


Abstract
Human single-stranded DNA binding protein 1 (hSSB1) plays a critical role in responding to DNA damage and maintaining genome stability. However, the regulation of hSSB1 remains poorly studied. Here, we determined that hSSB1 acetylation at K94 mediated by the acetyltransferase p300 and the deacetylases SIRT4 and HDAC10 impaired its ubiquitin-mediated degradation by proteasomes. Moreover, we demonstrated that the hSSB1-K94R mutant had reduced cell survival in response to DNA damage by radiation or chemotherapy drugs. Furthermore, the p300/CBP inhibitor C646 significantly enhanced the sensitivity of cancer cells to chemotherapy drugs, and a positive correlation between hSSB1 and p300 level was observed in clinical colorectal cancer samples. Acetylation, a novel regulatory modification of hSSB1, is crucial for its function under both physiological and pathological conditions. This finding supports the notion that the combination of chemotherapy drugs with acetylation inhibitors may benefit cancer patients.
INTRODUCTION
The genome constantly encounters DNA damage induced by environmental hazards, replication errors and other genotoxic stresses. DNA double strand breaks (DSBs) are among the most cytotoxic events. If not correctly repaired, they may induce genomic instability and tumorigenesis (1–3). It is therefore critical for cells to efficiently detect, transduce and repair the break to prevent the accumulation of damage. There are two main DSB repair mechanisms: non-homologous end joining (NHEJ) and homologous recombination (HR) (4). The repair mechanism used depends on the nature of the cell and the phase of the cell cycle. NHEJ is used during the entire cell cycle, whereas HR usually occurs in the S or G2 phase of the cell cycle (5,6) and is critical for the maintenance of genome stability because of its precise repair of DNA DSBs (7,8). One of the first events during HR is the recruiting of the heterotrimeric MRE11, RAD50 and NBS1 (MRN) repair complex to the DSB site (9), which is essential for the full activation of ataxia telangiectasia mutated (ATM) kinase. Activation of ATM in turn phosphorylates NBS1 at S343 and enhances the MRN complex nuclease activity to generate single-stranded DNA (ssDNA) (10,11). The ssDNA can bind ssDNA-binding proteins that play critical roles in DNA replication, recombination and repair (12).
Human replication protein A (RPA) is the most extensively studied human single-stranded DNA-binding protein (SSB). It has three subunits: RPA70, RPA32 and RPA14 (13). RPA is generally believed to be the major SSB protein in eukaryotic cells and plays critical roles in DNA replication and DNA repair pathways (14). However, the oligomeric structure of human RPA has no similarities to the bacterial SSBs (15). Recently, the novel human ssDNA-binding protein human single-stranded DNA binding protein 1 (hSSB1) was found to have better structural similarity to bacterial SSBs than RPA (16). hSSB1 is a 211 amino acid protein containing an N-terminal oligonucleotide/oligosaccharide-binding (OB) fold domain. hSSB1 also exists as a member of a heterotrimeric complex called the SOSS complex (sensor of ssDNA), which consists of hSSB1, INTS3 and C9ORF80 (17). Following DNA damage, hSSB1 quickly relocates to DNA damage sites where it is phosphorylated by ATM. hSSB1 can also augment both ATM kinase activity and the ATM-dependent cell cycle checkpoint activation (16). Mechanistically, hSSB1 directly binds with NBS1 and is required for efficient MRN complex recruitment to DSB sites (15,18). hSSB1 has recently been shown to recognize the linkage of each ADP ribose unit in poly(ADP ribose) (PAR) in response to DNA damage, as the OB fold domain of hSSB1is a PAR binding domain, promoting its recruitment to the DNA damage sites (19). In addition, our group has shown that hSSB1 plays a crucial role in the cell cycle and response to DNA damage by modulating p53 and p21 (20,21). The E3 ligase FBXL5 promotes the ubiquitin-proteasome mediated degradation of hSSB1 (22). However, the regulation of hSSB1 itself has been little explored. In this study, we provide evidence that acetylation of hSSB1 regulates its functions in both physiological and pathological conditions.
MATERIALS AND METHODS
Cells and reagents
HeLa, HEK293T and U2OS cells were maintained in Dulbecco's modified Eagle's medium (Life Technologies) supplemented with 10% fetal bovine serum (Life Technologies) with 5% CO2 at 37°C.
Plasmid construction
p300 truncation plasmids were a gift from Prof. Xiaolong Liu (Institute of Biochemistry and Cell Biology, SIBS, CAS). FLAG-SIRTs and FLAG-HDACs were gifts from Prof. Binhua P. Zhou (University of Kentucky). All other transient ectopic expression vectors were constructed using the pCDNA3.1 vector (Invitrogen). The p300 HAT dead mutation is SY (1396,1397) to RR.
Western blot analysis
Cells were lysed in RIPA lysis buffer (50 mM Tris–HCl, 150 mM NaCl, 5 mM EDTA, 0.5% Nonidet P-40 and a protease and phosphatase inhibitor cocktail (Calbiochem)). Proteins were separated by sodium dodecyl sulphate-polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to 0.45 μM PVDF membranes (Millipore). The immunoblots were processed according to standard procedures using primary antibodies directed to p300 (BD, 554215), CBP (CST, 4772), GAPDH (CST, 2118), HA (CST, 3724), Flag (CST, 14793), Hsp70 (Santa Cruz, sc-24), p53 (Santa Cruz, sc-6243), c-Myc (Santa Cruz, sc-40), hSSB1 (Bethyl, A301–938A), NBS1 (Bethyl, A300–187), tubulin (Bioworld, BS1482M) and ac-K (abnova, PAB10348).
Immunoprecipitation
HEK293T cells transfected with the indicated plasmids were lysed in RIPA lysis buffer and then centrifuged at 12 000 rpm for 30 min. The supernatants were first incubated with anti-FLAG-agarose or anti-HA-agarose (Sigma Chemical Co.) overnight at 4°C, and the precipitates were washed five times with RIPA lysis buffer.
RNAi treatment
The oligonucleotide sequences targeting hSSB1, p300 and CBP mRNA were as follows: hSSB1: 5′-CGACGGAGACCUUUGUGAAdTdT;
p300: GCACGAACTAGGAAAGAAA, CGACTTACCAGATGAATTA, GCACAAATGTCTAGTTCTT; CBP: CGGCACAGCCTCTCAGTCA, GGAGCCATCTAGTGCATAA, GGAACTAGAACAAGAAGAA. Transfection was performed according to the manufacturer's instructions using Lipofectamine RNAiMAX transfection reagent (Invitrogen) and 50 nM siRNA.
Cell survival assays
Cells were transfected twice with control siRNA or siRNAs specifically targeting hSSB1. Twenty-four hours after the second transfection, cells (1 × 103) were split and transferred into 60 mm dishes. Cells were incubated for 24 h before they were treated with IR or camptothecin (CPT) as indicated. The medium was replaced 24 h later and cells were then incubated for 14 days. Resulting colonies were fixed and stained with Coomassie blue.
Apoptosis assay
U2OS cells were seeded onto 6-well plates for 24 h and then treated with etoposide (Eto, 25 μM) or C646 (10 μM) for 24 h to induce apoptosis. Cells were then collected using trypsin without EDTA, washed with PBS, subjected to annexinV-EGFP and propidium iodide staining as the manufacturer's recommendations (KeyGen Biotec), and analyzed by flow cytometry.
CCK8 assay
The CCK8 assay is based on the detection of dehydrogenase activity in the viable cells. U2OS cells were seeded into 96-well plates for 24 h, treated with C646 (10 μM) and either Eto (10 μM), adriamycin (ADR, 0.5 μM) or CPT (0.5 μM) for 48 h, then incubated with CCK8 for 2 h. The CCK8 activity of cells was detected by 450 nm absorbance.
Immunohistochemistry
This procedure was approved by the Institutional Review Board of Sun Yat-sen University, and written informed consent was obtained from the patients prior to sample collection. The 196 colorectal cancer samples were retrospectively acquired from the surgical pathology archives of Sun Yat-sen University Cancer Center. Slides were dried overnight at 37°C, dewaxed in xylene, rehydrated through a graded alcohol series and immersed in 3% hydrogen peroxide for 20 min to block endogenous peroxidase activity. Antigen retrieval was performed with EDTA (pH 8) in a microwave oven for 15 min. The slides were incubated with 10% normal goat serum at room temperature for 30 min to reduce nonspecific binding. Subsequently, the slides were incubated with antibodies against hSSB1 (Bethyl, IHC-00403), p300 (Bethyl, IHC-00028) for 1 h at 37°C. After the slides were rinsed five times with PBS (pH 7.4) for 10 min, primary antibody detection was achieved using a secondary antibody (Dako) for 1 h at room temperature. The slides were stained with 3,3-diaminobenzidine after being washed in PBS again. Finally, the sections were counterstained with Mayer's hematoxylin, dehydrated and mounted. PBS was used in place of the primary antibody as a negative control. A brown particle in the nucleus was considered positive labeling. To evaluate hSSB1 and p300 staining, a semi-quantitative scoring criterion was used in which both staining intensity and positive areas were recorded. A staining index (values 0–12), which was obtained as the product of intensity of positive staining (weak, 1; moderate, 2; strong, 3) and the proportion of immunopositive cells of interest (0%, 0; <10%, 1; 10–50%, 2; 51–80%, 3; >80%, 4), was calculated. Finally, the cases were classified into two different groups: low expression (score 0–6) and high expression (score 8–12). Statistical analysis was performed using SPSS version 16 (SPSS Inc., Chicago, IL, USA). The association between hSSB1 and p300 abundance was assessed using χ2 tests.
RESULTS
hSSB1 is acetylated by p300 in cells
hSSB1 plays crucial roles in the DNA damage response (16), and our recent work has demonstrated that hSSB1 directly binds p300 and promotes its acetyltransferase activity on p53 (21). This interaction between hSSB1 and p300 prompted us to test whether hSSB1 is also acetylated by p300. First, as shown in Figure 1A, acetylation of endogenous hSSB1 was clearly detected in an immunoprecipitation (IP) complex using anti-hSSB1 in cells. This acetylation was specific for hSSB1 and not for hSSB2 (Figure 1B), although hSSB1 has high similarity to hSSB2 and both hSSB1 and hSSB2 can interact with INTS3 and C9ORF80 to form the SOSS complex (17). Second, as shown in Figure 1C, the acetylation of hSSB1 was increased by cotransfection with either p300 or CBP but not with the acetyltransferases PCAF, Tip60 and GCN5. Moreover, the acetylation of hSSB1 was dramatically decreased by knockdown of p300 but not of CBP, suggesting that p300 is the dominant acetyltransferase for hSSB1 in HEK293T cells (Figure 1D, Supplementary Figure S1). Third, the p300/CBP inhibitor C646 consistently reduced the hSSB1 acetylation level in a dose-dependent manner (Figure 1E). Wild-type p300 but not its dead mutant form was capable of increasing the acetylation of hSSB1 (Figure 1F). Collectively, our results demonstrate that hSSB1 is acetylated by the acetyltransferase p300 in HEK293T cells.
Figure 1.
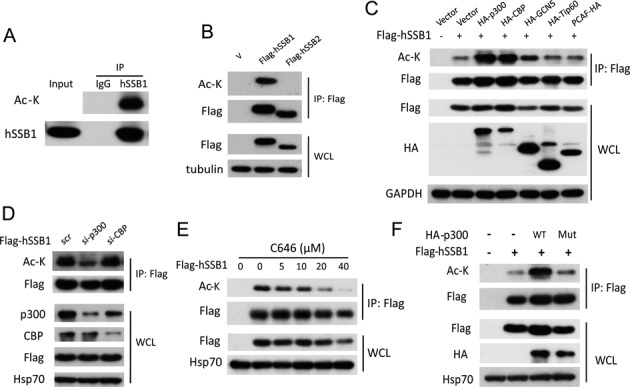
p300 acetylates hSSB1 in vivo. (A) HEK293T cell extracts were immunoprecipitated by an hSSB1 antibody and then detected by an anti-ac-K antibody. (B) HEK293T cells transfected with FLAG-hSSB1 or FLAG-hSSB2 plasmids for 48 h were lysed and analyzed using the anti-FLAG antibody followed by western blotting analysis. (C) HEK293T cells were cotransfected with FLAG-hSSB1 and HA-tagged acetyltransferases for 48 h, lysed and immunoprecipitated using an anti-FLAG antibody followed by western blotting analysis. (D) HEK293T cells were transfected with p300 or CBP siRNAs for 24 h and transfected with the FLAG-hSSB1 plasmid 24 h later to detect the hSSB1 acetylation levels. (E) FLAG-hSSB1 transfected HEK293T cells were treated with the p300/CBP inhibitor C646 for 12 h and then analyzed as in (A). (F) HEK293T cells were cotransfected with wild type (WT) or the acetyltransferase dead p300 mutant with FLAG-hSSB1 for 48 h and analyzed by IP and western blotting.
hSSB1 is acetylated by p300 at lysine 94 in cells
We next sought to determine which hSSB1 lysine (K) residue is acetylated by p300. Given that hSSB1 has 211 amino acids and only 13 lysine residues, we generated 13 hSSB1 mutants by individually altering each K into arginine (R), and the acetylation level in these mutants was monitored. As shown in Figure 2A, the acetylation of the hSSB1-K94R mutant was dramatically decreased, whereas the acetylation was only marginally altered in the other hSSB1 mutants. Indeed, the acetylation of hSSB1 at K94 in cells was further confirmed by mass spectrometry, as the acetylated K94 peptide was observed using this method (Figure 2B). Furthermore, as shown in Figure 2C, p300 could no longer increase the acetylation of hSSB1-K94R, illustrating that hSSB1 K94 is the p300 acetylation site in cells.
Figure 2.
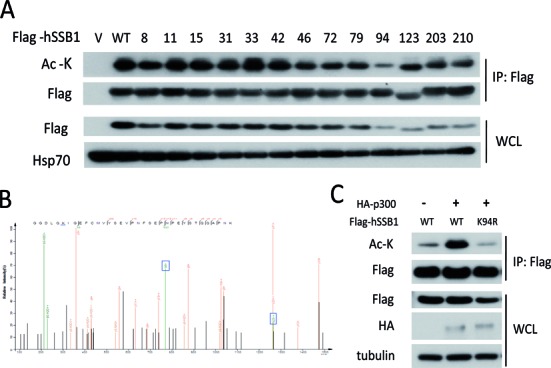
Identification of the lysine 94 acetylation site through site mutation and mass spectrometry. (A) HEK293T cells were transfected with FLAG-hSSB1 mutated into arginine at the indicated lysine residue for 48 h and then analyzed by IP and western blotting. (B) FLAG-hSSB1 were overexpressed in HEK293T cells, purified by FLAG-agarose and isolated through SDS-PAGE. The hSSB1 band on the SDS-PAGE was analyzed by ESI-QUAD-TOF mass spectrometry. (C) hSSB1 WT or K94R mutant plasmids were cotransfected with p300 for 48 h and acetylation levels were analyzed by western blotting.
The OB fold domain of hSSB1 and the CH3/HAT domain region of p300 mediated their interaction in cells
Our recent work has shown that hSSB1 interacts with p300 at endogenous levels (21). We therefore tried to map their interaction regions. As expected, the complex of FLAG-tagged hSSB1 with HA-tagged p300 was reciprocally detected in cells by IP using anti-FLAG or anti-HA (Figure 3A). Next, as shown in Figure 3B and C, five fragments of p300 were generated as previously described (23). hSSB1 was divided into two domains, the OB fold domain and the C terminal domain (20,21). As shown in Figure 3D, the p300–4 fragment containing amino acids 1410–1942, which has been shown to bind ThPOK (23), was responsible for the binding of p300 with hSSB1. As shown in Figure 3E, hSSB1 bound p300 through its OB fold domain containing K94. Furthermore, the K94R mutation diminished its interaction with p300 (Supplementary Figure S2). Thus, the interaction between hSSB1 and p300 in cells is mediated by the OB fold domain of hSSB1 and the CH3/HAT domain region of p300.
Figure 3.
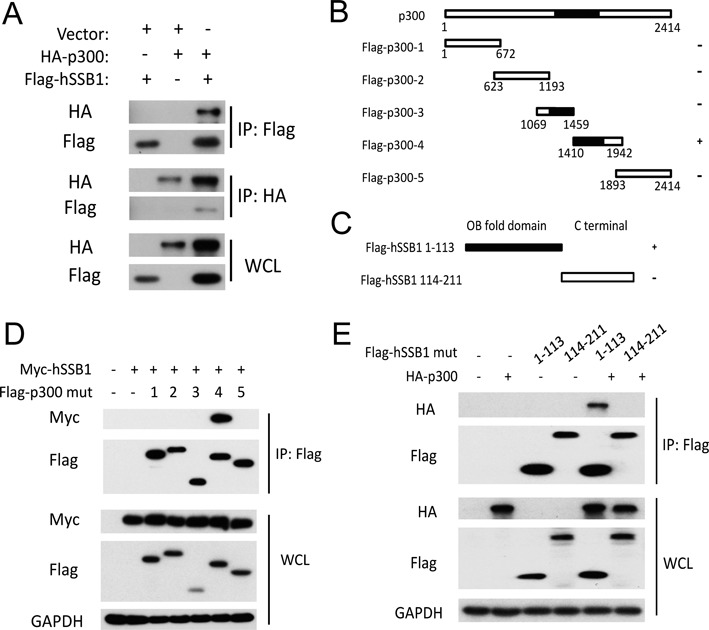
The hSSB1 OB fold domain interacts with the p300 HAT domain. (A) HEK293T cells transfected with the indicated plasmids for 48 h were lysed with RIPA buffer. IP using anti-HA or anti-FLAG antibodies and western blotting with the indicated antibody were performed. (B) Schematic description of the p300 domains. (C) Schematic description of the hSSB1domains. (D) HEK293T cells cotransfected with Myc-hSSB1 and p300 mutants as indicated for 48 h were lysed with RIPA buffer and then analyzed by IP and western blotting. (E) HEK293T cells cotransfected with HA-p300 and hSSB1 1–113 or 114–211 mutants as indicated for 48 h were lysed with RIPA buffer and analyzed by IP and western blotting.
Acetylation of hSSB1 was synergistically regulated by SIRT4 and HDAC10 in cells
Next, we asked which deacetylase was involved in the acetylation of hSSB1 at K94, as recent studies have shown that the acetylation of proteins such as SKP2, FOXO3a and p53 is a dynamic process reversed by histone deacetylases (HDACs) or/and SIRTs (24–26). Trichostatin A (TSA), an inhibitor for many HDACs, and nicotinamide (NAM), a SIRT inhibitor, were used. As shown in Figure 4A, both TSA and NAM alone increased the acetylation of hSSB1. This acetylation was significantly enhanced when these two inhibitors were combined, indicating that at least two different deacetylases are capable of deacetylating hSSB1. We next cotransfected each of the known deacetylases into cells with hSSB1. As shown in Figure 4B and C, hSSB1 acetylation was dramatically decreased by overexpressing either SIRT4 or HDAC10 in cells. More importantly, overexpression of SIRT4 or HDAC10 decreased the acetylation of wild-type hSSB1 to a similar level as hSSB1-K94R (Figure 4D). Taken together, these results strongly argue that SIRT4 and HDAC10 are responsible for deacetylating the K94 acetylation mediated by p300 in cells.
Figure 4.
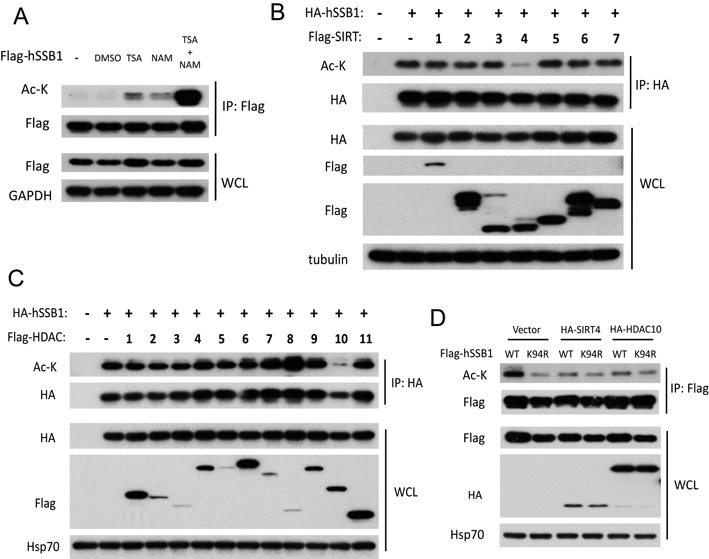
SIRT4 and HDAC10 deacetylate hSSB1. (A) HEK293T cells were transfected with FLAG-hSSB1 for 36 h, treated with TSA (2 μM) or NAM (5 mM) as indicated for 12 h, then lysed and analyzed by IP and western blotting. (B) hSSB1 was cotransfected with a series of FLAG-SIRTs into HEK293T cells for 48 h, then lysed and analyzed by HA-agarose IP and western blotting. (C) hSSB1 was cotransfected with a series of FLAG-HDACs into HEK293T cells for 48 h, then lysed and analyzed by HA-agarose IP and western blotting. (D) hSSB1 WT or K94R mutation plasmids were cotransfected with SIRT4 or HDAC10 for 48 h and acetylation levels were analyzed by western blotting.
Acetylation at hSSB1 K94 may enhance its protein stability by antagonizing its ubiquitination
Given that hSSB1 is regulated by the ubiquitin-proteasome system and is stabilized in response to DNA damage (16,20), acetylation may promote protein stability by antagonizing ubiquitination (27,28). We were curious to explore whether the acetylation of hSSB1 could affect its ubiquitination and stability. As shown in Figure 5A and B, the acetylation deficient mutant hSSB1-K94R had a much shorter half-life than wild-type hSSB1. Consistently, more ubiquitination was observed with the hSSB1-K94R mutant than with wild-type hSSB1 (Figure 5C), indicating that the K94 acetylation of hSSB1 may inhibit its ubiquitination and protect it from proteasomal degradation. Indeed, as shown in Figure 5D, hSSB1 ubiquitination was enhanced by C646 but was diminished by the combination of TSA and NAM, reinforcing the idea that acetylation of hSSB1 at K94 may positively regulate hSSB1 stability by inhibiting its ubiquitination.
Figure 5.
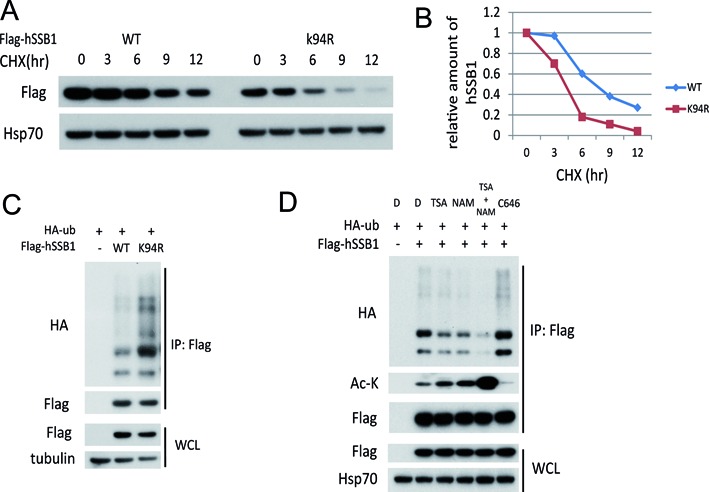
Acetylation of lysine 94 stabilizes hSSB1. (A) HEK293T cells transfected with FLAG-hSSB1 WT or K94R mutation plasmids for 36 h were incubated with 20 μg/ml cycloheximide (CHX) for the indicated periods and then analyzed by western blotting. (B) Quantitation of hSSB1 protein levels during degradation based on (A). (C) HEK293T cells were transfected with the indicated plasmids for 48 h and subjected to IP using an anti-FLAG antibody followed by western blot analysis. (D) HEK293T cells transfected with the indicated plasmids for 36 h were treated with TSA (2 μM), NAM (5 mM) or C646 (10 μM) for 12 h, subjected to IP using an anti-FLAG antibody and followed by western blot analysis.
The hSSB1 acetylation at K94 is increased in response to DNA damage
Next, we asked whether the K94 acetylation of hSSB1 played roles in the DNA damage response. Strikingly, the acetylation of hSSB1 increased in response to DNA damage induced by Eto in a dose- and time-dependent manner (Figure 6A and B). Second, this enhancement of hSSB1 acetylation by DNA damage was abolished by the p300/CBP inhibitor C646 (Figure 6C). The enhancement of hSSB1 acetylation was abolished in the hSSB1-K94R mutant, indicating that K94 is the dominant acetylation site of hSSB1 under DNA damage (Figure 6D).
Figure 6.
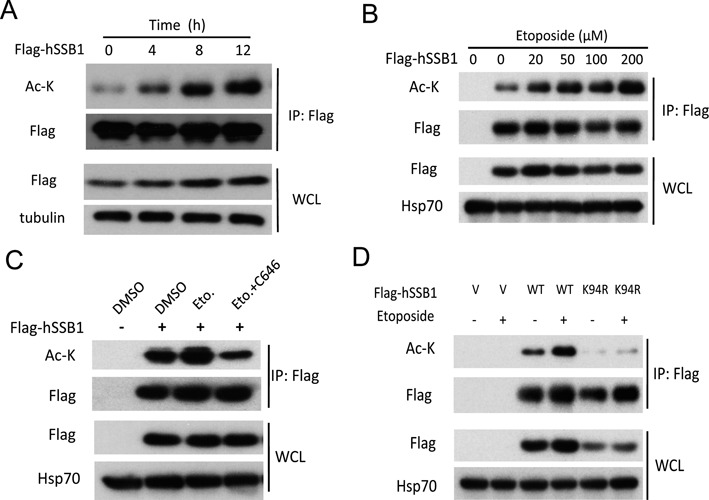
hSSB1 lysine 94 acetylation increases in response to DNA damage. (A, B) FLAG-hSSB1 transfected HEK293T cells were treated with Eto for the indicated periods and concentrations, then analyzed by western blotting. (C) HEK293T cells transfected with FLAG-hSSB1 for 36 h were treated with Eto (100 μM) singly or in combination with C646 (10 μM) for 12 h, then analyzed by IP and western blotting. (D) hSSB1 WT or K94R mutation plasmids were transfected into HEK293T cells for 36 h followed by Eto (100 μM) for 12 h and analyzed by IP and western blotting.
The hSSB1 acetylation at K94 mediated by p300 has biological significance
Finally, we determined the biological significance of hSSB1 acetylation at K94 mediated by p300, as knockdown of hSSB1 enhanced the sensitivity of HeLa cells to radiotherapy and chemotherapy (16). As shown in Figure 7A and B, using a colony formation assay, knockdown of endogenous hSSB1 by siRNA dramatically decreased cell survival under different doses of ionization radiation (IR) and CPT, a commonly used chemotherapeutic drug. Survival was largely rescued by reintroducing wild-type hSSB1 but not the hSSB1-K94R mutant. Furthermore, the genome instability induced by hSSB1 knockdown was rescued by wild-type hSSB1 but not the K94R mutant (Figure 7C and D). However, the recruitment of hSSB1 to laser-induced DNA damage sites showed no difference between WT and K94R (Supplementary Figure S3), indicating that hSSB1 acetylation is not essential for its recruitment to sites of DNA damage. These results further support the notion that the acetylation of hSSB1 at K94 may affect its biological functions in response to DNA damage. Furthermore, as shown in Figure 7E, C646 greatly increased the apoptosis induced by Eto. The sensitivities of cancer cells to several commonly used chemotherapy drugs, including Eto, CPT and ADR, were also increased by C646 in a CCK8 assay (Figure 7F). Most importantly, a positive correlation between p300 and hSSB1 was detected in colorectal cancer samples (Figure 7G and H), supporting the cellular result that p300 stabilized hSSB1.
Figure 7.
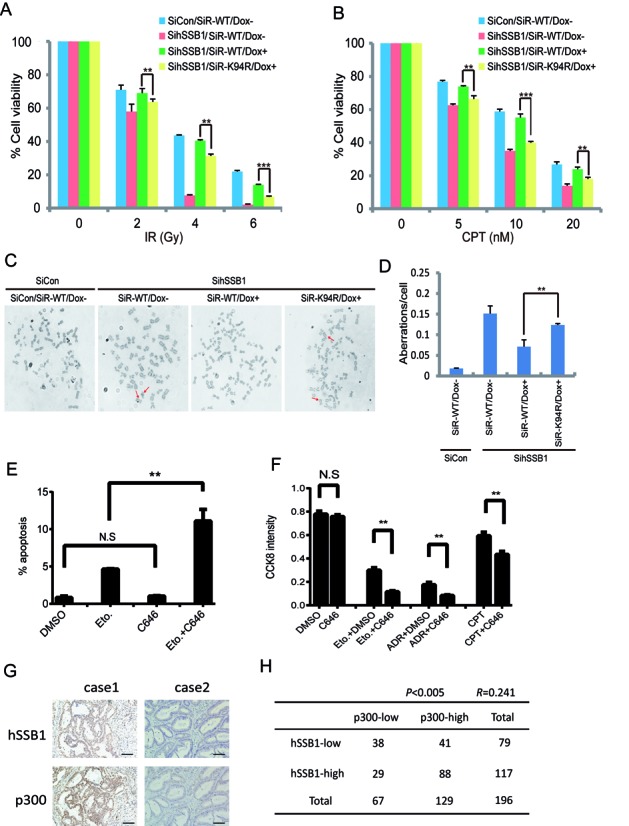
Inhibiting lysine 94 acetylation enhances cancer cell sensitivity to DNA damage agents. (A, B) Cells that express siRNA-resistant wild-type hSSB1 or its point mutant under the control of a tetracycline-inducible promoter were generated. The resulting cell lines were transfected twice with control or hSSB1 siRNAs. Following IR or CPT treatment, cells were permitted to grow for 14 days before staining. The experiments were performed in triplicate. The results shown are averages of three independent experiments. Bars indicate the SEM. **P < 0.01, ***P < 0.001, Student's t test. (C, D) Quantification of chromosomal aberrations in hSSB1-depleted HeLa cells expressing wild-type hSSB1 or hSSB1-K94R. The average of two experiments is shown. At least 50 cells were counted in each experiment. Bars indicate the SEM. **P < 0.01, Student's **t test. (E) U2OS cells were treated with either Eto (25 μM), C646 (10 μM) or combination for 24 h. The cells were then trypsinized and the percentage of apoptotic cells was detected by flow cytometry (n = 3), bars indicate the SEM. **P < 0.01, Student's t test. (F) U2OS cells were treated with C646 (10 μM) combined with Eto (10 μM), ADR (0.5 μM) or CPT (0.5 μM) for 48 h and the CCK8 activity of the cells was detected (n = 3), bars indicate the SEM. **P < 0.01, Student's t test. (G) Immunohistochemistry for hSSB1 and p300 was performed using 196 clinical colorectal cancer samples as described in the Materials and Methods section. Representative images of hSSB1 and of p300 expression levels in the same colorectal cancer sample. Case 1, both hSSB1 and p300 high expression. Case 2, both hSSB1 and p300 low expression. Scale bars: 100 μm. (H) A positive correlation was observed between hSSB1 and p300 in the 196 clinical colon cancer samples (P < 0.005, χ2 tests. R: Spearman correlation coefficient).
DISCUSSION
In this report, we have provided the first evidence that hSSB1 is acetylated by p300 at K94. This acetylation is increased in response to DNA damage and in turn stabilizes hSSB1 by impairing its ubiquitination.
hSSB1 is structurally close to the bacterial SSB, indicating it is an evolutionarily conserved protein. The ATM kinase phosphorylates hSSB1 at threonine (T) 117 in response to DNA damage, and this phosphorylation is required for hSSB1 stabilization (16). Here, we have shown that hSSB1 is acetylated by p300 at K94, which is enhanced by DNA damage and in turn stabilizes hSSB1 by impairing its ubiquitination (Figures 1, 2, 5, 6). These results indicate that both hSSB1 phosphorylation at T117 and acetylation at K94 have similar roles in regulating its protein stability in response to DNA damage. However, these modifications had no effect on each other under such conditions (data not shown). The recently revealed structure of the hSSB1-IntS3-C9ORF80 complex (29) suggests that K94 contributes to the stability of hSSB1, which is supported by our finding that the hSSB1-K94R mutant is more easily degraded than wild-type hSSB1 (Figure 4A and B). In addition, K94 locates in the OB fold domain of hSSB1, and this domain mediates both protein–DNA and the protein–protein interactions. We speculate that hSSB1 acetylation may affect its DNA binding ability. Interestingly, the acetylation of hSSB2, the close homologue of hSSB1, was undetectable in cells (Figure 1B), indicating that the regulation of hSSB1 is more complicated than that of hSSB2. This is consistent with previous reports showing that hSSB1 is the major partner of IntS3 and C9ORF80 in response to DNA damage (19).
p300, first identified as one of the major cellular proteins that interact with adenoviral E1A transforming protein (30), functions as a scaffold, a bridge and an acetyltransferase to regulate a diversity of cellular events and pathways. For example, p300 acetylates SKP2 at both K68 and K71, promoting its cytoplasmic translocation and inhibiting its degradation by APC/CCdh1. The cytoplasmic SKP2 promotes the degradation of E-cadherin to increase the mobility of cancer cells (24). It is noteworthy that the relationship between p300 and SKP2 is reciprocal. p300 acetylates SKP2 and modulates its localization and activity, while SKP2 interacts with the p300 CH1 and CH3 domains that also bind p53. Consequently, overexpression of SKP2 disturbs the acetylation and activation of p53 (31). Our results show that the relationship between hSSB1 and p300 is also reciprocal. hSSB1 interacts with both p53 and p300 and facilitates the acetylation of p53 (21), while p300 acetylates hSSB1 to regulate its stability and function under normal conditions and in response to DNA damage, as shown here. These results indicate that the regulation among hSSB1, p300 and p53 is tightly coordinated.
In addition, p300 has been reported to participate in the DNA damage response. The combination of the p300/CBP inhibitor C646 with cisplatin enhanced the apoptosis of the WM35 melanoma cell line (32). ATM phosphorylates p300 at serine 106, which in turn promotes the recruitment of NBS1 in response to DNA damage (33). Our results showed that p300 acetylates hSSB1 at K94, which is increased under DNA damage (Figures 1, 2, 6). This is the first report of p300 involvement in DNA damage through its acetyltransferase activity to modulate substrates such as hSSB1 and p53. Knockdown of hSSB1 enhances the sensitivity of cancer cells to radiotherapy and chemotherapy (16), and cells with the hSSB1-K94R mutation showed a higher sensitivity than those with wild-type hSSB1 (Figure 7A and B). The combination of C646 with chemotherapy drugs more efficiently promoted cancer cell apoptosis and inhibited proliferation (Figure 7E and F). More importantly, p300 and hSSB1 expression are positively correlated in the colorectal cancer samples (Figure 7G and H).
In summary, hSSB1 is both phosphorylated by ATM and acetylated by p300. The latter is antagonized by both SIRT4 and HDAC10 to prevent its proteasomal degradation in response to DNA damage and consequently enhance the DNA damage checkpoints, as proposed in Figure 8.
Figure 8.
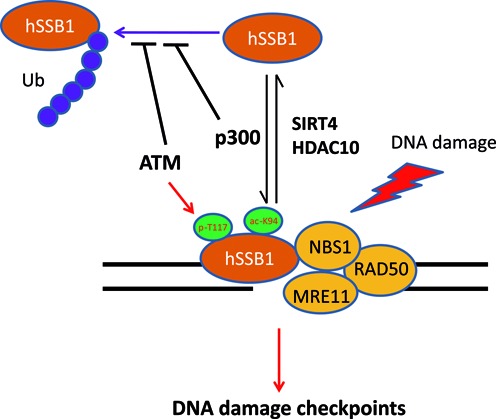
A proposed model for hSSB1 regulation in response to DNA damage. hSSB1 is phosphorylated by ATM and acetylated by p300, which is antagonized by both SIRT4 and HDAC10 to prevent proteasomal degradation, to recruit NBS1 and other molecules in response to DNA damage, and consequently to enhance the DNA damage checkpoints.
Supplementary Material
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Key project (2013ZX10002008005 to T.K.); National Nature Science Foundation in China (NSFC) (81125015 to T.K.); 973 project (2012CB967000 to T.K.). Funding for open access charge: 2013ZX10002008005 (to T.K.).
Conflict of interest statement. None declared.
REFERENCES
- 1.Bartek J., Lukas J. DNA damage checkpoints: from initiation to recovery or adaptation. Curr. Opin. Cell Biol. 2007;19:238–245. doi: 10.1016/j.ceb.2007.02.009. [DOI] [PubMed] [Google Scholar]
- 2.Bartkova J., Horejsi Z., Koed K., Kramer A., Tort F., Zieger K., Guldberg P., Sehested M., Nesland J.M., Lukas C., et al. DNA damage response as a candidate anti-cancer barrier in early human tumorigenesis. Nature. 2005;434:864–870. doi: 10.1038/nature03482. [DOI] [PubMed] [Google Scholar]
- 3.Hoeijmakers J.H. Genome maintenance mechanisms for preventing cancer. Nature. 2001;411:366–374. doi: 10.1038/35077232. [DOI] [PubMed] [Google Scholar]
- 4.Lukas J., Bartek J. Watching the DNA repair ensemble dance. Cell. 2004;118:666–668. doi: 10.1016/j.cell.2004.09.006. [DOI] [PubMed] [Google Scholar]
- 5.Matsumoto Y. [Smart choice between two DNA double-strand break repair mechanisms] Igaku Butsuri. 2014;34:57–64. [PubMed] [Google Scholar]
- 6.Kakarougkas A., Jeggo P.A. DNA DSB repair pathway choice: an orchestrated handover mechanism. Br. J. Radiol. 2014;87:20130685. doi: 10.1259/bjr.20130685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Liu T., Huang J. Quality control of homologous recombination. Cell. Mol. Life Sci. 2014;71:3779–3797. doi: 10.1007/s00018-014-1649-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Renkawitz J., Lademann C.A., Jentsch S. Mechanisms and principles of homology search during recombination. Nat. Rev. Mol. Cell Biol. 2014;15:369–383. doi: 10.1038/nrm3805. [DOI] [PubMed] [Google Scholar]
- 9.D'Amours D., Jackson S.P. The Mre11 complex: at the crossroads of dna repair and checkpoint signalling. Nat. Rev. Mol. Cell Biol. 2002;3:317–327. doi: 10.1038/nrm805. [DOI] [PubMed] [Google Scholar]
- 10.Jazayeri A., Balestrini A., Garner E., Haber J.E., Costanzo V. Mre11-Rad50-Nbs1-dependent processing of DNA breaks generates oligonucleotides that stimulate ATM activity. EMBO J. 2008;27:1953–1962. doi: 10.1038/emboj.2008.128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lim D.S., Kim S.T., Xu B., Maser R.S., Lin J., Petrini J.H., Kastan M.B. ATM phosphorylates p95/nbs1 in an S-phase checkpoint pathway. Nature. 2000;404:613–617. doi: 10.1038/35007091. [DOI] [PubMed] [Google Scholar]
- 12.Iftode C., Daniely Y., Borowiec J.A. Replication protein A (RPA): the eukaryotic SSB. Crit. Rev. Biochem. Mol. Biol. 1999;34:141–180. doi: 10.1080/10409239991209255. [DOI] [PubMed] [Google Scholar]
- 13.Wold M.S. Replication protein A: a heterotrimeric, single-stranded DNA-binding protein required for eukaryotic DNA metabolism. Ann. Rev. Biochem. 1997;66:61–92. doi: 10.1146/annurev.biochem.66.1.61. [DOI] [PubMed] [Google Scholar]
- 14.Oakley G.G., Patrick S.M. Replication protein A: directing traffic at the intersection of replication and repair. Front. Biosci. 2010;15:883–900. doi: 10.2741/3652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Richard D.J., Savage K., Bolderson E., Cubeddu L., So S., Ghita M., Chen D.J., White M.F., Richard K., Prise K.M., et al. hSSB1 rapidly binds at the sites of DNA double-strand breaks and is required for the efficient recruitment of the MRN complex. Nucleic Acids Res. 2011;39:1692–1702. doi: 10.1093/nar/gkq1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Richard D.J., Bolderson E., Cubeddu L., Wadsworth R.I., Savage K., Sharma G.G., Nicolette M.L., Tsvetanov S., McIlwraith M.J., Pandita R.K., et al. Single-stranded DNA-binding protein hSSB1 is critical for genomic stability. Nature. 2008;453:677–681. doi: 10.1038/nature06883. [DOI] [PubMed] [Google Scholar]
- 17.Huang J., Gong Z., Ghosal G., Chen J. SOSS complexes participate in the maintenance of genomic stability. Mol. Cell. 2009;35:384–393. doi: 10.1016/j.molcel.2009.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Richard D.J., Cubeddu L., Urquhart A.J., Bain A., Bolderson E., Menon D., White M.F., Khanna K.K. hSSB1 interacts directly with the MRN complex stimulating its recruitment to DNA double-strand breaks and its endo-nuclease activity. Nucleic Acids Res. 2011;39:3643–3651. doi: 10.1093/nar/gkq1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhang F., Chen Y., Li M., Yu X. The oligonucleotide/oligosaccharide-binding fold motif is a poly(ADP-ribose)-binding domain that mediates DNA damage response. Proc. Natl Acad. Sci. U.S.A. 2014;111:7278–7283. doi: 10.1073/pnas.1318367111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Xu S., Feng Z., Zhang M., Wu Y., Sang Y., Xu H., Lv X., Hu K., Cao J., Zhang R., et al. hSSB1 binds and protects p21 from ubiquitin-mediated degradation and positively correlates with p21 in human hepatocellular carcinomas. Oncogene. 2011;30:2219–2229. doi: 10.1038/onc.2010.596. [DOI] [PubMed] [Google Scholar]
- 21.Xu S., Wu Y., Chen Q., Cao J., Hu K., Tang J., Sang Y., Lai F., Wang L., Zhang R., et al. hSSB1 regulates both the stability and the transcriptional activity of p53. Cell Res. 2013;23:423–435. doi: 10.1038/cr.2012.162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chen Z.W., Liu B., Tang N.W., Xu Y.H., Ye X.Y., Li Z.M., Niu X.M., Shen S.P., Lu S., Xu L. FBXL5-mediated degradation of single-stranded DNA-binding protein hSSB1 controls DNA damage response. Nucleic Acids Res. 2015;42:11560–11569. doi: 10.1093/nar/gku876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhang M., Zhang J., Rui J., Liu X. p300-mediated acetylation stabilizes the Th-inducing POK factor. J. Immunol. 2010;185:3960–3969. doi: 10.4049/jimmunol.1001462. [DOI] [PubMed] [Google Scholar]
- 24.Inuzuka H., Gao D., Finley L.W., Yang W., Wan L., Fukushima H., Chin Y.R., Zhai B., Shaik S., Lau A.W., et al. Acetylation-dependent regulation of Skp2 function. Cell. 2012;150:179–193. doi: 10.1016/j.cell.2012.05.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang F., Chan C.H., Chen K., Guan X., Lin H.K., Tong Q. Deacetylation of FOXO3 by SIRT1 or SIRT2 leads to Skp2-mediated FOXO3 ubiquitination and degradation. Oncogene. 2012;31:1546–1557. doi: 10.1038/onc.2011.347. [DOI] [PubMed] [Google Scholar]
- 26.Lee J.T., Gu W. SIRT1: regulator of p53 deacetylation. Genes Cancer. 2013;4:112–117. doi: 10.1177/1947601913484496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhao S., Xu W., Jiang W., Yu W., Lin Y., Zhang T., Yao J., Zhou L., Zeng Y., Li H., et al. Regulation of cellular metabolism by protein lysine acetylation. Science. 2010;327:1000–1004. doi: 10.1126/science.1179689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yu H., Shao Y., Gao L., Zhang L., Guo K., Wu C., Hu X., Duan H. Acetylation of sphingosine kinase 1 regulates cell growth and cell-cycle progression. Biochem. Biophys. Res. Commun. 2012;417:1242–1247. doi: 10.1016/j.bbrc.2011.12.117. [DOI] [PubMed] [Google Scholar]
- 29.Ren W., Chen H., Sun Q., Tang X., Lim S.C., Huang J., Song H. Structural basis of SOSS1 complex assembly and recognition of ssDNA. Cell Rep. 2014;6:982–991. doi: 10.1016/j.celrep.2014.02.020. [DOI] [PubMed] [Google Scholar]
- 30.Harlow E., Whyte P., Franza B.R. Jr, Schley C. Association of adenovirus early-region 1A proteins with cellular polypeptides. Mol. Cell. Biol. 1986;6:1579–1589. doi: 10.1128/mcb.6.5.1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kitagawa M., Lee S.H., McCormick F. Skp2 suppresses p53-dependent apoptosis by inhibiting p300. Mol. Cell. 2008;29:217–231. doi: 10.1016/j.molcel.2007.11.036. [DOI] [PubMed] [Google Scholar]
- 32.Yan G., Eller M.S., Elm C., Larocca C.A., Ryu B., Panova I.P., Dancy B.M., Bowers E.M., Meyers D., Lareau L., et al. Selective inhibition of p300 HAT blocks cell cycle progression, induces cellular senescence, and inhibits the DNA damage response in melanoma cells. J. Invest. Dermatol. 2013;133:2444–2452. doi: 10.1038/jid.2013.187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jang E.R., Choi J.D., Lee J.S. Acetyltransferase p300 regulates NBS1-mediated DNA damage response. FEBS Lett. 2011;585:47–52. doi: 10.1016/j.febslet.2010.11.034. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.