


Abstract
Genome-wide expression profiling has revolutionized biomedical research; vast amounts of expression data from numerous studies of many diseases are now available. Making the best use of this resource in order to better understand disease processes and treatment remains an open challenge. In particular, disease biomarkers detected in case–control studies suffer from low reliability and are only weakly reproducible. Here, we present a systematic integrative analysis methodology to overcome these shortcomings. We assembled and manually curated more than 14 000 expression profiles spanning 48 diseases and 18 expression platforms. We show that when studying a particular disease, judicious utilization of profiles from other diseases and information on disease hierarchy improves classification quality, avoids overoptimistic evaluation of that quality, and enhances disease-specific biomarker discovery. This approach yielded specific biomarkers for 24 of the analyzed diseases. We demonstrate how to combine these biomarkers with large-scale interaction, mutation and drug target data, forming a highly valuable disease summary that suggests novel directions in disease understanding and drug repurposing. Our analysis also estimates the number of samples required to reach a desired level of biomarker stability. This methodology can greatly improve the exploitation of the mountain of expression profiles for better disease analysis.
INTRODUCTION
Gene expression studies use expression profiles of cases and controls to understand a disease by identifying genes and pathways that differ in their expression between the two groups. This methodology has become ubiquitous in biomedical research, and is often combined with additional information of either the patients or the genes to interpret the results (1–7). However, these analyses suffer from several limitations: the discovered biomarkers often have low reproducibility, and are difficult to interpret biologically and especially clinically (8,9).
A promising direction for increasing robustness is by integration of many gene expression datasets. The difficulty here is in creating a common denominator of multiple studies, often conducted using different platforms under diverse experimental conditions and tissues. Huang et al. (10) used 9169 gene expression samples, each associated with a set of disease terms of the Unified Medical Language System (UMLS). UMLS, and similar databases such as Disease Ontology (DO), provide ontology of disease terms organized in a hierarchy that models dependencies among diseases (11,12). The authors presented an algorithm that predicts a set of disease terms for each gene expression sample (10). Schmid et al. analyzed 3030 samples of one platform and predicted their UMLS terms using similarity-based analysis (13). Lee et al. used >14 000 profiles of one microarray technology to predict the tissue of a sample (14). While these studies reported good prediction quality, they have some limitations. First, data of only one or two expression platforms were analyzed, limiting the data used and the applicability of the results. Second, in Huang et al. and in Lee et al. the mapping of samples to their disease terms was done automatically, inevitably introducing mapping errors (10). Third, the predictor in (10) can be applied on new patient samples only if a set of new control samples accompanies them. Fourth, while the prediction performance of the classifiers was far from random there is still substantial room for improvement. Finally, many biomarker sets are hard to interpret biomedically, which hampers their adoption in clinics.
To improve interpretability, several classification methods that integrate different biological data were suggested. For example, combining patient gene expression profiles and protein–protein interaction data or pathway information was demonstrated to improve disease classification accuracy or biological interpretability in some studies (1–7,15,16). However, other studies reported no significant improvement when utilizing network data (1,17). Moreover, in some cases the contribution of the additional gene data was very mild, which questions the benefit from interpreting these models. Other methods for biomarker discovery used prior knowledge on genes to extract differential genes of similar functionality. As another example, Ciriello et al., integrated gene expression profiles, methylation profiles, and single nucleotide polymorphism (SNP) data from 3299 cancer patients to construct a hierarchical structure of the patients, and used it to detect novel biomarkers of cancer subtypes (18).
The main goal of this study is integration of numerous heterogeneous expression profiles to produce reliable results that could be used as a starting point for novel biomedical insights. We focus on identification of the main genes that are specifically differential in a disease of interest and putting them in the context of interactions, mutations, and drugs. To be able to produce such overviews in a meaningful way we developed a four-step procedure (Figure 1). Each step is essential to obtain reliable results. First, we manually annotated more than 14 000 gene expression profiles from 175 datasets to produce a compendium called ADEPTUS (Annotated Disease Expression Profiles Transformed into a Unified Suite). ADEPTUS covers 13 314 microarray samples from GEO and 1526 RNA-Seq samples from TCGA. To overcome study and sample heterogeneity, each sample was normalized using a non-parametric rank-based method. Samples were manually annotated with the most relevant disease terms in DO. Second, as a quality assurance step we tested different multi-label classification algorithms. Two key issues here were: (i) performing leave-dataset-out cross validation to reduce bias of unknown covariates (e.g. batch effects), and (ii) showing that standard performance measures produce over-optimistic results, and rectifying this by introduction of more stringent classification measures. Using our measures, classification performance was very high for 24 diseases, mostly cancer subtypes. Limiting the data to a single platform improved the performance for six additional diseases.
Figure 1.
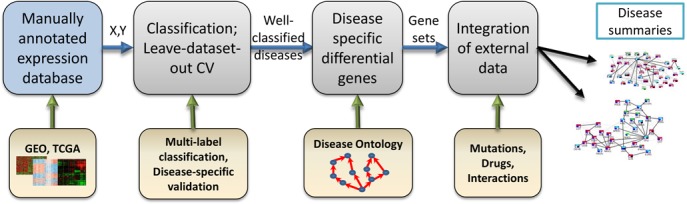
Study workflow overview. Step 1: Assembly of the ADEPTUS database: expression profiles from public sources were normalized and manually annotated. Step 2: Classification methods were used to identify well-classified diseases while avoiding over-optimistic results due to tissue and batch effects. Step 3: Disease-specific biomarker detection using the Disease Ontology structure. Step 4: Integration with other biomedical data produces gene-centric, disease-specific overview with therapeutic potential.
Third, we detect disease-specific differentially expressed genes, by accounting for the diversity of non-disease samples and the relationships among diseases. We demonstrate a shortcoming in the integration of multiple datasets of a single disease: without using alongside it data of other diseases, such analysis might find genes of general disease phenotypes that are not specific to the target disease. Our method was designed to overcome this difficulty. We demonstrate the robustness of our method, and also achieve estimates for the number of datasets and samples required to improve stability of biomarker detection. Functional enrichment analysis shows that the detected gene sets recapitulate known hallmarks of the diseases. Finally, for three cancer types we produce a network that highlights the molecular modification in the disease. This is done by an integrative analysis of the discovered differential genes with information on somatic mutations, drug targets, and gene interactions. We show that our results detect well known disease genes and treatments, and even suggest new indications of several known drugs.
MATERIALS AND METHODS
The expression profile compendium
We constructed a large compendium of expression profiles generated using different technologies, and manually annotated the diseases attributed to each profile (Supplementary Figure S1A). The compendium, called ADEPTUS, contains 174 gene expression studies from GEO (19), each with at least 20 samples. Overall, ADEPTUS covers 13 314 samples from 17 different microarray technologies, and 1526 RNA-Seq samples from TCGA (20). See Supplementary Text regarding using even larger compendia. For each study we used the preprocessed expression matrix given in the database. Each sample was either labeled as ‘case’ and manually assigned a set of the relevant DO terms based on the its textual description, or labeled as ‘control’. To allow cross-validation on whole datasets, we kept only DO terms that were represented by at least five different datasets in our compendium. This resulted in 48 disease terms.
Single sample gene scores
To allow joint analysis across platforms, expression profiles were transformed to rank-based scores (2,21) (Supplementary Figure S1A, see ‘Materials and Methods’ section). Given a gene expression profile of a single sample S in which k genes were measured, we ranked the genes by their expression levels g1, g2, g3,…, gk (with g1 having the highest level), and assigned a score to each gene based on its rank: WS(gi) = ie−i/k. See Supplementary Text for details.
The final compendium can be summarized as two matrices (Supplementary Figure S1A): A binary (samples × diseases) matrix Y where Ys,d = 1 if sample s is annotated with disease d, and a real-valued (samples × genes) matrix X where Xs,g = WS(g).
Multi-label classification
In multi-label classification each sample can belong to multiple true classes (e.g. cancer and lung cancer) (22,23). A sample can be predicted to have several labels and the sum over the predicted label probabilities need not be 1. Recent multi-label classification approaches (22,24,25) can be partitioned into two types: problem transformation and algorithm adaptation (23). See Supplementary Text for details. Here we used the label power-set (LP) transformation method, which defines for each sample a categorical class variable by concatenation of the sample's original labels (26). We also used the Bayesian correction (BC) adaptation method, which uses the known label hierarchy to correct errors after learning an independent single binary classifier for each label (10,27). Linear SVM (28,29) and random forest (30) were used as the binary classifiers.
Somatic mutation data
We analyzed the raw data of known somatic mutations from COSMIC (31). These data contained associations between genes and tumor samples. We kept only associations to non-silent mutations in coding regions that were also marked as ‘confirmed somatic mutations’. The result was 559 727 gene-tumor associations, covering a total of 43 517 tumor samples and 20 332 genes. We then assigned genes to tumor sites by calculating a hyper-geometric (HG) p-value for the overlap between the samples that had a mutation in the gene and the samples from the site. The p-values were FDR corrected for multiple testing and only significant associations were kept (q ≤ 0.05).
Gene–drug associations
Gene–drug associations were taken from DrugBank (32). Only approved drugs were used.
Network visualization and functional genomics
Network visualization was done using Cytoscape (33) and the Cytoscape application enhancedGraphics (34). Enrichment analysis in Cytoscape was done using BiNGO (35). GeneMania (36) was used to generate networks of a selected gene set. EXPANDER (37) was used for enrichment analysis of all discovered gene sets.
Validation of the multi-label classifier on RNA-Seq data
To test the performance of a multi-label classifier that was trained using the microarray samples, on the RNA-Seq samples, we transformed each RNA-Seq sample to gene weighted ranks. We then performed quantile normalization on all samples together. That is, we created a matrix whose rows are the samples including both the microarray samples and the RNA-Seq samples. The columns were the genes covered by the microarray data and the matrix values were the weighted ranks. Quantile normalization was performed to ensure that rows in the matrix would have similar distributions. This is crucial as any classifier assumes that the tested data and the training data are similarly distributed. Finally, the classifier was tested by computing its predictions on the rows of the RNA-seq samples.
Testing how biomarker stability depends on the amount of data
To test how the stability of our approach depends on the number of datasets used, we focused on DO term ‘organ system cancer’, which had 46 datasets in the compendium, of which 16 were not assigned to any sub-disease. To measure stability, we (i) randomly selected from these 46 datasets two disjoint subsets A and B of k datasets each, (ii) ran our pipeline and obtained biomarkers on each subset separately and (iii) measured the Jaccard score and the significance of the overlap between the two biomarkers. This process was repeated with k ranging from 5 to 23. As background controls, we added half of the remaining 128 non-’organ system cancer’ datasets to A and the rest to B. We rejected sets generated in step (1) if the total numbers of samples in A and B differed by more than 20%.
RESULTS
We collected and curated a large compendium of gene expression profiles from diverse diseases and developed and tested several approaches for classifying patient samples originating from each disease. For those diseases whose classification was validated successfully we developed specific biomarker genes and summarized them in the context of protein interaction, mutation and drug target data. Figure 1 shows an outline of our approach.
A curated gene expression compendium
We constructed a large compendium of >14 000 expression profiles generated using 18 different technologies. Each profile was designated as case or control and cases were manually assigned DO terms. The compendium, called ADEPTUS, contains 174 gene expression microarray studies and 1526 RNA-Seq samples. It covers 48 DO terms, including many cancer subtypes, obesity, neurodegenerative diseases and cardiovascular disease (see Supplementary Figure S2). Each sample was rank-normalized to allow comparison of samples from different technologies (see ‘Materials and Methods’ section). We observed that following rank normalization, the correlation between samples from different platforms (preprocessed using different methods) is high, see Supplementary Text.
Classification
We conducted a systematic analysis of classification methods in order to identify diseases in which the expression signal was consistent across datasets. The main components of the analysis were (i) utilization and comparison of several multi-label classification algorithms, (ii) leave-dataset-out cross validation to overcome technology and batch effects and (iii) a careful examination of the results in each disease separately in order to avoid over-optimistic conclusions due to tissue effects.
The classifiers
Samples in the compendium can have multiple related disease labels (e.g. hematologic cancer and ALL). Classification of the samples can be addressed in such situation as a multi-label classification problem. In that problem a sample can be classified into several disease terms, and the sum of its label probabilities need not be 1. See Supplementary Text for full details and references. We first analyzed the 13 314 microarray samples. We tested three multi-label classification approaches: (i) Single: learning a classifier for each disease separately, (ii) LP: classification using multiclass algorithms on the label power-set of the training data (22,23) and (iii) BC: Bayesian correction of single-label classifiers (10,27). All three approaches rely on a binary ‘base classifier’. See Supplementary Text for details. We tested support vector machines (SVM) (28,29) and random forest (RF) (30) as the base classifier.
Three sample categories for each disease
The common practice when testing a classifier is to train and evaluate its performance in a binary setting, separating the samples into the cases versus all the rest. However, this is problematic when the data come from many diseases: When classifying one disease, samples that come from other disease studies would typically originate from different tissues, and thus may be easier to separate from the cases based on tissue characteristics, irrespective of the disease. On the other hand, controls in the same study will typically originate from the same tissue, or the same patient, and match the cases in sex and edge distribution. As a result, they would be biologically more similar to the cases and harder to classify. For that reason, for each disease we chose to define three types of samples: (i) positives: patients with the disease; (ii) negatives: control samples originating from the same studies as the positive samples and (iii) background controls (BGCs): all other samples. Thus, a classifier that performs well in separating the positives from the rest may actually provide poor separation of the positives from the negatives (see Supplementary Figure S3).
Cross-validation
We wanted to test the classification quality on samples that are completely unrelated to those used for training, and possibly from different technologies. This would also reduce the risk of batch effects. For this purpose, we used leave-datasets-out cross-validation (LDO-CV): In each cross-validation round 15 complete datasets were put aside, a classifier was learned on the rest of the data and then tested on the left-out datasets. The output of each classifier is a matrix P, where Pij is the probability that sample i has disease j (computed when it was in the left-out test set during the LDO-CV).
Evaluation criteria
For each disease (i.e. taking a specific column of P) we calculated three scores: (i) PN-ROC: the area under the ROC curve (AUC-ROC) comparing positives and negatives (ii), PB-ROC: AUC-ROC comparing the positives to the BGCs and (iii) a meta-analysis statistical significance score, based on Stouffer's method (38), for separation of positives and negatives within datasets (see Supplementary Text), denoted as SMQ (Study-based Meta-analysis Q-value). These three scores provide complementary evaluation criteria.
Comparing classifiers
The performance of the classifiers is shown in Figure 2A. We designated a disease well-classified if its PB-ROC and PN-ROC scores exceeded 0.7 and its SMQ was significant (<0.05). See Supplementary Text for further explanation on the thresholds. Single-SVM and SVM-BC had the highest average PN-ROC (0.69). Notably, all classifiers had high standard deviation across diseases (0.175 < σ < 0.19). As expected, the PB-ROC scores were higher than the PN-ROC scores, indicating that obtaining separation between positives and BGCs is an easier task. Overall, Single-SVM performed second in PN-ROC, only slightly below the best algorithm (BC-SVM), and achieved the highest number of well-classified diseases (24). Moreover, when changing the ROC threshold, the SVM-based algorithms consistently outperformed the rest in terms of the number of well-classified diseases. For these reasons, and since the single-SVM classifier is simpler, we used this classifier in all subsequent analyses.
Figure 2.
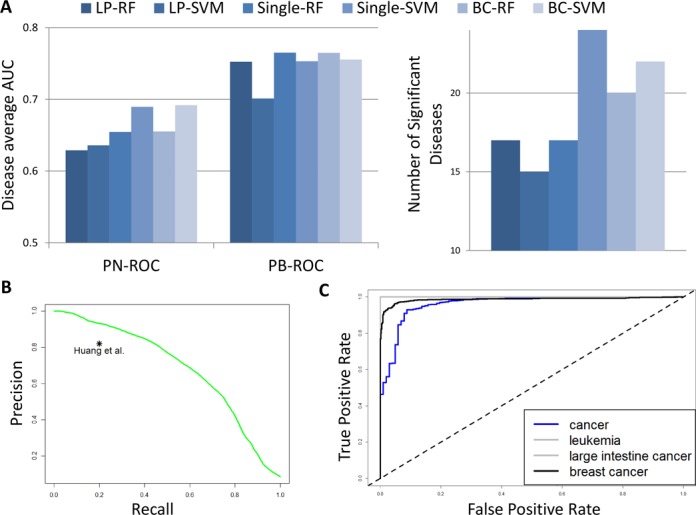
Multi-label classification performance. For each classifier we calculated for each disease the area under the ROC curve comparing positives and negatives (PN-ROC) and the area under the ROC curve comparing positives and background controls (PB-ROC). (A) Average performance of the classifiers. Left: Each bar shows the average ROC-AUC over all diseases. LP: label power-set, BC: Bayesian correction, single – single-class classifier; RF: random forest, SVM: support vector machine. Right: The number of disease terms that had both PN-ROC and PB-ROC at least 0.7 and were found significant in the SMQ test. (B) The global precision-recall curve of the single-SVM classifier. This analysis measures the overall agreement between the predicted probabilities of sample-disease association and the known labels. The point represents the performance of (18). (C) The Single-SVM classifier performance on a test set of 1526 RNA-Seq samples. The ROC score for both leukemia and large intestine cancer is 1.
Comparison to extant algorithms
We calculated a global precision-recall curve, also known as a micro-AUC score in learning (22): we treated P and Y as a set of pairs (Yij, Pij), and used Pij to rank all pairs. This ranking was then used to calculate a precision-recall curve, see Figure 2B. The AUPR was 0.68. Both precision and recall were much higher than in (10): we achieved 93% precision (compared to 82%) at 20% recall, and 44% recall (compared to 20%) at 82% precision (Figure 2B).
Testing classification on samples from a new technology
As an additional validation, we used the 1526 RNA-Seq samples in ADEPTUS. We trained the Single-SVM classifier using all microarray samples and tested its performance on the RNA-Seq samples. The RNA-Seq test data contained 918 breast cancer samples, 102 control breast biopsies, 182 intestinal cancer samples, 173 leukemia samples, and 151 samples from other cancer types. Given the classifier for each disease, we calculated the ROC curve comparing the disease samples to all other RNA-Seq samples, see Figure 2C. All ROC scores were >0.96. Note that in these data only the breast cancer samples had direct negative samples. This can explain why these ROC scores are much higher than those obtained in the cross validation. Nevertheless, our classifier correctly assigned the cancer to the patients even though the samples were from a technology that was not used at all in the training.
The validation confirms that our classifiers perform well across technologies and platforms. Our analysis produced successful classification for 24 diseases, most of which are cancer subtypes (see Figure 3). It may be possible that the other diseases were less well classified due to loss of information in the rank normalization. To test this, for each of those diseases we reran the LDO analysis process above using only samples from one platform, choosing the platform that had the largest number of the disease datasets. Profiles underwent standard quantile normalization, which retains more of the original signal than the weighted ranking needed when combining data across platforms. The results show that classification performance can be improved for some of these diseases by narrowing down the analysis to one platform, see Supplementary Text. For example, analyzing separately six datasets of ‘musculoskeletal system disease’ that used the same platform (GPL96, Affymetrix 133A), the classifier achieved a ROC score of 0.84.
Figure 3.
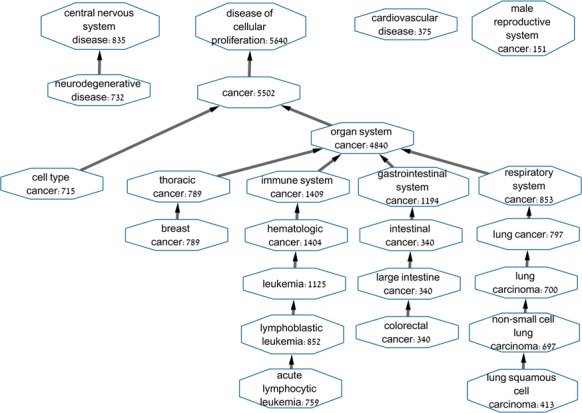
The 24 well-classified diseases. For each node, the Disease Ontology term and the number of positive samples are shown. Edges mark ‘is-a’ relation in the DO hierarchy.
Detecting disease-specific differential genes
In order to identify genes that are specifically differential in a particular disease, we used the three-way partition of the samples for that disease, and calculated for each gene the PN-ROC, PB-ROC, and SMQ scores. Note that here the distance from 0.5 (in either direction) indicates how informative a gene is. For simplicity, for each ROC score x we report here max (x, 1 – x), and indicate whether the gene is up- or down-regulated. Figure 4A and B shows two differential expression patterns. For TP53 in cancer, the positives are up-regulated compared to both negatives and BGCs (PN- and PB-ROC ≥ 0.65, SMQ ≤ 2.22E–10). For IFNGR1 in ALL, IFNG1 is up-regulated in positives compared to negatives (PN-ROC = 0.675, SMQ = 0.001) but down-regulated compared to BGCs (PB-ROC = 0.7). Although the PN-ROC and PB-ROC scores are computed by comparing the positives to two disjoint sample groups, we observed high correlation between them across different diseases (0.46 ± 0.15). For example, in cancer, most differential genes showed the same direction of change in positives versus negatives and in positives versus BGCs, and only a few showed different directions as in Figure 4B.
Figure 4.
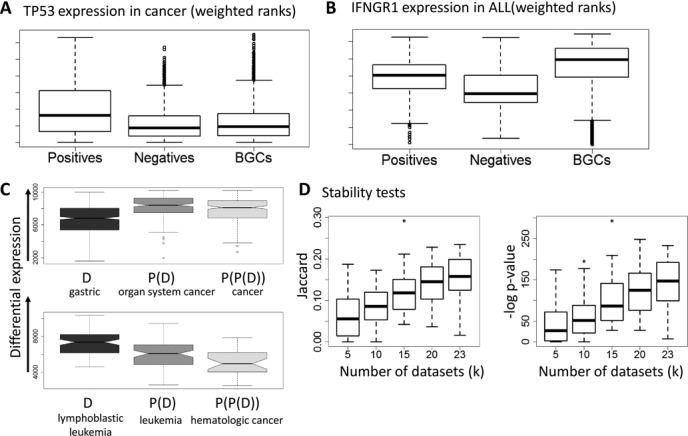
Expression patterns, specificity and robustness. (A) Expression of TP53 in cancer. (B) Expression of IFNGR1 in ALL. The y-axis represents the weighted rank of a gene, where higher ranks have better values. The boxplots show the expression distribution of the three sample cohorts: positives, negatives, and BGCs. TP53 is over-expressed in cancer compared to both negatives and BGCs. IFNGR1 is up-regulated compared to negatives, but down-regulated compared to BGCs. (C) Ranking of gastric cancer biomarker genes and of biomarkers for more general cancers on gastric cancer datasets. Plots for biomarkers of more general diseases (parent and grandparent nodes in the Disease Ontology) are further to the right. General cancer genes are ranked higher, indicating that analysis of these datasets alone will not discover the gastric cancer specific genes. (D) Testing stability. The plots show the overlap between solutions obtained using two disjoint sets of k disease datasets each, as a function of k. Each boxplot shows the distribution of the overlap scores for a specific k over 50 repeats.
We designate a gene as specific to a disease D if both of its ROC scores are ≥0.65 and it has SMQ score ≤0.05. (We chose the ROC threshold more permissively here since some diseases had only few genes with ROC > 0.7). Note that this approach is highly stringent in that we remove genes with a significant q-value whose differential signal is not intense enough. This process produces an initial set of potential genes for D, but can leave high overlap between gene sets of related DO terms. To make sure that a selected gene G is indeed specifically differential in D, D was considered only if it is a leaf or it has at least three datasets whose most specific annotation is D (i.e. samples in them were assigned to D but not to any of its children). In that case we re-calculated the SMQ score using these datasets only. If G was found significant in that test, this indicates that G is differential in D even when we exclude the samples of its sub-diseases. This test markedly reduces the overlap between related DO terms, see Supplementary Text. The resulting disease-specific gene set is designated the disease biomarker. These sets are provided in Supplementary Table S1.
Selection of the biomarker can also be done as part of classifier training. We compared the classification with both gene sets and the results were similar. We preferred determining the biomarker by the procedure described here, as it focuses on genes that are differential and directly addresses the redundancy between related diseases.
Biomarker specificity and robustness
We evaluated the specificity of the disease biomarker sets that we obtained. In each dataset we ranked all the genes by their differential expression score (the difference in the mean rank based score between the cases and the controls) and then computed the median rank of each gene across all the datasets of each disease (see Supplementary Text). Focusing on the datasets of a particular disease, we computed the ranks of its biomarker set, the ranks of the biomarker set of the parent disease, and of the grandparent disease, when available. We expected that a specific biomarker should show higher ranks on its disease data than the biomarker of the more general parent and grandparent disease. The results are summarized in Supplementary Figure S4 and Figure 4C. For most diseases, e.g. lymphoblastic leukemia (Figure 4C), the ranks of the disease gene sets are significantly higher than those of their ancestors. However, in gastrointestinal cancer (Figure 4C), the biomarker sets of the ancestors (organ system cancer and cancer) have much higher ranks (p < 1E–21). Hence, analyzing gastric cancer datasets without expression profiles of BGCs would lead to preferring general cancer genes over genes that are specific to gastric cancer.
A key problem in disease classification has been low overlap between biomarker gene sets obtained in different studies (39). We therefore tested how the stability of our biomarkers depends on the number of datasets used for learning. We focused on the disease term ‘organ system cancer’ because it had a large number of usable datasets (46, of which 16 were not assigned to any sub-disease). We computed biomarker sets twice based on disjoint data, and measured the overlap between the sets. This was repeated with the number of training datasets ranging from 5 to 23 (see Materials and Methods). The results (Figure 4D and Supplementary Figure S5) show that the overlap is highly significant when k > 10. Importantly, stability increases roughly linearly as a function of the number of datasets in the range we could test. We therefore fit a linear regression model to this trend and estimated the required numbers to achieve higher stability, assuming the linear trend continues. At 46 datasets and 4258 samples (all of the 46 datasets available for that disease) the predicted Jaccard score is 0.29 (expected p < 1E–270). Increasing the numbers to 100 datasets and 10 000 samples is expected to improve the Jaccard score to roughly 0.6.
Functional analysis rediscovers known disease factors and suggests novel ones
For each disease, the set of biomarker genes was partitioned by their differential expression compared to negatives and BGCs (compare Figure 4A and B) and each subgroup was tested for functional and pathway enrichment (see Materials and Methods). The results are summarized in Supplementary Table S2. Overall, the results validated our analysis by rediscovering known disease factors. In cancer the enriched biological processes included well known hallmarks of cancer such as cell cycle regulation, DNA replication, P53 signaling, chromosome organization and cell proliferation (40). In neurodegenerative disorders, the results included oxidative phosphorylation, Alzheimer's disease and Parkinson's disease. In lymphoblastic leukemia, primary immunodeficiency was down-regulated both compared to negatives and BGCs, whereas lymphocyte differentiation and V(D)J recombination were up-regulated. In gastrointestinal cancer, several pathways were down-regulated compared to negative samples, including the calcium signaling pathway and fatty acid metabolism. Interestingly, the latter is up-regulated compared to BGCs, indicating that this pathway's expression level in gastrointestinal cancer is reduced but not to the full extent manifested in other unrelated tissue.
We also performed network-based analysis of the identified cancer-specific gene set. This set contained 258 genes, of which 222 were up-regulated in cancer both compared to negatives and to BGCs. Figure 5 shows the two main connected components formed when connecting this gene set with the protein–protein interactions (PPI) from IntAct (41). The first component contains 14 genes including TP53 as the main hub. The second contains 64 genes. Surprisingly, two down-regulated cytoskeleton related genes, NDEL1 and GABARAPL1, connect two up-regulated sub-modules of this connected component. Functional analysis of these two sub-modules revealed that the first is composed of 12 mitosis-related genes (p = 2.5E–22), whereas the second is related to cell cycle and DNA replication (e.g. the MCM complex, p = 1.2E–10). Thus, the mitosis related sub-module is up-regulated but its ability to form physical interactions with cytoskeleton related factors is impaired, which suggests differential rewiring of the replication pathway in cancer. Such cellular modifications might cause instability and mitosis defects through impairment of cellular morphogenesis (42).
Figure 5.
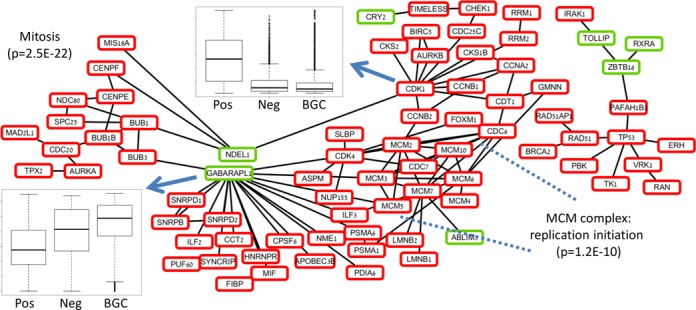
The main connected components of protein-protein interaction network of the cancer-specific differential genes. Up-regulated genes in cases versus negatives and BGCs are in red, down-regulated genes are in green. The large connected component (left) can be separated into two up-regulated sub-modules by removing the down-regulated genes. The down regulated genes are related to cytoskeleton, whereas the sub-modules contain mitosis, replication, and cell cycle genes. The small connected component (right) also contains mainly up-regulated genes, and has TP53 as the main hub.
Integration with information on SNPs and drugs reveals therapeutic potential
In order to interpret our biomarkers, we integrated them with external databases to produce an overview of the molecular changes in a specific cancer and suggest potential consequences to therapy. We used COSMIC (31) for association between genes and cancer types based on occurrence of somatic mutations in coding regions (see Materials and Methods), Drugbank (32) to mark druggable genes, and GeneMania (36) for genetic interactions (GIs) and PPIs between the genes. We tested in detail three examples: lung cancer, ALL, and colorectal cancer. In each case we focused only on genes that (1) were differential in the disease or one of its ancestor DO terms, and (2) either are targets of known drugs or the gene was found associated with the disease in COSMIC.
Lung cancer
Part of the network of lung cancer, containing the two largest connected components in the PPI network, is shown in Figure 6A. The network shows TP53 as a main hub. TP53 and most of its PPI neighbors are differential in cancer but are not specifically differential in lung cancer. Two neighbors of TP53 - TOP2A and HSPA5, however, are up-regulated but are not associated to the disease based on mutations. Interestingly, TOP2A (topoisomerase) is a target of multiple cancer-related inhibitory drugs such as Teniposide, and Valrubicin. In another PPI-based connected component of the network, the hub is DDR1, a key player in communication of cells with their microenvironment (43). It interacts with up-regulated collagen related genes COL5A2, COL11A1 and COL3A1 (44). DDR1, which is not covered by the major pathway databases KEGG (45), Reactome (46), NCI (47) and Biocarta (47), is specifically up-regulated in lung cancer and also associated to lung cancer based on mutations. In addition, this gene is a target of Imatinib, a drug used for treatment of leukemia and gastric cancers (48,49) caused by the bcr-abl1 translocation and by cKit mutations, respectively. In summary, the network highlights two main differential hubs (TP53 and DDR1) and additional connected genes, some of which could be targeted by known cancer drugs.
Figure 6.
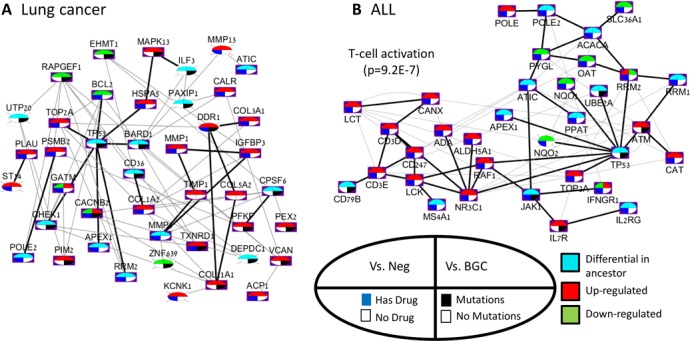
A network overview of the biomarkers in lung cancer and ALL. Each network shows genes that (i) were found differential specifically in the disease or in a more general disease that contains it according to the DO database, and (ii) have a drug targeting them, or were found to be associated with the disease according to the COSMIC database. Black edges are PPIs, and gray edges are GIs. Each node shows four features of a gene: (i) differential pattern compared to negatives, (ii) differential pattern compared to BGCs, (iii) whether a targeting drug exists and (iv) if the gene was associated to lung cancer according to COSMIC. Nodes without a purple background are genes that are not associated with any pathway in KEGG, Reactome, NCI, or Biocarta. (A) Lung cancer. The initial network (top left) contained 89 genes. The two largest connected components in the PPI network are shown. The GeneMANIA analysis added COL5A2 and TMP1 to the network. (B) ALL. The original network contained 136 genes and 424 edges. The figure focuses on the largest PPI connected component.
ALL
In the ALL network (Figure 6B), the largest PPI-based connected component has TP53 as a hub, connected to genes that are specifically up-regulated in ALL such as ATM and TOP2A. An up-regulated sub-module of the network is enriched with to T-cell activation genes (p = 9.2E−7), which were not found to be associated with leukemia according to COSMIC. However, some of the genes are targets of well known drugs of leukemia sub-diseases, such as ADA (Pentostatin, inhibition - lymphoproliferative malignancies) and LCK (Dasatinib and Ponatinib - chronic myeloid leukemia, ALL) (50–53). Interestingly, NR3C1, a glucocorticoid receptor transcription factor that promotes inflammatory responses, has high degree and is also connected to TP53. This gene is a target of 39 drugs, including both agonists and antagonists (32). In summary, the network reveals two main functional areas in the PPI network: the module surrounding the TP53 hub, and the T-cell sub-module. Both are differential in the disease. In addition, the network captures known related genes and treatments.
Colorectal cancer
As the initial network was large (see Supplementary Table S3) we focused only on up-regulated genes with PN-ROC > 0.8 (Supplementary Figure S6). The result was 27 genes interconnected by 30 GIs, and only one PPI. All GIs were from (54), representing gene pairs that are expected to share similar biological functions (55). The network is enriched with genes related to detection of mechanical stimulus (p = 2.11E−6). JUN, the main hub, is related to angiogenesis and to positive regulation of endothelial cell development. The network also contains three druggable genes associated with intestinal cancer based on the mutation data: SLC12A2, GABBR1, and CACNA1D. Interestingly, the drugs that target these genes are not known cancer drugs. For example, CACNA1D is a target of 13 inhibitory drugs related mainly to hypertension treatment (e.g. Felodipine, Israpidine) (56). In summary, our results suggest an up-regulated gene module in colorectal cancer and a possible link between colorectal cancer and other factors related to hypertension and psychological stress.
DISCUSSION
In this study, we present a novel approach for producing reliable disease-specific biomarkers that are readily interpretable, especially in terms of their clinical potential. To be able to do this, we first compiled and manually curated a very large collection of gene expression profiles spanning many studies from multiple diseases, called ADEPTUS. Each sample was normalized separately based on its weighted ranks, in order to allow joint analysis of samples from different technologies and studies, at the expense of some loss of information. Importantly, it also allows the use of a biomarker to classify a single new sample. Future studies could apply other non-parametric approaches that process the raw expression data and do not preserve the measured gene ranking, e.g. Barcode (57) or SCAN (58). ADEPTUS can be readily used to test novel multi-label classification algorithms, and it can be utilized alongside other data (expression or other) in future studies.
We utilized the compendium for improved disease classification. In contrast to previous studies, in our analysis the simple single-classifier approach outperformed more sophisticated methods. A possible explanation is that our analysis used fewer labels compared to other studies (since we only addressed diseases with at least five datasets), and therefore had fewer dependencies among them.
A key insight of our study is the risk of misleadingly optimistic performance when classifying multi-disease data. We showed that one must treat the non-disease samples as two distinct categories: negatives (non-disease samples from studies of the same disease) and background controls (samples from studies of other diseases), and evaluate the performance against each subgroup separately. The good classification results validated the approach and the data quality and allowed us to focus subsequent analyses on well-classified diseases. Our method reached substantially higher classification performance than (10) (e.g. 22% improvement in recall). However, performance is not directly comparable because in (10) fewer samples were used, and samples were limited to just two microarray platforms, the classifier did not predict the control class, and more diseases were tested.
Having identified 24 well-classified diseases, we set out to identify disease-specific genes in each of them using the DO structure, the three-way partition of the samples, and meta-analysis significance. This analysis reduced the overlap between gene sets of related diseases. Reassuringly, the discovered gene sets included established disease factors. While we focused on disease-specific genes, future studies could potentially use our database to search for genes with a similar expression pattern across different cancer types.
The issue of robustness in disease biomarker discovery has been troubling the community for quite some time (59–62). It has two aspects: good predictive power when biomarkers from one study are tested on a different cohort from an independent study, and reproducibility of the same biomarker gene set in independent studies. While the predictive power has been typically high, reproducibility remains low. Domany and colleagues estimated that for breast cancer prognosis prediction, thousands of samples will be needed in order to achieve 50% overlap between two such sets (39). Our study sheds additional light on this issue. It shows that reproducibility of the detected biomarkers improves as the number of disease datasets and samples in the training set grows. When the number of datasets available for a disease is at least 10, our analysis produces biomarker sets that are significantly overlapping on disjoint subsets of the data. Using the whole compendium, the expected Jaccard score for overlap is 0.3 (p < E−250) for the most represented disease category. In fact, with over 4200 samples for the organ systems cancer category, robustness is less than predicted by the model of (39). This can be attributed in part to factors that were not taken into account in that model, e.g. batch effects of different studies and technologies. Overall, our results imply that in order to further improve robustness and reproducibility, future studies should aim to increase the number of datasets and samples, while making judicious use of data on other diseases to guarantee specificity.
The final step of our approach involved integration of our results with information from external databases: somatic mutations in cancer, drug–gene associations, and protein interactions. For each tested disease, we summarized all this information and our results in a network. These networks provide a bird's eye view of the disease-specific genes, their relations and properties, and thus point to new therapeutic potential. Such an overview can serve as a starting point for considering novel therapeutics, such as drug repositioning that exploits approved genes for new treatments, or multi-drug treatments, in which several drugs are used to target different aspects of the biological network.
While our approach is effective, it has several limitations that future studies can address. We tested only 48 diseases since we included in the compendium only diseases that had at least five datasets with at least 20 samples each, in order to allow reliable cross validation on whole datasets. In addition, we analyzed only ∼15 000 gene expression profiles, a modest fraction of the human profiles in GEO, since we required manual curation of the disease terms for each profile (automatic curation had unsatisfactory quality). We view our work as a proof of concept: with some more effort of a team of curators, all available large databases can be curated and the same methodology can be applied for their analysis. Second, our multi-platform integration proved beneficial for half of the tested diseases, and most well-classified diseases were related to cancer. Nevertheless, neurodegenerative disorders and cardiovascular disease were well classified as well. In addition, we showed that narrowing down the analysis to a single platform can improve the performance in other disease terms. The low performance in some of the diseases could be due to several reasons: (i) low number of non-cancer datasets, (ii) integration of a large number of platforms, (iii) limitations of using methods that rank genes by their expression levels, (iv) inexistence of gene expression based robust classifier and (v) the tested disease might be too broadly defined (e.g. ‘disease of anatomical entity’).
AVAILABILITY
The data used in this study and the R code developed are available at http://acgt.cs.tau.ac.il/adeptus, together with a tutorial for using them.
Supplementary Material
Acknowledgments
The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Author Contribution: D.A. and R.S. conceived the study. D.A. and T.H. assembled the database. D.A., T.H., S.I. and R.S. analyzed the data. D.A., S.I. and R.S. wrote the paper.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Israel Science Foundation [317/13]; IDEA grant from the Dotan Center in Hemato-Oncology; D.A. is grateful to the Azrieli Foundation for the award of an Azrieli Fellowship; Edmond J. Safra Center for Bioinformatics at Tel Aviv University (to D.A.); Israeli Center of Research Excellence (I-CORE), Gene Regulation in Complex Human Disease, Center No. 41/11. Funding for open access charge: Israeli Center of Research Excellence (I-CORE), Gene Regulation in Complex Human Disease, Center No. 41/11; IDEA grant from the Dotan Center in Hemato-Oncology.
Conflict of interest statement. None declared.
REFERENCES
- 1.Lavi O., Dror G., Shamir R. Network-induced classification kernels for gene expression profile analysis. J. Comput. Biol. 2012;19:694–709. doi: 10.1089/cmb.2012.0065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Yang X., Regan K., Huang Y., Zhang Q., Li J., Seiwert T.Y., Cohen E.E., Xing H.R., Lussier Y.A. Single sample expression-anchored mechanisms predict survival in head and neck cancer. PLoS Comput. Biol. 2012;8:e1002350. doi: 10.1371/journal.pcbi.1002350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lee E., Chuang H.Y., Kim J.W., Ideker T., Lee D. Inferring pathway activity toward precise disease classification. PLoS Computat. Biol. 2008;4:e1000217. doi: 10.1371/journal.pcbi.1000217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Altschuler G.M., Hofmann O., Kalatskaya I., Payne R., Sui S.J.H., Saxena U., Krivtsov A.V., Armstrong S.A., Cai T.X., Stein L., et al. Pathprinting: An integrative approach to understand the functional basis of disease. Genome Med. 2013;5:68. doi: 10.1186/gm472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Xu M., Kao M.C.J., Nunez-Iglesias J., Nevins J.R., West M., Zhou X.J. An integrative approach to characterize disease-specific pathways and their coordination: a case study in cancer. BMC Genomics. 2008;9(Suppl. 1):S12. doi: 10.1186/1471-2164-9-S1-S12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ulitsky I., Krishnamurthy A., Karp R.M., Shamir R. DEGAS de novo discovery of dysregulated pathways in human diseases. PLoS One. 2010;5:e13367. doi: 10.1371/journal.pone.0013367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ideker T., Sharan R. Protein networks in disease. Genome Res. 2008;18:644–652. doi: 10.1101/gr.071852.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rung J., Brazma A. Reuse of public genome-wide gene expression data. Nat. Rev. Genet. 2013;14:89–99. doi: 10.1038/nrg3394. [DOI] [PubMed] [Google Scholar]
- 9.Ioannidis J.P., Allison D.B., Ball C.A., Coulibaly I., Cui X., Culhane A.C., Falchi M., Furlanello C., Game L., Jurman G., et al. Repeatability of published microarray gene expression analyses. Nat. Genet. 2009;41:149–155. doi: 10.1038/ng.295. [DOI] [PubMed] [Google Scholar]
- 10.Huang H., Liu C.C., Zhou X.J. Bayesian approach to transforming public gene expression repositories into disease diagnosis databases. Proc. Natl. Acad. Sci. U.S.A. 2010;107:6823–6828. doi: 10.1073/pnas.0912043107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bodenreider O., Nelson S.J., Hole W.T., Chang H.F. Beyond synonymy: exploiting the UMLS semantics in mapping vocabularies. Proc. Annu. Symp. AMIA. 1998:815–819. [PMC free article] [PubMed] [Google Scholar]
- 12.Schriml L.M., Arze C., Nadendla S., Chang Y.W., Mazaitis M., Felix V., Feng G., Kibbe W.A. Disease Ontology: a backbone for disease semantic integration. Nucleic Acids Res. 2012;40:D940–D946. doi: 10.1093/nar/gkr972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schmid P.R., Palmer N.P., Kohane I.S., Berger B. Making sense out of massive data by going beyond differential expression. Proc. Natl. Acad. Sci. U.S.A. 2012;109:5594–5599. doi: 10.1073/pnas.1118792109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lee Y.S., Krishnan A., Zhu Q., Troyanskaya O.G. Ontology-aware classification of tissue and cell-type signals in gene expression profiles across platforms and technologies. Bioinformatics. 2013;29:3036–3044. doi: 10.1093/bioinformatics/btt529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Papachristoudis G., Diplaris S., Mitkas P.A. SoFoCles: feature filtering for microarray classification based on gene ontology. J. Biomed. Inform. 2010;43:1–14. doi: 10.1016/j.jbi.2009.06.002. [DOI] [PubMed] [Google Scholar]
- 16.Subramanian A., Tamayo P., Mootha V.K., Mukherjee S., Ebert B.L., Gillette M.A., Paulovich A., Pomeroy S.L., Golub T.R., Lander E.S., et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. U.S.A. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Staiger C., Cadot S., Gyorffy B., Wessels L.F., Klau G.W. Current composite-feature classification methods do not outperform simple single-genes classifiers in breast cancer prognosis. Front. Genet. 2013;4:289. doi: 10.3389/fgene.2013.00289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ciriello G., Miller M.L., Aksoy B.A., Senbabaoglu Y., Schultz N., Sander C. Emerging landscape of oncogenic signatures across human cancers. Nat. Genet. 2013;45 doi: 10.1038/ng.2762. 1127-U1247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Parkinson H., Kapushesky M., Shojatalab M., Abeygunawardena N., Coulson R., Farne A., Holloway E., Kolesnykov N., Lilja P., Lukk M., et al. ArrayExpress - a public database of microarray experiments and gene expression profiles. Nucleic Acids Res. 2007;35:D747–D750. doi: 10.1093/nar/gkl995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cancer Genome Atlas Research Network. Weinstein J.N., Collisson E.A., Mills G.B., Shaw K.R., Ozenberger B.A., Ellrott K., Shmulevich I., Sander C., Stuart J.M. The Cancer Genome Atlas Pan-Cancer analysis project. Nat. Genet. 2013;45:1113–1120. doi: 10.1038/ng.2764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yang X., Bentink S., Scheid S., Spang R. Similarities of ordered gene lists. J. Bioinform. Computat. Biol. 2006;4:693–708. doi: 10.1142/s0219720006002120. [DOI] [PubMed] [Google Scholar]
- 22.Zhang M.L., Zhou Z.H. A review on multi-label learning algorithms. IEEE Trans. Knowl. Data En. 2014;26:1819–1837. [Google Scholar]
- 23.Tsoumakas G., Spyromitros-Xioufis E., Vilcek J., Vlahavas I. MULAN: a java library for multi-label learning. J. Mach. Learn. Res. 2011;12:2411–2414. [Google Scholar]
- 24.Madjarov G., Kocev D., Gjorgjevikj D., Dzeroski S. An extensive experimental comparison of methods for multi-label learning. Pattern Recognit. 2012;45:3084–3104. [Google Scholar]
- 25.Vens C., Struyf J., Schietgat L., Dzeroski S., Blockeel H. Decision trees for hierarchical multi-label classification. Mach. Learn. 2008;73:185–214. [Google Scholar]
- 26.Sucar L.E., Bielza C., Morales E.F., Hernandez-Leal P., Zaragoza J.H., Larranaga P. Multi-label classification with Bayesian network-based chain classifiers. Pattern Recognit. Lett. 2014;41:14–22. [Google Scholar]
- 27.Barutcuoglu Z., Schapire R.E., Troyanskaya O.G. Hierarchical multi-label prediction of gene function. Bioinformatics. 2006;22:830–836. doi: 10.1093/bioinformatics/btk048. [DOI] [PubMed] [Google Scholar]
- 28.Scholkopf B.S.P., Smola A., Vapnik V. Prior knowledge in support vector kernels. Advances in Neural information processings systems. 1998;10:640–646. [Google Scholar]
- 29.Vapnik V.N. Statistical Learning Theory. NY: Wiley; 1998. [DOI] [PubMed] [Google Scholar]
- 30.Breiman L. Random forests. Mach. Learn. 2001;45:5–32. [Google Scholar]
- 31.Forbes S.A., Bindal N., Bamford S., Cole C., Kok C.Y., Beare D., Jia M.M., Shepherd R., Leung K., Menzies A., et al. COSMIC: mining complete cancer genomes in the Catalogue of Somatic Mutations in Cancer. Nucleic Acids Res. 2011;39:D945–D950. doi: 10.1093/nar/gkq929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Law V., Knox C., Djoumbou Y., Jewison T., Guo A.C., Liu Y.F., Maciejewski A., Arndt D., Wilson M., Neveu V., et al. DrugBank 4.0: shedding new light on drug metabolism. Nucleic Acids Res. 2014;42:D1091–D1097. doi: 10.1093/nar/gkt1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Smoot M.E., Ono K., Ruscheinski J., Wang P.L., Ideker T. Cytoscape 2.8: new features for data integration and network visualization. Bioinformatics. 2011;27:431–432. doi: 10.1093/bioinformatics/btq675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Morris J.H., Kuchinsky A., Ferrin T.E., Pico A.R. enhancedGraphics: a Cytoscape app for enhanced node graphics. F1000Research. 2014;3:147. doi: 10.12688/f1000research.4460.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Maere S., Heymans K., Kuiper M. BiNGO: a Cytoscape plugin to assess overrepresentation of gene ontology categories in biological networks. Bioinformatics. 2005;21:3448–3449. doi: 10.1093/bioinformatics/bti551. [DOI] [PubMed] [Google Scholar]
- 36.Montojo J., Zuberi K., Rodriguez H., Kazi F., Wright G., Donaldson S.L., Morris Q., Bader G.D. GeneMANIA Cytoscape plugin: fast gene function predictions on the desktop. Bioinformatics. 2010;26:2927–2928. doi: 10.1093/bioinformatics/btq562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shamir R., Ulitsky I., Maron-Katz A., Shavit S., Sagir D., Linhart C., Elkon R., Tanay A., Sharan R., Shiloh Y. Expander: from expression microarrays to networks and functions. Nature Protoc. 2010;5:303–322. doi: 10.1038/nprot.2009.230. [DOI] [PubMed] [Google Scholar]
- 38.Hedges L.V., Olkin I. Statistical Methods for Meta-analysis. Orlando: Academic Press; 1985. [Google Scholar]
- 39.Ein-Dor L., Zuk O., Domany E. Thousands of samples are needed to generate a robust gene list for predicting outcome in cancer. Proc. Natl. Acad. Sci. U.S.A. 2006;103:5923–5928. doi: 10.1073/pnas.0601231103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hanahan D., Weinberg R.A. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 41.Kerrien S., Alam-Faruque Y., Aranda B., Bancarz I., Bridge A., Derow C., Dimmer E., Feuermann M., Friedrichsen A., Huntley R., et al. IntAct–open source resource for molecular interaction data. Nucleic Acids Res. 2007;35:D561–D565. doi: 10.1093/nar/gkl958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hall A. The cytoskeleton and cancer. Cancer Metast. Rev. 2009;28:5–14. doi: 10.1007/s10555-008-9166-3. [DOI] [PubMed] [Google Scholar]
- 43.Hilton H.N., Stanford P.M., Harris J., Oakes S.R., Kaplan W., Daly R.J., Ormandy C.J. KIBRA interacts with discoidin domain receptor 1 to modulate collagen-induced signalling. BBA-Mol. Cell Res. 2008;1783:383–393. doi: 10.1016/j.bbamcr.2007.12.007. [DOI] [PubMed] [Google Scholar]
- 44.Xu H., Raynal N., Stathopoulos S., Myllyharju J., Farndale R.W., Leitinger B. Collagen binding specificity of the discoidin domain receptors: binding sites on collagens II and III and molecular determinants for collagen IV recognition by DDR1. Matrix Biol. 2011;30:16–26. doi: 10.1016/j.matbio.2010.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kanehisa M., Goto S. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 2000;28:27–30. doi: 10.1093/nar/28.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Croft D., O'Kelly G., Wu G., Haw R., Gillespie M., Matthews L., Caudy M., Garapati P., Gopinath G., Jassal B., et al. Reactome: a database of reactions, pathways and biological processes. Nucleic Acids Res. 2011;39:D691–D697. doi: 10.1093/nar/gkq1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Schaefer C.F., Anthony K., Krupa S., Buchoff J., Day M., Hannay T., Buetow K.H. PID: the Pathway Interaction Database. Nucleic Acids Res. 2009;37:D674–D679. doi: 10.1093/nar/gkn653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Benjamin R.S., Blanke C.D., Blay J.Y., Bonvalot S., Eisenberg B. Management of gastrointestinal stromal tumors in the imatinib era: Selected case studies. Oncologist. 2006;11:9–20. doi: 10.1634/theoncologist.11-1-9. [DOI] [PubMed] [Google Scholar]
- 49.Vigneri P., Wang J.Y.J. Induction of apoptosis in chronic myelogenous leukemia cells through nuclear entrapment of BCR-ABL tyrosine kinase. Nat. Med. 2001;7:228–234. doi: 10.1038/84683. [DOI] [PubMed] [Google Scholar]
- 50.Chen X., Ji Z.L., Chen Y.Z. TTD: Therapeutic Target Database. Nucleic Acids Res. 2002;30:412–415. doi: 10.1093/nar/30.1.412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhu F., Han B.C., Kumar P., Liu X.H., Ma X.H., Wei X.N., Huang L., Guo Y.F., Han L.Y., Zheng C.J., et al. Update of TTD: Therapeutic Target Database. Nucleic Acids Res. 2010;38:D787–D791. doi: 10.1093/nar/gkp1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lindauer M., Hochhaus A. Dasatinib. Rec. Results Cancer Res. 2010;184:83–102. doi: 10.1007/978-3-642-01222-8_7. [DOI] [PubMed] [Google Scholar]
- 53.Jackson R.C., Leopold W.R., Ross D.A. The biochemical pharmacology of (2′-R)-chloropentostatin, a novel inhibitor of adenosine-deaminase. Adv. Enzyme Regul. 1986;25:125–139. doi: 10.1016/0065-2571(86)90011-7. [DOI] [PubMed] [Google Scholar]
- 54.Lin A., Wang R.T., Ahn S., Park C.C., Smith D.J. A genome-wide map of human genetic interactions inferred from radiation hybrid genotypes. Genome Res. 2010;20:1122–1132. doi: 10.1101/gr.104216.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Baryshnikova A., Costanzo M., Kim Y., Ding H.M., Koh J., Toufighi K., Youn J.Y., Ou J.W., San Luis B.J., Bandyopadhyay S., et al. Quantitative analysis of fitness and genetic interactions in yeast on a genome scale. Nat. Methods. 2010;7 doi: 10.1038/nmeth.1534. 1017-U1110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sinnegger-Brauns M.J., Huber I.G., Koschak A., Wild C., Obermair G.J., Einzinger U., Hoda J.C., Sartori S.B., Striessnig J. Expression and 1,4-dihydropyridine-binding properties of brain L-type calcium channel isoforms. Mol. Pharmacol. 2009;75:407–414. doi: 10.1124/mol.108.049981. [DOI] [PubMed] [Google Scholar]
- 57.McCall M.N., Jaffee H.A., Zelisko S.J., Sinha N., Hooiveld G., Irizarry R.A., Zilliox M.J. The Gene Expression Barcode 3.0: improved data processing and mining tools. Nucleic Acids Res. 2014;42:D938–D943. doi: 10.1093/nar/gkt1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Piccolo S.R., Withers M.R., Francis O.E., Bild A.H., Johnson W.E. Multiplatform single-sample estimates of transcriptional activation. Proc. Natl. Acad. Sci. U.S.A. 2013;110:17778–17783. doi: 10.1073/pnas.1305823110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Chuang H.Y., Lee E., Liu Y.T., Lee D., Ideker T. Network-based classification of breast cancer metastasis. Mol. Syst. Biol. 2007;3:140. doi: 10.1038/msb4100180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kim K., Zakharkin S.O., Allison D.B. Expectations, validity, and reality in gene expression profiling. J. Clin. Epidemiol. 2010;63:950–959. doi: 10.1016/j.jclinepi.2010.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ein-Dor L., Kela I., Getz G., Givol D., Domany E. Outcome signature genes in breast cancer: is there a unique set? Bioinformatics. 2005;21:171–178. doi: 10.1093/bioinformatics/bth469. [DOI] [PubMed] [Google Scholar]
- 62.Michiels S., Koscielny S., Hill C. Prediction of cancer outcome with microarrays: a multiple random validation strategy. Lancet. 2005;365:488–492. doi: 10.1016/S0140-6736(05)17866-0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.