


Abstract
Recent releases of genome three-dimensional (3D) structures have the potential to transform our understanding of genomes. Nonetheless, the storage technology and visualization tools need to evolve to offer to the scientific community fast and convenient access to these data. We introduce simultaneously a database system to store and query 3D genomic data (3DBG), and a 3D genome browser to visualize and explore 3D genome structures (3DGB). We benchmark 3DBG against state-of-the-art systems and demonstrate that it is faster than previous solutions, and importantly gracefully scales with the size of data. We also illustrate the usefulness of our 3D genome Web browser to explore human genome structures. The 3D genome browser is available at http://3dgb.cs.mcgill.ca/.
INTRODUCTION
Biological motivation
The release of the first draft of the human genome (1,2) announced the beginning of a data era in genomics. Gathering information does no longer appear as a major bottleneck in molecular biology studies. However, by contrast, storing and mining the massive amounts of data generated by genomics studies becomes increasingly difficult.
The development of efficient computing infrastructures to store and retrieve genomic information is thus an essential part of the discovery pipeline in genomic research. The UCSC genome browser (3,4) and the Ensembl genome browser (5,6) were among the first systems specifically developed to address this issue and opened the access of sequencing data to the whole scientific community.
Since then, the complexity and the nature of the data themselves has changed. In particular, the primary structure of genomes does no longer appear to contain all the information required to decipher our genetic code. Indeed, recent studies suggest that the three-dimensional (3D) structure of genomes is essential to understand some regulatory mechanisms (7).
3D structures and Hi-C data sets of Human (8,9) or Yeast genomes (10) are now available. Viewers have been developed to visualize complete genome structures (11) and Hi-C data annotations have been integrated in classical genome browsers (12). However, to date, there is no scalable solution to query simultaneously primary and tertiary genome structures. Moreover, unlike classical human genome browsers, these viewers are currently only available as Operating System (OS)-specific standalone applications that are not embedded into Web browsers.
In this paper, we aim to address these challenges and develop a complete solution for storing and analyzing 3D genomic data. More specifically, we develop a new database system for storing and querying 3D genomic data, and a lightweight 3D genome browser for real-time visualization and exploration of 3D genome structures. We emphasize that the development of efficient database architectures is key for the success of a novel generation of 3D genome browsers.
Database technology
There has been substantial related work on storing and querying 3D data in the context of astronomy, remote-sensing, neurology or more broadly for the storage of spatio-temporal data. Existing approaches can be broadly categorized along three axes:
Index-based versus cluster-based. Most existing work builds a 3D index over the raw data, but does not attempt to physically reorganize the raw data (index-based) so that co-queried objects are stored near each other (cluster-based).
Adaptive versus non-adaptive. Adaptive systems adjust storage structures based on the query size and/or the data. They generally require less tuning and can typically handle a broader range of data sets than non-adaptive systems.
On-line versus off-line. On-line systems change their storage representation as data arrives, whereas off-line systems assume that the indexed data does not change frequently and must recompute their storage layout from scratch as data arrives.
The ‘classic’ database structure for indexing objects along multiple dimensions is the R-Tree (13). Unlike 3DBG, R-Trees do not per se cluster data and are optimized for accessing arbitrary spatial objects, rather than large amounts of data organized along 3D trajectories. Of course, it is possible to attempt to physically co-locate (cluster) objects in the same R-Tree rectangle together on disk. Even so, as R-Trees consider nested bounding rectangles to index the objects, it is very likely that if there is much data within a small area, there will be large overlaps in these bounding rectangles, resulting in many I/Os to answer any query.
There have been many optimizations to R-Trees for spatio-temporal data, including TB-Trees (14) and SEB-Trees (15). TB-Trees are optimized R-Tree indices with special support for temporal predicates. They also do not deal well with very long 3D trajectories that tend to have very large bounding rectangles, and can include a high number of I/Os per lookup. SEB-Trees segment space and time, but are not specifically designed for indexing trajectories. Research on TB-trees and SEB-trees does not explicitly discuss how to cluster data, and both are non-adaptive (i.e. they do not reorganize previously added pages as new data arrives).
To address the concern with very large 3D meshes or trajectories, several systems have proposed segmenting the trajectories to reduce the sizes of bounding boxes and group portions of trajectories that are near each other in space together on disk. Rasetic et al. (16) propose splitting trajectories into a number of sub-trajectories, and then indexing those segments in an R-Tree. They propose a formal model for the number of I/Os needed to evaluate a query, and use a dynamic programming algorithm to minimize the I/O for an average query size. 3DBG also includes an algorithm for optimally splitting 3D genomic meshes and their associated metadata, but in addition physically clusters those segments rather than just indexing them.
SETI (17) also advocates a segmentation-based approach like 3DBG. It segments incoming 3D meshes/trajectories into sub-trajectories, groups them into a collection of ‘spatial partitions’ and then runs queries over just the spatial partitions that are most relevant to a given query. The principal differences between 3DBG and SETI are that: (i) the SETI paper does not describe how the size or geometry of partitions is selected, or whether it changes as inserts occur, which is a key contribution of 3DBG, and (ii) SETI does not discuss metadata storage and clustering, read-optimized operations or scalability features.
PIST (18) focuses on indexing individual points rather than 3D meshes. PIST is similar in spirit to 3DBG in that it attempts to optimally partition a collection of points into a variable-sized grid according to the density of the data and query size using a quad-tree like data structure. Unlike 3DBG, PIST is off-line (i.e. it does not adapt to new data being added dynamically).
A number of other systems, such as STRIPES (19), use a dual transformed space to index meshes or trajectories. While such indices are very compelling when indexing the future positions of moving objects, they are known to be suboptimal for answering historical or ad hoc queries (20).
Spatial clustering has been extensively studied (21–23). These approaches focus on generic methods to extract cluster information from large collections of ad hoc data points. Our clustering problem is more specific, since we deal with series of points ordered along 3D genes, and more importantly on the (potentially large) metadata associated to the 3D models.
Contribution
In this paper, we introduce a complete efficient and scalable database system to query genomes in space. The system includes two components: (i) a database 3DBG to store and query the 3D genomic data, and (ii) a web-based genome browser 3DGB to visualize and navigate 3D genome structures. As far as we are aware, 3DBG is the first database system that is online, adaptive and cluster-based. We designed 3DBG to optimize the speed of searching and accessing genomic annotations from their 3D spatial coordinates in genome structures. We also develop a lightweight 3D genome viewer 3DGB that is fully embedded in Web browsers and accessible to any web user who wishes to browse and query 3D genome structures.
Our system aims to foster the discovery of spatial relationships between genomic elements and accelerate the large-scale analysis of space-dependent regulatory mechanisms. Here, we map data from the 1000 genomes project (24) and experimental Chip-Seq data (4) onto most recent 3D models of the Human genome (8,9), and use 3DBG to mine these data. We benchmark 3DBG against state-of-the-art systems, and demonstrate that our database system is faster than previous solutions, and more importantly that it scales better with the size of data. We also illustrate the usefulness of our system and use our 3D genome Web browser to explore the 3D neighborhood of the retinoblastoma gene (RB1) and identify potentially interesting genetic relationships between retinoblastoma and sleep disorders.
Our system is freely available at https://github.com/mavlyutovrus/3d_genome_browser and a sample deployment of our 3D genome explorer is accessible at http://3dgb.cs.mcgill.ca/.
MATERIALS AND METHODS
3D genome database
We have implemented a fully functional database based on YARN (http://hadoop.apache.org/docs/current/hadoop-yarn/hadoop-yarn-site/YARN.html), currently one of the most promising Big Data processing framework available. 3DBG takes advantage of the lower-level distributed filesystem of YARN (HDFS) to store the data chunks over large clusters of commodity machines. Our system is based on three main components:
a 3D client, which allows the user to navigate through the 3D genomic space. It dynamically retrieves high-resolution 3D data and genomic metadata from the rest of the system as the user moves through the 3D space and makes queries about certain 3D regions.
a sparse, adaptive 3D index, which dictates how genomic metadata associated to contiguous regions in the 3D space are co-located in the distributed filesystem. The 3D index translates the 3D query posed by the user into a series of data chunks that have to be retrieved from the distributed filesystem.
immutable data chunks that compactly store genomic data and metadata in the distributed filesystem.
Figure 1 gives an overview of our database. We implemented our own indices and ancillary data structures to optimize all operations, and bypass the Hadoop NameNode whenever possible to reduce the end-to-end latency of the queries. We do not rely on higher-level Hadoop data structures such as those offered by SpatialHadoop (25) or Impala (http://www.cloudera.com/content/cloudera/en/products-and-services/cdh/impala.html), since these higher-level structures negatively impact the performance of on-line queries. Along similar lines, we do not directly use large-scale batch-processing features a la MapReduce, since they would introduce unreasonably high latencies in our context, but could take advantage of such functionalities for off-line operations such as batch updates or complex analytics.
Figure 1.
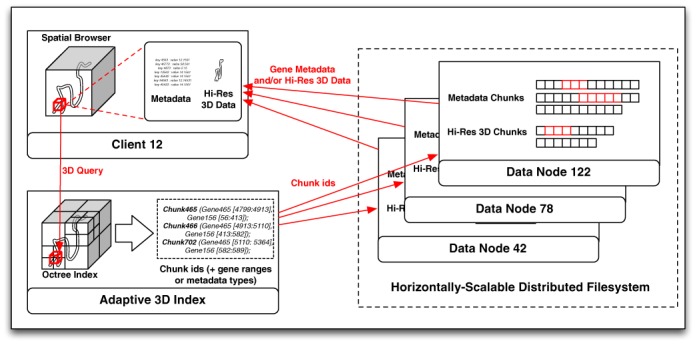
Overall architecture of 3DBG.
A detailed description of each component of our database, as well as an explanation on our query insertion and query execution techniques, is available as supplementary material. The full codebase of our current implementation is available online at https://github.com/mavlyutovrus/3d_genome_browser.
Data and web services
We describe the data stored in our database and the syntax of web queries to access them. Additional instructions and scripting packages for javascript users can be found at http://3dgb.cs.mcgill.ca/scripting/.
3D structures
Currently, three complete models of human 3D genome structures are stored in our database. We retrieve these data from (9,11) and describe them in Table 1. It is worth noting that (9) provides individual structures for each chromosome, but no global relative arrangement of all chromosomes. For this reason, we provide independently each chromosome structure.
Table 1. Origin and description of the 3D models stored in 3DBG.
The structures are interpolated with a finite number of points. This number of points depends of the resolution of the model and therefore varies from one model to another.
The volume encompassing the genome structure is segmented in 3D cubic cells. Spatial queries use the coordinates of a cube (starting and ending positions on the x, y and z axis) as an input and return the coordinates of the interpolation points modeling the DNA chains contained within this cell. In particular, it allows us to identify the ranges of DNA subsequences within this volume.
The syntax of a query is http://1kgenome.exascale.info/<mode>?xstart=<x1>& xend=<x2>&ystart=<y1>¥d=<y2>&zstart=<z1>&zend=<z2>, where <mode> should be replaced by js_test to query the model from (11), or 3D to query the model from (9). <x1> to <z2> indicate the spatial coordinate of the cell. Queries to the structure issued from (9) should also include the chromosome number and the type of the cell (normal or leukemia). In that case, an example of a full query could be http://1kgenome.exascale.info/3d?chr=19&m=normal& xstart=1& xend=2&zstart=1&zend=2& ystart=1¥d=2. The output is represented as an array of arrays, which represent contiguous chains within the volume.
Nucleotide sequences
We use GRCh38 assembly of the human genome from the UCSC genome browser as our reference human sequence (3,4). Nucleotide sequences can be accessed from their chromosomic location. The syntax of a query is http://1kgenome.exascale.info/range?start=<start>&end=<end>&chrid=<chr>, where <start> and <end> are the first and last index of the subsequence of interest, and <chr> is the chromosome number (N.B.: X and Y chromosomes are identified using letters X and Y instead of numbers).
Single nucleotide polymorphism
We store the Single Nucleotide Polymorphism (SNP) data from the 1000 Genomes Project (24). Web users can retrieve SNPs data within a specific range of a chromosome with the following query http://1kgenome.exascale.info/js_snp?chr=<chr>& start=<start>&end=<end>, where <start> and <end> are the first and last index of the subsequence of interest, and <chr> is the chromosome number.
A query returns an array of arrays showing information for each individual SNP found within this interval. This information is represented as a 4-tuple including the SNP position, the SNP ID and the two alleles.
Experimental ChIP-Sequencing data
We recorded experimental ChIP-Sequencing (Chip-Seq) data from the ENCODE project (26) stored in the UCSC genome browser (3,4). These data help us to identify transcription factors binding sites (TFBS).
ChIP-Seq data can be retrieved with a query to http://1kgenome.exascale.info/chipseq?chr=<chr>& start=<start>&end=<end>&celline=<cellid>, where the variables <start> and <end> are the first and last index of the subsequence of interest, <chr> is the chromosome number, and <cellid> is the cell line from which we obtained the experimental data. It is worth noting that in practice, Chip-Seq data may not always be available for all 3D structures models and cell types.
The output of such query is an array of 7-tuples that contain basic information on the Chip-Seq data. A 7-tuple stores the chromosome number, starting and ending index of the Chip-Seq peak, the transcription factor name, a normalized value (ranging from 1 to 1000) indicating the magnitude of the binding, the cell lines with similar TFBS and a list of SNPs occurring in this binding site.
Determining single nucleotide 3D coordinates
A key feature of a system for querying genomes in space is its capacity to directly access the 3D coordinates of any nucleotide. However, 3D genome structures are often modeled with (sparse) discrete sets of points corresponding to enzyme cut sites. In that case, it is useful to directly access the closest cut site (in each strand direction) of a model.
This information is accessible with a web query to http://1kgenome.exascale.info/chr_pos?chrid=<chr>&bp=<index>&m=<mode>, where <chr> is the chromosome number and <index> is the sequence index of the nucleotide. The variable <mode> should be set at ‘normal’ to query the GM06990 cell data or ‘leukemia’ to query the leukemia cell data (N.B.: these key words are subject to change for more precise acronyms with the addition of new cell types). This argument can be simply ignored if the user wishes to query the K562 data. The query returns an array of triplets indicating the 3D coordinate of the closest interpolation points.
3D genome Web visualization interface
Web users can access and visualize data stored in our servers via a GUI accessible at http://3dgb.cs.mcgill.ca/. Our client, based on dynamic Javascript, mostly allows the user to navigate through the 3D structure in real-time, fetching genomic data as well as high-resolution 3D meshes representing the DNA backbone from the server. It runs on most common web browsers (Firefox and Chrome). This contrasts with previous viewers that were implemented as standalone applications for specific operating systems. The source code of our browser is freely available at https://github.com/mavlyutovrus/3d_genome_browser.
Before starting to explore 3D genome structures, the users must select a model. The front page of 3DGB allows users to select which model they wish to use. Currently, three complete 3D data sets have been implemented in the database. The first is a simulation of the complete diploid human genome by Asbury et al. (11), while the second and third ones are recent reconstruction of individual chromosomes by Trieu and Cheng (9) for normal B-cells (GM06990) and acute lymphoblastic leukemia cells, respectively. New models will be added to the database as they appear in the literature.
Once a model is selected, users access a search engine that enables them to directly request specific genomic locations (i.e. chromosome number and position), target genes or arbitrary spatial coordinates. Queries re-direct the users to a 3D structure viewer pointing at the desired location. From there, they can explore and navigate the genome structure in real-time. The web client downloads all genomic and structural data in the neighborhood of the query location. More data are dynamically loaded when the user travels in the 3D space. This allows a smooth exploration of the 3D genome structure on any computing device. A screenshot of the 3D genome browser is presented in Figure 2.
Figure 2.
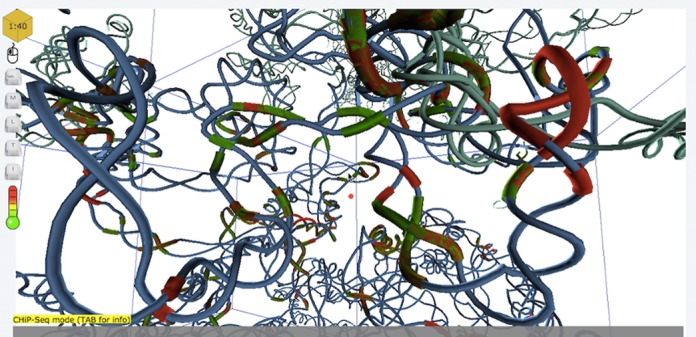
Sample screenshot of 3DGB.
Web tools
The viewer implements multiple features allowing its users to access and visualize Human genome data stored in the database. At the core of 3DGB resides our ability to define and query a 3D neighborhood, and thus to identify potential spatial relationship between genomic elements. In our viewer, a cursor of points at the center of a box representing a neighborhood to be explored. This neighborhood is represented by the red box in Figure 2. The size of the box is adjustable (by scrolling up or down), allowing the users to tune the range of spatial relationships. Once a volume has been selected (directly from a query or following an exploration of the genome structure), the user can retrieve and download the list of all SNPs located within that box, or use hyperlinks to directly access detailed information stored on the NCBI databases (27) for each individual SNP. In addition, it is also possible to access the list of all genes present in the query cell.
Alternatively, the users can switch to a linear mode. In that case, the neighborhood of the query position is defined as a sequence interval. It is equivalent to the viewing frame used in classical (i.e. one dimensional) genome browsers. This mode also allows the users to retrieve all SNPs present in this one-dimensional neighborhood.
The third mode enables the users to highlight transcription factor binding sites in the viewer. TFBSs are represented as colored regions of the DNA chain. The color indicates the intensity of the Chip-Seq experiment (green for low and red for high). The user can access detailed information about the Chip-Seq data by clicking on the TFBS region, or access UCSC genome browser records through a hyperlink.
We linked 3DGB to a 2D genome map viewer (28) to help users navigating the genome. Upon request, users can open a secondary window that displays a linear representation of the genome. The latter highlights the closest 2D genomic position to the center of the current 3D cell. The two windows are dynamically linked. A change of 3D coordinates or 2D position in either window updates automatically the position/coordinates in the other one.
Finally, we also implemented a distance calculation tool that enables the users to automatically determine the physical distance between two points in space. We intentionally did not use physical units, but instead rely on the model coordinates. Indeed, the determination of physical distances requires to interpret experimental data and make approximations which are often subject of discussions. By contrast, we believe that arbitrary units allow the users to estimate relative differences and leave them the freedom to interpret the experimental data used to obtain the 3D model.
Visualization of custom genotyping data
An important feature of our viewer is to enable users to map their private genotyping data onto reference 3D architectures, and allow them to visualize the data within our browser. This functionality is intended to provide users with tools to identify geometrical dependencies in custom genotyping data sets. The query interface allows users to upload a local file containing genotyping data. In order to prevent any formatting issues, we implemented a program to validate and convert most standard genotyping data file.
Once uploaded, the users can browse and query the 3D genome as described above. In addition to the reference data stored in our database, the users can now access simultaneously the reference SNPs collected from (24) together with those stored in the local file. To prevent any privacy issues, user data are stored locally and not transmitted to our server. A similar solution has been adopted by the UCSC genome browser (29).
RESULTS
Experimental setting
To evaluate the performance of our system, we used sequence read alignments of chromosome 11 available from the 1000 Genomes project (24). This data consists of short (around 100 bases) DNA sequence reads, mapped onto the Human reference genome. We used ∼1.5 billions of records, which constitute 250GB of raw data.
All data have been stored in a cluster (Hadoop version 2.2.0) of 10 machines. Worker nodes were commodity machines with Quad-Core Intel i7-2600 CPUs @ 3.40GHz, 8GB of DDR3-1600 RAM, 500GB Serial ATA HDD, running Ubuntu 12.04.2 LTS. The index node was similar, but with 16GB RAM. The replication factor was set to 3.
The main metric we take into account is response time (latency). As a matter of fact, execution time depends on the amount of records to be returned. In our context, we considered simple, uniform and fixed-size cube queries returning from 100 to 1000 records.
Benchmarking against the PostGIS database
The performance of storage systems can be characterized by their speed to access the data (i.e. by the average time needed to execute a query) and the influence of the size of the output on the time required for returning a response. In this section, we evaluate the performance of 3DBG compared to the PostGIS database (http://postgis.net/) to store and query 3D neighborhoods of a genome.
We uploaded in the database a data set of ∼1.5 GB (gigabytes) that contains the 3D coordinates of reference points of a simulated model of the human genome (11). All these positions were indexed in the database using the spatial index (the description of PostGIS's spatial index can be found at http://revenant.ca/www/postgis/workshop/indexing.html). Then, we measured the speed of reaching the data through the Java application using the PostGIS JDBC driver, and the influence of the size of the output (results of query) on the processing time.
In our experiments, we queried for all different reference points available in the model from (11), and called the database to get all points that were stored in the cube centered around the current reference point, with a constant edge size (100, 200, 300 and 400 base units). Our results are shown in Figures 3 and 4.
Figure 3.
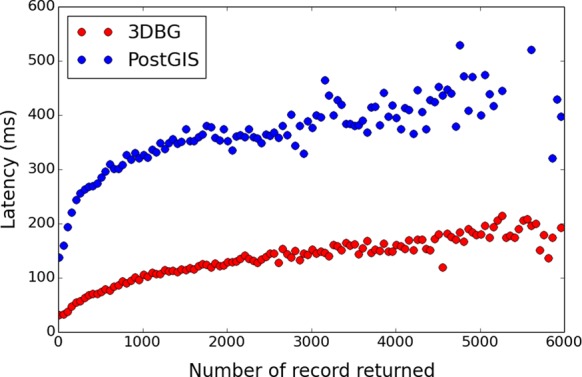
Comparison of 3DBG and PostGIS query latencies. The x-axis shows the number of records returned and the y-axis shows the latency in milliseconds (ms). Red dots are 3DBG data and blue dots PostGIS data.
Figure 4.
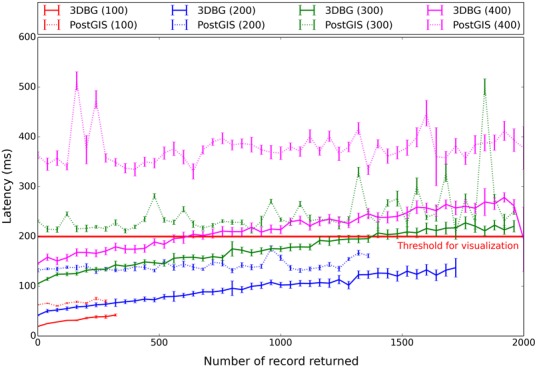
Dependencies of 3DBG and PostGIS latencies with query size. The x-axis shows the number of records returned and the y-axis shows the latency in milliseconds (ms). 3DBG data are represented with full lines, and PostGIS data with dotted lines. The colors of the curves are associated with the different sizes of the query (edge sizes of the cube varying from 100 to 400 base units). The latency threshold for real-time visualization (200 ms) is indicated with a horizontal red line.
Figure 3 shows the speed of accessing the data. Here, PosGIS has on average a query execution time well above 300 ms, and thus well above the time to gracefully retrieve and visualize data dynamically for on-line 3D browsing. By contrast, when we run the same experiment with 3DBG, the access time is clearly below this threshold. This observation demonstrates that 3DBG performs satisfactorily to visualize the 3D space at high resolution, while the latency of standard solutions such as PosGIS is too high, even for relatively small data sets.
Next, for each edge size (size of the neighborhood delimited by the cube query), we plot in Figure 4 the relation between the processing time (i.e. latency), the size of the neighborhood that we wish to explore and the number of records returned. Experiments were repeated five times to obtain the variances. Here again, we observe that PostGIS yields unsatisfactory latencies, which rapidly grow as we retrieve more data and increase the size of the query. In particular, PostGIS latencies for large queries (edge size of 400 units) exceed the threshold required for real-time visualization (∼200 ms), while 3DBG performs satisfactorily. This is an important aspect because a comfortable browsing (volume and resolution of data retrieved) requires large query (in our 3D genome browser 3DGB, we use cubes of 400 units for the 3D structure from (11)). Finally, it is worth noting that the data returned by 3DBG are also already sorted, which is not the case for PostGIS.
Benchmarking against the Hbase database
To compare the performance of our back-end solution with Hbase (http://hbase.apache.org/), we installed an Hbase cluster (version 0.96.1) on our experimental infrastructure. We also split all data according to our index for HBase, but used a standard HBase database rather than our own chunk storage. The caching for the Hbase cluster was switched off to ensure valid results. Figure 5 shows the results. As can be observed, the execution times of our system are much lower than those of HBase. 3DBG outperforms HBase for relatively small queries (left of the graph), thus ensuring a smooth navigation from the client side. Overall, both systems scale gracefully, thanks to the indexing and clustering offered by our adaptive 3D index.
Figure 5.
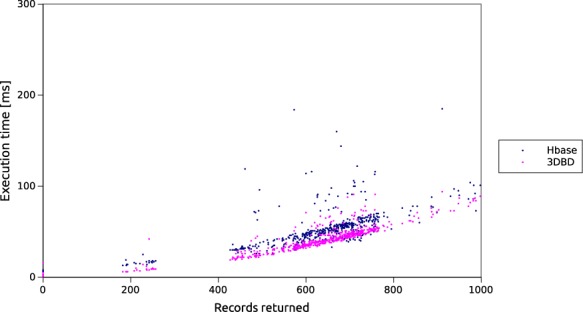
Comparison of 3DBG and HBase query latencies. The x-axis shows the number of record returned and the y-axis the latency in milliseconds (ms).
Using 3DGB to explore the 3D neighborhood of a gene
We illustrate the usefulness of 3DGB with an exploration of the 3D neighborhood of the Retinoblastoma 1 gene (RB1)—a tumor suppressor gene that has been associated with many types of cancer. This experiment does not necessarily intend to provide new insights into RB1 regulatory mechanisms, but aims to demonstrate what type of novel information can be obtained with the use of 3DGB.
We started our investigation by exploring a 3D neighborhood centered on the promoter of the RB1 gene in the 3D structure of chromosome 13 in normal B-cells (GM06990) (9). We retrieved the list of SNPs found in the promoter region of RB1, and in other DNA strands that are not in the immediate sequence neighborhood of RB1 promoter.
Distribution of SNPs in the 3D neighborhood of RB1
In addition to the promoter region, we found three other strands in the spatial vicinity of RB1: S1 (44762907, 45379923), S2 (57184305, 57531250) and S3 (58747059, 59087738). These strands are located in a radius R = 0.2 of RB1 transcription start site, which corresponds approximately to 88 Å.
A total of 1199 SNPs were identified in this 3D neighborhood, for which we retrieve their associated phenotype from (6). A complete list of these SNPs with associated phenotypes is available in the supplementary data. As expected we identified many SNPs related to various types of cancer. However, another interesting finding has been to detect the occurrence of one SNP (rs10492604) related to sleep disorders in the strand S3. Importantly, we found only two SNPs related to sleep disorders in the whole chromosome. Moreover, with a distance of 230 Å from RB1 transcription start site (R = 0.53), this other SNP (rs10492507) is also in the vicinity of RB1 gene.
Previous studies have identified that children with hereditary retinoblastoma have also an increased risk of developing trilateral retinoblastoma (30). Trilateral retinoblastoma is the combination of retinoblastoma (usually bilateral) and pineoblastoma (a tumor in the brain's pineal gland). The pineal gland secretes multiple hormones (including melatonin) that are implicated in the regulation of sleep patterns in seasonal and circadian rhythms (31).
Although our finding does not imply any causation, it suggests possible interesting genetic relationships between retinoblastoma and sleep disorders. The scripts used in this experiment are available at http://3dgb.cs.mcgill.ca/scripting.html.
DISCUSSION
We presented 3DBG, a novel storage paradigm and database system to store and query genomic data in a 3D space, and developed a lightweight 3D genome browser to visualize and navigate these data from any internet browser.
We compared 3DBG to existing systems and demonstrated that our technology enables us to significantly lower the latency of spatial queries. Importantly, we also showed that our system scales gracefully when handling more data. This technology aims to develop the infrastructure needed to mine big data sets generated by new large-scale genomic studies, and to prepare the next generation of genome browsers.
Although this paper focuses on the technical description of the database system and the evaluation of its performances, we designed 3DBG to permit complex queries in the 3D space. In particular, we also aim to use our system to extract spatial relationship between genomic elements in genome-scale studies, for example using efficient batch-oriented operations a la MapReduce on top of our data chunks (implementing such features is easy, as our whole system is based on the lower levels of the Hadoop/Yarn stack). An example of such queries could be to retrieve all pairs of enhancers/promoters that are co-localized in the the 3D genome structure.
Finally, even though we did not specifically tailor 3DBG to optimize the storage space, 3DBG is already at least as efficient as existing systems. Further versions of the database will integrate compression techniques in order to reduce the space requirements and further reduce query latencies.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Genome Canada and Genome Québec (Bioinformatics and Computational Biology competition, in part); Canadian Institutes of Health Research [CIHR BOP-130836 to J.W. and M.B.]; Natural Sciences and Engineering Research Council of Canada Discovery [NSERC RGPIN 386596-10 to J.W.]; Swiss National Science Foundation [200021_143649 to PCM]. Funding for open access charge: Genome Canada/CIHR funding.
Conflict of interest statement. None declared.
REFERENCES
- 1.Lander E.S., Linton L.M., Birren B., Nusbaum C., Zody M.C., Baldwin J., Devon K., Dewar K., Doyle M., FitzHugh W., et al. Initial sequencing and analysis of the human genome. Nature. 2001;409:860–921. doi: 10.1038/35057062. [DOI] [PubMed] [Google Scholar]
- 2.Venter J.C., Adams M.D., Myers E.W., Li P.W., Mural R.J., Sutton G.G., Smith H.O., Yandell M., Evans C.A., Holt R.A., et al. The sequence of the human genome. Science. 2001;291:1304–1351. doi: 10.1126/science.1058040. [DOI] [PubMed] [Google Scholar]
- 3.Kent W.J., Sugnet C.W., Furey T.S., Roskin K.M., Pringle T.H., Zahler A.M., Haussler D. The human genome browser at UCSC. Genome Res. 2002;12:996–1006. doi: 10.1101/gr.229102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Karolchik D., Barber G.P., Casper J., Clawson H., Cline M.S., Diekhans M., Dreszer T.R., Fujita P.A., Guruvadoo L., Haeussler M., et al. The UCSC Genome Browser database: 2014 update. Nucleic Acids Res. 2014;42:D764–D770. doi: 10.1093/nar/gkt1168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hubbard T., Barker D., Birney E., Cameron G., Chen Y., Clark L., Cox T., Cuff J., Curwen V., Down T., et al. The Ensembl genome database project. Nucleic Acids Res. 2002;30:38–41. doi: 10.1093/nar/30.1.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Flicek P., Amode M.R., Barrell D., Beal K., Billis K., Brent S., Carvalho-Silva D., Clapham P., Coates G., Fitzgerald S., et al. Ensembl 2014. Nucleic Acids Res. 2014;42:D749–D755. doi: 10.1093/nar/gkt1196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mercer T.R., Edwards S.L., Clark M.B., Neph S.J., Wang H., Stergachis A.B., John S., Sandstrom R., Li G., Sandhu K.S., et al. DNase I-hypersensitive exons colocalize with promoters and distal regulatory elements. Nat. Genet. 2013;45:852–859. doi: 10.1038/ng.2677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lieberman-Aiden E., van Berkum N.L., Williams L., Imakaev M., Ragoczy T., Telling A., Amit I., Lajoie B.R., Sabo P.J., Dorschner M.O., et al. Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science. 2009;326:289–293. doi: 10.1126/science.1181369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Trieu T., Cheng J. Large-scale reconstruction of 3D structures of human chromosomes from chromosomal contact data. Nucleic Acids Res. 2014;42:e52. doi: 10.1093/nar/gkt1411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Duan Z., Andronescu M., Schutz K., McIlwain S., Kim Y.J., Lee C., Shendure J., Fields S., Blau C.A., Noble W.S. A three-dimensional model of the yeast genome. Nature. 2010;465:363–367. doi: 10.1038/nature08973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Asbury T.M., Mitman M., Tang J., Zheng W.J. Genome3D: a viewer-model framework for integrating and visualizing multi-scale epigenomic information within a three-dimensional genome. BMC Bioinformatics. 2010;11:444. doi: 10.1186/1471-2105-11-444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhou X., Lowdon R.F., Li D., Lawson H.A., Madden P. A.F., Costello J.F., Wang T. Exploring long-range genome interactions using the WashU Epigenome Browser. Nat. Methods. 2013;10:375–376. doi: 10.1038/nmeth.2440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Guttman A. Proceedings of the 1984 ACM SIGMOD international conference on Management of data (SIGMOD '84) NY: ACM; 1984. R-Trees: a dynamic index structure for spatial searching; pp. 47–57. [Google Scholar]
- 14.Pfoser D., Jensen C.S., Theodoridis Y. Novel approaches to the indexing of moving object trajectories. In: Abbadi AE, Brodie ML, Chakravarthy S, Dayal U, Kamel N, Schlageter G, Whang K-Y, editors. San Francisco, CA: Morgan Kaufmann Publishers Inc; 2000. pp. 395–406. [Google Scholar]
- 15.Song Z., Roussopoulos N. SEB-tree: an approach to index continuously moving objects. In: Chen M-S, Chrysanthis PK, Sloman M, Zaslavsky A.B., editors. London: Springer-Verlag; 2003. pp. 340–344. [Google Scholar]
- 16.Rasetic S., Sander J., Elding J., Nascimento M.A. VLDB Endowment; 2005. A trajectory splitting model for efficient spatio-temporal indexing; pp. 934–945. [Google Scholar]
- 17.Prasad V., Adam C., Everspaugh C., Patel J.M. Indexing large trajectory data sets with SETI. 2003. http://www-db.cs.wisc.edu/cidr/cidr2003/program/p15.pdf http://www-db.cs.wisc.edu/cidr/cidr2003/program/p15.pdf
- 18.Botea V., Mallett D., Nascimento M.A., Sander J. PIST: an efficient and practical indexing technique for historical spatio-temporal point data. GeoInformatica. 2008;12:143–168. [Google Scholar]
- 19.Patel J.M., Chen Y., Chakka V.P. NY: ACM; 2004. STRIPES: an efficient index for predicted trajectories; pp. 635–646. [Google Scholar]
- 20.Porkaew K., Lazaridis I., Mehrotra S. Querying mobile objects in spatio-temporal databases. In: Jensen CS, Schneider M, Seeger B, Tsotras VJ, editors. London: Springer-Verlag; 2001. pp. 59–78. [Google Scholar]
- 21.Ankerst M., Breunig M.M., Kriegel H.-P., Sander J. NY: ACM; 1999. OPTICS: ordering points to identify the clustering structure; pp. 49–60. [Google Scholar]
- 22.Wang W., Yang J., Muntz R.R. STING: a statistical information grid approach to spatial data mining. In: Jarke M, Carey MJ, Dittrich KR, Lochovsky FH, Loucopoulos P, Jeusfeld MA, editors. San Francisco, CA: Morgan Kaufmann Publishers Inc; 1997. pp. 186–195. [Google Scholar]
- 23.Zhang T., Ramakrishnan R., Livny M. BIRCH: an efficient data clustering method for very large databases. In: Widom J, editor. NY: ACM; 1996. pp. 103–114. [Google Scholar]
- 24.1000 Genomes Project Consortium. Abecasis G.R., Altshuler D., Auton A., Brooks L.D., Durbin R.M., Gibbs R.A., Hurles M.E., McVean G.A. A map of human genome variation from population-scale sequencing. Nature. 2010;467:1061–1073. doi: 10.1038/nature09534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Eldawy A., Mokbel M.F. A demonstration of spatialhadoop: an efficient mapreduce framework for spatial data. Proc. VLDB Endowment. 2013;6:1230–1233. [Google Scholar]
- 26.ENCODE Project Consortium. The ENCODE (ENCyclopedia Of DNA Elements) project. Science. 2004;306:636–640. doi: 10.1126/science.1105136. [DOI] [PubMed] [Google Scholar]
- 27.Sayers E.W., Barrett T., Benson D.A., Bolton E., Bryant S.H., Canese K., Chetvernin V., Church D.M., DiCuccio M., Federhen S., et al. Database resources of the National Center for Biotechnology Information. Nucleic Acids Res. 2011;39:D38–D51. doi: 10.1093/nar/gkq1172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Medina I., Salavert F., Sanchez R., de Maria A., Alonso R., Escobar P., Bleda M., Dopazo J. Genome Maps, a new generation genome browser. Nucleic Acids Res. 2013;41:W41–W46. doi: 10.1093/nar/gkt530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Haeussler M., Raney B.J., Hinrichs A.S., Clawson H., Zweig A.S., Karolchik D., Casper J., Speir M.L., Haussler D., Kent W.J. Navigating protected genomics data with UCSC Genome Browser in a Box. Bioinformatics. 2015;31:764–766. doi: 10.1093/bioinformatics/btu712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.de Jong M.C., Kors W.A., de Graaf P., Castelijns J.A., Kivelä T., Moll A.C. Trilateral retinoblastoma: a systematic review and meta-analysis. Lancet Oncol. 2014;15:1157–1167. doi: 10.1016/S1470-2045(14)70336-5. [DOI] [PubMed] [Google Scholar]
- 31.Macchi M.M., Bruce J.N. Human pineal physiology and functional significance of melatonin. Front. Neuroendocrinol. 2004;25:177–195. doi: 10.1016/j.yfrne.2004.08.001. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.