

Abstract
Acute myeloid leukemia (AML) is characterized by a high relapse rate that has been attributed to the quiescence of leukemia stem cells (LSCs), which renders them resistant to chemotherapy. However, this hypothesis is largely supported by indirect evidence and fails to explain the large differences in relapse rates across AML subtypes. To address this, bone marrow aspirates from 41 AML patients and five healthy donors were analyzed by high-dimensional mass cytometry. All patients displayed immunophenotypic and intracellular signaling abnormalities within CD34+CD38low populations and several karyotype and genotype-specific surface marker patterns were identified. The immunophenotypic stem and early progenitor cell populations from patients with clinically favorable core-binding factor AML demonstrated a five-fold higher fraction of cells in S-phase compared to other AML samples. Conversely, LSCs in less clinically favorable FLT3-ITD AML exhibited dramatic reductions in S-phase fraction. Mass cytometry also allowed direct observation of the in vivo effects of cytotoxic chemotherapy.
Keywords: Acute Myeloid Leukemia, Cell Cycle, Mass Cytometry, Leukemia Stem Cell, FLT3, Core-Binding Factor AML
Introduction
Acute myeloid leukemia (AML) includes molecularly and biologically distinct subtypes of disease in which clonal populations of abnormal stem and progenitor cells give rise to a large population of proliferative myeloid blasts and other immature cell types (1). It has long been appreciated that recurrent cytogenetic and molecular abnormalities delineate distinct AML subtypes with differing biologic and clinical characteristics (2, 3). It is also now well established that early stem and progenitor cell subsets (leukemia stem cells; LSCs) within the abnormal myeloid cell compartment contain the leukemia initiating activity of AML and likely mediate clinical relapse following therapy (4–6). Although LSCs have been studied extensively in recent years, the relationship between the specific properties of LSCs and the clinical features of different disease subtypes remains poorly understood.
LSCs are hypothesized to mediate relapse on the basis of their relative quiescence (7, 8) and protective interactions with the bone marrow niche (9). The evidence for the quiescence of AML stem cells is largely indirect, however. Most studies of human AML cells have been performed on samples treated in vitro or ex vivo with chemotherapy agents that kill bone marrow cells in S-phase, followed by the demonstration that surviving quiescent cells initiate disease in immunocompromised mice. Other studies have demonstrated that murine hematopoietic stem cells (HSCs) are generally quiescent in vivo; however, even in normal HSC populations 50% of the cells will enter S-phase within 6 days and >90% enter S-phase within 30 days (10). A more important weakness of this hypothesis is the marked differences in chemotherapy responsiveness of different AML disease subtypes. Chemotherapy alone cures 60 – 79% of patients with core-binding factor AML (patients with t(8:21), inv(16), or t(16;16) cell karyotypes) (11, 12), whereas patients with FLT3-ITD mutated AML have a 3–5 year overall survival (OS) rate of 20% – 30% (3, 13) in spite of high rates of initial remission. This suggests that the resistance of LSCs to chemotherapy depends on the disease subtype; however, the molecular mechanisms that underlie these differences are largely unknown.
Recent years have witnessed an explosion in the genetic characterization of human malignancies in general and in AML in particular (14, 15). The complete genomic sequencing of hundreds of AML samples has now been completed, and these analyses have demonstrated that the relatively small number of well-characterized mutations in AML (e.g., FLT3, NPM1, cKit, CEBPa) exist in a complex landscape of hundreds of other mutations (14, 15). These studies have yielded promising new molecular targets for AML treatment, but how these mutations relate to the functional behaviors of these cells is not well understood. Furthermore, analysis of the largest cohort of sequenced AML samples revealed more than 100 unique combinations of known or suspected oncogenic mutations among 200 AML patients (14). This complexity suggests that personalized treatment approaches cannot simply target all of the mutations present in a given patient’s leukemia and that an understanding of how each mutation contributes to leukemia cell function (and which must be targeted in a given patient) will be required to fully realize the therapeutic promise of the knowledge obtained through genomic studies.
As a first step in addressing these issues, we report a mass cytometry-based approach for performing high-dimensional functional profiling of AML. The primary aim of this study was to directly measure the immunophenotypic, cell cycle, and intracellular signaling properties of AML cells that were processed and fixed immediately after bone marrow aspiration to preserve their in vivo biologic properties. Mass cytometry was utilized to perform the first high-dimensional characterization of cell cycle state and basal intracellular signaling across major immunophenotypic cell subsets of AML patient samples. This approach was facilitated by the recent developments of methodologies for the assessment of cell cycle state by mass cytometry (16) and barcoding techniques that allow multiple samples to be stained and analyzed with high precision (17, 18). The combination of these techniques enabled a unique characterization of the in vivo cell cycle and signaling states of immunophenotypically distinct AML cell populations across a variety of common AML disease subtypes and yielded insights into the mechanisms of chemotherapy response in AML patients.
Results
Immediate sample collection and barcoded staining resulted in consistent immunophenotypic and functional measurements by mass cytometry
Bone marrow aspirates were collected from 35 AML patients (18 newly diagnosed, 11 relapsed/refractory, one patient with relapsed myeloid sarcoma, and five patients with AML in complete remission (CR) at the time of sample collection), four patients with acute promyelocytic leukemia (APL), two patients with high-risk myelodysplastic syndromes (MDS; both transformed to AML within 60 days of biopsy), and five healthy donors (46 total biopsy samples). The clinical characteristics of the patients are listed in Supplementary Table 1.
Two 39-antibody staining panels (with 23 surface markers and two intracellular markers common between them) were utilized for analysis (Supplementary Table 2). To ensure the consistency and accuracy of mass cytometric analysis, samples were collected immediately after bone marrow aspiration (<1 min), maintained at 37 °C prior to fixation, and frozen at −80 °C until the time of analysis. Samples were barcoded in groups of 20 to allow simultaneous antibody staining and mass cytometric analysis (17, 18). These protocols produced highly reproducible measurements of surface markers across replicates of the normal samples with an average coefficient of variation (CV) of 15.4%, with the majority of antibodies (39/45) having CVs of less than 20% (Supplementary Table 2) (17). Average CVs were similar for both surface proteins (15.7%) and intracellular functional markers (14.4%). Most samples had been analyzed by clinical flow cytometry as part of routine diagnostic testing; blast antigen expression patterns determined by flow cytometry and by mass cytometry were comparable (Supplementary Table 3). These data are consistent with prior studies (19–21) and confirmed that mass cytometry can be used with a high degree of reproducibility and accuracy for the analysis of AML clinical samples.
Distribution of cells across developmental stages is AML subtype specific
To perform immunophenotypic analysis of the mass cytometry data, both traditional gating and high dimensional SPADE clustering were performed using 19 of the surface markers common to both staining panels (Supplementary Table 2). The resulting SPADE analysis of the normal bone marrow was consistent across all of the healthy donors; an example from one healthy donor is shown in Figure 1 and Supplementary Figure 1. SPADE clustering yielded cell groupings that corresponded to commonly defined immunophenotypic subsets across normal hematopoietic development. Both SPADE clustering (Figure 2A) and manual gating (Figure 2B and 2C; Supplementary Figure 2) demonstrated that patients with core-binding factor mutations (CBF-AML; n=5; t(8;21), inv(16), and t(16;16) karyotypes) and those with adverse-risk karyotypes (ARK-AML; n=6) had the highest prevalence of immature immunophenotypes, particularly hematopoietic stem cells (HSC; lin−CD34+CD38lowCD45RA−CD90+CD33low) and multipotent progenitor cells (MPPs; lin−CD34+CD38lowCD45RA−CD90−CD33low). The fractions of these two populations were increased more than 50-fold in CBF-AML and ARK-AML patients samples compared to healthy donors (p<0.002, in all comparisons). By contrast, patients with normal karyotype AML (NK-AML; with or without FLT3 mutation; n=17) and APL (n=4) exhibited much smaller increases of approximately three to ten-fold in the HSC and MPP populations. The HSC and MPP populations of both CBF-AML and ARK-AML samples were significantly increased compared to these populations in NK-AML or APL (p values ranging from 0.0014 to 0.039; except for ARK-AML MPP vs. APL MPP where p=0.067). These findings demonstrate that high dimensionality of mass cytometry can detect unique patterns of cell development that can yield novel potentially diagnostic information.
Figure 1.

SPADE plots of normal bone marrow sample #6. SPADE clustering was performed on all samples (normal and AML) simultaneously to generate a single tree structure for all samples. All of the cell events from each sample were then mapped to the common tree structure. Each node of the SPADE tree is colored for the median expression of the indicated markers from low (blue) to high (red). The size of each node is correlated to the fraction of cells mapping to the node; however, a minimum size was enforced for most nodes to allow visualization of node color. Immunophenotypic grouping of nodes was performed manually on the basis of the median marker expression level of each node, and based on analysis of the relevant biaxial plots (e.g., CD38 vs. CD34).
Figure 2.

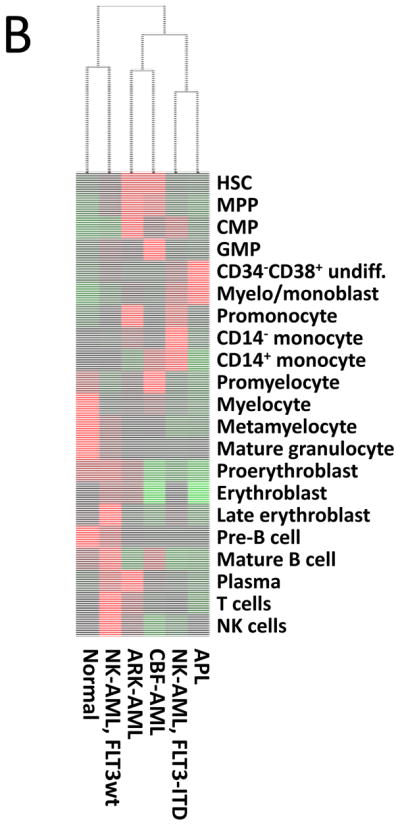

Different AML samples demonstrate karyotype and genotype-specific patterns of cell distribution across developmental populations. A) SPADE tree colored for the fraction of total cells in each node from lowest (blue) to highest (red). The size of each node is correlated to the fraction of cells mapping to the node; however, a minimum size was enforced for most nodes to allow visualization of node color. B) Heat map demonstrating the relative frequency of cells in each of the indicated manually-gated immunophenotypic subsets averaged across the patients in the indicated karyotypic and genotypic groups. C) The frequency of cells in the indicated (manually-gated) stem and progenitor cell compartments for patients of each AML subtype. Error bars indicate standard errors of the means. *The 14 replicate normal samples came from 5 donors.
High-resolution analysis reveals karyotype- and genotype-specific patterns of abnormal surface marker expression
The simultaneous measurement of up to 28 markers (staining panel A), allowed analysis of aberrant marker expression in each immunophenotypic population at a high resolution. Surface marker expression in AML samples was evaluated against normal samples by first gating cells from the normal samples into developmental immunophenotypic subsets based on standard surface markers. Each population from each of the normal and AML samples was then compared across the 28 surface markers as shown in Figure 3A. Since the normal and AML samples were stained and analyzed in the same tubes simultaneously, levels of each surface maker in each of 35 immunophenotypic populations of each patient sample could be reliably assessed. The summed number of markers (of the 28) with aberrant expression levels was calculated for each gated immunophenotypic population in each patient sample (Figure 3B). Immunophenotypic aberrancies were detected at every stage of myeloid development spanning from HSCs to mature myeloid populations in all AML samples. In the majority of patients, aberrancies were also detected in most non-myeloid immunophenotypic populations (including B, T, and NK cells), suggesting that the AML clone has wide-ranging effects in the bone marrow.
Figure 3.


Systematic detection of multiple small aberrancies defines large karyotype and genotype-specific immunophenotypic changes across hematopoiesis in patients with AML. A) Method for defining aberrant marker expression. All immunophenotypic gates were defined on the basis of the normal samples and the same gates were applied to the each of the AML samples. Once each population was gated, the median expression of each of the 28 surface markers was extracted and compared to the median expression in the 14 samples from five healthy donors for each gated population. AML samples that were outside 2-fold the total variance of the normal samples were considered to be aberrant for that marker (CD45 is shown as an example). B) The total number of aberrant markers (of 28 measured markers) was summed for each population and each patient. Each box is colored for the number of the 28 markers that was aberrant for each patient (rows) in each gated immunophenotypic population (columns). The color scale ranges from green indicating no aberrant marker expression to the highest numbers of aberrancies colored red. The exact number of aberrant markers expressed (of the 28 tested) is printed in each box. The high rates of aberrancy observed in the pre-B cell population may be due in part to contamination of this gate with dimly CD19-positive malignant myeloid cells due to the limited number of markers defining this immunophenotypic subset (CD19 and CD10) and to the relatively dim staining of these antibodies in normal cells.
In addition to analysis of the total number of immunophenotypic aberrancies, specific aberrancies were also detected in patients with different AML subtypes. These were particularly informative in the immunophenotypic hematopoietic stem and progenitor cell (HSPC) compartment (lin−CD34+CD38low) where strong trends were observed across the different AML subsets (as shown in Figure 4A and Supplementary Table 4). FLT3-ITD+ NK-AML samples (n=11) were characterized by increased expression of CD7, CD33, CD123, CD45, CD321, and CD99, and decreased expression of CD34, CD117, and CD38 relative to expression in healthy patient samples. FLT3wt NK-AML samples (n=6) were characterized by increased expression of CD99 and decreased expression of CD71, CD47, CD34, and CD45. ARK-AML samples (n=6) were characterized by increased expression of CD99 and decreased expression of CD47. All p values were significant (ranging from 0.02 to 4.5×10−7). Expression of CD99 was elevated in most of the AML samples (30/36) but was normal in six samples including two of the three samples with t(8;21) karyotypes. Each of the 36 leukemia samples displayed immunophenotypic abnormalities within the HSPC gated population (3.6 abnormalities on average).
Figure 4.


AML subtypes are characterized by specific marker aberrances in CD34+CD38low cells. A) Median expression level of the indicated markers in the total CD34+CD38low population, each data point represents the median expression level of one patient sample or one of the 14 sample aliquots from the five healthy donors. B) Upper panels: Biaxial plots of HLA-DR vs. CD33 in the gated CD34+CD38low subset from three healthy donors. Lower panels: Biaxial plots of patient samples show that HLA-DR was either abnormally low (in APL samples) or abnormally high (AML sample #22) in immunophenotypically abnormal cells as compared to residual immunophenotypically normal cells (green circles).
In nine samples, unambiguous separation into immunophenotypically normal and abnormal cell populations was observed among the cells in this gate. Interestingly, among these samples, some markers were aberrantly high in abnormal cells from patients with one disease subtype and aberrantly low in the abnormal cells of another. For example, HLA-DR was extremely high in the abnormal HSPCs of sample #22 (MLL rearrangement) and aberrantly low in the abnormal cells of all four APL samples relative to immunophenotypically normal cells from the same patients (green circles; Figure 4B). The high-resolution immunophenotypic analysis enabled by mass cytometry thus allowed both the detection of surface marker abnormalities at all stages of hematopoietic development in AML samples and also the separation of immunophenotypically normal and abnormal stem cell populations in many patients.
Unique high-dimensional immunophenotypic patterns within the HSPC population characterize different AML subtypes
In order to simultaneously view and compare the entire aberrant expression pattern within the HSPC populations as a function of AML subtype, a visualization tool called viSNE was utilized. viSNE employs a non-linear, iterative process of single cell alignment to minimize the multidimensional distance between events and represents the separation of cell events in a two-dimensional map (19). The HSPC population (CD34+CD38low) of each sample was analyzed by viSNE utilizing 19 surface markers. The viSNE patterns from the five healthy donors were consistent (Figure 5). The AML and APL samples all displayed patterns divergent from normal with notably few cells falling within the viSNE space populated by the CD34+CD38low cells from healthy donors. Separate, but distinct, patterns could be seen for the two samples with inv(16) karyotypes, two of the three samples with a t(8;21) karyotype, and 10 of the 11 NK-AML samples with FLT3-ITD mutations (of note, the one FLT3-ITD+ patient lacking this common pattern, AML#20, did not harbor a FLT3-ITD mutation at disease relapse six months later). The HSPCs of the four patients with APL (which all had FLT3-ITD mutations) and two of the three samples with FLT3 tyrosine kinase domain mutations (FLT3-TKD) also exhibited a pattern similar to that of the FLT3-ITD+ NK-AML patients. No clear patterns were observed across the patients with FLT3wt NK-AML or with ARK-AML; however, each sample was clearly different from the normal HSPCs of the healthy donor samples. These differences were statistically significant in at least one tSNE dimension (as described in Supplementary Methods).
Figure 5.

viSNE analysis of CD34+CD38low subset reveals distinct karyotype and genotype-specific immunophenotypic patterns in high-dimensional space. Each sample was analyzed by viSNE (up to 5,000 sampled events per individual) using 19 dimensions (Supplementary Table 2). Two gates encompassing the vast majority of normal CD34+CD38low events are shown for reference. The AML subtype is indicated for each sample. Each cell event is colored for it’s expression level of CD38 from blue (0 ion counts) to red (approximately 40 ion counts). Red cell events still fall within the CD34+CD38low gate and demonstrated dim CD38 expression.
To confirm that high dimensional differences observed in the viSNE analysis could group samples based on AML subtype, the gated CD34+CD38low subset from each sample was analyzed by binning samples into 100 bins using a K-means clustering of the expression levels of the 19 markers used in the viSNE analysis. This independent analysis produced a hierarchical grouping of the samples based on the pairwise correlation of the distribution of cells across 100 multi-dimensional bins. This grouped all of the normal bone marrow samples into a single branch of the dendrogram (Supplementary Figure 3). Ten of the 11 FLT3-ITD+ NK-AML samples were grouped with two of the FLT3-ITD+ APL samples and two of the FLT3-TKD+ NK-AML samples into a separate branch. As in the viSNE analysis, AML#20 was dissimilar from the other FLT3-ITD+ NK-AML samples. Sample AML#18 (ARK-AML, FLT3-ITD+) was also not in this branch of the dendrogram. A z-transform of the correlation of each sample to the normal samples revealed that all AML and APL patient samples were statistically different from normal (p<0.01). Notably, the one sample from a patient in CR, who remains free of disease at this time (>2 years from the time of biopsy), was not significantly different from normal (CR#2; p=0.10). The three patients who had achieved CR or complete response with incomplete count recovery (CRi) at the time of sampling but ultimately demonstrated shorter survivals (112–329 days), were all significantly different from normal (CR#5, p=0.00088; CR#6, p=0.0084; CR#7, p=0.00061) despite having no morphologic evidence of AML at the time of bone marrow biopsy. These results demonstrate that the high-dimensional analysis of HSPC immunophenotype can accurately group together distinct AML disease subtypes, and is potentially diagnostic of certain AML subtypes such as FLT3-ITD+ NK-AML.
AML subtypes are characterized by distinct patterns of cell cycle distribution
Given the important role of the proliferative rate in chemosensitivity, each immunophenotypic population was analyzed for cell cycle state (16). Bone marrow aspirates from AML patients and healthy donors were exposed to iodo-deoxyuridine (IdU) immediately after collection (<1 min after aspiration) and incubated for 15 min at 37 °C followed by immediate fixation and storage. This method allowed for the closest possible estimation of the in vivo cell cycle status of the bone marrow cells. Consistent with data from animal models, healthy human bone marrow samples exhibited the highest S-phase fractions in early committed progenitor populations: early erythroblasts (50–60% S-phase), pre-B cells (14.4%), myelo-monoblasts (18.3%), promonocytes (12.4%), and promyelocytes (22.1%; Figure 6A and 6B). Normal immunophenotypic HSPC populations exhibited relatively low S-phase fractions, with HSCs exhibiting an S-phase fraction of 3.72%, MPPs exhibiting an S-phase fraction of 5.97%, and an overall S-phase fraction of 6.58% for all lin− CD34+CD38low cells (Figure 6A and 6B).
Figure 6.




Karyotype and genotype-specific patterns of S-phase fraction in AML. A) SPADE trees of representative patients of the indicated AML subtypes. Nodes are colored for the frequency of S-phase cells in each node from lowest (blue) to highest (red). B) AML cells exhibit a lower S-phase fraction than immunophenotypically similar normal cells. C) Stem and progenitor cell populations from patients with CBF-AML exhibit a higher S-phase fraction than immunophenotypically similar cells from patients with other AML subtypes (APL samples excluded from analysis). D) Stem and progenitor cell populations from patients with FLT3-ITD+ NK-AML exhibit a lower S-phase fraction than cell from patients with other AML subtypes (APL samples and patients with FLT3-TKD mutations excluded from analysis). Colored boxes group immunophenotypic populations: HSPC, hematopoietic stem and progenitor cells; B, blasts (immunophenotypic); Mono, monocyte lineage cells; Gran, granulocyte lineage; RBC, red blood cell lineage; B-Cell, B cell lineage. Error bars indicate standard errors.
Strikingly, the combined analysis of all AML samples demonstrated that, on average, AML cells of any given immunophenotypic subset exhibited a lower S-phase fraction than normal cells of that immunophenotypic subset (Figure 6B). Furthermore, the S-phase fraction of AML cells followed a similar developmental pattern as immunophenotypically similar normal cells with proliferation increasing from HSCs (2.2% S-phase) to a peak in immunophenotypic myelo-monoblast cells (5.2% S-phase) and promyelocytes (8.0% S-phase), and decreasing to nearly zero by the completion of immunophenotypic differentiation (Figure 6A and 6B).
The high resolution of mass cytometry allowed a detailed comparison of S-phase fraction sizes between AML disease subtypes. This demonstrated that the immunophenotypic HSPC populations of CBF-AML samples exhibited a significantly higher S-phase fraction than the corresponding populations in the other AML samples (p values 0.0002 to 0.007; Figures 6A and 6C). This effect appeared to be specific to the HSPCs (myelo-monoblast cell S-phase fractions were not significantly different) and is consistent with greater chemotherapy sensitivity of patients with CBF-AML, particularly during consolidation therapy (when HSPCs/LSCs would be expected to be enriched).
To further corroborate the association of S-phase fraction with clinical response to chemotherapy, the S-phase fractions of immunophenotypic HSPC populations from FLT3-ITD+ patients, who are unlikely to be cured with chemotherapy alone, were compared to all other AML samples. FLT3-ITD+ NK-AML stem and progenitor populations had significantly lower S-phase fractions than the other AML samples, with S-phase fractions as low as 0.25% for FLT3-ITD+ HSC and 0.29% for FLT3-ITD+ MPP populations. The differences were significant for all of the HSPC populations (p values 0.001 to 0.01; Figures 6A and 6D). These dramatic differences in fractions of S-phase cells were not observed in more mature myeloid cells (myelo-monoblast population S-phase fraction did not differ significantly; p=0.57). In both CBF-AML and FLT3-ITD+ NK-AML samples, the observed cell cycle differences in the HSPCs were supported by differences in levels of Ki67 and PCNA positivity, well-established markers of proliferation (data not shown). Thus, the direct quantification of the cell cycle state in HSPCs from chemotherapy sensitive and resistant AML subtypes provides strong evidence of its pivotal role in clinical response.
Phospho-flow analysis reveals AML subtype-specific intracellular signaling correlated with immunophenotypic aberrancy
In order to relate additional functional phenotypes to immunophenotypic aberrancies, intracellular signaling was measured in each immunophenotypic population of all samples. Consistent with previous findings (22, 23), FLT3-ITD+ samples had significantly higher levels of pSTAT5 (2.5–6 fold relative to normal) across all immunophenotypically-defined myeloid populations (Supplementary Figure 4A). This was not the case for HSPC populations in the other leukemia subtypes where pSTAT5 levels were decreased compared to normal (p=0.008 for FLT3-ITD+ AML vs. normal; p=3×10−5 for FLT3-ITD+ AML vs. all other AML). Additionally, levels of pSTAT5 were also significantly higher in the FTL3-ITD+ APL samples. A similar trend was observed for MAPKAPK2 phosphorylation, which was consistently higher in myeloid cell subsets of FLT3-ITD+ NK-AML samples (significant for all FLT3-ITD+ myeloid populations except for the CMP/GMP gate where p=0.058; Supplementary Figure 4B). Almost all AML subtypes exhibited higher levels of pERK relative to normal (an average increase of 4-fold; p<0.05 for all populations; Supplementary Figure 4C). Conversely, phosphorylation of 4E-BP1 was lower than normal across all AML subtypes and in all gated myeloid populations with the exception of the metamyelocyte and mature granulocyte populations of patients with FLT-ITD+ AML (Supplementary Figure 4D).
As aberrant intracellular signaling could be detected as early as the HSC/MPP population, the multidimensional viSNE plots of individual CD34+CD38low cells from each sample were analyzed for phosphoprotein expression level to verify that the abnormal signaling was found in the immunophenotypically abnormal cells. This analysis is shown in Supplementary Figure 5A–B. AML cells in the distinct regions of the viSNE plot associated with the FLT3-ITD+ immunophenotype appear to exhibit higher activation of pSTAT5 and pMAPKAPK2. In samples from patients with other AML subtypes, cells with either abnormally high or low levels of pSTAT5 or pMAPKAPK2 were also more likely to appear outside regions of the viSNE plot populated by the CD34+CD38low cells of healthy donors (Supplementary Figure 5A–B). Analysis of other measured signaling molecules in the CD34+CD38low population is shown in Supplementary Figure 6A–I. Thus, mass cytometry analysis identified immunophenotypically aberrant HSPCs in AML patients and allowed visualization of abnormal intracellular signaling states in these same cells.
High-dimensional analysis of AML samples enabled direct assessment of chemotherapy response
To assess the feasibility of using mass cytometry to monitor chemotherapy responses, samples from ten of the patients who were receiving oral hydroxyurea (HU; to control elevated blast count) were compared to samples from the 23 patients not receiving HU. Consistent with the expected reduction in cellular deoxyribonucleic acid pools induced by HU, samples from treated patients exhibited 80–90% suppression of IdU incorporation relative to untreated patients (Figure 7A and 7B). However, HU treatment had minimal effect on the fraction of cells in S-phase across the myeloid immunophenotypic populations. A significant decrease was observed only in the GMP (3.44% vs. 1.25% S-phase cells; p=0.016) and promyelocyte populations (10.1% vs. 2.98% S-phase cells; p=0.0013; Figure 7C). The CD34+CD38low cell populations were least affected by HU treatment; there was no significant difference in S-phase fraction between samples from patients treated with HU and those who were not (2.72% vs. 2.86%; p=0.21), in spite of the >6-fold difference IdU incorporation in S-phase cells from these populations (median counts of 129 vs. 19.8, p=4×10−6; Figure 7B and 7C). Additionally, the fraction of actively cycling cells (indicated by phosphorylated Rb) was unchanged or increased in all cell populations from patients treated with HU (Figure 7D).
Figure 7.




In vivo response to hydroxyurea (HU) was readily detectable and distinct from the response of leukemia cell lines in vitro. A) IdU incorporation and Ki-67 staining of representative untreated and HU-treated AML samples demonstrates that treatment leads to a reduction in IdU incorporation with minimal change in S-phase fraction. B) IdU incorporation is significantly decreased in the S-phase cells of all immunophenotypic populations in samples from patients being treated with IdU at the time of their bone marrow biopsy. (HSC and MPP populations were combined for this analysis given the low numbers of S-phase cells in these rare cell populations.) C) HU treatment does not significantly decrease S-phase fraction in most immunophenotypic populations. D) Fraction of pRb+ cells does not decrease in response to HU treatment. Colored boxes group immunophenotypic populations: HSPC, hematopoietic stem and progenitor cells; B, blasts (immunophenotypic); Mono, monocyte lineage cells; Gran, granulocyte lineage; RBC, red blood cell lineage; B-Cell, B cell lineage. Error bars indicate standard errors.
Levels of apoptosis (as indicated by cleaved PARP) did not differ in any of the immunophenotypic cell populations of patients treated with HU (Supplementary Figure 7). While in contrast with the complete S-phase arrest and cell cycle exit commonly observed when leukemic cell lines are treated with HU in vitro (24), the analyses of patient cells shown here are much more consistent with the modest clinical effects observed in AML patients receiving HU in the clinic (25). This discrepancy suggests that cultured leukemic cell lines may represent only a subpopulation of AML differentiation states (e.g. promyelocytes) and that in vivo functional assays using mass cytometry will enable a more comprehensive understanding how patients respond to leukemia treatment.
Discussion
High-dimensional cytometry enables characterization of the in vivo functional properties of malignant cells
The incredible genetic and developmental heterogeneity of human malignancies creates critical questions in the study of many cancers. To what extent do oncogenic mutations disrupt developmental signaling programs in cells? Do these mutations lead to reproducible aberrant developmental patterns? To what extent is cell cycle state driven by these different trajectories? Answers to these questions could help target therapies for both de-bulking of tumors and elimination of cells with stem cell like capacities.
In this study, high-dimensional cytometric characterization of minimally manipulated samples from AML patients provided a wide range of information regarding the in vivo functional properties of malignant cells. Combined immunophenotypic analysis and assessment of cell function demonstrated several advantages. First, in all samples we observed immunophenotypically abnormal cells at all developmental stages through analyses of combinations of common immunophenotypic makers. Second, the characteristics of individual AML cells could be understood within the context of their developmental state, allowing comparisons to be performed after controlling for changes mediated by differentiation. Thus, more sensitive comparisons between different leukemia patient samples could be performed by using immunophenotypically-gated subpopulations than would be possible if the total cell populations or blast cell populations were compared (this was particularly true for the S-phase fraction, H3K9ac, pMAPKAPK2). This enabled the detection of AML subtype-specific changes in cell cycle state and surface marker expression among rare immunophenotypic HSPC populations that would be obscured by traditional analysis of total cell populations. These findings are consistent with a recent study of gene expression analysis that demonstrated improved prognostic accuracy when analysis was performed based on the differential expression patterns between AML populations and developmentally similar normal cells (26). Third, the integration of measurements of cell cycle and intracellular signaling enabled the real-time assessment of HU response and demonstrated that the chemotherapy response of AML cells in vivo is different in many ways to the in vitro response of cultured AML cell lines (Figure 7), a difference that may be due to the inability of cell culture systems to model the differentiation that occurs in malignant cell clones in vivo.
Distinct subtype-specific immunophenotypic and functional properties of AML LSCs
The immunophenotypic patterns of LSCs differed in samples from patients with different AML disease subtypes; these cell surface markers might represent targets for antibody-mediated therapies. Previous studies of aberrant surface markers on AML LSCs yielded inconsistent results, which may stem from the incorrect assumption that all AML LSCs have a similar phenotype (5, 27). In the patient cohort analyzed here, no one marker was specific for the presumed LSCs of all patients. Strong subtype-specific trends were apparent, however, particularly in FLT3-ITD+ NK-AML patients in whom immunophenotypic HSPCs exhibited characteristic increases in CD33 in 8 of 11 patients and CD123 in all 11 patients (a finding also observed in all FLT3-TKD+ samples and three of four FLT3-ITD+ APL samples; Figure 4A and Supplementary Table 4). Consistent with previous reports (28), CD99 was the most consistently elevated marker in the LSCs of AML samples in general (30 of 36 samples). CD99 levels were normal, however, in six patients, including a majority of those with the t(8;21) karyotype.
These specific patterns were even more pronounced in the 19-dimensional viSNE and binning analyses of the CD34+CD38low population, which accurately grouped the FLT3-ITD+ AML samples on the basis of immunophenotypic information alone (Figure 5 and Supplementary Figure 3). The observation that the only FLT3-ITD+ NK-AML sample that did not have this HSPC phenotype had lost the FLT3-ITD mutation at relapse suggests that the HSPC immunophenotype might be more accurate than binary genetic testing in certain clinical situations. These findings greatly extend previous reports of other subtype-specific immunophenotypes such as aberrantly increase in CD7 in patients with double CEBPα mutations (29) and increased CD56 in patients with t(8;21) karyotype (findings also observed in this study; Supplementary Table 4). High-dimensional mass or fluorescent cytometry approaches should therefore be able to rapidly identify at least some AML subtypes with high accuracy if internal staining controls are employed and analysis is restricted to the most predictive sub-gated populations (i.e., CD34+CD38low).
Critically, the AML subtype-specific immunophenotypic changes observed were detected using four independent analyses to define the developmental subpopulations: manual gating by standard surface markers (gates defined solely on the basis of the normal samples), 19-dimensional SPADE clustering, viSNE analysis, and a K-means-based cell binning approach. The immunophenotypic populations within each AML sample with the greatest number of immunophenotypic aberrancies also had: (i) the greatest expansion relative to the normal population frequency (Figures 2 and 3), (ii) the largest abnormalities in cell cycle state (Figure 6), (iii) and aberrant intracellular signaling (Supplementary Figures 4–6 and Supplementary Table 4). Taken as a whole, these observations indicate that high-dimensional analysis provides qualitatively different information from that obtained using standard diagnostic approaches and should enhance classification of malignant hematologic diseases.
High-dimensional cytometry enables assessment of AML LSC capabilities in vivo
To date, the functional properties of human leukemia stem cells have been almost exclusively studied in vitro or in the setting of murine transgenic (30) or xenograft models (4, 31–33). Although these groundbreaking studies led to the current understanding of stem cell biology and the complex clonal structure of AML, several important issues remain unresolved. Studies in which large series of patient samples have been engrafted into immunocompromised mice consistently demonstrated that engraftment capacity differs significantly across AML disease subtypes and may not reliably capture the LSC activity of all patients (31–33). Additionally, the engraftment process has been shown to be cell cycle dependent (34), greatly complicating the use of xenograft models for the study LSC proliferation. The protocol utilized in this study circumvented these limitations by collecting whole bone marrow samples from AML patients immediately at the bedside, and stringently limiting sample manipulation prior to fixation. While it remains possible that changes in cell immunophenotype or functional properties could have occurred in the 15–20 minutes between bone marrow aspiration and sample fixation (a delay required for incubation with IdU (16)), this report represents the closest measurement of the in vivo functional properties of AML cells possible with current technology of which we are aware.
One limitation of the mass cytometry technology is that the cells studied are destroyed during measurement, thereby precluding subsequent functional analyses. As a result, stem and progenitor cells had to be identified on the basis of previously established immunophenotypic criteria. This is reasonable given that the identification of stem cells was performed primarily on the basis of very well established immunophenotypic criteria (lin−CD34+CD38low)(4), and a variety of methods for immunophenotypic identification were employed yielding similar results (manual gating, SPADE clustering (35), and viSNE analysis(19)). Although controversy persists regarding the leukemia initiating potential of partially differentiated leukemia progenitor cell populations (30, 36), the majority of comparative studies of LSC activity have demonstrated that CD34+CD38low leukemia cells have a similar or greater capacity to xenograft AML than other more mature populations (9, 30, 36–39). Even in AML subtypes in which CD34− LSCs are present (40), CD34+ LSCs engraft leukemia more efficiently than CD34− LSCs (41), and are enriched in patients at relapse (6). Most relevant to the hypothesis of LSC-mediated AML relapse is the consistent finding that increased residual CD34+CD38− cells are predictive of leukemia recurrence (5, 6). Finally, the LSC populations gated based on immunophenotypic criteria in this study exhibited functional properties similar to normal HSPCs (particularly S-phase fraction, H3K9ac and pATM; Figure 6, Supplementary Figure 8, and Table 4). Regardless of assumptions about stem cell immunophenotype, the data presented here demonstrate that patients with CBF-AML do not exhibit any identifiable immature cell fraction with a lower than normal S-phase fraction, whereas samples from AML patients with FLT3-ITD mutations had immature cell populations with very low S-phase fractions that could potentially mediate disease relapse.
AML subtype-specific LSC cell cycle patterns correlate with the known responses to chemotherapy
The data reported here indicate that the cell cycle properties of immunophenotypic LSCs depend on AML disease subtype. Though preliminary, these results have important implications for the understanding of consolidation chemotherapy, which primarily serves to eliminate residual stem and early progenitor cell populations. Currently, high-dose, single-agent cytarabine is one of the most commonly employed consolidation chemotherapy regimens in the treatment of AML, largely based on its superior efficacy for the treatment of core-binding factor AML (42). The cytotoxic effect of cytarabine, however, is almost exclusively restricted to cells in S-phase of the cell cycle (43). Thus, the finding that the S-phase fraction of LSCs in CBF-AML patient samples is approximately 5-fold higher than that in samples from patients with other AML subtypes (Figure 6C) is consistent with the known responsiveness of this patient subset.
Conversely, despite relatively high rates of initial response to chemotherapy, patients with FLT3-ITD+ AML are not commonly cured with consolidation chemotherapy alone (13). The data presented in this report also fit well this observation: The LSCs in FLT3-ITD+ patients would be expected to be resistant to cytarabine-based treatment by virtue of their low S-phase fraction (allowing disease relapse). Interestingly, the much more common myelo-monoblast cells in FLT3-ITD+ AML patients exhibited the same S-phase fraction as samples from other AML subtypes and would thus be expected to be comparably sensitive to cytotoxic therapy (allowing for the initial responses generally observed in these patients; Figure 6D).
Unique to this study is the ability to directly compare individual immunophenotypic populations in AML to the same populations in normal human bone marrow. This yielded the surprising finding that the S-phase fraction of AML cells in vivo is actually lower than normal cells of the same developmental stage. While our results vis-à-vis cell cycle are consistent with a variety of prior investigations, they are not conventionally appreciated in clinical practice. Our ability to detect these differences appears to require rapid processing of samples, as we have anecdotally observed changes in cell cycle markers and basal signaling after as little as an hour ex vivo. Halogenated uridine analogs have previously been utilized in the study of both normal murine bone marrow (10, 44), and human leukemia myeloid cell populations (45) in vivo, and our results yielded S-phase fractions that are consistent with these studies.
While the sample size of this study allowed detection of highly significant differences between common AML subtypes, it remains small relative to the high degree of genomic complexity of AML and somewhat over-representative of patients with higher-risk subtypes. In the 24 samples from this cohort that were also tested for an expanded panel of somatic mutations, several additional mutations were detected. Of particular note was the common occurrence of DNMT3a mutations in FLT3-ITD+ NK-AML patients (14, 15, 46), observed in four of the seven FLT3-ITD+ NK-AML patients from this cohort (Supplementary Table 1). Due to the sample size, it was not possible to determine the additive effect of DNMT3a mutation in FLT3-ITD+ NK-AML (though the three patients with wild-type DNMT3a all exhibited similar immunophenotypes and low S-phase fractions). Mass cytometric analysis of a much larger patient cohort should allow the functional consequences of these complex genetic interactions to be addressed.
In vivo functional profiling of AML to guide design of novel therapeutic strategies
This study provides an example of how characterization of the in vivo functional properties of LSCs can potentially inform the development of novel therapeutic strategies. The finding that FLT3-ITD+ AML LSCs have a significantly decreased S-phase fraction relative to other AML subtypes suggests that single-agent, high-dose cytarabine is unlikely to be a curative consolidation therapy for these patients. As such, these results also suggest two immunophenotypic targets, CD33 and CD123, could be exploited to treat these cells in a cell cycle-independent manner with antibody drug conjugates (ADCs). Consistent with this hypothesis are the results of the phase III ALFA-0701 AML clinical trial (46, 47) that utilized fractionated treatment with the CD33-directed ADC gemtuzumab ozogamycin (GO) in both induction and consolidation therapy. This trial demonstrated a significant improvement in overall survival (estimated 2-year OS of 36% vs. 64%; p=0.023) specifically among NK-AML patients with FLT3-ITD mutations who received GO. In addition to explaining this observed benefit of CD33 targeting, the results reported here also predict that the addition of a CD123-targeted drug might further enhance LSC clearance and improve the survival of patients with FLT3-ITD+ AML.
Finally, this study establishes that high-dimensional cytometry can potentially be used to monitor disease treatment in “real time” and proximal to the acquisition of clinical material. Detection of aberrant signaling in FLT3-ITD+ AML and measurement of cell cycle effects of HU treatment also demonstrate that monitoring the responses of patients with hematologic malignancies to both targeted and cytotoxic therapies is clinical feasible. This would be particularly useful for therapies with incompletely understood mechanisms of action and those that target LSCs or other rare cell populations. Since mass cytometry allows the simultaneous assessment of many bone marrow cell types, the efficacy of immunotherapy approaches (e.g., chimeric antigen receptor T cells) could also be monitored. As understanding of the underlying genetic complexity of hematologic malignancies expands, real-time, functional measurement of therapy response will be increasingly important for understanding which of the many genetic lesions present in these diseases represent the most relevant targets for therapeutic intervention and how to best utilize the novel agents designed to target them.
Methods
Antibodies
Antibodies, manufacturers, and concentrations are listed in Supplementary Table 2. Antibody staining was performed in two overlapping panels, as indicated. Primary antibody transition metal-conjugates were either purchased or conjugated using 200-μg antibody lots combined with the MaxPAR antibody conjugation kit (Fluidigm Sciences, Toronto, Canada) according to the manufacturer’s instructions. Following conjugation, antibodies were diluted to 100x working concentration in Candor PBS Antibody Stabilization solution (Candor Bioscience GmbH, Wangen, Germany) and stored at 4 °C. Antibody coefficients of variation (CVs) were calculated by comparing the replicate analyses of aliquots of the healthy donor samples.
Human samples
Fresh bone marrow aspirates were collected immediately (<1 minute) after aspiration into heparinized tubes containing IdU (at a final concentration of approximately 20 μM) and incubated at 37 °C (16). After 15 minutes, samples were fixed using a fixation/stabilization buffer (SmartTube, Palo Alto, CA, USA), according to the manufacturer’s instructions and then frozen at −80 °C for up to 36 months prior to analysis by mass cytometry. Samples were collected from patients at Stanford University Hospital who were undergoing routine diagnostic bone marrow aspiration and provided informed consent to donate a portion of the sample for tissue banking as part of a protocol approved by the Stanford University Institutional Review Board in accordance with the Declaration of Helsinki. The clinical characteristics of each patient are shown in Supplementary Table 1. Healthy control samples were obtained from Allcells (Emoryville, CA, USA) using the same protocol. The exception was healthy donor sample #1; this sample was incubated for ~1 hour with IdU at 37 °C. All healthy cell samples were collected into 15 μM IdU. Bone marrow cell samples were thawed just prior to analysis in a 4 °C water bath, and red cells were lysed using a hypotonic lysis buffer (SmartTube). Cells were then washed twice in cell staining medium (CSM; 1xPBS with 0.5% bovine serum albumin and 0.02% sodium azide) at room temperature.
Antibody staining
Prior to antibody staining, mass tag cellular barcoding was performed as previously described, (17, 18) and full details are provided in Supplementary Methods. Barcoded cells were incubated with surface marker antibodies in 2 mL of CSM for 50 minutes with continuous mixing. Cells were washed twice with CSM, and then surface antibodies were fixed in place by a 15-minute incubation with 1.5% paraformaldehyde (PFA; Electron Microscopy Sciences, Hatfield, PA, USA). Cells were pelleted by centrifugation and resuspended with vortexing in ice-cold methanol. After a 15-minute incubation at −20 °C, cells were washed twice with CSM prior to incubation with antibodies against intracellular signaling proteins for 50 minutes at room temperature as previously described (48).
Mass cytometry
After completion of antibody staining, cells were washed twice with CSM and then incubated overnight (staining panel A) or for 36 hours (staining panel B) in PBS with a 1:5000 dilution of the iridium intercalator pentamethylcyclopentadienyl-Ir(III)-dipyridophenazine (Fluidigm Sciences, Toronto, Canada) and 1.5% paraformaldehyde. Excess intercalator was then removed with one CSM wash and two washes in pure water. Cells were then resuspended in pure water at approximately 1 million cells per mL and mixed with mass standard beads (Fluidigm Sciences). Cell events were acquired on the CyTOF mass cytometer (Fluidigm Sciences) at an event rate of 100–300 events per second with instrument-calibrated dual-count detection (49). Noise reduction was used, a cell length of 10–90, and lower convolution threshold of 200.. After data acquisition, the mass bead signal was used to correct short-term signal fluctuation during the course of each experiment and bead events were removed. (50). Approximately 240,000 cell events were collected for each sample in each of the two staining panels (480,000 total events per sample; Supplementary Table 5).
Immunophenotypic aberrancy analysis
Aberrant immunophenotype analysis was performed by first gating the normal populations into developmental immunophenotypic subsets on the basis of standard surface markers (as in Supplementary Figure 9). AML cells in each population were then compared to the normal samples across the 28 surface markers. Since the normal populations and AML samples were all stained and analyzed in the same tube simultaneously, the same gates were used to identify immunophenotypic populations from each of the AML samples. The median expression level of each maker in each gated population from each patient sample was then calculated. As the 14 replicate normal samples analyzed came from only five healthy donors, AML sample aberrancy was defined conservatively as an AML sample median expression level greater than or less than the median of the similar healthy bone marrow population plus or minus two times the absolute variance of the healthy control samples. For example, over-expression of CD33 in the multipotent progenitor (MPP) population of AML sample #23 was determined as follows: AML#23MPP median CD33 expression > normal sampleMPP median CD33 expression + 2 * absolute variance of normal sampleMPP CD33 expression. This process was repeated for each of the other measured surface markers in each of 35 gated immunophenotypic populations in every AML sample. The summed number of markers with aberrant expression patterns was calculated for each gated immunophenotypic population from each patient sample.
Supplementary Material
Significance.
The mechanisms underlying differences in relapse rates across AML subtypes are poorly understood. This study suggests that known chemotherapy sensitivities of common AML subsets are mediated by cell cycle differences among LSCs and provides a basis for using in vivo functional characterization of AML cells to inform therapy selection.
Acknowledgments
Financial Support: GKB was supported by a developmental research grant from the Stanford Cancer Center; GPN: NIH/NCI grants: U19 AI057229, 1U19AI100627, U54 CA149145, 5U54CA143907, 1R01CA130826, 5R01AI073724, R01 GM109836, R01CA184968, 1R01NS089533, P01 CA034233, R33 CA183654, R33 CA183692, 41000411217, 201303028, N01-HV-00242, HHSN268201000034C, HHSN272201200028C, HHSN272200700038C; CIRM DR1-01477; Department of Defense OC110674, 11491122; FDA: HHSF223201210194C-FDA:BAA-12-00118; and the Bill and Melinda Gates Foundation (GF12141-137101; OPP1113682). G.PN. is also supported by the Rachford and Carlota A. Harris Endowed Professorship.
Abbreviations
- AML
Acute Myeloid Leukemia
- LSCs
Leukemia Stem Cells
- HSCs
Hematopoietic Stem Cells
- OS
Overall Survival
- CV
Coefficient of Variation
- IdU
Iodo-deoxyuridine
- CSM
Cell Staining Medium
- PFA
Paraformaldehyde
- MPP
Multipotent Progenitor
- CR
Complete Response
- APL
Acute Promyelocytic Leukemia
- CBF-AML
Core Binding Factor AML
- ARK-AML
Adverse Risk Karyotype AML
- NK-AML
Normal Karyotype AML
- HSPC
Hematopoietic Stem and Progenitor Cell
- HU
Hydroxyurea
- ADC
Antibody Drug Conjugate
- GO
Gemtuzumab Ozogamycin
Footnotes
Conflicts of Interest: G.K.B. and G.P.N. have provided paid consulting to Fluidigm Sciences, G.P.N. has equity ownership of Fluidigm Sciences. Other authors disclose no potential conflicts of interest.
References
- 1.Estey E, Dohner H. Acute myeloid leukaemia. Lancet. 2006;368:1894–907. doi: 10.1016/S0140-6736(06)69780-8. [DOI] [PubMed] [Google Scholar]
- 2.Grimwade D, Walker H, Oliver F, Wheatley K, Harrison C, Harrison G, et al. The importance of diagnostic cytogenetics on outcome in AML: analysis of 1,612 patients entered into the MRC AML 10 trial. The Medical Research Council Adult and Children’s Leukaemia Working Parties. Blood. 1998;92:2322–33. [PubMed] [Google Scholar]
- 3.Mrózek K, Marcucci G, Nicolet D, Maharry KS, Becker H, Whitman SP, et al. Prognostic Significance of the European LeukemiaNet Standardized System for Reporting Cytogenetic and Molecular Alterations in Adults With Acute Myeloid Leukemia. Journal of Clinical Oncology. 2012;30:4515–23. doi: 10.1200/JCO.2012.43.4738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bonnet D, Dick JE. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat Med. 1997;3:730–7. doi: 10.1038/nm0797-730. [DOI] [PubMed] [Google Scholar]
- 5.van Rhenen A, Moshaver B, Kelder A, Feller N, Nieuwint AW, Zweegman S, et al. Aberrant marker expression patterns on the CD34+CD38- stem cell compartment in acute myeloid leukemia allows to distinguish the malignant from the normal stem cell compartment both at diagnosis and in remission. Leukemia. 2007;21:1700–7. doi: 10.1038/sj.leu.2404754. [DOI] [PubMed] [Google Scholar]
- 6.Gerber JM, Smith BD, Ngwang B, Zhang H, Vala MS, Morsberger L, et al. A clinically relevant population of leukemic CD34+CD38– cells in acute myeloid leukemia. Blood. 2012;119:3571–7. doi: 10.1182/blood-2011-06-364182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Terpstra W, Ploemacher RE, Prins A, van Lom K, Pouwels K, Wognum AW, et al. Fluorouracil selectively spares acute myeloid leukemia cells with long-term growth abilities in immunodeficient mice and in culture. Blood. 1996;88:1944–50. [PubMed] [Google Scholar]
- 8.Guan Y, Gerhard B, Hogge DE. Detection, isolation, and stimulation of quiescent primitive leukemic progenitor cells from patients with acute myeloid leukemia (AML) Blood. 2003;101:3142–9. doi: 10.1182/blood-2002-10-3062. [DOI] [PubMed] [Google Scholar]
- 9.Ishikawa F, Yoshida S, Saito Y, Hijikata A, Kitamura H, Tanaka S, et al. Chemotherapy-resistant human AML stem cells home to and engraft within the bone-marrow endosteal region. Nat Biotech. 2007;25:1315–21. doi: 10.1038/nbt1350. [DOI] [PubMed] [Google Scholar]
- 10.Cheshier SH, Morrison SJ, Liao X, Weissman IL. In vivo proliferation and cell cycle kinetics of long-term self-renewing hematopoietic stem cells. Proceedings of the National Academy of Sciences of the United States of America. 1999;96:3120–5. doi: 10.1073/pnas.96.6.3120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schlenk RF, Benner A, Krauter J, Büchner T, Sauerland C, Ehninger G, et al. Individual Patient Data–Based Meta-Analysis of Patients Aged 16 to 60 Years With Core Binding Factor Acute Myeloid Leukemia: A Survey of the German Acute Myeloid Leukemia Intergroup. Journal of Clinical Oncology. 2004;22:3741–50. doi: 10.1200/JCO.2004.03.012. [DOI] [PubMed] [Google Scholar]
- 12.Burnett AK, Hills RK, Milligan D, Kjeldsen L, Kell J, Russell NH, et al. Identification of Patients With Acute Myeloblastic Leukemia Who Benefit From the Addition of Gemtuzumab Ozogamicin: Results of the MRC AML15 Trial. Journal of Clinical Oncology. 2011;29:369–77. doi: 10.1200/JCO.2010.31.4310. [DOI] [PubMed] [Google Scholar]
- 13.Chou S-C, Tang J-L, Hou H-A, Chou W-C, Hu F-C, Chen C-Y, et al. Prognostic implication of gene mutations on overall survival in the adult acute myeloid leukemia patients receiving or not receiving allogeneic hematopoietic stem cell transplantations. Leukemia Research. 2014;38:1278–84. doi: 10.1016/j.leukres.2014.08.012. [DOI] [PubMed] [Google Scholar]
- 14.Genomic and Epigenomic Landscapes of Adult De Novo Acute Myeloid Leukemia. New England Journal of Medicine. 2013;368:2059–74. doi: 10.1056/NEJMoa1301689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Patel JP, Gönen M, Figueroa ME, Fernandez H, Sun Z, Racevskis J, et al. Prognostic Relevance of Integrated Genetic Profiling in Acute Myeloid Leukemia. New England Journal of Medicine. 2012;366:1079–89. doi: 10.1056/NEJMoa1112304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Behbehani GK, Bendall SC, Clutter MR, Fantl WJ, Nolan GP. Single-cell mass cytometry adapted to measurements of the cell cycle. Cytometry A. 2012;81:552–66. doi: 10.1002/cyto.a.22075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Behbehani GK, Thom C, Zunder ER, Finck R, Gaudilliere B, Fragiadakis GK, et al. Transient partial permeabilization with saponin enables cellular barcoding prior to surface marker staining. Cytometry A. 2014;85:1011–9. doi: 10.1002/cyto.a.22573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zunder ER, Finck R, Behbehani GK, Amir E-aD, Krishnaswamy S, Gonzalez VD, et al. Palladium-based mass tag cell barcoding with a doublet-filtering scheme and single-cell deconvolution algorithm. Nat Protocols. 2015;10:316–33. doi: 10.1038/nprot.2015.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Amir el AD, Davis KL, Tadmor MD, Simonds EF, Levine JH, Bendall SC, et al. viSNE enables visualization of high dimensional single-cell data and reveals phenotypic heterogeneity of leukemia. Nat Biotechnol. 2013;31:545–52. doi: 10.1038/nbt.2594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bendall SC, Simonds EF, Qiu P, Amir el AD, Krutzik PO, Finck R, et al. Single-cell mass cytometry of differential immune and drug responses across a human hematopoietic continuum. Science. 2011;332:687–96. doi: 10.1126/science.1198704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Han L, Qiu P, Zeng Z, Jorgensen JL, Mak DH, Burks JK, et al. Single-cell mass cytometry reveals intracellular survival/proliferative signaling in FLT3-ITD-mutated AML stem/progenitor cells. Cytometry Part A. 2015;87:346–56. doi: 10.1002/cyto.a.22628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Obermann EC, Arber C, Jotterand M, Tichelli A, Hirschmann P, Tzankov A. Expression of pSTAT5 predicts FLT3 internal tandem duplications in acute myeloid leukemia. Ann Hematol. 2010;89:663–9. doi: 10.1007/s00277-009-0890-8. [DOI] [PubMed] [Google Scholar]
- 23.Seedhouse CH, Pallis M, Grundy M, Shang S, Russell NH. FLT3-ITD expression levels and their effect on STAT5 in AML with and without NPM mutations. Br J Haematol. 2009;147:653–61. doi: 10.1111/j.1365-2141.2009.07901.x. [DOI] [PubMed] [Google Scholar]
- 24.Darzynkiewicz Z, Halicka HD, Zhao H, Podhorecka M. Cell Synchronization by Inhibitors of DNA Replication Induces Replication Stress and DNA Damage Response: Analysis by Flow Cytometry. In: Banfalvi G, editor. Cell Cycle Synchronization. Humana Press; 2011. pp. 85–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Oberoi S, Lehrnbecher T, Phillips B, Hitzler J, Ethier M-C, Beyene J, et al. Leukapheresis and low-dose chemotherapy do not reduce early mortality in acute myeloid leukemia hyperleukocytosis: A systematic review and meta-analysis. Leukemia Research. 2014;38:460–8. doi: 10.1016/j.leukres.2014.01.004. [DOI] [PubMed] [Google Scholar]
- 26.Rapin N, Bagger FO, Jendholm J, Mora-Jensen H, Krogh A, Kohlmann A, et al. Comparing cancer vs normal gene expression profiles identifies new disease entities and common transcriptional programs in AML patients. Blood. 2014;123:894–904. doi: 10.1182/blood-2013-02-485771. [DOI] [PubMed] [Google Scholar]
- 27.Taussig DC, Pearce DJ, Simpson C, Rohatiner AZ, Lister TA, Kelly G, et al. Hematopoietic stem cells express multiple myeloid markers: implications for the origin and targeted therapy of acute myeloid leukemia. Blood. 2005;106:4086–92. doi: 10.1182/blood-2005-03-1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kang LC, Dunphy CH. Immunoreactivity of MIC2 (CD99) and terminal deoxynucleotidyl transferase in bone marrow clot and core specimens of acute myeloid leukemias and myelodysplastic syndromes. Archives of pathology & laboratory medicine. 2006;130:153–7. doi: 10.5858/2006-130-153-IOMCAT. [DOI] [PubMed] [Google Scholar]
- 29.Lin LI, Chen CY, Lin DT, Tsay W, Tang JL, Yeh YC, et al. Characterization of CEBPA mutations in acute myeloid leukemia: most patients with CEBPA mutations have biallelic mutations and show a distinct immunophenotype of the leukemic cells. Clin Cancer Res. 2005;11:1372–9. doi: 10.1158/1078-0432.CCR-04-1816. [DOI] [PubMed] [Google Scholar]
- 30.Cozzio A, Passegué E, Ayton PM, Karsunky H, Cleary ML, Weissman IL. Similar MLL-associated leukemias arising from self-renewing stem cells and short-lived myeloid progenitors. Genes & Development. 2003;17:3029–35. doi: 10.1101/gad.1143403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pearce DJ, Taussig D, Zibara K, Smith L-L, Ridler CM, Preudhomme C, et al. AML engraftment in the NOD/SCID assay reflects the outcome of AML: implications for our understanding of the heterogeneity of AML. Blood. 2006;107:1166–73. doi: 10.1182/blood-2005-06-2325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rombouts WJ, Martens AC, Ploemacher RE. Identification of variables determining the engraftment potential of human acute myeloid leukemia in the immunodeficient NOD/SCID human chimera model. Leukemia. 2000;14:889–97. doi: 10.1038/sj.leu.2401777. [DOI] [PubMed] [Google Scholar]
- 33.Monaco G, Konopleva M, Munsell M, Leysath C, Wang R-Y, Jackson CE, et al. Engraftment of Acute Myeloid Leukemia in NOD/SCID Mice Is Independent of CXCR4 and Predicts Poor Patient Survival. Stem Cells. 2004;22:188–201. doi: 10.1634/stemcells.22-2-188. [DOI] [PubMed] [Google Scholar]
- 34.Schubert M, Herbert N, Taubert I, Ran D, Singh R, Eckstein V, et al. Differential survival of AML subpopulations in NOD/SCID mice. Exp Hematol. 2011;39:250–63. e4. doi: 10.1016/j.exphem.2010.10.010. [DOI] [PubMed] [Google Scholar]
- 35.Qiu P, Simonds EF, Bendall SC, Gibbs KD, Bruggner RV, Linderman MD, et al. Extracting a cellular hierarchy from high-dimensional cytometry data with SPADE. Nat Biotech. 2011;29:886–91. doi: 10.1038/nbt.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sarry J-E, Murphy K, Perry R, Sanchez PV, Secreto A, Keefer C, et al. Human acute myelogenous leukemia stem cells are rare and heterogeneous when assayed in NOD/SCID/IL2Rγc-deficient mice. The Journal of Clinical Investigation. 2011;121:384–95. doi: 10.1172/JCI41495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Krivtsov AV, Figueroa ME, Sinha AU, Stubbs MC, Feng Z, Valk PJM, et al. Cell of origin determines clinically relevant subtypes of MLL-rearranged AML. Leukemia. 2013;27:852–60. doi: 10.1038/leu.2012.363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lapidot T, Sirard C, Vormoor J, Murdoch B, Hoang T, Caceres-Cortes J, et al. A cell initiating human acute myeloid leukaemia after transplantation into SCID mice. Nature. 1994;367:645–8. doi: 10.1038/367645a0. [DOI] [PubMed] [Google Scholar]
- 39.Blair A, Hogge DE, Sutherland HJ. Most Acute Myeloid Leukemia Progenitor Cells With Long-Term Proliferative Ability In Vitro and In Vivo Have the Phenotype CD34+/CD71–/HLA-DR–. Blood. 1998;92:4325–35. [PubMed] [Google Scholar]
- 40.Taussig DC, Vargaftig J, Miraki-Moud F, Griessinger E, Sharrock K, Luke T, et al. Leukemia-initiating cells from some acute myeloid leukemia patients with mutated nucleophosmin reside in the CD34(−) fraction. Blood. 2010;115:1976–84. doi: 10.1182/blood-2009-02-206565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Martelli MP, Pettirossi V, Thiede C, Bonifacio E, Mezzasoma F, Cecchini D, et al. CD34+ cells from AML with mutated NPM1 harbor cytoplasmic mutated nucleophosmin and generate leukemia in immunocompromised mice. Blood. 2010;116:3907–22. doi: 10.1182/blood-2009-08-238899. [DOI] [PubMed] [Google Scholar]
- 42.Bloomfield CD, Lawrence D, Byrd JC, Carroll A, Pettenati MJ, Tantravahi R, et al. Frequency of prolonged remission duration after high-dose cytarabine intensification in acute myeloid leukemia varies by cytogenetic subtype. Cancer research. 1998;58:4173–9. [PubMed] [Google Scholar]
- 43.Sampath D, Rao VA, Plunkett W. Mechanisms of apoptosis induction by nucleoside analogs. Oncogene. 2003;22:9063–74. doi: 10.1038/sj.onc.1207229. [DOI] [PubMed] [Google Scholar]
- 44.Passegue E, Wagers AJ, Giuriato S, Anderson WC, Weissman IL. Global analysis of proliferation and cell cycle gene expression in the regulation of hematopoietic stem and progenitor cell fates. J Exp Med. 2005;202:1599–611. doi: 10.1084/jem.20050967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Raza A, Preisler H, Lampkin B, Yousuf N, Tucker C, Peters N, et al. Biological significance of cell cycle kinetics in 128 standard risk newly diagnosed patients with acute myelocytic leukaemia. British Journal of Haematology. 1991;79:33–9. doi: 10.1111/j.1365-2141.1991.tb08003.x. [DOI] [PubMed] [Google Scholar]
- 46.Renneville A, Abdelali RB, Chevret S, Nibourel O, Cheok M, Pautas C, et al. Clinical impact of gene mutations and lesions detected by SNP-array karyotyping in acute myeloid leukemia patients in the context of gemtuzumab ozogamicin treatment: results of the ALFA-0701 trial. Oncotarget. 2014;5:916–32. doi: 10.18632/oncotarget.1536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Castaigne S, Pautas C, Terre C, Raffoux E, Bordessoule D, Bastie JN, et al. Effect of gemtuzumab ozogamicin on survival of adult patients with de-novo acute myeloid leukaemia (ALFA-0701): a randomised, open-label, phase 3 study. Lancet. 2012;379:1508–16. doi: 10.1016/S0140-6736(12)60485-1. [DOI] [PubMed] [Google Scholar]
- 48.Krutzik PO, Nolan GP. Intracellular phospho-protein staining techniques for flow cytometry: monitoring single cell signaling events. Cytometry A. 2003;55:61–70. doi: 10.1002/cyto.a.10072. [DOI] [PubMed] [Google Scholar]
- 49.Ornatsky OI, Kinach R, Bandura DR, Lou X, Tanner SD, Baranov VI, et al. Development of analytical methods for multiplex bio-assay with inductively coupled plasma mass spectrometry. J Anal At Spectrom. 2008;23:463–9. doi: 10.1039/b710510j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Finck R, Simonds EF, Jager A, Krishnaswamy S, Sachs K, Fantl W, et al. Normalization of mass cytometry data with bead standards. Cytometry A. 2013;83:483–94. doi: 10.1002/cyto.a.22271. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.