direct interaction of avrainvillamide with Cys528 of Crm1.
We additionally observe that the ability of avrainvillamide to bind to Crm1 and prevent its binding to NES-containing proteins appears to disrupt the interaction between Crm1 and Thr199-phosphorylated NPM1 at key points in the cell-cycle, resulting in unregulated centrosome duplication. Simultaneously, avrainvillamide causes an increase in the amount of Thr199-phosphorylated NPM1, likely by inhibiting the action of PP1β on NPM1. Since Thr199 phosphorylation targets NPM1 to nuclear speckles,47 avrainvillamide-mediated increases in phospho-NPM1 may indirectly displace NPM1 from the nucleolus. Given the structural and functional importance of NPM1 in the nucleolus and nucleus,7 this finding suggests a potential for global disruption of cellular structure and function by avrainvillamide. Indeed, cells exposed to lethal doses of avrainvillamide displayed nuclear and nucleolar abnormalities in OCI-AML2, OCI-AML3, and HCT-116 cells (Figures 3, 6B). It is important to note, however, that avrainvillamide does not appear to displace NPM1 from the nucleolus at lower doses or shorter time points (Figures 3, 4). Rather, it seems that extended avrainvillamide treatment results in an accumulation of Thr199-phosphorylated NPM1, eventually depleting NPM1 in the nucleolus. Thus, the observed effects of avrainvillamide on the subcellular localization of NPMc+ variants may represent a sublethal phenotype, while the effects of avrainvillamide on the relative levels of Thr199-phosphorylated NPM1 and on the localization of Thr199-phosphorylated NPM1 during mitosis may help explain avrainvillamide-induced antiproliferation in human cancer cells. Taken together, our data suggest that avrainvillamide affects cell biology both by directly binding NPM1 and Crm1 as well as by inhibiting the association of these proteins with certain native cellular partners.
Methods
Complete experimental procedures are available within the Supporting Information.
Supplementary Material
Acknowledgments
We thank J. Vaughan for assistance with immunofluorescence microscopy and S. Trauger for assistance with mass spectroscopy. Financial support from the National Institutes of Health (Grant CA047148) is gratefully acknowledged. This material is based upon work supported by a National Science Foundation Graduate Research Fellowship under Grant Nos. DGE0946799 and DGE1144152 (H.M.), by a Singapore Agency of Science Technology and Research (A*STAR) Postdoctoral Fellowship (K. P. C.), and by grants from the Bergen Medical Research Foundation (V. A.), the Norwegian Cancer Society with Solveig, Ole Lunds Legacy, and the Western Regional Norwegian Authority (Helse Vest) (B. T. G.).
Footnotes
Supporting Information: Supporting results and discussion, methods, Supporting Information Figures 1–8, spectra, and references. This material is available free of charge via the Internet at http://pubs.acs.org.
Notes: The authors declare no competing financial interest.
References
- 1.Wang W, Budhu A, Forgues M, Wang XW. Temporal and spatial control of nucleophosmin by the Ran–Crm1 complex in centrosome duplication. Nat Cell Biol. 2005;7:823–830. doi: 10.1038/ncb1282. [DOI] [PubMed] [Google Scholar]
- 2.Ma Z, Kanai M, Kawamura K, Kaibuchi K, Ye K, Fukusawa K. Interaction between ROCK II and nucleophosmin/B23 in the regulation of centrosome duplication. Mol Cell Biol. 2006;26:9016–9034. doi: 10.1128/MCB.01383-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Maggi LB, Kuchenruether M, Dadey DY, Schwope RM, Grisendi S, Townsend RR, Pandolfi PP, Weber JD. Nucleophosmin serves as a rate-limiting nuclear export chaperone for the mammalian ribosome. Mol Cell Biol. 2008;28:7050–7065. doi: 10.1128/MCB.01548-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Okuwaki M, Matsumoto K, Tsujimoto M, Nagata K. Function of nucleophosmin/B23, a nucleolar acidic protein, as a histone chaperone. FEBS Lett. 2001;506:272–276. doi: 10.1016/s0014-5793(01)02939-8. [DOI] [PubMed] [Google Scholar]
- 5.Namboodiri VM, Akey IV, Schmidt-Zachmann MS, Head JF, Akey CW. The structure and function of Xenopus NO38-core, a histone chaperone in the nucleolus. Structure. 2004;12:2149–2160. doi: 10.1016/j.str.2004.09.017. [DOI] [PubMed] [Google Scholar]
- 6.Emmott E, Hiscox JA. Nucleolar targeting: the hub of the matter. EMBO Rep. 2009;10:231–238. doi: 10.1038/embor.2009.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Amin MA, Matsunaga S, Uchiyama S, Fukui K. Depletion of nucleophosmin leads to distortion of nucleolar and nuclear structures in HeLa cells. Biochem J. 2008;415:345–351. doi: 10.1042/BJ20081411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Szebeni A, Olson MO. Nucleolar protein B23 has molecular chaperone activities. Protein Sci. 1999;8:905–912. doi: 10.1110/ps.8.4.905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kurki S, Peltonen K, Latonen L, Kiviharju TM, Ojala PM, Meek D, Laiho M. Nucleolar protein NPM interacts with HDM2 and protects tumor suppressor protein p53 from HDM2-mediated degradation. Cancer Cell. 2004;5:465–475. doi: 10.1016/s1535-6108(04)00110-2. [DOI] [PubMed] [Google Scholar]
- 10.Di Fiore PP. Playing both sides: nucleophosmin between tumor suppression and oncogenesis. J Cell Biol. 2008;182:7–9. doi: 10.1083/jcb.200806069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Grisendi S, Mecucci C, Falini B, Pandolfi PP. Nucleophosmin and cancer. Nat Rev Cancer. 2006;6:493–505. doi: 10.1038/nrc1885. [DOI] [PubMed] [Google Scholar]
- 12.Grisendi S, Bernardi R, Rossi M, Cheng K, Khandker L, Manova K, Pandolfi PP. Role of nucleophosmin in embryonic development and tumorigenesis. Nature. 2005;437:147–153. doi: 10.1038/nature03915. [DOI] [PubMed] [Google Scholar]
- 13.Cheng K, Grisendi S, Clohessy JG, Majid S, Bernardi R, Sportoletti P, Pandolfi PP. The leukemia-associated cytoplasmic nucleophosmin mutant is an oncogene with paradoxical functions: Arf inactivation and induction of cellular senescence. Oncogene. 2007;26:7391–7400. doi: 10.1038/sj.onc.1210549. [DOI] [PubMed] [Google Scholar]
- 14.Li J, Sejas DP, Burma S, Chen DJ, Pang Q. Nucleophosmin suppresses oncogene-induced apoptosis and senescence and enhances oncogenic cooperation in cells with genomic instability. Carcinogenesis. 2007;28:1163–1170. doi: 10.1093/carcin/bgm025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bonetti P, Davoli T, Sironi C, Amati B, Pelicci PG, Colombo E. Nucleophosmin and its AML-associated mutant regulate c-Myc turnover through Fbw7 gamma. J Cell Biol. 2008;182:19–26. doi: 10.1083/jcb.200711040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li Z, Boone D, Hann SR. Nucleophosmin interacts directly with c-Myc and controls c-Myc-induced hyper-proliferation and transformation. Proc Natl Acad Sci U S A. 2008;105:18794–18799. doi: 10.1073/pnas.0806879105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pianta A, Puppin C, Franzoni A, Fabbro D, Di Loreto C, Bulotta S, Deganuto M, Paron I, Tell G, Puxeddu E, Filetti S, Russo D, Damante G. Nucleophosmin is overexpressed in thyroid tumors. Biochem Biophys Res Commun. 2010;397:499–504. doi: 10.1016/j.bbrc.2010.05.142. [DOI] [PubMed] [Google Scholar]
- 18.Falini B, Nicoletti I, Martelli MF, Mecucci C. Acute myeloid leukemia carrying cytoplasmic/mutated nucleophosmin (NPMc+ AML): biologic and clinical features. Blood. 2007;109:874–885. doi: 10.1182/blood-2006-07-012252. [DOI] [PubMed] [Google Scholar]
- 19.Falini B, Mecucci C, Tiacci E, Alcalay M, Rosati R, Pasqualucci L, La Starza R, Diverio D, Colombo E, Santucci A. Cytoplasmic nucleophosmin in acute myelogenous leukemia with a normal karyotype. New Engl J Med. 2005;352:254–266. doi: 10.1056/NEJMoa041974. [DOI] [PubMed] [Google Scholar]
- 20.Jain P, Kantarjian H, Patel K, Faderl S, Garcia-Manero G, Benjamini O, Borthakur G, Pemmaraju N, Kadia T, Daver N. Mutated NPM1 in patients with acute myeloid leukemia in remission and relapse. Leukemia Lymphoma. 2014;55:1337–1344. doi: 10.3109/10428194.2013.840776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hingorani K, Szebeni A, Olson MO. Mapping the functional domains of nucleolar protein B23. J Biol Chem. 2000;275:24451–24457. doi: 10.1074/jbc.M003278200. [DOI] [PubMed] [Google Scholar]
- 22.Liu QR, Chan PK. Formation of nucleophosmin/B23 oligomers requires both the amino- and the carboxyl-terminal domains of the protein. Eur J Biochem. 1991;200:715–721. doi: 10.1111/j.1432-1033.1991.tb16236.x. [DOI] [PubMed] [Google Scholar]
- 23.Nishimura Y, Ohkubo T, Furuichi Y, Umekawa H. Tryptophans 286 and 288 in the C-terminal region of protein B23.1 are important for its nucleolar localization. Biosci Biotechnol Biochem. 2006;66:2239–2242. doi: 10.1271/bbb.66.2239. [DOI] [PubMed] [Google Scholar]
- 24.Grummitt CG, Townsley FM, Johnson CM, Warren AJ, Bycroft M. Structural consequences of nucleophosmin mutations in acute myeloid leukemia. J Biol Chem. 2008;283:23326–23332. doi: 10.1074/jbc.M801706200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Falini B, Bolli N, Shan J, Martelli MP, Liso A, Pucciarini A, Bigerna B, Pasqualucci L, Mannucci R, Rosati R, Gorello P, Diverio D, Roti G, Tiacci E, Cazzaniga G, Biondi A, Schnittger S, Haferlach T, Hiddemann W, Martelli MF, Gu W, Mecucci C, Nicoletti I. Both carboxy-terminus NES motif and mutated tryptophan(s) are crucial for aberrant nuclear export of nucleophosmin leukemic mutants in NPMc+ AML. Blood. 2006;107:4514–4523. doi: 10.1182/blood-2005-11-4745. [DOI] [PubMed] [Google Scholar]
- 26.Ranganathan P, Yu X, Na C, Santhanam R, Shacham S, Kauffman M, Walker A, Klisovic R, Blum W, Caligiuri M, Croce CM, Marcucci G, Garzon R. Preclinical activity of a novel CRM1 inhibitor in acute myeloid leukemia. Blood. 2012;120:1765–1773. doi: 10.1182/blood-2012-04-423160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Walker CJ, Oaks JJ, Santhanam R, Neviani P, Harb JG, Ferenchak G, Ellis JJ, Landesman Y, Eisfeld AK, Gabrail NY, Smith CL, Caligiuri MA, Hokland P, Roy DC, Reid A, Milojkovic D, Goldman JM, Apperley J, Garzon R, Marcucci G, Shacham S, Kauffman MG, Perrotti D. Preclinical and clinical efficacy of XPO1/CRM1 inhibition by the karyopherin inhibitor KPT-330 in Ph+ leukemias. Blood. 2013;122:3034–3044. doi: 10.1182/blood-2013-04-495374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Etchin J, Sun Q, Kentsis A, Farmer A, Zhang ZC, Sanda T, Mansour MR, Barcelo C, McCauley D, Kauffman M, Shacham S, Christie AL, Kung AL, Rodig SJ, Chook YM, Look AT. Antileukemic activity of nuclear export inhibitors that spare normal hematopoietic cells. Leukemia. 2013;27:66–74. doi: 10.1038/leu.2012.219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Liu Q, Jiang Q, Zhang C. A fraction of Crm1 locates at centrosomes by its CRIME domain and regulates the centrosomal localization of pericentrin. Biochem Biophys Res Commun. 2009;384:383–388. doi: 10.1016/j.bbrc.2009.04.154. [DOI] [PubMed] [Google Scholar]
- 30.Okuda M. The role of nucleophosmin in centrosome duplication. Oncogene. 2002;21:6170–6174. doi: 10.1038/sj.onc.1205708. [DOI] [PubMed] [Google Scholar]
- 31.Okuda M, Horn HF, Tarapore P, Tokuyama Y, Smulian AG, Chan P, Knudsen ES, Hofmann IA, Snyder JD, Bove KE, Fukasawa K. Nucleophosmin/B23 is a target of CDK2/cyclin E in centrosome duplication. Cell. 2000;103:127–140. doi: 10.1016/s0092-8674(00)00093-3. [DOI] [PubMed] [Google Scholar]
- 32.Tokuyama Y, Horn HF, Kawamura K, Tarapore P, Fukasawa K. Specific phosphorylation of nucleophosmin on Thr(199) by cyclin-dependent kinase 2-cyclin E and its role in centrosome duplication. J Biol Chem. 2001;276:21529–21537. doi: 10.1074/jbc.M100014200. [DOI] [PubMed] [Google Scholar]
- 33.Wulff JE, Siegrist R, Myers AG. The natural product avrainvillamide binds to the oncoprotein nucleophosmin. J Am Chem Soc. 2007;129:14444–14451. doi: 10.1021/ja075327f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Qi W, Shakalya K, Stejskal A, Goldman A, Beeck S, Cooke L, Mahadevan D. NSC348884, a nucleophosmin inhibitor disrupts oligomer formation and induces apoptosis in human cancer cells. Oncogene. 2008;27:4210–4220. doi: 10.1038/onc.2008.54. [DOI] [PubMed] [Google Scholar]
- 35.Jian Y, Gao Z, Sun J, Shen Q, Feng F, Jing Y, Yang C. RNA aptamers interfering with nucleophosmin oligomerization induce apoptosis of cancer cells. Oncogene. 2009;28:4201–4211. doi: 10.1038/onc.2009.275. [DOI] [PubMed] [Google Scholar]
- 36.Dückert H, Pries V, Khedkar V, Menninger S, Hübel K, Ziegler S, Kumar K, Waldmann H. Natural product-inspired cascade synthesis yields modulators of centrosome integrity. Nat Chem Biol. 2012;8:179–184. doi: 10.1038/nchembio.758. [DOI] [PubMed] [Google Scholar]
- 37.Quentmeier H, Martelli M, Dirks W, Bolli N, Liso A, MacLeod R, Nicoletti I, Mannucci R, Pucciarini A, Bigerna B. Cell line OCI/AML3 bears exon-12 NPM gene mutation-A and cytoplasmic expression of nucleophosmin. Leukemia. 2005;19:1760–1767. doi: 10.1038/sj.leu.2403899. [DOI] [PubMed] [Google Scholar]
- 38.Huang M, Thomas D, Li MX, Feng W, Majeti R, Mitchell BS. Role of cysteine 288 in nucleophosmin cytoplasmic mutations: sensitization to toxicity induced by arsenic trioxide and bortezomib. Leukemia. 2013;27:1970–1980. doi: 10.1038/leu.2013.222. [DOI] [PubMed] [Google Scholar]
- 39.Kudo N, Matsumori N, Taoka H, Fujiwara D, Schreiner EP, Wolff B, Yoshida M, Horinouchi S. Leptomycin B inactivates CRM1/exportin 1 by covalent modification at a cysteine residue in the central conserved region. Proc Natl Acad Sci U S A. 1999;96:9112–9117. doi: 10.1073/pnas.96.16.9112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Plafker K, Macara IG. Facilitated nucleocytoplasmic shuttling of the Ran binding protein RanBP1. Mol Cell Biol. 2000;20:3510–3521. doi: 10.1128/mcb.20.10.3510-3521.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Love DC, Sweitzer TD, Hanover JA. Reconstitution of HIV-1 rev nuclear export: independent requirements for nuclear import and export. Proc Natl Acad Sci U S A. 1998;95:10608–10613. doi: 10.1073/pnas.95.18.10608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yu LR, Zhu Z, Chan KC, Issaq HJ, Dimitrov DS, Veenstra TD. Improved titanium dioxide enrichment of phosphopeptides from HeLa cells and high confident phosphopeptide identification by cross-validation of MS/MS and MS/MS/MS spectra. J Proteome Res. 2007;6:4150–4162. doi: 10.1021/pr070152u. [DOI] [PubMed] [Google Scholar]
- 43.Sarek G, Jaerviluoma A, Moore HM, Tojkander S, Vartia S, Biberfeld P, Laiho M, Ojala PM. Nucleophosmin phosphorylation by v-cyclin-CDK6 controls KSHV latency. PLOS Pathol. 2010;6(3):e1000818. doi: 10.1371/journal.ppat.1000818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Adon AM, Zeng X, Harrison MK, Sannem S, Kiyokawa H, Kaldis P, Saavedra HI. Cdk2 and Cdk4 regulate the centrosome cycle and are critical mediators of centrosome amplification in p53-null cells. Mol Cell Biol. 2010;30:694–710. doi: 10.1128/MCB.00253-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Olsen JV, Vermeulen M, Santamaria A, Kumar C, Miller ML, Jensen LJ, Gnad F, Cox J, Jensen TS, Nigg EA, Brunak S, Mann M. Quantitative phosphoproteomics reveals widespread full phosphorylation site occupancy during mitosis. Sci Signal. 2010;3:ra3. doi: 10.1126/scisignal.2000475. [DOI] [PubMed] [Google Scholar]
- 46.Lin CY, Tan BC, Liu H, Shih C, Chein K, Lin C, Yung BY. Dephosphorylation of nucleophosmin by PP1β facilitates pRB binding and consequent E2F1-dependent DNA repair. Mol Biol Cell. 2010;21:4409–4417. doi: 10.1091/mbc.E10-03-0239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tarapore P, Shinmura S, Suzuki H, Tokuyama Y, Kim S, Mayeda A, Fukasawa K. Thr199 phosphorylation targets nucleophosmin to nuclear speckles and represses pre-mRNA processing. FEBS Lett. 2006;580:399–409. doi: 10.1016/j.febslet.2005.12.022. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.