


Abstract
Most delivery systems for small interfering RNA therapeutics depend on endocytosis and release from endo-lysosomal compartments. One approach to improve delivery is to identify small molecules enhancing these steps. It is unclear to what extent such enhancers can be universally applied to different delivery systems and cell types. Here, we performed a compound library screen on two well-established siRNA delivery systems, lipid nanoparticles and cholesterol conjugated-siRNAs. We identified fifty-one enhancers improving gene silencing 2–5 fold. Strikingly, most enhancers displayed specificity for one delivery system only. By a combination of quantitative fluorescence and electron microscopy we found that the enhancers substantially differed in their mechanism of action, increasing either endocytic uptake or release of siRNAs from endosomes. Furthermore, they acted either on the delivery system itself or the cell, by modulating the endocytic system via distinct mechanisms. Interestingly, several compounds displayed activity on different cell types. As proof of principle, we showed that one compound enhanced siRNA delivery in primary endothelial cells in vitro and in the endocardium in the mouse heart. This study suggests that a pharmacological approach can improve the delivery of siRNAs in a system-specific fashion, by exploiting distinct mechanisms and acting upon multiple cell types.
INTRODUCTION
Interfering with gene expression has long been proposed as a potential therapeutic strategy. The combination of potent RNAi therapeutics and innovative delivery strategies has opened new opportunities to efficiently silence disease-associated genes at therapeutically relevant doses. Numerous delivery systems, such as viruses (1), liposomes (2), polycationic polymers (3), conjugates (4,5), and lipid nanoparticles (LNPs) (6–11), are now being used to deliver siRNAs in vivo. Advances in the development of these delivery technologies have enabled the entry of numerous systemic RNAi products into the clinic (12,13).
Nevertheless, existing delivery systems for siRNA delivery may still be further improved and particularly efficient systemic delivery to extra-hepatic cells and tissues remains a challenge (6,14–15). Delivery is a multistep process consisting of targeting to the appropriate tissue and cell types, cellular uptake and escape of the siRNAs from the endosomes into the cytosol for loading on the RNA-induced silencing complex (RISC) (16). Recently, significant emphasis has been placed on the targeting step and some solutions have emerged (17,18). Most notably, efficient systemic delivery to hepatocytes has been achieved by combining multivalent GalNAc ligands with advanced siRNA chemistry (19). However, improving uptake and especially release of siRNA from unproductive intracellular compartments remains a challenge for many other tissues and cell types (6,15).
Recently, high throughput screening strategies have been applied to improve the composition (20–22) and physicochemical properties (23) of siRNA delivery systems. This approach has also been used to rapidly define the optimal conditions for efficient transfection (24). An alternative approach is to identify chemical compounds that enhance the efficiency of existing siRNA delivery systems (25,26). However, to what extent such approach is a viable strategy remains to be determined. First, some chemical compounds could improve delivery of oligonucleotides by interfering with endocytic uptake, endosomal acidification and progression of cargo along the degradative pathway, as in the case of choloroquine or bafilomycin (27,28). This would increase the residence time of siRNAs in early endosomes leading to a higher probability of escape before degradation in late endosomes and lysosomes. However, such enhancers may not have sufficient potency and also induce high cell toxicity given the essential function of the endocytic system in cell homeostasis, signaling and metabolism (29–31). Second, identifying compounds that enhance the escape of oligonucleotides from endosomes remains a challenge. This is because, with the exception of single molecule detection, the fluorescence microscopy methods do not have the adequate sensitivity and resolution to detect the few hundreds of molecules in the cytosol that are necessary for gene silencing (6,32–33). Several approaches have been used to circumvent such a limit in the sensitivity of detection such as the use of high doses of fluorescently labeled oligonucleotides (34), or unspecific markers like fluorescently labeled Dextran (26) or the colocalization with endosomal markers (26,35–37). However, these indirect approaches do not faithfully reveal the true state of siRNA escape from endosomes into the cytosol within the therapeutic concentration range. Therefore, more quantitative and higher resolution methods are necessary to assess the mode of action of oligonucleotide delivery enhancers under physiological conditions. Third, for compounds acting upon the endocytic system, it is unclear whether they can be active across multiple delivery systems or exhibit system-specificity and, fourth, whether they can enhance delivery in multiple cell types or rather display a narrow range of cell specificity.
Here, we screened a small molecule library aimed to improve the efficiency of gene silencing of two siRNA delivery systems, LNPs and cholesterol-conjugated siRNAs (Chol-siRNAs). Interestingly, comparison of the compounds identified from the two screens indicated that the majority were specific for either delivery system. By applying a combination of high resolution fluorescence microscopy and electron microscopy we found that the compounds have different modes of action, either acting upon the delivery system itself or upon the cellular machinery to either enhance uptake or increase endosomal escape. Finally, the compounds were also effective in physiologically relevant cell types, including cells that usually are refractory to delivery such as primary fibroblasts and hepatocytes in vitro, and endothelial cells in vivo.
MATERIALS AND METHODS
Animals
All animal studies were conducted in accordance with German animal welfare legislation and in strict pathogen-free conditions in the animal facility of the Max Planck Institute of Molecular Cell Biology and Genetics, Dresden, Germany. Protocols were approved by the Institutional Animal Welfare Officer (Tierschutzbeauftragter), and necessary licenses were obtained from the regional Ethical Commission for Animal Experimentation of Dresden, Germany (Tierversuchskommission, Landesdirektion Dresden). All procedures used in animal studies conducted at Alnylam Pharmaceuticals, Cambridge, U.S.A. were approved by the Institutional Animal Care and Use Committee and were consistent with local, state and federal regulations as applicable.
siRNA modification and formulation into lipid nanoparticles
The siRNAs used in this study target GFP (eGFP plasmid, Clonetech). The procedure used to produce LNP-siRNA, LNP-siRNA-alexa647 and LNP-siRNA-gold were extensively described previously (6).
Cholesterol conjugates were made as described previously (38).
Cell culture and cell lines
GFP-HeLa cells (39) were cultured in DMEM media complemented with 10% FBS and 1% penicillin-streptomycin at 37°C and 5% CO2. Primary human fibroblasts (GM00041), obtained from Coriell Institute, were cultured and infected with Rab5-GFP as previously described (40). Primary mouse hepatocytes and endothelial cells were obtained from GFP-lifeact transgenic mice (41), following previously described isolation and culture protocols (42,43). When required the cells were seeded on 24 (for electron microscopy analysis) or 96 (for fluorescence microscopy analysis) well plates.
High throughput screening
GFP-HeLa cells were seeded in 96 or 384 well plates. Cells were transfected with a mixture of LNP-siRNA (5 nM) pre-incubated for 16 h with the compounds (10 μM) or DMSO (MOCK). The library contains 45 567 diverse compounds with a subset of kinase inhibitors (75 compounds), FDA approved drugs (∼1000 compounds), pure natural compounds (∼400 compounds) and compounds selected on drug-like criteria (∼44 000 compounds). After 5h, the cells were washed and incubated with fresh media and fixed with PFA 4% 72 h after transfection. Nuclei were stained with DAPI and the cells were imaged (at least 25 fields per conditions) with a Perkin Elmer Operetta automated microscope (TDS, MPI-CBG, Dresden) and analyzed with Acapella and MotionTracking software (44). Similar procedures were applied for Cholesterol conjugated-siRNA (250 nM) except that the compounds (10 μM) or DMSO (MOCK) were not pre-incubated but freshly added to the cells. All transfections (LNPs and Chol-siRNA) were performed in serum containing media to mimic blood flow conditions. The chemical structures of the enhancers are shown in the Supplementary Figure S1A–C.
We determined the mean GFP intensity within the segmented nuclei instead of the total GFP intensity per field to exclude false positives of GFP reduction caused by variations in cell number (either due to toxicity or decreased proliferation). The toxicity of the compounds based on cell number is shown in Supplementary Figure S2. We also used defined thresholds to select the enhancers. Compounds were considered as enhancers when they improved GFP silencing by at least 20%. In addition, compounds that reduced cell number by more than 35% were considered as toxic and, thus, excluded from the rest of the analysis. Few compounds that improved the Chol-siRNA silencing efficiently (#26, #29, #30, #41), but with a toxicity value slightly above the threshold, were retained. The rationale was that varying their concentration allows finding a window where they are active but non-toxic.
Importantly, to control for non-specific silencing, in addition to un-treated (UT) and DMSO treated conditions, we verified that the compounds did not decrease the GFP intensity when incubated alone (i.e. without the delivery system) with the GFP-expressing cells. All compounds showing a reduction in GFP mean intensity within the segmented nuclei without addition of LNPs or Chol-siRNAs were excluded from the hit list. We also tested whether the enhancers could transfect naked (non-formulated) siRNAs, to identify potential transfection reagents. We identified two compounds that have this property.
Knock-down assay
HeLa GFP cells, Rab5-GFP human primary fibroblasts and GFP–lifeact primary mouse hepatocytes were transfected with LNP-siRNA formulation preincubated or not with the compounds (following similar procedures as in the primary screen). After 72h, the cells were fixed with PFA 4% (pH 7.2 in phosphate buffer) for 20 min at room temperature. After washing, nuclei were labeled with Dapi and cytosol with SytoBlue. Acquisition and analysis of images (at least 25 fields per conditions) were done on an ArrayscanVTI with TwisterII automated wide field microscope (TDS, MPI-CBG, Dresden).
Uptake assay
For the in vitro uptake assay, cells were transfected either with LNP-siRNA-alexa647 or with cholesterol conjugated-siRNA-alexa647 treated or not with the compounds. Then, cells were fixed and stained as for the knock-down assay. Images were acquired on a Perkin Elmer Opera automated confocal microscope (TDS, MPI-CBG, Dresden) and analyzed on MotionTracking software (http://motiontracking.mpi-cbg.de) as previously described (6).
To determine the endocytic pathway used by LNPs or Chol-siRNAs to enter the cell, we performed a depletion of key endocytic machinery as previously described (6).
For the in vivo uptake assay, LNP-siRNA-alexa647, treated or not with BADGE, were injected in the heart cavity of sacrificed mice. Then the hearts were collected, washed extensively in PBS and fixed with PFA 4% overnight at 4°C. Tissues were sliced on cryostat after OCT embedding and nuclei were stained with Dapi. Then, sections were mounted with mowiol and coverslip designed for high resolution observation. Images (at least 15 fields per conditions) were acquired on an Olympus Fluoview 1000 laser scanning confocal microscope (light microscopy facility, MPI-CBG, Dresden) equipped with an Olympus UPlanSApo 60x 1.35 Oil immersion objective. Images were analyzed on MotionTracking.
Determination of the mechanism of action
Two pilot screens were performed either by pre-incubating the compounds with the delivery systems overnight prior to adding them to the cells (pre-incubation condition), or by adding the compounds together with the delivery system directly to the cells (direct incubation condition). The pilot screens revealed that the pre-incubation condition increased the number of hits for LNPs but not for Chol-siRNAs. Therefore, we performed the full primary screen under the pre-incubation condition for LNPs and under the direct incubation condition for Chol-siRNAs. Since, all the identified enhancers for LNPs exert their effect with an overnight pre-incubation, a secondary screen was performed to determine which compounds are able to improve silencing under direct incubation condition.
From these two screens, we were able to distinguish compounds that improved GFP down-regulation by acting most probably on the LNPs from those that were not. In addition, we determined the compounds that act on the uptake or on the siRNA release. For this, we analyzed the uptake of alexa647-labeled siRNAs (incorporated in LNPs or cholesterol-conjugated) under pre-incubation (compounds that act on delivery systems) or direct incubation condition (compounds that act on cells). Compounds that significantly increased the amount of siRNA-alexa647 were considered as acting on uptake. Compounds that did not affect or reduce the amount of intracellular siRNA were considered as acting on siRNA endosomal release.
Electron microscopy
Morphological experiments were analyzed in a blind fashion using a code that was not broken until the quantitation was completed.
For electron microscopy analysis, HeLa cells were transfected with LNP-siRNA-gold and fixed with 2.5% glutaraldehyde (in phosphate buffer) overnight. Then, cells were post-fixed in ferrocyanide reduced osmium as previously described (45). Cells were dehydrated in increasing bath of ethanol for 10 min, infiltrates with mixture of ethanol and epon (3:1 and 1:3) and pure epon for 1h. After epon polymerization overnight at 60°C, the 24 well plates were broken and pieces of epon were glued on epon sticks. 70–50 nm sections were then cut and stained with uranyl acetate and lead citrate following classical procedure. Supermontages of 100 images were randomly collected at 11000x magnification on a Tecnai 12 TEM microscope (FEI) (electron microscopy facility, MPI-CBG, Dresden) and the stitching of the images was achieved by using the open access software Blendmont (Boulder Laboratory, University of Colorado, USA).
To quantify the total uptake as well as the ratio of structures labeled versus unlabeled in a reliable manner, a stereological approach based on randomly distributed crosses was applied allowing relative loading index calculation and normalization of the number of structures counted (46). To quantify the ratio of siRNA escape from endosomes, we developed a plugin for automatically counting the total number of gold particles per montage. Images were processed by performing morphological bottom-hat filtering on the grayscale input image (47). The structuring element used for this was a circle of a radius bigger than the object of interest (radius 4). Following this, we performed image equalization to the interval [0;1] and thresholding with a threshold set at 0.3. The binarized images were then analyzed by the watershed transform to split contiguous gold particles. A last post-processing step was performed to remove uncertain gold particles (particles having the average intensity value less than 5 standard deviations of the median intensity value in the whole image). Then, for a set of images, the number of particles were automatically counted and manually counted with an error rate determined to be <1% confirming that our procedure succeeded in correctly identifying gold particles. Finally, the procedure was applied to determine the total number of gold particles in the images. In addition, the number of gold particles was counted manually within the cytoplasm based on morphological recognition. For this analysis, we selected three pieces of epon containing cells incubated for 6 h with LNP-siRNA-gold. For each piece, we cut several sections that were collected on eight grids. Among these eight grids covering a large portion of the cells, we randomly picked three grids. Five super-montaging, at random places but in areas containing cells, were made for each grid. The super-montaging covered approximately two cells. In each experiment we analyzed approximately 45 super-montaging corresponding to about 90 cells per condition. We analyzed three independent experiments, amounting to ∼270 cells per condition. For each condition we counted automatically at least 100 000 siRNA-gold particles.
Quantitative multiparametrics image analysis
Quantitative multi-parametric image analysis was performed in two sequential rounds of calculations. In the first round, aiming at the identification of fluorescent vesicles, the image intensity was fitted by a sum of powered Lorenzian functions (48). The coefficients of those functions were then used to describe the features of individual objects (e.g. intracellular position relative to the nucleus, size, intensity, total vesicular intensity, etc.). Additionally, nuclei and cells were identified by a pipeline involving several operations from morphological image analysis (47). Briefly, nuclei were found by a maximum-entropy based local thresholding and cells by a region growing algorithm based on the watershed transform. In the second round, a set of statistics was extracted from the distributions of the endosome parameters measured in the first round. Statistical filters based on the mean intensity of the fitted object were then applied to remove the background and the unspecific staining (using control image with secondary antibody alone). This set of values, that quantitatively describes the fluorescence information of every channel in the image, has been used for comparing the different conditions as previously described (44).
Co-localization analysis was performed by assessing the percentage of overlapping objects. Object ‘A’ in channel ‘1’ is considered to co-localize to object ‘B’ in channel ‘2’ if the integral intensity profile of A overlaps to the one of B more than a user-defined percentage threshold, here set to 40%. Co-localization was calculated both by number (percentage of LNP vesicles that are positive for LAMP) and by intensity volume (percentage of LNP amount in the LAMP-positive compartment). The described approach is more powerful than classical correlation and pixel co-occurrence analyses, since it allows us to (i) discriminate between background and foreground (object) fluorescence and (ii) interpret the results in terms of percentage of structures that are localized to objects in another channel of interest.
Statistics
Data were expressed as the mean ± standard error of the mean (SEM). Statistical analysis was determined using ANOVA test followed by Student T-test test. The two-tailed Pvalue were added within the figure or the figure legends.
RESULTS
LNPs and cholesterol conjugate siRNA delivery systems have different uptake mechanisms
We aimed at improving the efficiency of two well-established siRNA delivery systems, lipid nanoparticles (LNPs) and cholesterol-conjugated siRNAs (Chol-siRNAs) (9,49–51) by performing a high throughput screen to discover small molecule delivery enhancers. Given that these two delivery systems differ fundamentally in composition, size and morphology, we hypothesized that their mechanism of action may differ significantly. Therefore, we first investigated their mechanism of uptake and endocytosis.
The siRNAs were labeled with Alexa Fluor 647 and formulated in LNPs (6) or conjugated to cholesterol. Internalization in HeLa cells could be visualized (Figure 1A) at concentrations sufficient for efficient silencing of targeted genes, in vitro (Figure 1B) as previously shown in vivo (38,42). Chol-siRNAs uptake behaved like free cholesterol (52), yielding both diffuse staining and a punctate pattern. In contrast, siRNAs encapsulated in LNPs only displayed a punctate pattern (Figure 1A). Such difference in properties may entail different mechanisms of association with the plasma membrane and cellular uptake. To test this, we depleted various regulatory components of the endocytic machinery and analyzed the effects on uptake. The internalization of cholesterol is thought to be mainly mediated by LDL receptor endocytosis upon interaction with serum lipoproteins (53,54). Consistent with this, we found that the uptake of Chol-siRNAs required mainly components of clathrin-mediated endocytosis (CME) (Figure 1C), in contrast to LNP which enter via both CME and macropinocytosis (6). Moreover, the uptake kinetics of Chol-siRNA and LNPs were markedly different. Chol-siRNA was internalized quickly and followed a Gaussian curve (Figure 1D). The decrease in intracellular Chol-siRNA over time can be explained by depletion of LDL receptor on the cell surface or increase in efflux as previously observed for cholesterol (55,56). The increased intracellular Chol-siRNA upon CAV1 knock down (Figure 1C) may be explained by the role of caveolin in the cholesterol efflux as recently shown (57). This process that avoids an excess of Chol-siRNA within the cell could have important impact on the silencing efficiency of this particular conjugate. In contrast, the uptake of LNPs was delayed in time and followed an exponential curve (Figure 1D), as shown previously (6). Altogether, these data indicate that the two siRNA delivery systems differ in the way they enter the endocytic pathway. Therefore, we rationalized that separate screens would need to be carried out for the two systems, as unique compounds may emerge for each.
Figure 1.
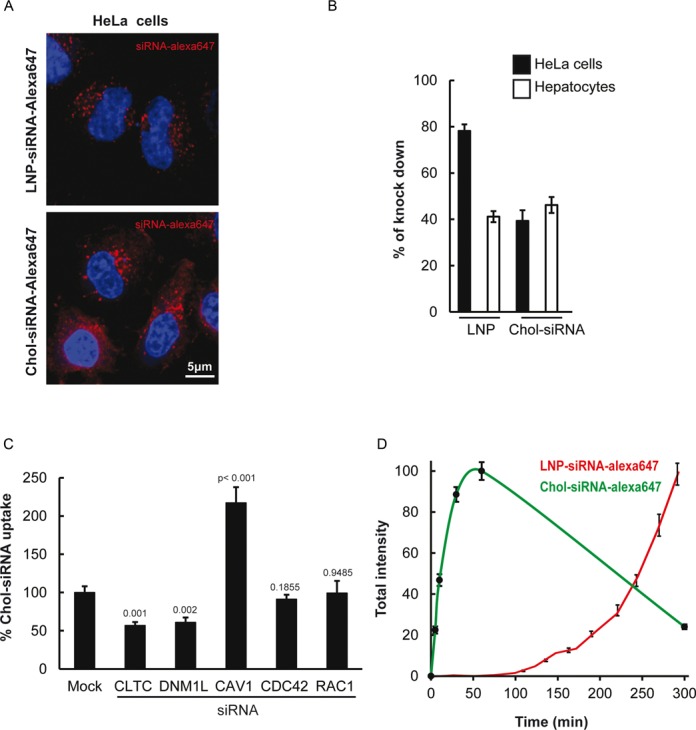
LNPs and cholesterol conjugates differ in uptake mechanism. (A) Images of HeLa cells after incubation with LNP-siRNA-alexa647 (top panels) or cholesterol conjugated-siRNA (Chol-siRNA)-alexa647 (bottom panels) for 5 h and 1 h, respectively. (B) Quantification of the percentage of GFP downregulation in HeLa GFP-expressing cells and primary mouse hepatocytes expressing Lifeact-GFP exposed to 40 nM of LNP-siRNA and 1 μM of Chol-siRNA (n = 3, mean ± SEM). (C) Chol-siRNAs-alexa647 uptake in HeLa cells after silencing of dynamin (DNM1L), clathrin light chain (CLTC), Caveolin (CAV1), CDC42 (for clathrin-independent endocytosis)) and RAC1 (Macropinocytosis). Mean ± SEM, n = 3, P-value relative to control. (D) Uptake kinetics of LNP-siRNA-alexa647 (40 nM, red curve) compared to Chol-siRNA-alexa647 (250 nM, green curve).
A high throughput chemical screen allows identification of compounds that improve siRNA delivery
To identify chemical compounds that improve siRNA delivery, we screened both delivery systems with the same library of compounds. We performed our primary screen in HeLa cells, a cell line easy to culture in a screenable format and which exhibits the key general features of the mammalian endocytic pathway. The cells constitutively express green fluorescent protein (GFP) for direct readout of RNAi-mediated silencing by quantitative fluorescence microscopy. To exclude chemicals that may affect GFP translation or nuclear localization, the compounds were also tested alone, i.e. in the absence of siRNA. In a second step, we validated the results by performing a secondary screen in primary cells, fibroblasts and hepatocytes, to assess the cross-activity between cell types and species. Our primary assay consisted of GFP-expressing HeLa cells transfected with suboptimal doses of either LNPs or Chol-siRNAs with an anti-GFP siRNA. We optimized the assay to obtain reduction in GFP expression of only about 20% as measured by quantitative fluorescence microscopy. As a pre-requisite for the screen, we verified that under the same conditions silencing could be boosted to higher than 80% by adding transfection reagent (see GFP-positive versus -negative cells in Figure 2A). This demonstrated that the amount of siRNA per se was not limiting, but rather cellular uptake and/or escape from the endolysosomal system was suboptimal. Therefore, the experimental conditions used were appropriate to bias the screen toward compounds, which could improve these specific steps.
Figure 2.
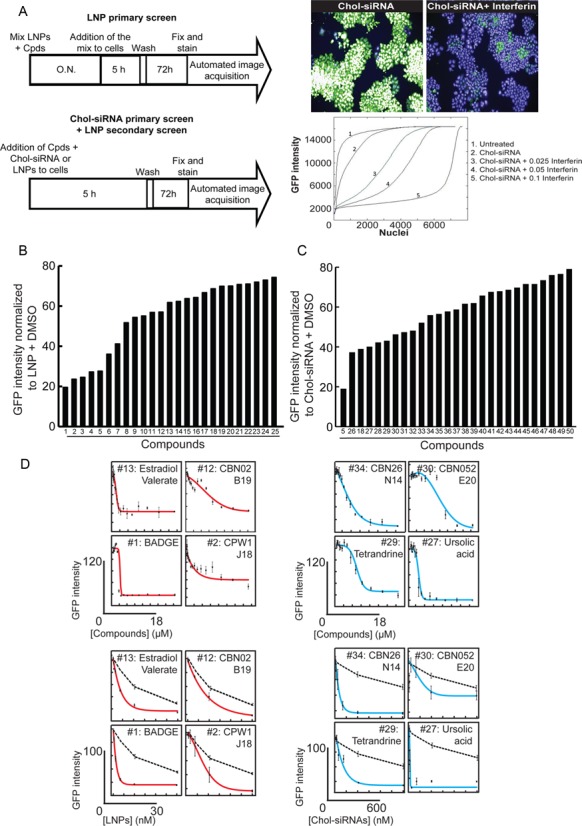
Enhancers of gene silencing identified by high throughput compound library screen for both delivery systems. (A) The workflow for the chemical screen (top panel) includes a simple image-based analysis readout (bottom left panels) by segmenting nuclei and analysing the distribution of the GFP intensity per nuclei. The doses for both delivery systems were selected such that potential improvements in activity could be detected over a wide range as confirmed by addition of increasing doses of interferin (bottom right panel). (B) GFP intensity in HeLa GFP cells transfected with LNPs and individual compounds identified as hits ([LNP-siRNA] = 5 nM and [Compounds] = 10 μM). The compounds structures can be found in Supplementary Figure S1A–C. (C) GFP intensity in HeLa GFP cells transfected with Chol-siRNA and individual compounds identified as hits ([Chol-siRNA] = 250 nM and [Compounds] = 10 μM). The compounds structures can be found in Supplementary Figure S1A–C. (D) IC50 from selected identified compounds. In the top panels, the concentration of siRNAs was fixed to 5 nM for LNPs (red lines) and to 250 nM for cholesterol-conjugates (blue lines), and the concentration of the compounds was increasing from 0 to 18 μM. In the bottom panels the concentration of the compounds was fixed to 10 μM, and those of the delivery systems were increasing from 0 to 30 nM for LNPs (red lines) and from 0 to 600 nM for cholesterol conjugate (blue lines). The black dotted lines represents the respective DMSO treated delivery systems The toxicity at various doses of compounds was determined based on cell numbers (Supplementary Figure S4) and could be considered as not very significant for all the compounds at 10 μM.
We initially performed two pilot screens on a small set of compounds, with or without overnight pre-incubation of the compounds with the delivery system prior to adding the mixtures to the cells. The pilot screens revealed that an overnight pre-incubation increased the number of hits for LNPs but not for Chol-siRNAs. Therefore, such a pre-incubation step was incorporated in the LNP screen, but not in the Chol-siRNA screen. Under these conditions, all the identified LNP enhancers will work with an overnight pre-incubation with the LNPs, whereas those improving Chol-siRNAs silencing will work upon direct contact with the cells. The scheme of the assay protocol used in the screen is shown in Figure 2A. After 5 h of incubating the cells with the compound (10 μM) and siRNA mixtures, the medium was replaced by fresh medium, and the cells were further incubated for 72 h. Thereafter, cells were fixed, nuclei were stained with Hoechst and the GFP expression was quantified on an automated fluorescence microscope. In order to exclude toxic compounds, enhancers were considered when the mean GFP intensity per cell decreased by > 20% but the number of cells (nuclei) decreased by <35%. Of the 45 567 compounds tested in both delivery systems, 25 and 28 compounds (see the structures provided on Supplementary Figure S1A–C) improved the silencing activity of LNPs and Chol-siRNAs, respectively (Figure 2B, C). Silencing efficiency reached up to 80% without (or with very limited) toxicity for the cells (Supplementary Figure S2A, B). Strikingly, only two compounds (#5 and #18) were found to improve silencing in both delivery systems. These compounds were also able to induce silencing with non-modified, commercially available siRNAs (Supplementary Figure S3), suggesting that they may act as general transfection reagents. Importantly, all other identified hits reduced GFP expression only in conjunction with their respective delivery systems. We examined the dose response of the hit compounds and determined their IC50 to be in the low micromolar range (Figure 2D), with very limited or no toxicity up to 10 μM (Supplementary Figure S4). Therefore, the screen identified chemical compounds that improve siRNA delivery specifically for each of the LNPs and Chol-siRNA delivery systems.
Compounds identified in a high throughput chemical screen efficiently improve siRNA delivery in primary cells
We previously demonstrated that different cell types differ in their siRNA delivery mechanism (6). Can the enhancers identified in the screen improve delivery in different cell types? To test this, we performed a screen in human primary fibroblasts and mouse primary hepatocytes. First, we tested all compounds identified with their respective siRNA delivery systems in the fibroblasts. We chose these particular cells because they internalize LNPs inefficiently and, consequently, are also poorly transfected (6). This screen revealed that 18% of the LNP hits (Figure 3A; #1, #4, #15 and #24) and 21% of the Chol-siRNA hits (Figure 3B; #5, #18, #28, #29 and # 47) identified in HeLa cells improved the efficiency of GFP silencing by 1.5- to 4-fold also in those cells. Some compounds (3 LNP and 3 Chol-siRNA hits; #11, #12, #20, #26, #32 and #39), however, demonstrated toxicity in the primary fibroblasts (Supplementary Figure S2C, D).
Figure 3.
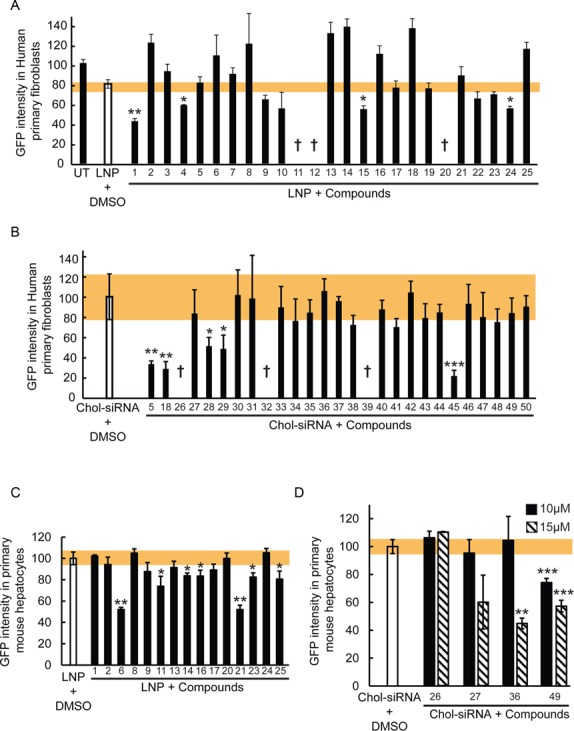
Various library compounds improve LNP- and cholesterol conjugate-based siRNA delivery and silencing. (A) Percentage of GFP intensity in human primary fibroblasts expressing Rab5-GFP after LNP-based silencing of GFP treated with the identified compound hits ([LNP-siRNA] = 5 nM and [Compounds] = 10 μM). UT = untreated, † = cytotoxic effect. Mean ± SEM, n = 3 (*P-value < 0.05, **P < 0.01). The toxicity was determined based on cell numbers (Supplementary Figure S2C). The yellow bar represents the SEM of the DMSO treated condition. (B) Percentage of GFP intensity in human primary fibroblasts expressing Rab5-GFP after Chol-siRNA-based silencing of GFP treated with the identified hits ([Chol-siRNA] = 250 nM and [Compounds] = 10 μM). † = cytotoxic effect. Mean ± SEM, n = 3 (*P-value < 0.05, **P < 0.01, ***P-value < 0.001). The toxicity was determined based on cell numbers (Supplementary Figure S2D). The yellow bar represents the SEM of the DMSO treated condition. (C) Percentage of GFP intensity in mouse primary hepatocytes expressing Lifeact-GFP after LNP-based silencing of GFP treated with the identified hits ([LNP-siRNA] = 5 nM and [Compounds] = 10 μM). Mean ± SEM, n = 2 (*P-value < 0.05, **P < 0.01). The yellow bar represents the SEM of the DMSO treated condition. (D) Percentage of GFP intensity in mouse primary hepatocytes expressing Lifeact-GFP after Chol-siRNA-based silencing of GFP treated with 10 μM (black bars) or 15 μM (striped bars) of the identified hits. Mean ± SEM, n = 2 (**P-value < 0.01, ***P < 0.001). The yellow bar represents the SEM of the DMSO treated condition.
Unlike fibroblasts, hepatocytes internalize LNPs efficiently. Therefore, we tested the compounds in mouse primary hepatocytes to determine whether they can enhance the silencing also in these cells. However, since these cells are not well suited for high throughput screening, we tested a random sample of the full set of hits, consisting of 15 hits from the LNP screen and 4 from the Chol-siRNA screen. Seven compounds (46%) from the LNP screen and 1 compound (25%) from the Chol-siRNA screen improved the silencing efficiency (Figure 3C, D; #6, #11, #14, #16, #21, #23, #25 and #49). However, increasing the concentration from 10 μM (used in the screen) to 15 μM improved the KD efficiency in hepatocytes, as shown for compounds #27, #36 and #49, suggesting that more compounds could be active at higher doses. Note that we did not observe hepatocyte toxicity at these doses (Supplementary Figure S2E, F). This analysis revealed that the compounds that improve silencing in fibroblasts are not efficient in hepatocytes and, conversely, those active in hepatocytes are inefficient in fibroblasts.
Altogether, our results demonstrate that some of the hits identified in HeLa cells were also active across different cell types, including primary cells with very different origin and characteristics.
Compounds can improve uptake or endosomal escape
The finding that there was little overlap between compounds acting on fibroblasts and hepatocytes may reflect a different mechanism of action of the compounds. Since the LNPs and Chol-siRNAs were directly added to the cells, the hits could either increase the uptake or improve the release of the siRNAs from intracellular compartments. To discriminate between the two scenarios, we analyzed the impact of the hits on the uptake of LNP-siRNA-alexa647 or Chol-siRNA-alexa647 in HeLa cells (Figure 4A). We found that 56% and 64% of the compounds were able to strongly increase the uptake of LNPs and Chol-siRNA, respectively (Figure 4B). This increase is exemplified by the higher intensity of fluorescent siRNAs in endosomes delivered via LNPs in cells incubated with BADGE (#1, Bisphenol A diglycidyl ether; 2,2′-[(1-methylethylidene)bis(4,1-phenyleneoxymethylene)]bis-oxirane) and via Chol-siRNAs in cells treated with compound #29 (Tetrandrine), compared with control (Figure 4A). Interestingly, 11 compounds out of 25 for the LNPs and 5 out of 14 for the Chol-siRNAs did not significantly increase cellular uptake (Figure 4B), as shown for the compound designated as #2 (CPW1-J18) and #35 (Lomatin) (Figure 4A). These compounds in all likelihood improve delivery by enhancing the intracellular release of the siRNA from endo-lysosomal compartments (see below). The uptake of LNP-siRNAs or Chol-siRNAs is a multi-step process that requires binding to the plasma membrane and/or to a specific receptor (LDLR for LNPs (6,8)) followed by internalization. Similarly, the release of siRNAs from endosomes requires the unpacking of the siRNAs from the particles before they can cross the endosomal membrane to reach the cytosol. The identified enhancers may act on one or several of these steps to improve silencing.
Figure 4.
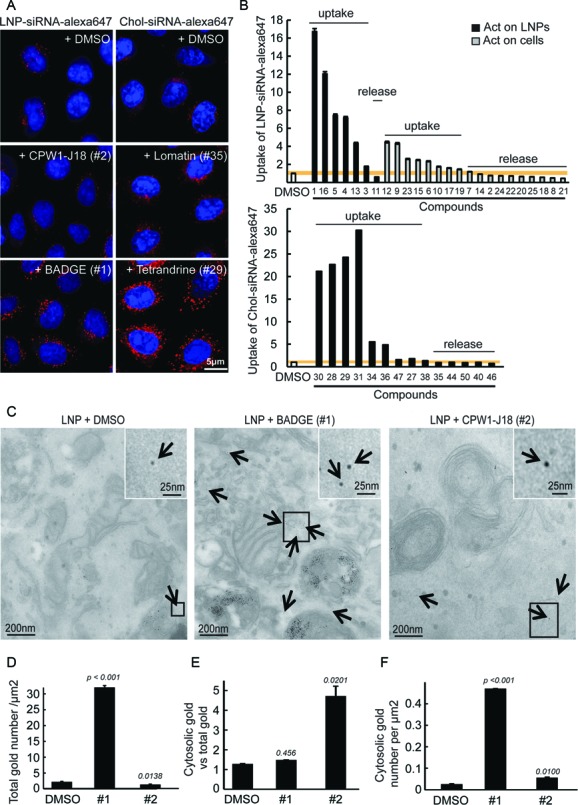
Mechanism of action of the compounds on LNP- and cholesterol conjugate-based siRNA delivery and silencing. (A) Images illustrating the uptake of LNP-siRNA-alexa647 (40 nM, 5 h incubation, left panel) and Chol-siRNA-alexa647 (250 nM, 5h incubation, right panel) treated with DMSO (top panels) or with the compounds (10 μM) that increased the uptake (bottom panels: BADGE and Tetrandrine) or not (middle panels: CPW1-J18 and Lomatin) in HeLa cells. (B) Quantitative analysis of LNP-siRNA-alexa647 (40 nM, 5 h incubation, top panel) and Chol-siRNA-alexa647 (250 nM, 5h incubation, bottom panel) uptake in cells treated with DMSO or compounds (10 μM). Compounds acting on the delivery systems are represented with black bars and those acting on the cells with gray bars. The compounds structure can be found in Supplementary Figure S1A–C. (C) LNP-siRNA-gold detected in HeLa cells in vitro, by EM ([LNP = 40 nM]; [compounds] = 10 μM). siRNA-gold were detected in the cytosol (arrows) or within several endocytic compartments. Magnified images (insets) permit appreciation of the cytosolic localization of siRNA-gold. (D) Automated quantification of the total number of siRNA-gold (representing the uptake) found per μm2 of cell section. Mean ± SEM, n = 3 (*P-value < 0.05, ***P < 0.001). (E) Semi-automated quantification of the ratio between the number of cytosolic siRNA-gold and the total number of siRNA-gold internalized (representing the percentage of siRNA escape). Mean ± SEM, n = 3 (*P-value < 0.05). (F) Semi-automated quantification of the number of cytosolic siRNA-gold per μm2 of cells. Mean ± SEM, n = 3 (*P-value < 0.05, ***P-value < 0.001).
To gain insights into the mode of action of the LNP enhancers, we tested whether they can improve silencing upon immediate contact with the cells as compared to an overnight pre-incubation. To this end, the silencing enhancers were either pre-incubated with the cells or were added to the cells concomitantly with the delivery systems. The rationale behind this experiment is that if a compound were acting primarily on the cell, we would expect its silencing enhancer effect to occur also when added to the cells prior to adding the siRNAs, whereas an action on the delivery platform would depend on a prior pre-incubation with the siRNAs. By this criterion, 28% of the enhancer compounds were active only when pre-incubated with the LNPs (Figure 4B; black bars) whereas the remaining 72% were active upon direct incubation with the cells (Figure 4B; gray bars). Interestingly, almost all compounds that improved silencing activity presumably by facilitating endo-lysosomal escape were active upon direct incubation with the cells, suggesting that they act upon the cellular machinery. We found that the enhancers that act only when pre-incubated with the LNPs are more active in increasing the uptake than those acting upon direct incubation with the cells (Figure 4B; black bars versus gray bars), suggesting that they may directly impact on the LNPs and facilitate their endocytosis.
To explore the mechanism whereby the compounds enhance the siRNA activity, we focused on two of the most active compounds, BADGE and CPW1-J18 impacting LNP-mediated delivery. We used the analytical electron microscopy methodology previously developed (6) to quantify the uptake and escape of LNPs encapsulating gold-labeled siRNA (Figure 4C). Among the LNP hits, BADGE and CPW1-J18 had the strongest positive effect of GFP down-regulation in their respective categories, e.g. improving uptake or endosomal release (Figure 4B and Supplementary Figure S5). This assay was limited to LNPs, since covalent attachment of gold particles to Chol-siRNA can be expected to substantially impact uptake and trafficking.
By using this approach, we first confirmed that BADGE increased the uptake of LNPs as indicated by the 15-fold increase in the amount of siRNA-gold internalized by the cells (Figure 4D). Moreover, the ratio of siRNA-gold in the cytosol versus the total amount internalized was not increased (Figure 4E). These results suggest that BADGE acts exclusively on the uptake rather than on the release of siRNAs from endosomes. In contrast, CPW1-J18 was found to slightly reduce the total amount of siRNAs-gold internalized (Figure 4D) while increasing the ratio of siRNA-gold in the cytosol versus the total amount internalized by ∼5-fold (Figure 4E). These results suggest that CPW1-J18 improves the release of siRNAs from endosomes. Overall, the total amount of cytosolic siRNA-gold was significantly more elevated with BADGE and CPW1-J18 treatment compared to DMSO (Figure 4F), as evidenced by the EM images (arrows in Figure 4C). Our results suggest that the compounds identified act via two different mechanisms, improving either uptake or endosomal escape.
Endosomal escape of siRNA can be enhanced via different mechanisms
We can envisage various mechanisms whereby the chemical compounds can facilitate endosomal escape. One possibility is that they prevent lysosomal acidification similar to choloroquine or bafilomycin and/or progression of cargo along the degradative pathway (28). Interestingly, hydroxychloroquine and bafilomycin were not hits in the screen, neither for LNPs nor Chol-siRNA. This discrepency could be explained either by the fact that the compound concentrations in the screens were different than those reported (58,59), or that they also inhibit endocytosis (60–61,6). An alternative possibility is that some compounds locally destabilize the endosomal membrane thus increasing leakage of content from the lumen (62,63).
To explore the effects of the compounds on the endosomal system, we performed a focused screen on the candidate compounds for endosomal escape at the same concentration as in the RNAi enhancement primary screen (10 μM) using an image-based assay that quantitatively measures the uptake of EGF and transferrin, as described (64). Quantitative multi-parametric image analysis (QMPIA) was applied for a phenotypic description of the effect of each compound on the endosomal network (endosome number, size, intracellular position, etc.) (44). We then compared the multi-parametric profiles across the compounds and with respect to hydroxychloroquine, because this compound has a characteristic phenotypic signature of blocked endosome acidification and maturation. The compounds had very different effects on the endosomal system (Figure 5). For LNP enhancers, the phenotypic profiles of the compounds could be grouped in four distinct categories. In the first category, the profiles correlated with that of hydroxychloroquine (Figure 5A), suggesting that these compounds have a similar inhibitory effect on endosome acidification and/or maturation. In contrast, the second category was anti-correlated (Figure 5B), ruling out such an inhibitory effect. The third, which contains only compounds #7, was neither correlated nor anti-correlated with the hydroxychloroquine profile (Figure 5C), suggesting that it affects endocytic trafficking but by a mechanism distinct from endosome maturation. The fourth category is very interesting because none of the compounds in this group were found to have a significant impact on cargo endocytosis and the endosomal parameters (Figure 5D). These enhancers therefore do not appear to have a strong influence on endosomal transport and are candidates for improving escape through destabilization of the endosomal membrane.
Figure 5.
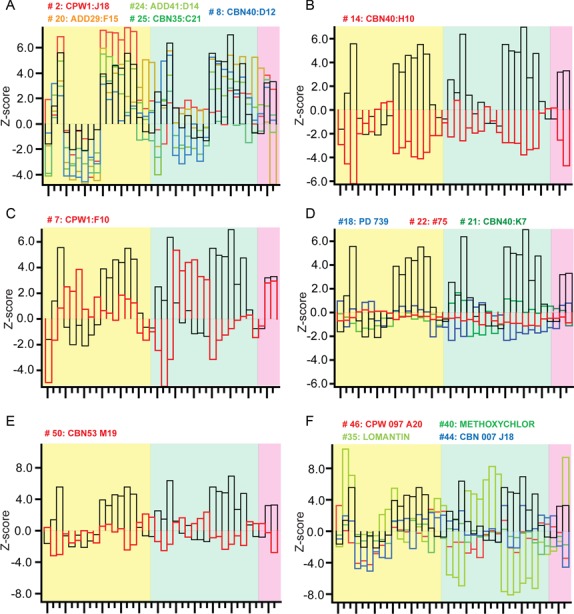
Comparison of multi-parametric profiles of the enhancers on EGF and transferrin endocytosis. HeLa cells were incubated with the enhancers (10 μM), allowed to internalize fluorescently labeled EGF and transferrin, imaged and analyzed as described (44,64). LNP enhancers had profiles correlated (A), anti-correlated (B) or not correlated (C) with the endocytic profile of hydroxychloroquine (Black bars) or (D) having very modest effects on endocytosis. The profiles of Chol-siRNA enhancers were anti-correlated (E) or not correlated (F) with hydroxychloroquine (Black bars). The yellow, green and pink background color represents the EGF parameters, the transferrin parameters and co-localizations parameters, respectively. The detailed list of parameters is presented in Supplementary Figure S7.
For the Chol-siRNA enhancers, we could distinguish only two categories, none of which was correlated with hydroxychloroquine. The first category was anti-correlated (Figure 5E) and the second had no correlation with the profile of hydroxychloroquine, suggesting again an action that is distinct from a block of acidification and/or endosomal maturation. Among these compounds, #35 (Lomatin), was particularly interesting because of its uncoupling between the effect on EGF and transferrin endocytosis (Figure 5F; light green bars). This uncoupling suggests that compound #35 act specifically on the endocytic/recycling pathway.
Altogether, these results demonstrate that the compounds have very distinct effects on the endosomal system and suggest that they enhance endosomal escape by different mechanisms.
BADGE treatment increases cell delivery and kinetics through LNP size reduction
To determine the mechanism of action underlying the enhanced uptake, we focused on BADGE (compound #1) since it had the strongest effect on LNP uptake. By EM negative staining analysis of LNPs treated with BADGE, we found that this compound had an effect on the nanoparticles themselves (Figure 6A), consistent with the predictions from the incubation analysis (Figure 4B). Indeed, this enhancer was capable of reducing the size of LNPs by ∼2-fold (Figure 6A). This reduction in size was associated with a dramatic acceleration of uptake kinetics (Figure 6B). Therefore, the activity of BADGE could result from the facilitation of the entry mechanism used by LNPs. We tested this hypothesis by interfering with different endocytic routes. Strikingly, we found that the uptake of LNPs exposed to BADGE is much less sensitive to the knock-down of clathrin, ARF-1 and RAC-1 compared to the control (Figure 6C), suggesting that the smaller LNPs are captured through a broader set of endocytic mechanisms.
Figure 6.
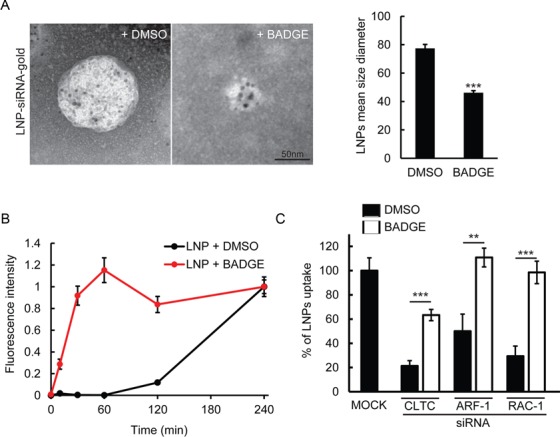
BADGE reduces the size of LNPs leading to increased uptake kinetics and exploitation of entry routes. (A) Visualization of LNP-siRNA-gold by electron microscopy after incubation with DMSO or BADGE (10 μM). LNPs mean size diameter quantification (right panel). Mean ± SEM, n = 3 (***P-value < 0.001). (B) Uptake kinetics of LNP-siRNA-alexa647 (40 nM) treated with DMSO (black curve) or BADGE (red curve, 10 μM). (C) Percentage of uptake of LNP-siRNA-alexa647 treated with DMSO (black columns) or BADGE (white columns) in HeLa cells after silencing of key regulators of CME (CLTC) and Macropinocytosis (ARF-1, RAC1) ([LNP = 40 nM]; [Compound] = 10 μM). Mean ± SEM, n = 3 (**P-value < 0.01, ***P-value < 0.001).
The finding that BADGE modifies the physicochemical properties of LNPs to improve uptake both in HeLa cells and primary fibroblasts in vitro, prompted us to test whether it can also enhance the delivery of siRNAs in other cell types in vitro and within a tissue. We focused on endothelial cells, which are important in a number of diseases and difficult to transfect with siRNAs. First, we looked at the effect of BADGE treatment on LNP-based siRNA delivery in primary endothelial cells (Figure 7A). We found that BADGE-treated LNPs were delivered twice as efficiently as control LNPs in these cells (Figure 7A, B). Consistently, this increase in uptake was accompanied by a similar (∼2-fold) increase in silencing activity, as determined by analyzing the GFP intensity in primary endothelial cells isolated from GFP-lifeact transgenic mice (Figure 7A bottom panel and Figure 7C). Second, to test whether BADGE could improve delivery to the endothelium in a whole tissue, we injected BADGE-treated LNPs into the heart of mice. LNPs pre-treated with BADGE were strongly captured by the endocardium cell layer as evidenced by the numerous fluorescent vesicles (Figure 7D). The quantification of the number of vesicles loaded with LNP-siRNA-alexa647 revealed that the treatment with BADGE increased the uptake in the endothelial cell in vivo by ∼14-fold (Figure 7E).
Figure 7.
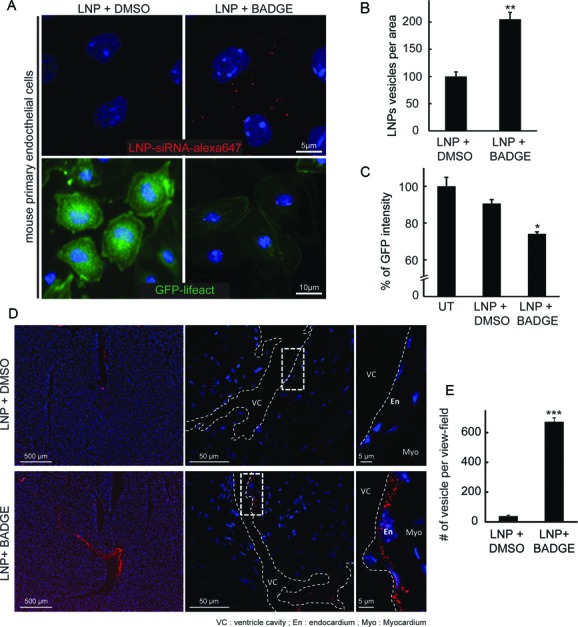
BADGE improves LNP-based siRNA delivery in endothelial cells in vitro and in vivo. (A) Uptake of LNP-siRNA-alexa647 (40 nM, 5 h incubation, top panels) and GFP downregulation (40 nM, 72 h after transfection, bottom panels) after treatment with 10 μM of DMSO (left panels) or BADGE (right panels) in mouse primary endothelial cells and primary endothelial cells from GFP-lifeact mice. (B) Quantification of the number of siRNA-alexa647 positive vesicle per area in mouse primary endothelial cells. Mean ± SEM, n = 3 (**P-value < 0.01). (C) Percentage of GFP intensity in primary endothelial cells isolated from GFP-lifeact mice after 72 h of transfection with LNP-siRNA treated with DMSO or BADGE. Mean ± SEM, n = 3 (*P-value < 0.05). (D) Uptake of LNP-siRNA-alexa647 treated with DMSO (top panels) or BADGE (bottom panels) injected into the heart of mice immediately after sacrifice and incubated for 10 min ([LNP = 40 nM]; [Compound] = 10 μM). Increasing magnifications evidenced the localization of the signal in the endocardium cell layer (En). VC = ventricle cavity, Myo = myocardium. (E) Quantitative analysis of the number of LNP-siRNA-alexa647 per view field in vivo after treatment with DMSO or BADGE. Mean ± SEM, n = 3 (***P-value < 0.001).
Altogether, our results demonstrate that performing a screen of chemical compounds in HeLa cells allowed the identification of small molecules that efficiently improve the silencing in several cell types having different properties and functions such as skin fibroblasts, hepatocytes and endothelial cells.
DISCUSSION
In this study, we performed a chemical library screen and identified compounds that improve the activity of siRNAs delivered to cells via LNPs and Chol-siRNAs. An interesting result was that we found fundamental differences in the cellular routes used by the two delivery systems, and most compounds identified acted upon one delivery system but not the other. Furthermore, using a combination of quantitative fluorescence light and electron microscopy we identified compounds that act on two distinct bottlenecks of siRNA delivery (6), the uptake of siRNAs by cells and their release from endosomes into the cytosol. We would like to emphasize that without these methods it is impossible to accurately determine the intracellular localization and fraction of siRNAs that could escape from the lumen of the endosomes into the cytosol without overloading cells with siRNA doses beyond the therapeutic range or using unspecific and biased ‘escape’ markers (26,34–37). This is because the efficacy of endosomal escape is typically very low even with the best delivery systems (6) and the number of molecules that are required to load onto the RISC complex for silencing is below the detection sensitivity of the conventional fluorescence microscopy (32,33). Furthermore, several compounds identified in the screen on HeLa cells could also improve siRNA delivery in other cell types, such as primary fibroblasts and hepatocytes, arguing that they act upon conserved molecular mechanisms. In particular, those compounds that enhance endosomal escape appear to exert their effects on the cells rather than the delivery systems. In addition, we demonstrated that compounds selected based on their mechanism of action in HeLa cells are able to enhance delivery in cell types usually refractory to transfection. Thus, our study provides proof of principle for the use of chemical screens to improve siRNA delivery in a system-dependent fashion.
By performing a secondary screen where the LNP enhancers were directly incubated with HeLa cells, i.e. without a pre-incubation, we could distinguish two classes of enhancers, one that required the pre-incubation and a second that did not. Our interpretation is that the compounds that required the pre-incubation step likely act on the LNP itself. The lack of efficiency when the enhancers are directly added to the cells is presumably due to the dilution in the medium. On the other hand, compounds that did not require the pre-incubation are less likely to modify the LNPs and expected to act primarily on the cells. Strongly supporting this interpretation, the analysis of BADGE, which requires overnight pre-incubation, revealed that it acts upon the structure of LNPs, reducing their size. There is an optimal size range of LNPs activity (65). Increasing particle size was found to reduce silencing in hepatocytes but increase it in antigen presenting cells (34). In principle, decreasing the size of the particles could improve the uptake by gaining access to more tissues and cellular entry routes. Recent technical developments using microfluidic mixing technologies have enabled the formulation of siRNAs in LNPs as small as 25 nm (66). However, such a reduction in size occurs at the expense of a decreased payload of siRNA (67). In our experiments, the mean number of gold-siRNA per LNP was similar in DMSO and BADGE-treated LNPs (7.77 ± 0.37 and 7.86 ± 0.5, respectively) (Supplementary Figure S6), arguing that the reduction in size is not due to fission but rather to particle compaction. This means that BADGE reduced the size of the LNPs without sacrificing the payload of siRNA. Interestingly, Dahlman and colleagues found that new polymeric nanoparticles were able to deliver siRNAs into endothelial cells rather than hepatocytes (68). The size of these particles was between 35 and 45 nm, in the range of LNPs modified by BADGE. Based on the chemical structure of BADGE, we can envisage a possible explanation for its activity in the compaction of the particles. BADGE could insert into the LNP lipid structure mediated by hydrophobic and stacking π-π-interactions due to the aromatic rings of BADGE and cis double bonds from the lipid (Supplementary Figure S8). Close proximity to the dimethyl amino head groups may even lead to lipid crosslinking through epoxide opening and alkylation of the amino groups. This would increase the density of positively charged quaternary head groups of the amino lipid, resulting in enhanced interactions with the negatively charged siRNA backbone and a more condensed LNP structure. Clearly, more work is necessary to determine the mechanism of action of BADGE and its impact on (i) the lipid composition of the LNPs, (ii) the LNP charge density, and (iii) the abilities of LNPs to get opsonized. Further analysis of BADGE and other compounds could thus lead to new approaches for the synthesis of LNPs of small and homogeneous size, but high siRNA payload and widespread bio-availability in an organism.
Other compounds, such as CPW1-J18, increased the escape of siRNAs from endosomes. First, these compounds also elicited their effect when administered to cells prior to the addition of the siRNAs, suggesting that they act upon the cells, presumably on the endosomal system. Second, they did not alter the efficacy of uptake. In the case of CPW1-J18, we measured an increase in the ratio of siRNA-gold in the cytosol versus the total amount internalized. To our knowledge, this is the first quantitative and direct evidence of enhanced release of siRNAs from the endosomal lumen into the cytosol. Although CPW1-J18 was originally developed as Rab GGTase inhibitor (69), its mode of action for siRNA delivery may be different. It has been proposed (70) that blocking endosomal maturation may increase the time window during which the siRNAs can be released from the endosomal lumen. Indeed, we previously demonstrated that the release of siRNAs delivered by LNP occurs in a specific stage of endosomal transport (6). Surprisingly however, neither hydroxychloroquine nor Bafilomycin were hits in our screen. Yet, they were active as judged by their multi-parametric profiles. One possibility is that inhibiting endosomal acidification per se and slower endosomal progression may not be sufficient to significantly increase delivery of the siRNAs by the particular systems used and under the stringency of our experimental conditions (low siRNA concentrations, compounds concentration, incubation, threshold of detection, etc). Another possibility is that chloroquine and bafilomycin improve siRNA release but concomitantly decrease in LNP uptake, as they are known to inhibit receptor mediated endocytosis (6,60–61). In contrast, the enhancers we identified do not significantly reduce LNP uptake as compared to DMSO (see Figure 4B). Undoubtedly, a preferred strategy would be to enhance escape of siRNAs from endosomes.
Analysis of the effect of the compounds on the endocytic system and on different cell types provided some important clues on their respective mechanisms of action. Using hydroxychloroquine as reference compound that alters endosomal maturation we could profile the compounds with respect to this mechanism. We found that several compounds had a phenotypic profile similar to hydroxychloroquine. On this basis, one could interpret that they may indeed alter endosomal acidification and/or maturation. However, the finding that such compounds were not active on all cell types tested speaks against this interpretation. For example, the activity of CPW1-J18 in fibroblast is modest (Figure 3), arguing that its mechanism of action may still differ from that of hydroxychloroquine, which is a lysosomotropic agent active on a wide spectrum of cells. Therefore, increasing the residence time in the endosomes may not be sufficient to significantly enhance the release of siRNAs into the cytosol and CPW1-J18 and similarly active compounds may have some additional effect. In addition, most compounds had a very different profile from hydroxychloroquine. Among those compounds, #35 (Lomatin) appeared to effect specifically endocytic/recycling cargo (as visualized by the specific alteration on Tf but not EGF trafficking). However, for LNPs none of the enhancers had a similar profile to #35. Strikingly, a group of enhancers did not have detectable effects on the endosomal system. These compounds are candidates for destabilizing endosomal membranes, as it has been proposed for larger cationic and amphipathic cell-permeating peptides (71,72), or for fusion with the limiting membrane as shown for intra-luminal vesicles (73). Altogether, our results suggest that enhancers may facilitate the escape of siRNAs from endosomes by a variety of mechanisms.
Interestingly, the vast majority of compounds active on one delivery system were not active on the other. This supports the idea that the compounds do not share a common mechanism but enhance siRNA delivery depending on the system. In contrast, enhancers within a delivery system did show an appreciable level of activity between different cells, but depending on the cell type. For example, 60% of the compounds (both for LNPs and Chol-siRNAs) increased uptake in fibroblasts, cells for which uptake is typically inefficient. In contrast, only 20% of the compounds increased uptake in hepatocytes, which are normally proficient in internalization. Therefore, the knowledge of the mode of action of the compounds may help predicting which cell type may be predisposed to the specific effect of a given compound on delivery.
The endocytic pathway is largely conserved between cells. Although most compounds identified in the screen improved delivery for only one delivery system, it is possible that the principle behind their mechanism of action may be applicable to multiple delivery systems of the same class and shared between different cell types. The compounds that improved the particular LNPs used here may also improve nanoparticles of different chemical composition. Likewise, those that improved the Chol-siRNAs might have similar effects on other siRNA-conjugate types. Nevertheless, several of the compounds identified were active on different primary cells but others were not. This suggests that there is considerable cell-type specificity for the enhancers in conjunction with the delivery system.
Future work will be required to determine the precise mode of action of the identified enhancers. Our results argue that more than one mechanism can be exploited and pose a number of questions. First, by which mechanism compounds like BADGE are able to reduce the size of the LNPs without reducing the siRNA payload? Second, which are the molecular targets of the compounds improving uptake? Third, by which mechanisms can siRNAs escape from endosomes? Clearly, the pharmacological approach needs further development to improve delivery across multiple delivery systems and cell types for therapeutic applications. However, there is another important aspect of this approach that may have more far reaching implications. It provides the possibility to learn from the mechanism of action of the compounds and establish common principles for improving the uptake and escape from endosomes. One possible approach is to directly measure the escape of siRNAs from the lumen of endosomes in an in vitro assay in the presence of the compounds (63,74). The assay could shed light on the membrane integrity (74), dependence on membrane fusion and transport. Understanding these mechanisms in more detail may provide the means of using a rational approach either to improve the existing or to design a new generation of delivery systems.
Supplementary Material
Acknowledgments
We acknowledge I. Patten, T. Galvez, E. Perini and K. Diamantara for discussions and comments on the manuscript. We thank J-M. Verbavatz and J. Peychl respectively for the management of the Electron Microscopy Facility and the Light Microscopy Facility. We acknowledge A. Pal for Rab5-GFP primary human fibroblasts preparation. We thank W. John and A. Muench-Wuttke from the Biomedical Service Facility for mouse care and injections. We are thanking Russian Scientific Fund (grant 14–34–00017) for financial support.
Author contributions: M.Z. conceived and directed the project. M.Z., J.G., P.P., A.Z., W.Q., A.A., M.A.M., M.B., C.A., K.F. and Y.K. designed the experiments. M.A.M. synthesized the siRNA-alexa647 and the siRNA-gold conjugates. A.A. formulated LNP. C.A. under the supervision of M.B. performed the High throughput screening. J.G., P.P. and C.A. performed the mode of action validation in HeLa and human fibroblasts. M.S. and C.A. performed the automated image acquisitions with the OPERA. G.M. under the supervision of Y.K. provided the quantitative multiparametrics image analysis and the statistics. S.S. under the supervision of J.G. performed mouse intra-cardiac injection of LNPs. J.G. performed the sections, staining and imaging of the heart. S.S. and P.A. under the supervision of A.Z. performed primary hepatocytes and primary endothelial cells preparation. J.G. developed the quantitative electron microscopy. G.M. developed the software for automated counting of gold number on electron microscopy images. M.Z. and J.G. wrote the manuscript and M.B, M.A.M., M.M., W.Q. and A.A. edited it.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Max Planck Society (MPG); DFG and Alnylam Pharmaceuticals; EMBO [ALTF298-2009 to J.G.]. Funding for open access charge: Max Planck Society (MPG).
Conflict of interest statement. W.Q., K.F., A.A., M.A.M. and M.M. are Alnylam Pharmaceuticals employees; M.Z. received funding from and was a consultant with Alnylam Pharmaceuticals.
REFERENCES
- 1.Sliva K., Schnierle B.S. Selective gene silencing by viral delivery of short hairpin RNA. Virol. J. 2010;7:248–258. doi: 10.1186/1743-422X-7-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Buyens K., De Smedt S.C., Braeckmans K., Demeester J., Peeters L., van Grunsven L.A., de Mollerat du Jeu X., Sawant R., Torchilin V., Farkasova K., et al. Liposome based systems for systemic siRNA delivery: stability in blood sets the requirements for optimal carrier design. J. Control. Release. 2012;158:362–370. doi: 10.1016/j.jconrel.2011.10.009. [DOI] [PubMed] [Google Scholar]
- 3.Castillo B., Bromberg L., Lopez X., Badillo V., Gonzalez Feliciano J.A., Gonzalez C.I., Hatton T.A., Barletta G. Intracellular Delivery of siRNA by Polycationic Superparamagnetic Nanoparticles. J. Drug Deliv. 2012;2012:218940–218951. doi: 10.1155/2012/218940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lopez-Davila V., Seifalian A.M., Loizidou M. Organic nanocarriers for cancer drug delivery. Curr. Opin. Pharmacol. 2012;12:414–419. doi: 10.1016/j.coph.2012.02.011. [DOI] [PubMed] [Google Scholar]
- 5.Nair J., Willoughby J., Chan A., Charisse K., Alam R.M., Wang Q., Hoekstra M., Kandasamy P., Kel'in A.V., Milstein S., et al. Multivalent N-Acetylgalactosamine-Conjugated siRNA Localizes in Hepatocytes and Elicits Robust RNAi-mediated Gene Silencing. J. Am. Chem. Soc. 2014;136:16958–16961. doi: 10.1021/ja505986a. [DOI] [PubMed] [Google Scholar]
- 6.Gilleron J., Querbes W., Zeigerer A., Borodovsky A., Marsico G., Schubert U., Manygoats K., Seifert S., Andree C., Stoter M., et al. Image-based analysis of lipid nanoparticle-mediated siRNA delivery, intracellular trafficking and endosomal escape. Nat. Biotechnol. 2013;31:638–646. doi: 10.1038/nbt.2612. [DOI] [PubMed] [Google Scholar]
- 7.Akinc A., Goldberg M., Qin J., Dorkin J.R., Gamba-Vitalo C., Maier M., Jayaprakash K.N., Jayaraman M., Rajeev K.G., Manoharan M., et al. Development of Lipidoid-siRNA Formulations for Systemic Delivery to the Liver. Mol. Ther. 2009;17:872–879. doi: 10.1038/mt.2009.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Akinc A., Querbes W., De S., Qin J., Frank-Kamenetsky M., Jayaprakash K.N., Jayaraman M., Rajeev K.G., Cantley W.L., Dorkin J.R., et al. Targeted delivery of RNAi therapeutics with endogenous and exogenous ligand-based mechanisms. Mol. Ther. 2010;18:1357–1364. doi: 10.1038/mt.2010.85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jayaraman M., Ansell S.M., Mui B.L., Tam Y.K., Chen J., Du X., Butler D., Eltepu L., Matsuda S., Narayanannair J.K., et al. Maximizing the potency of siRNA lipid nanoparticles for hepatic gene silencing in vivo. Angew. Chem. Int. Ed. Engl. 2012;51:8529–8533. doi: 10.1002/anie.201203263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Love K.T., Mahon K.P., Levins C.G., Whitehead K.A., Querbes W., Dorkin J.R., Qin J., Cantley W., Qin L.L., Racie T., et al. Lipid-like materials for low-dose, in vivo gene silencing. Proc. Nat. Acad. Sci. U.S.A. 2010;107:1864–1869. doi: 10.1073/pnas.0910603106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zimmermann T.S., Lee A.C., Akinc A., Bramlage B., Bumcrot D., Fedoruk M.N., Harborth J., Heyes J.A., Jeffs L.B., John M., et al. RNAi-mediated gene silencing in non-human primates. Nature. 2006;441:111–114. doi: 10.1038/nature04688. [DOI] [PubMed] [Google Scholar]
- 12.Kanasty R., Dorkin J.R., Vegas A., Anderson D. Delivery materials for siRNA therapeutics. Nat. Mater. 2013;12:967–977. doi: 10.1038/nmat3765. [DOI] [PubMed] [Google Scholar]
- 13.Coelho T., Adams D., Silva A., Lozeron P., Hawkins P.N., Mant T., Perez J., Chiesa J., Warrington S., Tranter E., et al. Safety and Efficacy of RNAi Therapy for Transthyretin Amyloidosis. N. Engl. J. Med. 2013;369:819–829. doi: 10.1056/NEJMoa1208760. [DOI] [PubMed] [Google Scholar]
- 14.Pei Y., Hancock P.J., Zhang H., Bartz R., Cherrin C., Innocent N., Pomerantz C.J., Seitzer J., Koser M.L., Abrams M.T., et al. Quantitative evaluation of siRNA delivery in vivo. RNA. 2010;16:2553–2563. doi: 10.1261/rna.2255810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Xu Y., Ou M., Keough E., Roberts J., Koeplinger K., Lyman M., Fauty S., Carlini E., Stern M., Zhang R., et al. Quantitation of physiological and biochemical barriers to siRNA liver delivery via lipid nanoparticle platform. Mol. Pharm. 2014;11:1424–1434. doi: 10.1021/mp400584h. [DOI] [PubMed] [Google Scholar]
- 16.Akinc A., Battaglia G. Exploiting endocytosis for nanomedicines. Cold Spring Harb. Perspect. Biol. 2013;5:a016980. doi: 10.1101/cshperspect.a016980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lee S.K., Siefert A., Beloor J., Fahmy T.M., Kumar P. Cell-specific siRNA delivery by peptides and antibodies. Methods Enzymol. 2012;502:91–122. doi: 10.1016/B978-0-12-416039-2.00005-7. [DOI] [PubMed] [Google Scholar]
- 18.Nielsen C., Kjems J., Sorensen K.R., Engelholm L.H., Behrendt N. Advances in targeted delivery of small interfering RNA using simple bioconjugates. Expert Opin. Drug. Deliv. 2014;11:791–822. doi: 10.1517/17425247.2014.896898. [DOI] [PubMed] [Google Scholar]
- 19.Prakash T.P., Graham M.J., Yu J., Carty R., Low A., Chappell A., Schmidt K., Zhao C., Aghajan M., Murray H.F., et al. Targeted delivery of antisense oligonucleotides to hepatocytes using triantennary N-acetyl galactosamine improves potency 10-fold in mice. Nucleic Acids Res. 2014;42:8796–8807. doi: 10.1093/nar/gku531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hibbitts A., Lieggi N., McCabe O., Thomas W., Barlow J., O'Brien F., Cryan S.A. Screening of siRNA nanoparticles for delivery to airway epithelial cells using high-content analysis. Ther. Deliv. 2011;2:987–999. doi: 10.4155/tde.11.73. [DOI] [PubMed] [Google Scholar]
- 21.Li L., Wang F., Wu Y., Davidson G., Levkin P.A. Combinatorial synthesis and high-throughput screening of alkyl amines for nonviral gene delivery. Bioconjug. Chem. 2013;24:1543–1551. doi: 10.1021/bc400158w. [DOI] [PubMed] [Google Scholar]
- 22.Zugates G.T., Anderson D.G., Langer R. High-throughput methods for screening polymeric transfection reagents. Cold Spring Harb. Protoc. 2013;11:1–14. doi: 10.1101/pdb.prot078634. [DOI] [PubMed] [Google Scholar]
- 23.Wang A., Marinakos S.M., Badireddy A.R., Powers C.M., Houck K.A. Characterization of physicochemical properties of nanomaterials and their immediate environments in high-throughput screening of nanomaterial biological activity. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 2013;5:430–448. doi: 10.1002/wnan.1229. [DOI] [PubMed] [Google Scholar]
- 24.Nabzdyk C.S., Chun M., Pradhan L., Logerfo F.W. High throughput RNAi assay optimization using adherent cell cytometry. J. Transl. Med. 2011;9:48–56. doi: 10.1186/1479-5876-9-48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ming X., Carver K., Fisher M., Noel R., Cintrat J.C., Gillet D., Barbier J., Cao C., Bauman J., Juliano R.L. The small molecule Retro-1 enhances the pharmacological actions of antisense and splice switching oligonucleotides. Nucleic Acids Res. 2013;41:3673–3687. doi: 10.1093/nar/gkt066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yang B., Ming X., Cao C., Laing B., Yuan A., Porter M.A., Hull-Ryde E.A., Maddry J., Suto M., Janzen W.P., et al. High-throughput screening identifies small molecules that enhance the pharmacological effects of oligonucleotides. Nucleic Acids Res. 2015;43:1987–1996. doi: 10.1093/nar/gkv060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cotten M., Langle-Rouault F., Kirlappos H., Wagner E., Mechtler K., Zenke M., Beug H., Birnstiel M.L. Transferrin-polycation-mediated introduction of DNA into human leukemic cells: stimulation by agents that affect the survival of transfected DNA or modulate transferrin receptor levels. Proc. Natl. Acad. Sci. U.S.A. 1990;87:4033–4037. doi: 10.1073/pnas.87.11.4033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bhattarai S.R., Muthuswamy E., Wani A., Brichacek M., Castaneda A.L., Brock S.L., Oupicky D. Enhanced gene and siRNA delivery by polycation-modified mesoporous silica nanoparticles loaded with chloroquine. Pharm. Res. 2010;27:2556–2568. doi: 10.1007/s11095-010-0245-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Settembre C., Fraldi A., Medina D.L., Ballabio A. Signals from the lysosome: a control centre for cellular clearance and energy metabolism. Nat. Rev. Mol. Cell Biol. 2013;14:283–296. doi: 10.1038/nrm3565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sorkin A., von Zastrow M. Endocytosis and signalling: intertwining molecular networks. Nat. Rev. Mol. Cell Biol. 2009;10:609–622. doi: 10.1038/nrm2748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Saftig P., Klumperman J. Lysosome biogenesis and lysosomal membrane proteins: trafficking meets function. Nat. Rev. Mol. Cell Biol. 2009;10:623–635. doi: 10.1038/nrm2745. [DOI] [PubMed] [Google Scholar]
- 32.Wei J., Jones J., Kang J., Card A., Krimm M., Hancock P., Pei Y., Ason B., Payson E., Dubinina N., et al. RNA-induced silencing complex-bound small interfering RNA is a determinant of RNA interference-mediated gene silencing in mice. Mol. Pharmacol. 2011;79:953–963. doi: 10.1124/mol.110.070409. [DOI] [PubMed] [Google Scholar]
- 33.Landesman Y., Svrzikapa N., Cognetta A. 3rd, Zhang X., Bettencourt B.R., Kuchimanchi S., Dufault K., Shaikh S., Gioia M., Akinc A., et al. In vivo quantification of formulated and chemically modified small interfering RNA by heating-in-Triton quantitative reverse transcription polymerase chain reaction (HIT qRT-PCR) Silence. 2010;1:16–29. doi: 10.1186/1758-907X-1-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Basha G., Novobrantseva T.I., Rosin N., Tam Y.Y., Hafez I.M., Wong M.K., Sugo T., Ruda V.M., Qin J., Klebanov B., et al. Influence of Cationic Lipid Composition on Gene Silencing Properties of Lipid Nanoparticle Formulations of siRNA in Antigen-Presenting Cells. Mol. Ther. 2011;19:2186–2200. doi: 10.1038/mt.2011.190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tamura A., Oishi M., Nagasaki Y. Enhanced cytoplasmic delivery of siRNA using a stabilized polyion complex based on PEGylated nanogels with a cross-linked polyamine structure. Biomacromolecules. 2009;10:1818–1827. doi: 10.1021/bm900252d. [DOI] [PubMed] [Google Scholar]
- 36.Akita H., Kogure K., Moriguchi R., Nakamura Y., Higashi T., Nakamura T., Serada S., Fujimoto M., Naka T., Futaki S., et al. Nanoparticles for ex vivo siRNA delivery to dendritic cells for cancer vaccines: programmed endosomal escape and dissociation. J. Control. Release. 2010;143:311–317. doi: 10.1016/j.jconrel.2010.01.012. [DOI] [PubMed] [Google Scholar]
- 37.Sakurai Y., Hatakeyama H., Sato Y., Akita H., Takayama K., Kobayashi S., Futaki S., Harashima H. Endosomal escape and the knockdown efficiency of liposomal-siRNA by the fusogenic peptide shGALA. Biomaterials. 2011;32:5733–5742. doi: 10.1016/j.biomaterials.2011.04.047. [DOI] [PubMed] [Google Scholar]
- 38.Soutschek J., Akinc A., Bramlage B., Charisse K., Constien R., Donoghue M., Elbashir S., Geick A., Hadwiger P., Harborth J., et al. Therapeutic silencing of an endogenous gene by systemic administration of modified siRNAs. Nature. 2004;432:173–178. doi: 10.1038/nature03121. [DOI] [PubMed] [Google Scholar]
- 39.Bramsen J.B., Laursen M.B., Nielsen A.F., Hansen T.B., Bus C., Langkjaer N., Babu B.R., Hojland T., Abramov M., Van Aerschot A., et al. A large-scale chemical modification screen identifies design rules to generate siRNAs with high activity, high stability and low toxicity. Nucleic Acids Res. 2009;37:2867–2881. doi: 10.1093/nar/gkp106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pal A., Severin F., Lommer B., Shevchenko A., Zerial M. Huntingtin-HAP40 complex is a novel Rab5 effector that regulates early endosome motility and is up-regulated in Huntington's disease. J. Cell Biol. 2006;172:605–618. doi: 10.1083/jcb.200509091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Riedl J., Flynn K.C., Raducanu A., Gartner F., Beck G., Bosl M., Bradke F., Massberg S., Aszodi A., Sixt M., et al. Lifeact mice for studying F-actin dynamics. Nat. Methods. 2010;7:168–169. doi: 10.1038/nmeth0310-168. [DOI] [PubMed] [Google Scholar]
- 42.Zeigerer A., Gilleron J., Bogorad R.L., Marsico G., Nonaka H., Seifert S., Epstein-Barash H., Kuchimanchi S., Peng C.G., Ruda V.M., et al. Rab5 is necessary for the biogenesis of the endolysosomal system in vivo. Nature. 2012;485:465–470. doi: 10.1038/nature11133. [DOI] [PubMed] [Google Scholar]
- 43.Limmer A., Ohl J., Kurts C., Ljunggren H.G., Reiss Y., Groettrup M., Momburg F., Arnold B., Knolle P.A. Efficient presentation of exogenous antigen by liver endothelial cells to CD8+ T cells results in antigen-specific T-cell tolerance. Nat. Med. 2000;6:1348–1354. doi: 10.1038/82161. [DOI] [PubMed] [Google Scholar]
- 44.Collinet C., Stoter M., Bradshaw C.R., Samusik N., Rink J.C., Kenski D., Habermann B., Buchholz F., Henschel R., Mueller M.S., et al. Systems survey of endocytosis by multiparametric image analysis. Nature. 2010;464:243–249. doi: 10.1038/nature08779. [DOI] [PubMed] [Google Scholar]
- 45.Karnovsky M.J. Proceedings of the Eleventh Annual Meeting of the American Society for Cell Biology. Vol. 284. 11th Am Soc Cell Biol New Orleans; 1971. p. 146. [Google Scholar]
- 46.Lucocq J.M. Efficient quantitative morphological phenotyping of genetically altered organisms using stereology. Transgenic Res. 2007;16:133–145. doi: 10.1007/s11248-006-9048-9. [DOI] [PubMed] [Google Scholar]
- 47.Soille P. Morphological Image Analysis: Principles and Applications. London: Springer-Verlag Telos; 1999. [Google Scholar]
- 48.Rink J., Ghigo E., Kalaidzidis Y., Zerial M. Rab conversion as a mechanism of progression from early to late endosomes. Cell. 2005;122:735–749. doi: 10.1016/j.cell.2005.06.043. [DOI] [PubMed] [Google Scholar]
- 49.Wolfrum C., Shi S., Jayaprakash K.N., Jayaraman M., Wang G., Pandey R.K., Rajeev K.G., Nakayama T., Charrise K., Ndungo E.M., et al. Mechanisms and optimization of in vivo delivery of lipophilic siRNAs. Nat. Biotechnol. 2007;25:1149–1157. doi: 10.1038/nbt1339. [DOI] [PubMed] [Google Scholar]
- 50.Frank-Kamenetsky M., Grefhorst A., Anderson N.N., Racie T.S., Bramlage B., Akinc A., Butler D., Charisse K., Dorkin R., Fan Y., et al. Therapeutic RNAi targeting PCSK9 acutely lowers plasma cholesterol in rodents and LDL cholesterol in nonhuman primates. Proc. Natl. Acad. Sci. U.S.A. 2008;105:11915–11920. doi: 10.1073/pnas.0805434105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Semple S.C., Akinc A., Chen J., Sandhu A.P., Mui B.L., Cho C.K., Sah D.W., Stebbing D., Crosley E.J., Yaworski E., et al. Rational design of cationic lipids for siRNA delivery. Nat. Biotechnol. 2010;28:172–176. doi: 10.1038/nbt.1602. [DOI] [PubMed] [Google Scholar]
- 52.Mukherjee S., Zha X., Tabas I., Maxfield F.R. Cholesterol distribution in living cells: fluorescence imaging using dehydroergosterol as a fluorescent cholesterol analog. Biophys. J. 1998;75:1915–1925. doi: 10.1016/S0006-3495(98)77632-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ikonen E. Cellular cholesterol trafficking and compartmentalization. Nat. Rev. Mol. Cell Biol. 2008;9:125–138. doi: 10.1038/nrm2336. [DOI] [PubMed] [Google Scholar]
- 54.Brown M.S., Goldstein J.L. A receptor-mediated pathway for cholesterol homeostasis. Science. 1986;232:34–47. doi: 10.1126/science.3513311. [DOI] [PubMed] [Google Scholar]
- 55.Brown M.S., Goldstein J.L. The SREBP pathway: regulation of cholesterol metabolism by proteolysis of a membrane-bound transcription factor. Cell. 1997;89:331–340. doi: 10.1016/s0092-8674(00)80213-5. [DOI] [PubMed] [Google Scholar]
- 56.Maxfield F.R., Tabas I. Role of cholesterol and lipid organization in disease. Nature. 2005;438:612–621. doi: 10.1038/nature04399. [DOI] [PubMed] [Google Scholar]
- 57.Chen Y.H., Lin W.W., Liu C.S., Hsu L.S., Lin Y.M., Su S.L. Caveolin-1 provides palliation for adverse hepatic reactions in hypercholesterolemic rabbits. PLoS One. 2014;9:e71862. doi: 10.1371/journal.pone.0071862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Simeoni F., Morris M.C., Heitz F., Divita G. Insight into the mechanism of the peptide-based gene delivery system MPG: implications for delivery of siRNA into mammalian cells. Nucleic Acids Res. 2003;31:2717–2724. doi: 10.1093/nar/gkg385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Luthman H., Magnusson G. High efficiency polyoma DNA transfection of chloroquine treated cells. Nucleic Acids Res. 1983;11:1295–1308. doi: 10.1093/nar/11.5.1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Pack D.W., Hoffman A.S., Pun S., Stayton P.S. Design and development of polymers for gene delivery. Nature reviews. Drug discovery. 2005;4:581–593. doi: 10.1038/nrd1775. [DOI] [PubMed] [Google Scholar]
- 61.Tietze C., Schlesinger P., Stahl P. Chloroquine and ammonium ion inhibit receptor-mediated endocytosis of mannose-glycoconjugates by macrophages: apparent inhibition of receptor recycling. Biochemical and biophysical research communications. 1980;93:1–8. doi: 10.1016/s0006-291x(80)80237-3. [DOI] [PubMed] [Google Scholar]
- 62.Starai V.J., Jun Y., Wickner W. Excess vacuolar SNAREs drive lysis and Rab bypass fusion. Proc. Natl. Acad. Sci. U.S.A. 2007;104:13551–13558. doi: 10.1073/pnas.0704741104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ohya T., Miaczynska M., Coskun U., Lommer B., Runge A., Drechsel D., Kalaidzidis Y., Zerial M. Reconstitution of Rab- and SNARE-dependent membrane fusion by synthetic endosomes. Nature. 2009;459:1091–1097. doi: 10.1038/nature08107. [DOI] [PubMed] [Google Scholar]
- 64.Sundaramurthy V., Barsacchi R., Samusik N., Marsico G., Gilleron J., Kalaidzidis I., Meyenhofer F., Bickle M., Kalaidzidis Y., Zerial M. Integration of chemical and RNAi multiparametric profiles identifies triggers of intracellular mycobacterial killing. Cell Host Microbe. 2013;13:129–142. doi: 10.1016/j.chom.2013.01.008. [DOI] [PubMed] [Google Scholar]
- 65.Akinc A., Goldberg M., Qin J., Dorkin J.R., Gamba-Vitalo C., Maier M., Jayaprakash K.N., Jayaraman M., Rajeev K.G., Manoharan M., et al. Development of Lipidoid-siRNA Formulations for Systemic Delivery to the Liver. Mol. Ther. 2009;17:872–879. doi: 10.1038/mt.2009.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Belliveau N.M., Huft J., Lin P.J., Chen S., Leung A.K., Leaver T.J., Wild A.W., Lee J.B., Taylor R.J., Tam Y.K., et al. Microfluidic Synthesis of Highly Potent Limit-size Lipid Nanoparticles for In Vivo Delivery of siRNA. Mol. Ther. Nucleic Acids. 2012;1:e37. doi: 10.1038/mtna.2012.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Tam Y.Y., Chen S., Cullis P.R. Advances in Lipid Nanoparticles for siRNA Delivery. Pharmaceutics. 2013;5:498–507. doi: 10.3390/pharmaceutics5030498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Dahlman J.E., Barnes C., Khan O.F., Thiriot A., Jhunjunwala S., Shaw T.E., Xing Y., Sager H.B., Sahay G., Speciner L., et al. In vivo endothelial siRNA delivery using polymeric nanoparticles with low molecular weight. Nat Nanotechnol. 2014;9:648–655. doi: 10.1038/nnano.2014.84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Tan K.T., Guiu-Rozas E., Bon R.S., Guo Z., Delon C., Wetzel S., Arndt S., Alexandrov K., Waldmann H., Goody R.S., et al. Design, synthesis, and characterization of peptide-based rab geranylgeranyl transferase inhibitors. J. Med. Chem. 2009;52:8025–8037. doi: 10.1021/jm901117d. [DOI] [PubMed] [Google Scholar]
- 70.Ma D. Enhancing endosomal escape for nanoparticle mediated siRNA delivery. Nanoscale. 2014;6:6415–6425. doi: 10.1039/c4nr00018h. [DOI] [PubMed] [Google Scholar]
- 71.Thoren P.E., Persson D., Esbjorner E.K., Goksor M., Lincoln P., Norden B. Membrane binding and translocation of cell-penetrating peptides. Biochemistry. 2004;43:3471–3489. doi: 10.1021/bi0360049. [DOI] [PubMed] [Google Scholar]
- 72.El-Andaloussi S., Holm T., Langel U. Cell-penetrating peptides: mechanisms and applications. Curr. Pharm. Des. 2005;11:3597–3611. doi: 10.2174/138161205774580796. [DOI] [PubMed] [Google Scholar]
- 73.Bissig C., Gruenberg J. ALIX and the multivesicular endosome: ALIX in Wonderland. Trends Cell. Biol. 2014;24:19–25. doi: 10.1016/j.tcb.2013.10.009. [DOI] [PubMed] [Google Scholar]
- 74.Bartz R., Fan H., Zhang J., Innocent N., Cherrin C., Beck S.C., Pei Y., Momose A., Jadhav V., Tellers D.M., et al. Effective siRNA delivery and target mRNA degradation using an amphipathic peptide to facilitate pH-dependent endosomal escape. Biochem. J. 2011;435:475–487. doi: 10.1042/BJ20101021. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.