

Abstract
The nosology of bullous lesions or equivalents (vesicles, erosions, and crusts) in patients with lupus erythematosus (LE) is rarely addressed.
The primary aim of this study was to draw up a precise phenotypic inventory of such skin lesions; the secondary objective was to assess a potential relationship between the different types of loss of epidermis and extracutaneous lupus manifestations.
We conducted a retrospective multicenter study including 22 patients with definite LE and bullous lesions or equivalents. All biopsies were reviewed. Patients were recruited in the dermatology departments of 6 centers. Patients were included if they met the diagnosis of systemic LE according to American College of Rheumatology and/or Systemic Lupus International Collaborating Clinics criteria or diagnosis of cutaneous LE based on classic clinical criteria and/or histological ascertainment of LE. Patients were recruited through clinician's memory and photographic collections.
Three clinico-pathological patterns could be individualized. First, toxic epidermal necrolysis (TEN)-like, sheet-like, skin detachment; sun-exposure, mild mucosal involvement, and dermal mucin deposition allow differential diagnosis with classical Lyell syndrome. Second, vesiculo-bullae and/or crusting occurring on typical lesions of subacute cutaneous lupus erythematosus or chronic cutaneous lupus erythematosus. Third, tense vesicles and/or blisters with an underlying neutrophilic dermatosis and a usual response to dapsone.
A careful analysis of 22 LE patients with epidermal detachment reveals 2 main pathomechanisms: a classic LE interface dermatitis, which can be hyperacute and lead to TEN-like skin detachment; and a neutrophilic dermatosis, with tense vesicles and/or blisters, including classic bullous LE.
INTRODUCTION
To date, the nosology of bullous lesions during lupus erythematosus (LE) remains poorly defined and often confusing.1,2 During the course of LE, bullous cutaneous lesions or equivalents, including vesicles, erosions, and/or crusts, can occur. Different pathogenetic mechanisms underlie the formation of such lesions, which can occur in heterogeneous groups of cutaneous lupus subtypes. However, their exact frequency in patients with LE is unknown, and most series devoted to cutaneous LE do not even mention them.3–8 If bullous systemic LE (SLE) has been the subject of numerous publications,9–15 bullous lesions or equivalents occurring on specific lesions of LE are less studied. Therefore, LE presenting as toxic epidermal necrolysis (TEN) was the subject of some publications, 16–18,23 but it is probably still largely underdiagnosed. A classification of vesiculobullous lesions in LE was published in 2004 by Ting et al.18 He divided the various types of vesicular or bullous lesions that can be encountered in patients with LE into those that have or do not have LE-specific pathology.
The aim of this study was to clarify clinical, histological, and immunopathological features of bullous skin lesions or any other form of loss of epidermis in a series of 22 patients with LE. Patients with LE and any form of skin detachment—vesicles, bullae, erosions, and crusts—were included in order to make a precise phenotypic inventory and better assess the pathogenesis of such skin lesions. Pragmatically, these lesions will be grouped under the term “loss of epidermis.” Another objective was to identify whether a relationship exists between the different types of loss of epidermis and extracutaneous lupus manifestations.
METHODS
We conducted a descriptive retrospective multicenter study on 22 patients who had developed vesicles, bullae, erosions, or crusts in the course of LE. Under French law, this type of retrospective study does not need approval of an institutional review board. Patients were recruited in the dermatology departments of 2 secondary referral centers (Pointe-à-Pitre and Colmar) and 4 tertiary referral centers (Lyon, Montpellier, Paris, and Strasbourg) in France.
Patients were included if they met the following criteria:
Diagnosis of SLE according to American College of Rheumatology (ACR) and/or Systemic Lupus International Collaborating Clinics (SLICC) criteria or diagnosis of cutaneous LE based on classic clinical criteria and/or histological ascertainment of LE.
Loss of epidermis as a direct consequence of LE except for those lesions resulting from a lupus-related thrombotic vasculopathy or the presence of antiphospholipid antibodies or porphyria cutanea tarda.
Patients’ recruitment was based on clinicians’ memory and/or review of photographic collections (from 1985 to 2012). In all patients, medical records were reviewed and relevant clinical data including age, sex, duration, distribution and morphology of skin lesions, history of LE, serologic data, medications at the time of diagnosis, and response to treatment were recorded. All biopsies were reviewed by 2 of us (CM-D and DL). Mean duration of follow-up time was 5 years (2 months–25 years).
RESULTS
The files of 22 patients with loss of epidermis in the course of LE were reviewed. Two of them have been reported previously.19,20
Clinical Findings
There were 16 women and 6 men. The average age for the onset of bullous or equivalent lesions was 52 years (7–79 years). The average age for the diagnosis of LE was 46 years. Bullous or equivalent lesions were the presenting manifestation LE in 9 of 22 patients. The individual lesions were flaccid blisters or vesicles, sheet-like detachment, erosions, crusts, tense blisters, or vesicles. Lesions were photodistributed in 7 of 22 patients. Mucous membrane involvement was seen in 7 of 22 patients (genital in 2 patients, oral in 6 patients, and conjonctival in 1 patient). Concomitant-specific LE lesions of either acute, subacute, or chronic type were seen in 14 patients. Thirteen of 22 patients had 4 or >4 ACR criteria and 17 had 4 or >4 SLICC criteria.
Histopathological Findings
The histopathological findings of these lesions were highly variable. Besides the typical lupus interface dermatitis, some patients had extensive epidermal necrosis, whereas others had a neutrophilic dermatosis. Fourteen of 22 patients had a typical interface dermatitis with varying degrees of vacuolar changes, basal cell necrosis, basement membrane thickening, epidermal atrophy, superficial and deep dermal lymphocytic infiltrate, and mucin deposition. Extensive necrosis of the epidermis was observed in 5 of 22 patients. Eight of 22 patients had a neutrophilic dermal infiltrate, forming papillary microabscesses in some patients, occupying all the upper dermis or even the whole dermis with important interstitial spreading in other patients, sometimes with leucocytoclasia but without vasculitis. Thus, patients could be grouped into 3 profiles shown in Table 1, Table 2, and Table 3.
TABLE 1.
Toxic Epidermal Necrolysis-Like Cutaneous Lupus Erythematosus

TABLE 2.
Vesiculobullous Annular Subacute Cutaneous Lupus Erythematosus and Vesiculobullous Chronic Cutaneous Lupus Erythematosus
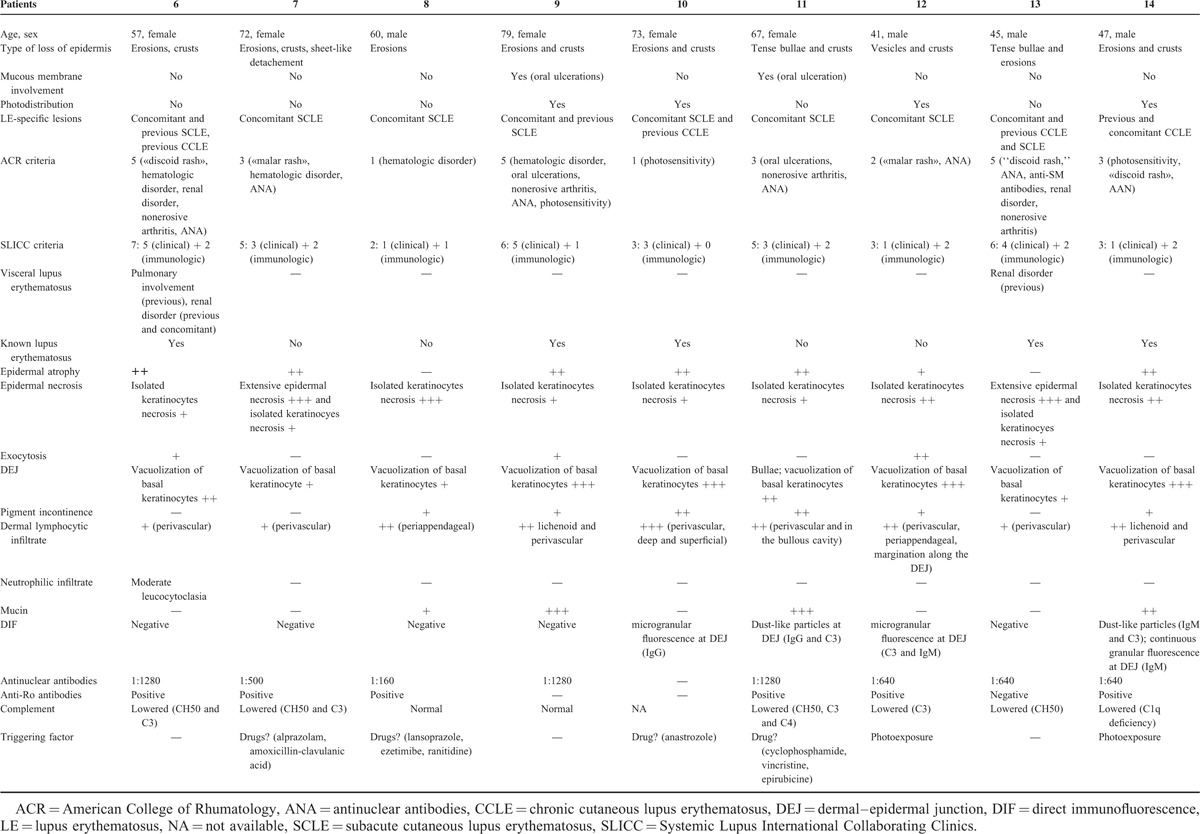
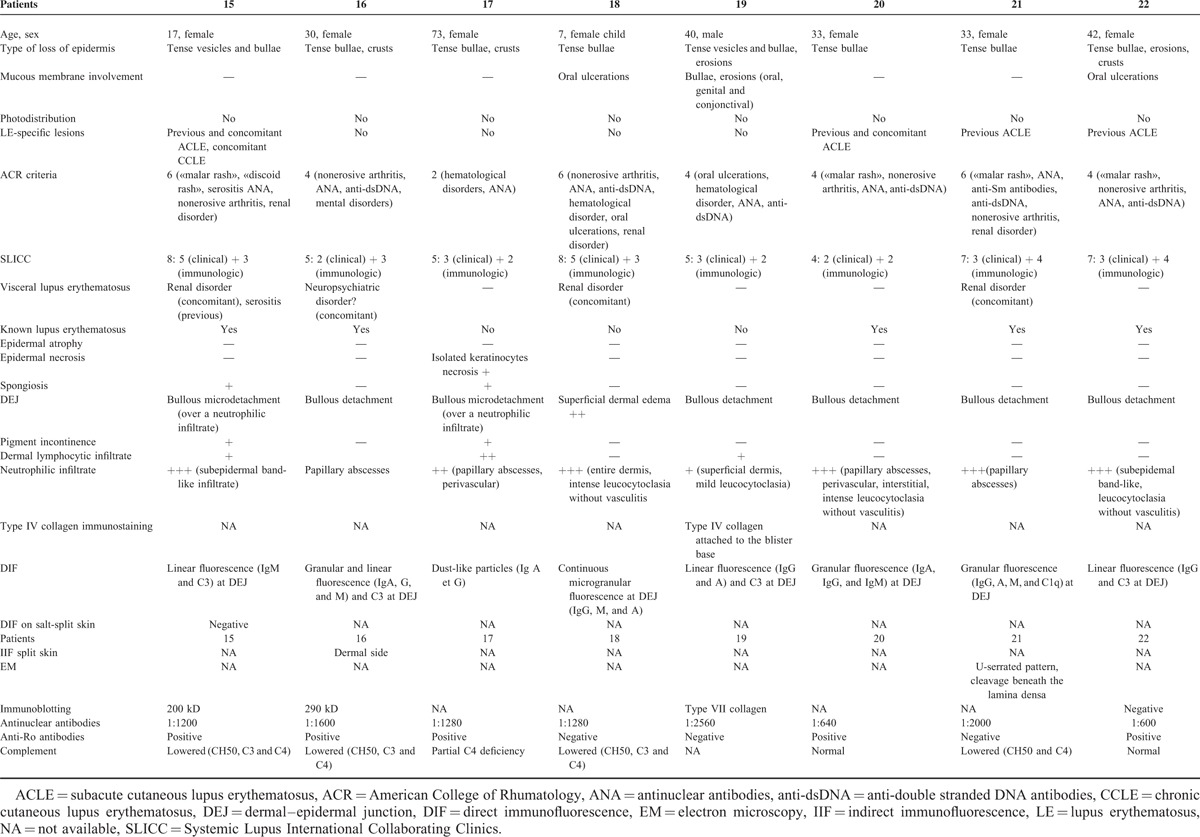
TABLE 3.
Bullous Neutrophilic Lupus Erythematosus
Group 1: TEN-Like LE
In the first group composed of 5 patients, the following common clinical features were observed: flaccid blisters (4/5), sheet-like detachments (4/5), erosions (5/5), and tense blisters (1/5). There was mild vulvar mucosal involvement in 1 patient and cheilitis in 2 of 5 patients. Lesions began on sun-exposed skin or were photodistributed in 4 of 5 patients. Only one of them had a history of previous subacute cutaneous lupus erythematosus (SCLE). Three of 5 patients met ACR criteria for SLE, and 4 of them met SLICC criteria. In 2 of 5 patients, no triggering drug was found. In 3 of 5 patients, sheet-like detachments were preceded by drug intake. These drugs were not classically associated with TEN (diacereine, sulbutiamin, docetaxel, cyclophosphamide, and trastuzumab) (Table 1, Figure 1).
FIGURE 1.
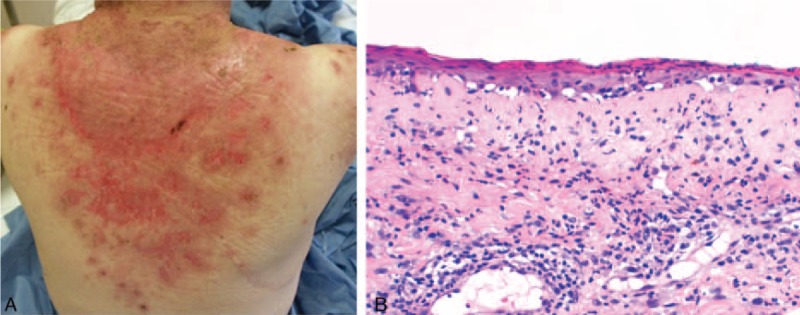
A case of TEN-like LE—Patient 1: (A) photodistributed erosions and crusts; (b) epidermal atrophy and vacuolization of the basal layer. TEN = toxic epidermal necrolysis.
At histological examination, there was an extensive epidermal necrosis in 4 of 5 patients. Basal vacuolization and isolated keratinocytes necrosis were seen in all patients and mucin deposition in 3 patients. Direct immunofluorescence revealed a granular fluorescence at dermal–epidermal junction (DEJ) in 2 of 5 patients, with IgG, IgM, and C3. These patients can be classified as having a TEN-like LE.
Group 2: Classic Cutaneous LE (Interface Dermatitis) With Loss of Epidermis
In the second group composed of 9 patients, the following common clinical features were observed: erosions (7/9), crusts (7/9), localized sheet-like detachment (1/9), tense bullae (2/9), and/or vesicles (1/9). These losses of epidermis arose on LE-specific lesions (SCLE in 8/9 and chronic cutaneous lupus erythematosus [CCLE] in 1/9). There was no mucosal involvement in 7 of 9 patients. Two had oral ulcerations. Lesions were photodistributed in 3 of 9 patients. Five of them had a history of cutaneous LE. Three patients of 9 met ACR criteria for SLE, and 5 of 9 met SLICC criteria (Table 2, Figure 2).
FIGURE 2.
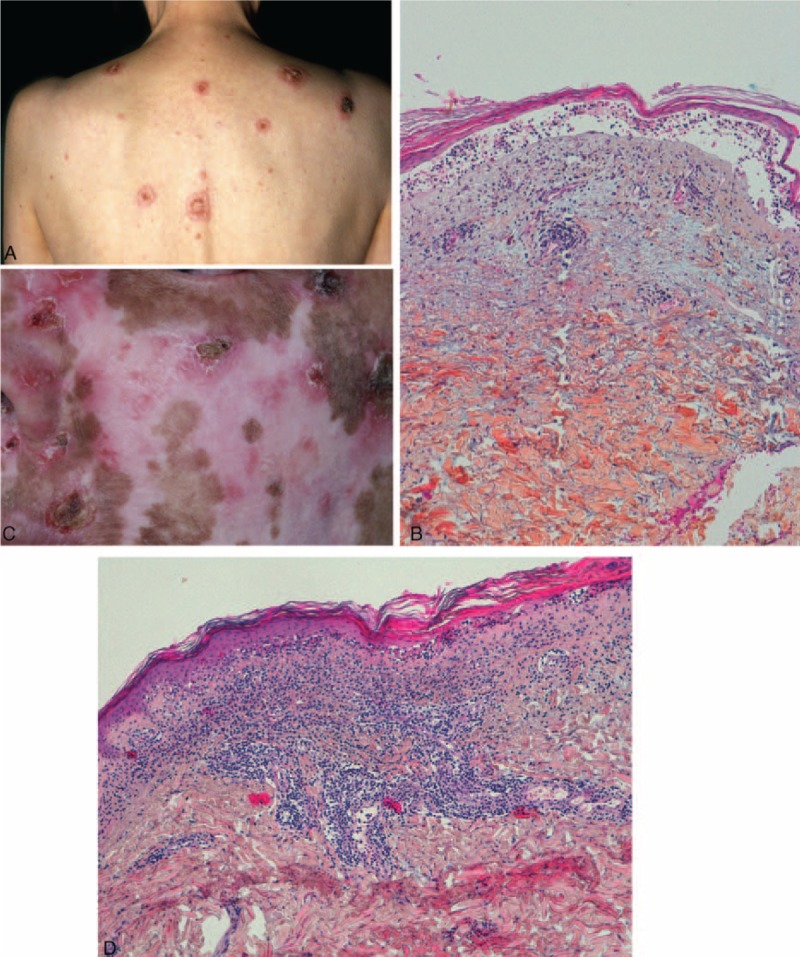
Cases of classic cutaneous LE with loss of epidermis: (A) patient 11—annular plaques centered by a crust; (B) patient 11—epidermal atrophy and dermo-epidermal blister; cavity filled with lymphocytes; (C) patient 14—erosions and crust on sun exposed skin; depigmented scars and atrophy; (D) patient 14—epidermal atrophy, interface dermatitis with vacuolization, lichenoid lympho-histiocytic infiltrate and mucin deposition.
At histopathological examination, there was epidermal atrophy (7/9), isolated keratinocyte necrosis (9/9) or extensive epidermal necrosis (2/9), vacuolization of basal keratinocytes (9/9), and dermo-epidermal detachment (1/9). A lymphocytic infiltrate was present. It could be lichenoid and/or distributed around the vessels or appendages. Mucin deposition was seen in 4 of 8 patients. Direct immunofluorescence revealed a granular fluorescence at DEJ in 2 of 8 patients and a dust-like particle pattern in 2 of 8 patients. These patients can be classified as having a vesiculobullous annular SCLE or CCLE.
Group 3: Neutrophilic Bullous LE
In the third group composed of 8 patients, the following common clinical features were observed: tense vesicles and bullae (2/8), tense bullae (5/8), crusts (3/8), and/or erosions (2/8). Three of 8 patients presented mucosal involvement (isolated oral ulcerations in 2 of 8 patients and oral, genital, and conjonctival ulcerations in 1 of 8 patients). Lesions were not photodistributed. They occurred on normal appearing or inflammatory skin. Only 2 patients had concomitant LE-specific lesions (acute cutaneous lupus erythematosus [ACLE] or CCLE). Four of 8 patients had no history of cutaneous LE. Seven of 8 patients met ACR criteria for SLE and all of them had at least 4 SLICC criteria. Among these 8 patients, 3 had concomitant glomerulonephritis (Table 3, Figure 3).
FIGURE 3.
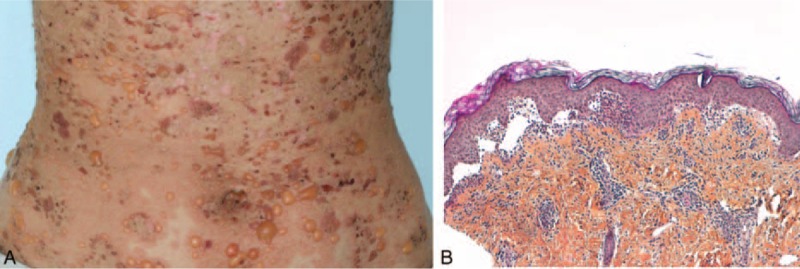
A case of neutrophilic bullous LE: patient 16—(A) tense blisters and crusts on inflammatory skin; (B) blisters and neutrophilic papillary microabscesses.
Histological examination showed either dermal–epidermal cleavage (in 7 of 8 patients) or superficial dermal edema (in 1 patient). A neutrophilic infiltrate was constant, with a pattern of papillary microabscesses in 4 of 8 patients, a dense subepidermal infiltrate in 3 of 8 patients or involving the entire dermis in 1 patient. LE-specific lesions, namely interface dermatitis, were absent in all 8 cases. Direct immunofluorescence examination revealed granular or linear immunoglobulin and C3 deposition at the DEJ (7/8) or a dust-like particles pattern (1/8). By immunoblotting, a 290 kD antigen was found in 2 patients and a 200 kD antigen was found in 1 patient. Immunoblotting was negative in 1 patient and was not performed in the remaining 4 of the 8 patients. These patients can be classified as having a bullous neutrophilic LE.
DISCUSSION
We report a series of 22 patients with loss of epidermis in the course of LE. The study of these patients led to distinguish 2 different pathological mechanisms that are likely to induce “loss of epidermis.” In the first group, skin surface alterations are related to a lupus-typical interface dermatitis at varying levels of intensity, which, if carried to the extreme, may cause TEN-like lesions. In the second group, the formation of bullae is underlaid by a neutrophilic dermal infiltrate; this group includes patients with classic bullous LE. To the best of our knowledge, no study has so far specifically addressed skin surface alterations in patients with LE. As the entry point of this study was purely morphologic, it is therefore relevant bedside, also in regards to treatment. Indeed, the erosive variants of DLE or SCLE are treated with antimalarials. In case of TEN-like LE, patients are usually hospitalized. As drug induction is not exceptional, a careful history is mandatory and every suspected drug must be interrupted. Photoprotection and appropriate skin care are essential and antimalarials are indicated. Finally, dapsone is the drug of choice for the neutrophilic variant of LE.
Patients’ recruitment mode (clinicians’ memory and/or review of photographic collections) constitutes a limitation in this study. Some data are missing due to the retrospective nature of the study. Thus, though the methodology of this study does not allow to draw any conclusion about the frequency of the different clinicopathological entities that we report, we nevertheless estimate that it is representative of the different types of surface alterations that are encountered in daily practice. First, because only experienced dermatologists participated in this study, and thus skin surface alterations would not go unnoticed; second, because in most participating centers, photos are taken from all patients with LE, and the systematic study of the photos provided a representative spectrum of the different clinical findings.
LE-Specific Vesiculobullous Skin Disease
When the lupus-typical interface dermatitis is particularly intense and acute, it can lead to epidermal necrosis, as in the course of TEN.16,17,23 This variant still is often not recognized as being LE21 and misdiagnosed as TEN. Ting et al18 distinguished 3 forms of TEN-like LE: TEN-like ACLE, in which the sheet-like cleavage of skin changes evolves rapidly from a preexisting photodistributed confluent or patchy erythema reaction that would otherwise be typical of localized or generalized ACLE; TEN-like SCLE, in which the sheet-like cleavage of skin changes evolves from otherwise typical photodistributed nonscarring annular or papulosquamous SCLE, in association with anti-Ro/SS-A or La/SS-B autoantibody production; TEN occurring in SLE patients with no conventional LE-specific skin lesions.
The diagnosis of TEN-like LE was made in 5 patients. All of them presented with an extensive epidermal necrosis and interface dermatitis. The presence or absence of anti-Ro is an element allowing the classification into one of the categories according to Ting. Four of our 5 patients had anti-Ro antibodies. The small size of these 2 groups does not allow retaining this finding as a determining factor. Drawing a distinction between subgroups of TEN-like LE seems irrelevant because the clinical features are similar in the 3 forms (flaccid bullae, vesicles, and sheet-like detachment and erosions) as well as histological features. It seems more didactic to group these 3 forms of TEN-like LE under the term “TEN-like hyperacute LE.” This diagnosis should be considered in any patient with sheet-like detachment when a photodistribution is noted, when mucous membrane involvement is discrete or absent, when antinuclear antibodies are present, or when mucin deposition is found in the biopsy specimen, particularly in the absence of high-risk drug intake. This entity remains probably underdiagnosed, as real TEN can also occur in SLE patients.22 It is important, however, to consider LE as a potential cause of acute syndrome of pan-epidermolysis, as are drug-induced Lyell syndrome or some fulminant cases of acute graft-versus-host disease (GVHD). Three of the 5 patients reported here with TEN-like LE had 4 ACR criteria (and 4 of them had 4 or more SLICC criteria), but lacked significant manifestations of visceral LE. Intravenous immunoglobulin therapy has been reported to be useful in TEN-like LE as well as in TEN and acute GVHD.4,23 TNF inhibitors have recently been reported to improve outcome in patients with TEN.24–28 These drugs are known to potentially induce LE and it is so far recommended not to administer them to patients with SLE. If their efficacy in patients with TEN should be confirmed, their use in patients with TEN-like LE should be carefully addressed.17
We also show here that classical LE lesions can evolve into “loss of epidermis,” through the same, but less acute mechanism. Histologically, there is a continuum between these different forms, supporting the notion of dermo-epidermal LE.29 Although oral mucosa ulcerations are a classic manifestation of LE, and a diagnostic criterion in both ACR and SLICC criteria, our knowledge of loss of epidermis in classic LE lesions is poor. The relatively few patients with bullous evolution in classic LE variant as compared to TEN-like LE is probably biased. Physicians more easily remembered the patients with TEN-like LE who were always hospitalized, often for a few weeks, whereas the other patients are mainly seen on an outpatient basis, and attention is not always paid to crusting or peripheral vesiculation. Vesiculobullous annular SCLE and vesiculobullous CCLE were mainly characterized by erosions and crusts, and more rarely by bullae and vesicles. These losses of epidermis occurred at the active advancing edge of LE skin lesions or at the center of plaques.
Neutrophilic Vesiculobullous Skin Disease in Patients With LE
In the second group of patients, histopathological evaluation revealed a neutrophilic infiltrate, often mimicking dermatitis herpetiformis (DH). No lupus-characteristic interface dermatitis was present in cutaneous biopsy specimens. These LE-nonspecific vesiculobullous skin diseases do not occur as an extension of the interface dermatitis that is characteristic of LE-specific skin disease. In these cases, all the criteria for a defined autoimmune bullous dermatosis must be searched. When no autoimmune bullous dermatosis, such as DH, can be nosologically characterized, patients can be classified as having a neutrophilic bullous LE. In these cases, antibodies directed against collagen VII are usually detected, similarly to patients with epidermolysis bullosa acquisita (EBA), though the spectrum of autoantibodies found in these patients can probably be expanded.15
An explanation of the co-occurrence of these diseases could be that the interface dermatitis of classical LE could lead to the exposure of multiple epidermal and dermal antigens and cause a sensitization against these antigens. This sensitization would lead to the production of autoantibodies responsible for the induction of autoimmune bullous dermatoses, either defined (eg, EBA, DH, linear IgA dermatosis, P200 pemphigoid, or bullous pemphigoid), or not defined, when the antigen is not characterized or when the essential criteria for the definition of these dermatoses are not met. According to this hypothesis, bullous neutrophilic LE associated with the presence of antibodies directed against collagen VII, considered as "EBA-like vesiculobullous LE” in Ting's classification, would be more an EBA secondary to LE than a subtype of neutrophilic LE.
Similarly, the individualization of “DH-like vesiculobullous LE” can be put into question. According to Ting, it is characterized by papillary microabscesses in combination with dense granular IgA and/or IgG deposits at the DEJ. It is necessary to differentiate the situation in which antitranglutaminase or antiendomysial antibodies are found, leading to the diagnosis of the DH, from the situation in which these antibodies are absent. In the latter case, in a patient with a history of LE (or in which LE is discovered on this occasion), the clinicopathological and immunopathological clinical picture can be considered as a bullous neutrophilic LE.
We could apply the same reasoning to other autoimmune bullous dermatoses such as linear IgA dermatosis or P200 pemphigoid occurring in patients with LE. But only half of the patients reported herein had previous LE lesions and thus this pathogenic hypothesis of exaggerated antigen exposition related to the interface dermatitis will not apply to them. We think that a subgroup of patients with LE is more prone to neutrophilic dermatoses in general including the different bullous variants.30,31 This is one more phenotypic dermatological presentation where the distinction between classic and neutrophilic LE is crucial.32–34 The correct recognition and diagnosis of the neutrophilic variant is critical, as treatment with dapsone will often allow complete control.
In patients with LE, we should definitely separate bullous lesions/loss of epidermis occurring in the setting of an interface dermatitis, from those occurring as a consequence of a neutrophilic dermatosis. The latter usually respond to dapsone, and can or cannot be immunopathologically characterized, whereas the former can either be a bullous variant of classic lupus lesions or, rarely, a life-threatening TEN-like acute dermatosis. Classic “bullous LE” is a dapsone-sensitive neutrophilic dermatosis, which probably encompasses different autoimmune bullous diseases. In the series reported here, the patients with neutrophilic bullous LE were those who had most frequently experienced associated significant renal involvement.
Acknowledgments
The authors thank the following dermatologists and pathologists for having provided us the slides for the histologic data: Luc Durand, MD (Montpellier, France), Brigitte Balme, MD (Lyon, France), Stéphane Barete, MD (Paris, France), Jean Sarrouy, MD (Point-à-Pitre, France), Marie-Claire Tortel, MD (Colmar, France). All these physicians gave permission to be named.
Footnotes
Abbreviations: ACLE = acute cutaneous lupus erythematosus, ACR = American College of Rheumatology, CCLE = chronic cutaneous lupus erythematosus, DEJ = dermal–epidermal junction, DH = dermatitis herpetiformis, EBA = epidermolysis bullosa acquisita, GVHD = graft versus host disease, LE = lupus erythematosus, SCLE = subacute cutaneous lupus erythematosus, TEN = toxic epidermal necrolysis.
The Study Group of Systemic Diseases in Dermatology (EMSED: Etude des Maladies Systémiques en Dermatologie): Didier BESSIS, Nadège CORDEL, Camille FRANCES, Dan LIPSKER
The authors have no funding and conflicts of interest to disclose.
REFERENCES
- 1.Mebazaa A, El Euch D, Sellami M, et al. Lupus érythémateux systémique vésiculo-bulleux. Rev Med Interne 2009; 30:88–89.18433941 [Google Scholar]
- 2.Itoi S, Tanemura A, Tsuji C, et al. A rare case of male bullous lupus erythematosus complicated with subsequent annular hypopigmentation. Case Rep Dermatol 2014; 6:91–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Biazar C, Sigges J, Patsinakidis N, et al. Cutaneous lupus erythematosus: first multicenter database analysis of 1002 patients from the European Society of Cutaneous Lupus Erythematosus (EUSCLE). Autoimmun Rev 2013; 12:444–454. [DOI] [PubMed] [Google Scholar]
- 4.Vera-Recabarren MA, García-Carrasco M, Ramos-Casals M, et al. Comparative analysis of subacute cutaneous lupus erythematosus and chronic cutaneous lupus erythematosus: clinical and immunological study of 270 patients. Br J Dermatol 2010; 162:91–101. [DOI] [PubMed] [Google Scholar]
- 5.Vera-Recabarren MA, García-Carrasco M, Ramos-Casals M, et al. Cutaneous lupus erythematosus: clinical and immunological study of 308 patients stratified by gender. Clin Exp Dermatol 2010; 35:729–735. [DOI] [PubMed] [Google Scholar]
- 6.Walling HW, Sontheimer RD. Cutaneous Lupus Erythematosus. Am J Clin Dermatol 2009; 10:365–381. [DOI] [PubMed] [Google Scholar]
- 7.Callen JP. Chronic cutaneous lupus erythematosus. Clinical, laboratory, therapeutic, and prognostic examination of 62 patients. Arch Dermatol 1982; 118:412–416. [DOI] [PubMed] [Google Scholar]
- 8.Wallace DJ, Pistiner M, Nessim S, et al. Cutaneous lupus erythematosus without systemic lupus erythematosus: clinical and laboratory features. Semin Arthritis Rheum 1992; 21:221–226. [DOI] [PubMed] [Google Scholar]
- 9.Christodoulou G, Powell M, Nguyen VH, et al. An atypical case of bullous systemic lupus erythematosus in a 16-year-old boy. Pediatr Dermatol 2014; 31:e164–e166. [DOI] [PubMed] [Google Scholar]
- 10.Ranario JS, Smith JL. Bullous lesions in a patient with systemic lupus erythematosus. J Clin Aesthet Dermatol 2014; 7:44–49. [PMC free article] [PubMed] [Google Scholar]
- 11.Liu KL, Shen JL, Yang CS, et al. Bullous systemic lupus erythematosus in a child responding to dapsone. Pediatr Dermatol 2014; 31:e104–e106. [DOI] [PubMed] [Google Scholar]
- 12.Maley A, Parker S. Bullous systemic lupus erythematosus in a patient with human immunodeficiency virus infection: a paradox of autoimmunity and immunodeficiency. Dermatol Online J 2014; 20: 9: [PubMed] [Google Scholar]
- 13.Contestable JJ, Edhegard KD, Meyerle JH. Bullous systemic lupus erythematosus: a review and update to diagnosis and treatment. Am J Clin Dermatol 2014; 15:517–524. [DOI] [PubMed] [Google Scholar]
- 14.Lourenço DMR, Cunha Gomes R, Aikawa NE, et al. Childhood-onset bullous systemic lupus erythematosus. Lupus 2014; 23:1422–1425. [DOI] [PubMed] [Google Scholar]
- 15.Chan LS, Lapiere JC, Chen M, et al. Bullous systemic lupus erythematosus with autoantibodies recognizing multiple skin basement membrane components, bullous pemphigoid antigen 1, laminin-5, laminin-6, and type VII collagen. Arch Dermatol 1999; 135:569–573. [DOI] [PubMed] [Google Scholar]
- 16.Monga B, Ghosh S, Jain V. Toxic epidermal necrolysis-like rash of lupus: a dermatologist's dilemma. Indian J Dermatol 2014; 59:401–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Napolitano M, Giampetruzzi AR, Didona D, et al. Toxic epidermal necrolysis-like acute cutaneous lupus erythematosus successfully treated with a single dose of etanercept: report of three cases. J Am Acad Dermatol 2013; 69:e303–e305. [DOI] [PubMed] [Google Scholar]
- 18.Ting W, Stone MS, Racila D, et al. Toxic epidermal necrolysis-like acute cutaneous lupus erythematosus and the spectrum of the acute syndrome of apoptotic pan-epidermolysis (ASAP): a case report, concept review and proposal for new classification of lupus erythematosus vesiculobullous skin lesions. Lupus 2004; 13:941–950. [DOI] [PubMed] [Google Scholar]
- 19.Boisnic S, Frances C, Foldes C, et al. Manifestations cutanées rares au cours du lupus systémique: lésions bulleuses. Ann Dermatol Venereol 1986; 113:930–933. [Google Scholar]
- 20.Lipsker D, Hauptmann G. Cutaneous manifestations of complement deficiencies. Lupus 2010; 19:1096–1106. [DOI] [PubMed] [Google Scholar]
- 21.Mahfouz A, Mahmoud AN, Ashfaq PA, et al. A case report of hydralazine-induced skin reaction: probable toxic epidermal necrolysis (TEN). Am J Case Rep 2014; 15:135–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ziemer M, Kardaun Sh, Liss Y, et al. Stevens-Johnson syndrome and toxic epidermal necrolysis in patients with lupus erythematosus: a descriptive study of 17 cases from a national registry and review of the literature. Br J Dermatol 2012; 166:575–600. [DOI] [PubMed] [Google Scholar]
- 23.Mandelcorn R, Shear NH. Lupus-associated toxic epidermal necrolysis: a novel manifestation of lupus? J Am Acad Dermatol 2003; 48:525–529. [DOI] [PubMed] [Google Scholar]
- 24.Gaitanis G, Spyridonos P, Patmanidis K, et al. Treatment of toxic epidermal necrolysis with the combination of infliximab and high-dose intravenous immunoglobulin. Dermatology 2012; 224:134–139. [DOI] [PubMed] [Google Scholar]
- 25.Zárate-Correa LC, Carrillo-Gómez DC, Ramírez-Escobar AF, et al. Toxic epidermal necrolysis successfully treated with infliximab. J Investig Allergol Clin Immunol 2013; 23:61–63. [PubMed] [Google Scholar]
- 26.Hunger RE, Hunziker T, Buettiker U, et al. Rapid resolution of toxic epidermal necrolysis with anti-TNF-alpha treatment. J Allergy Clin Immunol 2005; 116:923–924. [DOI] [PubMed] [Google Scholar]
- 27.Paradisi A, Abeni D, Bergamo F, et al. Etanercept therapy for toxic epidermal necrolysis. J Am Acad Dermatol 2014; 71:278–283. [DOI] [PubMed] [Google Scholar]
- 28.Wojtkiewicz A, Wysocki M, Fortuna J, et al. Beneficial and rapid effect of infliximab on the course of toxic epidermal necrolysis. Acta Derm Venereol 2008; 88:420–421. [DOI] [PubMed] [Google Scholar]
- 29.Lipsker D. Classification of specific cutaneous manifestations in patients with lupus erythematosus: a time for change? The concept of dermal lupus erythematosus. Dermatology 2006; 212:324–326. [DOI] [PubMed] [Google Scholar]
- 30.Tobón GJ, Toro CE, Bravo JC, et al. Linear IgA bullous dermatosis associated with systemic lupus erythematosus: a case report. Clin Rheumatol 2008; 27:391–393. [DOI] [PubMed] [Google Scholar]
- 31.Kurano TL, Lum CA, Izumi AK. The association of dermatitis herpetiformis and systemic lupus erythematosus. J Am Acad Dermatol 2010; 63:892–895. [DOI] [PubMed] [Google Scholar]
- 32.Lipsker D, Saurat JH. Neutrophilic cutaneous lupus erythematosus. At the edge between innate and acquired immunity? Dermatology 2008; 216:283–286. [DOI] [PubMed] [Google Scholar]
- 33.Kieffer C, Cribier B, Lipsker D. Neutrophilic urticarial dermatosis: a variant of neutrophilic urticaria strongly associated with systemic disease. Report of 9 new cases and review of the literature. Medicine (Baltimore) 2009; 88:23–31. [DOI] [PubMed] [Google Scholar]
- 34.Gusdorf L, Bessis D, Lipsker D. Lupus erythematosus and neutrophilic urticarial dermatosis: a retrospective study of 7 patients. Medicine (Baltimore) 2014; 93:e351. [DOI] [PMC free article] [PubMed] [Google Scholar]