

Abstract
Investigating the mechanisms of action (MOAs) of bioactive compounds and the deconvolution of their cellular targets is an important and challenging undertaking. Drug resistance in model organisms such as S. cerevisiae has long been a means for discovering drug targets and MOAs. Strains are selected for resistance to a drug of interest, and the resistance mutations can often be mapped to the drug’s molecular target using classical genetic techniques. Here we demonstrate the use of next generation sequencing (NGS) to identify mutations that confer resistance to two well-characterized drugs, benomyl and rapamycin. Applying NGS to pools of drug-resistant mutants, we develop a simple system for ranking single nucleotide polymorphisms (SNPs) based on their prevalence in the pool, and for ranking genes based on the number of SNPs that they contain. We clearly identified the known targets of benomyl (TUB2) and rapamycin (FPR1) as the highest-ranking genes under this system. The highest-ranking SNPs corresponded to specific amino acid changes that are known to confer resistance to these drugs. We also found that by screening in a pdr1Δ null background strain that lacks a transcription factor regulating the expression of drug efflux pumps, and by pre-screening mutants in a panel of unrelated anti-fungal agents, we were able to mitigate against the selection of multi-drug resistance (MDR) mutants. We call our approach “Mutagenesis to Uncover Targets by deep Sequencing, or “MUTseq”, and show through this proof-of-concept study its potential utility in characterizing MOAs and targets of novel compounds.a
Introduction
Phenotypic screening provides a powerful mechanism for identifying compounds with novel mechanisms of action (MOAs). Such compounds can become therapeutic leads themselves, or can be used to illuminate new druggable targets. According to one survey, of the 50 first-in-class small molecule drug approvals with novel molecular MOAs from 1999-2008, 28 were discovered through phenotypic screens while only 17 were discovered from target-based programs[1]. The major drawback with phenotypic approaches is that there is no general method for identifying the molecular targets of active compounds. The most common approaches to target ID have involved biochemical purification and affinity-based methods, which often require the costly and time-consuming synthesis of covalently immobilized derivatives. Notable exceptions include the recent application of deep sequencing technology to pinpoint drug resistance mutations in HCT-116 cells[2] and the use of mass spectrometry to identify targets based on their stability to proteolysis in the presence of ligand[3].
Although relatively uncommon in higher eukaryotes, genetic methods have been used for some time to facilitate target ID in simpler systems, especially fungi and bacteria. The budding yeast S. cerevisiae, in particular, is an excellent model system for the study of the mechanisms of action of small molecules due to the relative ease with which it can be manipulated genetically and the high degree of conservation in basic cellular processes between yeast and higher eukaryotes (reviewed in[4]). Indeed, a variety of genome-wide tools have been developed for investigating small molecule MOAs in S. cerevisiae. These include the use of barcoded deletion strains to identify chemical-genetic interactions[5-8], high-copy expression libraries to identify phenotypic suppressors[9-11], and high-throughput complementation strategies using heterologous expression of barcoded open reading frames(ORFs)[12]. More recently, an ultra-diverse, barcoded “variomic” library containing thousands of alternate alleles for every yeast gene was used to identify drug resistance alleles which can point directly to drug targets[13]. Using these strategies, not only primary targets, but also “off-target” activities and alternate modes of action for a number of drugs have been identified in yeast[14].
In contrast to techniques that rely on genomic libraries of yeast strains and expression constructs, point mutations that confer drug resistance can be mapped directly to a drug’s molecular target. This strategy was used, for example, to identify the targets of rapamycin, TOR1 and TOR2 in yeast[15-17], which ultimately helped to confirm the important homolog mTOR in humans[18]. In addition, targets of the antifungal compounds LY214352 (dihydroorotate dehydrogenase) [19] and UK-118005 (RNA Pol III)[20] were identified by cloning drug resistance genes. In general, classical genetic techniques are employed to characterize specific drug resistance mutations in yeast[20] [21]. These methods require genetic crosses and the cloning of large numbers of mutant alleles of genes, and also a plentiful supply of compound, which, in many cases, may be in limited supply and/or difficult to synthesize.
Next-generation sequencing (NGS) technology has made whole-genome sequencing a viable alternative to traditional genetic mapping approaches. Mutations that confer drug resistance can be pinpointed by simply comparing sequence reads of compound-resistant strains to those of the parental strain. Genes or pathways that display an enrichment in new mutations represent potential targets. For example, the target of a new anti-tuberculosis drug was identified by whole-genome sequencing of resistant clones[22], and NGS approaches have been used to identify mutations responsible for echinocandin resistance in Candida galbrata[23]. Deep sequencing was also used to identify mutations that confer resistance to oxidative stress in S. cerevisiae[24].
Although these studies point toward whole-genome sequencing as an attractive approach for characterizing drug resistant mutants in S. cerevisiae, the specific application of NGS toward the identification of small molecule targets has not been reported. Here we describe the use of NGS to identify drug targets in yeast using a straightforward approach that does not involve the use of tagged genomic libraries or require downstream genetic manipulations. We found that screening for resistance mutants in a pdr1Δ deletion strain minimized the selection of multi-drug resistant (MDR) mutants, and demonstrate that MDR false positives can be further limited by performing cross-resistance screens of candidate mutants in a panel of unrelated drugs. We identified the known targets of benomyl and rapamycin using this approach (Figure 1), and show that NGS offers an orthogonal technique to other chemical genetic approaches available for studying small molecule MOAs in yeast.
Figure 1. Benomyl and rapamycin are well-studied antifungals.
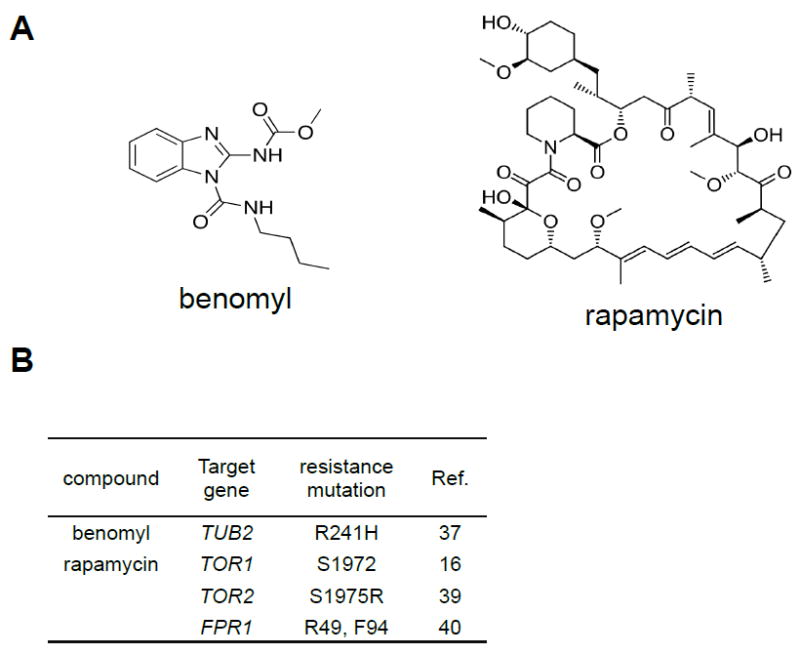
(A) Structures of benomyl and rapamycin.
(B) A list of the genes that are known to encode the proteins that are targeted by benomyl and rapamycin. Also listed are residues that when modulated have been shown previously to confer resistance or inhibit drug binding.
Methods
Resistant Mutant selection
The pdr1Δ strain used for this study was created via homologous recombination from the background strain, BY4741, a derivative of S288C[25](see Supplemental Information for genotype). A preliminary growth study was conducted for both benomyl and rapamycin to determine an optimal drug screening concentration. YPD plates containing 1X, 5X, 10X, 20X, and 40X the IC50 of each compound (IC50 = 30 μM for benomyl; 25 nM for rapamycin) were inoculated with ~107 pdr1Δ cells and incubated for two days at 30°C. The lowest concentration at which less than five colonies were observed was chosen as the dose for the selection of resistant mutants. The optimal selection concentrations for benomyl and rapamycin were determined to be 150 μM and 0.25 μM, respectively. To mutagenize cells with ethyl methanesulfonate (EMS), 1 mL of an overnight culture (~108 cells/mL) of pdr1Δ was added to a 1.5 mL microcentrifuge tube and pelleted by centrifugation. The supernatant was discarded and the pellet was resuspended in sterile water. The cells were pelleted again and then resuspended in 1 mL of 0.1 M sodium phosphate buffer at pH 7. Next, 30 μL of EMS was added to the EMS sample tube and the tube was vortexed for 15 s and then incubated with inversion at 30° C for 1 h. After incubation, the cells were pelleted and resuspended in 200 μL of 5% sodium thiosulfate to quench the remaining EMS, and then transferred to a clean tube. This thiosulfate wash step was repeated for a total of three times. After the final wash the pellet was resuspended in 1 mL of water, plated in 100 μl aliquots (~107 cells) onto 10 plates containing the selection dose determined above and incubated at 30° C for 2 days. As a control, a second aliquot of cells treated identically except for the omission of EMS was selected for resistance in an attempt to discover spontaneous drug-resistant mutants.
Confirmation of resistance and MDR cross-resistance screening
EMS-treated and spontaneously resistant mutants from the initial selection were confirmed by re-streaking onto YPD/agar plates containing compound, along with the parental starting strain to serve as a non-viable control. Mutants that yielded colonies within 3 days were considered resistant and evaluated further in a multi-drug resistance cross-screen. This screen was performed using the 384 halo assay as previously reported [26]. Overnight cultures of resistant mutants were seeded in YPD top-agar at an OD600 of 0.06 and poured into OmniTrays. After the agar solidified, lethal doses of sixteen known anti-fungal compounds (see Supporting Information for a list of the anti-fungals used), dissolved in DMSO, were pinned into the agar. Plates were incubated at 30° C overnight and then analyzed using an optical density plate reader to quantify growth inhibition by assigning each anti-fungal a ‘halo score’ for that particular resistant strain.
Genomic DNA preparation
Mutants chosen for sequencing were grown overnight in 10 mL YPD liquid at 30°C. To pool samples, cultures of individual mutants were diluted to equal ODs and equal amounts of each strain were mixed to give a final volume of 10 mL. Cells were pelleted by centrifugation, resuspended in 1 mL of sterile water and transferred to a 1.5 mL microcentrifuge tube. The cells were pelleted again and resuspended in 200 μl of lysis buffer (1% SDS, 2% Triton X 100, 100 mM NaCl, 10 mM Tris pH 8, 1 mM EDTA). Approximately 3 g of acid-washed glass beads and 200 μl of phenol:chloroform:isoamyl alcohol (25:24:1) were added to the resuspended cells, which were then vortexed for 3 min. An additional 200 μl of TE was added to the tube and the mixture was centrifuged for 5 min. Following centrifugation, 350 μl of the aqueous (top) layer was carefully removed with a pipette and transferred to a new tube with 1 mL of cold 100% ethanol. The DNA was allowed to precipitate at -20° for at least 1 h (at most, overnight) and then centrifuged for 10 min to pellet the DNA. The DNA pellet was then resuspended in 400 μl of TE and 30 μg of RNase A was added and allowed to incubate at 37° C for 2 h. The sample was then extracted with 400 μl of chloroform:isoamyl alcohol (24:1). Following centrifugation, 350 μl of the aqueous layer was removed and placed into a new tube containing 1 mL of cold 100% ethanol and allowed to precipitate for at least 1 h at 20° C. After this second precipitation the DNA was pelleted and washed twice with 70% ethanol. The DNA pellet was air-dried at room temperature for 10 m and then resuspended in ultra pure water (80-100 μl). The quality and quantity of all samples were checked by gel electrophoresis.
Whole-genome DNA sequencing of yeast cells
For NGS, high-molecular weight genomic DNA (gDNA) was obtained from pdr1Δ benomyl and rapamycin resistant samples as described above. For the DNA library prep, 500 ng of gDNA was first sheared down to 300-400 bp using the Covaris S2 (Woburn, Massachusetts) according to the manufacture’s recommendations. A target insert size of 300-400bp was then size-selected using an automated electrophoretic DNA fractionation system, LabChip XT (Caliper Life Sciences, Hopkinton, Massachusetts). Paired-end sequencing libraries were prepared using Illumina’s TruSeq DNA Sample Preparation Kit (San Diego, CA). Following DNA library construction, samples were quantified using the Agilent Bioanalyzer per manufacturer’s protocol (Santa Clara, CA). DNA libraries were sequenced using the Illumina HiSeq 2000 in one flow cell lane with sequencing paired-end read length at 2 × 100 bp. Reads were de-multiplexed using CASAVA (version 1.8.2).
Sequencing data analysis
Using the software tool Bowtie 2[27], we mapped the raw Illumina sequence data (as .fastq files representing all paired-end reads) from the drug-resistant mutants, as well as the parental strain pdr1Δ, to the most current S. cerivisiae reference genome assembly (sacCer3; April 2011). Sequencing was performed at a depth of 116 and 100 for the paired reads, and these were trimmed to 70 bases each for the mapping. We kept only the uniquely mapping reads to generate .bam files for each sample, including the parental strain. We found that 95% of the genome was covered by at least one read. These reads were then filtered to include only those that were inside the 1%-tile and 96%-tile in the read-depth distribution (see Supporting Information S1). We applied the genome analysis toolkit GATK[28, 29] to the .bam files from each mapped sample to produce SNP calls relative to the sacCer3 reference genome. In order to generate SNP calls, the mapped files were processed using the GATK software to generate VCF files, using the following quality filters for calling SNPs: MQ < 30; FS > 60; ReadPosRankSum < -8.0. For each drug-resistant sample, we subtracted those SNPs that were also found in the pdr1Δ parental sample.
Results
Use of pdr1 deletion strain as parental strain for mutant selection
In an earlier study, we had identified a number of novel cytotoxic compounds in S. cerevisiae[30] and had set out to identify their targets by selecting and sequencing drug-resistant mutants. In the first of these studies, we selected eight spontaneous mutants that were resistant to the drug of interest but remained sensitive to a panel of unrelated antifungal compounds. We anticipated that screening for cross-resistance against a diverse panel of unrelated drugs would allow us to eliminate any mutants that acquired resistance through multi-drug resistance (MDR) mechanisms, e.g., through up-regulation of drug efflux pumps or xenobiotic metabolism. Eight of the most promising drug-resistant mutants were selected based on a) resistance to the drug of interest and b) lack of resistance to the cross-screening panel. Sequencing the eight mutants plus the parental strain using NGS (SOLID) technology showed that, despite our efforts to eliminate MDR mutants, all eight of the resistant strains carried a mutation in the multidrug resistance gene PDR1. The PDR1 gene encodes a transcription factor that regulates the expression of multi-drug resistance genes including drug efflux pumps in the PDR family. A variety of point mutations in PDR1 are known to confer the MDR phenotype[31] and are clustered in distinct regions within PDR1. All eight of the pdr1 point mutations in our drug-resistant samples also clustered in these regions (data not shown).
Different point mutations in PDR1 are known to confer unique patterns of drug resistance, possibly due to the effect of each point mutation on the expression of specific ABC transporters[32]. Thus, while the drug resistance in these mutants appeared to be specific to our drug of interest, this specificity was probably due to the particular efficiency with which the drug was effluxed compared to the other drugs in the cross-screening panel, and not to a mechanism of resistance related to the drug’s specific molecular target. These observations prompted us to select for resistance mutations in a pdr1Δ genetic background, which would not only eliminate pdr1 mutations as sources of drug resistance, but would also make the yeast more drug sensitive in general. This enhanced sensitivity would allow us to use less compound in the selection experiments and could help to minimize other off-target effects. We next set out to test these hypotheses using two drugs whose targets are well established in yeast, benomyl and rapamycin.
Selection and cross-screening of benomyl- and rapamycin- resistant mutants
Equal aliquots of EMS-treated and -untreated pdr1Δ cells were plated onto YPD/agar with an optimal lethal dose of either benomyl or rapamycin, determined based on preliminary growth experiments with the parental strain (See Methods). 65 benomyl resistant colonies were isolated from the EMS-treated cells, whereas no resistant colonies arose from the non-EMS-treated cells. All 65 EMS-derived benomyl resistant mutants formed substantial colonies when subjected to a second round of selection on benomyl media, while the parental control strain produced no colonies. Rapamycin selection yielded only six resistant colonies, one from EMS-treated cells and four from non-EMS-treated cells. In a second round of selection on rapamycin plates, all six mutants formed normal sized colonies while the parental formed none.
Using an automated yeast halo assay that we had developed previously[33], we screened the benomyl- and rapamycin-resistant mutants for multi-drug resistance in the presence of 14 antifungal drugs representing a variety of MOA classes (Figure 2). Each mutant was seeded in agar and poured into a 384-well-format “omni-tray”, and DMSO stock solutions of benomyl, rapamycin, and the 14 drugs in the cross-resistance panel were pin-transferred to each mutant tray. Mutants were chosen for sequencing based on two criteria: 1) They showed no discernable halo for the drug of interest (benomyl or rapamycin); and 2) On average, they were as sensitive as the parental control to the 14-drug panel. Based on the above criteria, 9 of the 65 benomyl-resistant mutants and 5 of the 6 rapamycin-resistant mutants were pooled and genomic DNA from the pools were prepared for sequencing using Illumina HiSeq 2000 (San Diego, Ca).
Figure 2. MDR cross resistance screen.
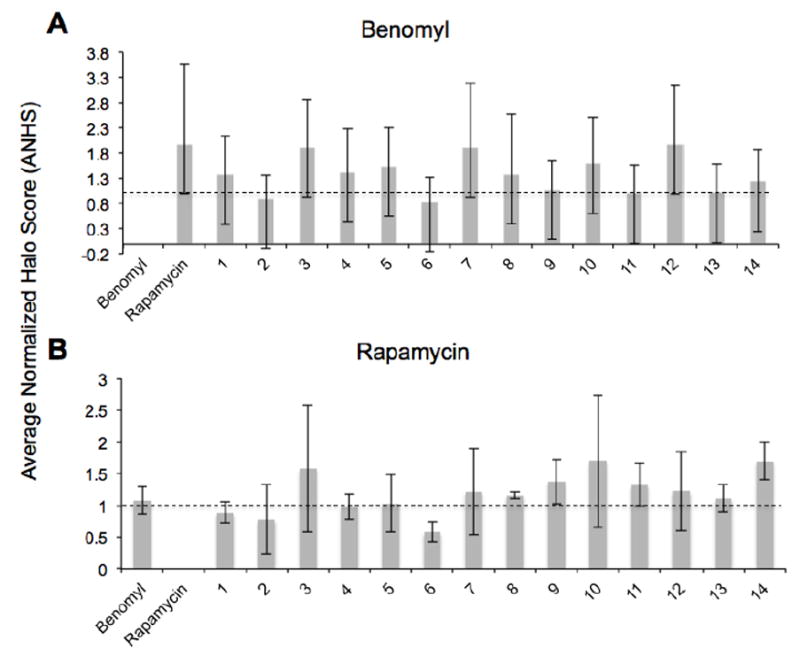
(A) Chart displaying the average normalized halo score (ANHS) of the nine benomyl resistant mutants for 16 antifungals, including benomyl (first entry). ANHS values below one (dotted line) indicate resistance, and those above the line indicate sensitivity. Benomyl ANHS values of zero corresponds to an IC50 > 2mM.
(B) Chart displaying the ANHS of the five rapamycin resistant mutants for 16 antifugals including rapamycin (second entry). Rapamycin ANHS value of zero corresponds to an IC50 value of > 1μM.
Sequencing of benomyl- and rapamycin- resistant mutants
We sequenced the pooled benomyl- and rapamycin-resistant mutants using the Illumina HiSeq 2000, and the reads were mapped onto the most current S. cerevisiae reference genome (sacCer3). The pool of 9 benomyl- and 5 rapamycin-resistant mutants were sequenced to average read depths of 211 and 199, respectively. When we compared the pdr1Δ sequence with that of the reference genome, ~20% of the 128 SNPs in the putatively haploid pdr1Δ appeared heterozygous. The detection of non-uniform SNPs in a haploid organism is consistent with reports from other genome-wide studies in yeast[24], in which spurious diploidization and transient polysomy has been known to occur during or prior to selection[34, 35]. Indeed, all of the SNPs that appeared heterozygous in the parental strain were also found at a similar allele frequency in the drug-selected pools. For this reason, all SNPs that were called in the parental strain were discarded, including at loci that appeared to be diploid.
After subtracting SNPs that were inherited from the parental strain, we obtained 1401 SNPs unique to the benomyl pool, averaging to ~156 SNPs per strain. The vast majority (97%) of these mutations were G-to-A and C-to-T transitions and distributed roughly evenly among the chromosomes. The number and type of mutations were consistent with previous reports on the base change frequency and specificity observed in EMS-treated yeast[36]. The SNPs were further filtered to remove synonymous and non-coding mutations, yielding a final list of 700 exonic SNPs mapping to 639 unique genes (Figure 3).
Figure 3. Benomyl pool SNP statistics.

(A) Matrix showing the base changes for all new SNPs (N=1401) in the benomyl pool.
(B) Mutations by region for all new SNPs in the benomyl pool.
(C) Distribution of exonic mutation types (N=1006).
For each SNP, an “allele frequency” (AF) was calculated as the proportion of total reads at that locus carrying the alternate allele. Since each mutant contributed roughly the same amount of DNA to the pool, the number of strains carrying a particular SNP within the pool, i.e., the “allele count” (AC), could be estimated using the GATK software package. We initially set out to determine the significance of obtaining a particular AC by estimating probabilities based on the known mutation frequency and effective EMS-sensitive genome, assuming a random distribution of SNPs among the 9 strains (see Supporting Information, statistical analysis). If all the SNPs in the pool were distributed randomly over the 9 genomes in the pool, we calculate that observing even one mutation shared by two or more strains in the pool would occur in about 1 in 5 experiments. Finding three or more strains with the same mutation would occur by chance in only 2 in 105 experiments. And yet we observed 127 SNPs with AC values of 2, and 20 SNPs with AC values of 3. Many of these mutations were synonymous or occurred in non-coding regions, suggesting that they were not selective, and were most likely due to variations in the amount of DNA introduced per strain or variations among strains during the amplification of the DNA. Nonetheless, of the 700 SNPs in the benomyl-resistant pool, the SNP with the highest AC (AC = 4) was a C-to-T transition located in TUB2, the gene that encodes benomyl’s known target, β-tubulin.
Multiple amino acid changes can confer drug resistance within the same target. Therefore, extending the allele count analysis to the gene level can, in principle, add another layer of confidence to the analysis by sidestepping the noise intrinsic to the calculated allele frequencies. For each of the 639 SNP-bearing genes from the benomyl pool, we created a new metric called the gene-level allele count (GL-AC), which represents the sum of the allele counts of all SNPs within a gene (see supplemental, information, statistical analysis, for formula). This gives us an upper bound on the number of strains with a mutation in a particular gene. The known benomyl target TUB2 ranks highest among all genes with a GL-AC score of 8, which means that as many as 8 of the 9 strains might have mutations in that gene. The next-highest ranked genes were two genes with GL-AC scores of 5 (Table 1).
Table 1. Sequencing results for benomyl pool.
Highest ranking genes based on (gene level allele count) GL-AC.
| gene | position | alternate reads/ total reads | AC | p-value (AC) | GL-AC | p-value (GL-AC) | mutation |
|---|---|---|---|---|---|---|---|
| TUB2 | chrVI:57056 | 105/223 | 4 | 1.20E-09 | 8 | 0.008 | R241C |
| chrVI:57371 | 69/250 | 3 | 1.80E-05 | P346S | |||
| chrVI:56403 | 17/214 | 1 | 1 | T23S | |||
| SCJ1 | chrXIII:695582 | 38/125 | 3 | 1.80E-05 | 5 | 0.65 | P78L |
| chrXIII:696377 | 18/186 | 1 | 1 | G343D | |||
| chrXIII:6964 | 22/180 | 1 | 1 | V359I | |||
| SPE4 | chrXII:433410 | 58/195 | 3 | 1.80E-05 | 5 | 0.65 | V106I |
| chrXII:433661 | 51/209 | 2 | 0.17 | S22N |
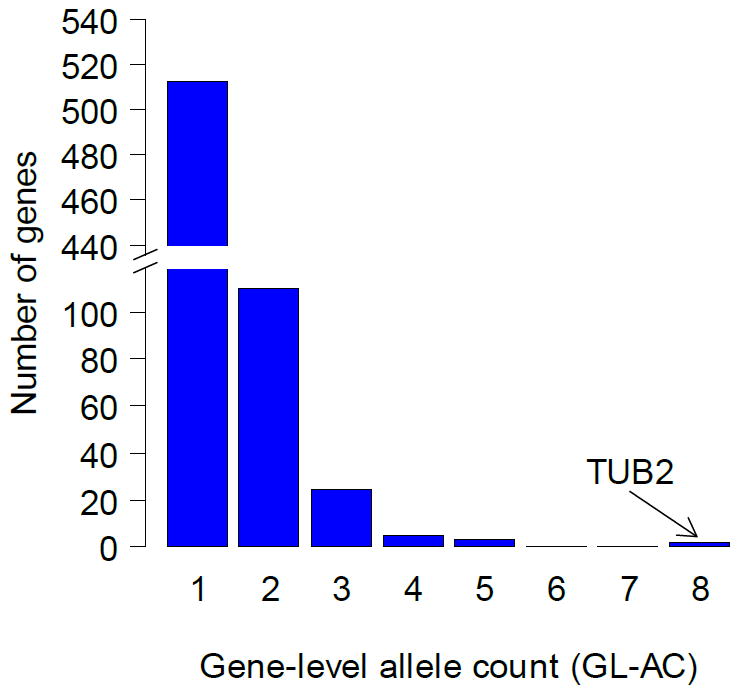
In the histogram of GL-AC scores presented in Figure 4, TUB2 stands out among the other genes with its GL-AC score of 8. To determine if the GL-AC scores we observe are evidence of selection, we estimated the probability of observing any genes with GL-AC scores as large or larger than the ones we observe under non-selective conditions (i.e., the GL-AC distribution that one would expect if the 9 strains had been selected at random from a pool of EMS-treated strains under nonselective conditions). We did this by simulating the distribution of GL-ACs under the assumption that the SNPs are distributed randomly over the genes, taking into account gene length and base pair composition (based on the specificity of EMS-treatment for G and C (Supporting Information, statistical analysis)). This simulation showed that without selective pressure, from a pool of 9 EMS-treated strains finding even one gene with a GL-AC of 8 or higher would occur in only 8 out of 1000 experiments. On the other hand, finding at least a single gene with a GL-AC of 5 or higher would occur in 650 out of 1000 experiments. Therefore, TUB2 is the only gene with a GL-AC value that is highly unlikely to have occurred by chance.
Figure 4. Distribution of gene level allele counts for benomyl pool for GL-AC ≥ 1.
For the 5 pooled rapamycin mutants, one was derived from an EMS-treated line and the other four were spontaneous mutants. The genomic DNA of the 5 mutants were pooled in equal amounts and sequenced in the same manner as described above, to an average read depth of 199. SNP calls were performed using the same parameters as for the benomyl pool, yielding 116 SNPs that were not inherited from the parent. After filtering out synonymous and non-coding SNPs, 50 exonic, non-synonymous SNPs remained (Figure 5), each mapping to a unique gene. All but one of these SNPs had allele frequencies near 1/5 as depicted in the histogram in figure 6. The only SNP with an AC > 1 was located in the gene FPR1 (AC=3), which encodes the yeast homolog of the human FK506-binding protein FKBP12, a well-known target of rapamycin (Table 2). Furthermore, there were two unique, non-parental alleles found at the same locus (an A-to-G and an A-to-T transition at position chrXIV:372100) proving that, at the very least, there were two strains harboring FPR1 SNPs. The probability of observing three or more strains with a mutation at the same base by chance is very low (p-value = 1.4 × 10-9) given the low level of mutations in the genome and the fact that there are only 5 strains in the pool (Table 2). The two SNPs represent different but similar amino acid changes: Phe43-to-Ile and Phe43-to- Leu, located near the rapamycin binding pocket in the crystal structure (PDB 1FKB)[38]. Fpr1p binds rapamycin with high affinity, forming a toxic complex that binds and inhibits target of rapamycin proteins Tor1 and Tor2. Indeed, TOR1 was among the 50 genes in the rapamycin-resistant pool that carried a mutation.
Figure 5. Rapamycin pool SNP statistics.
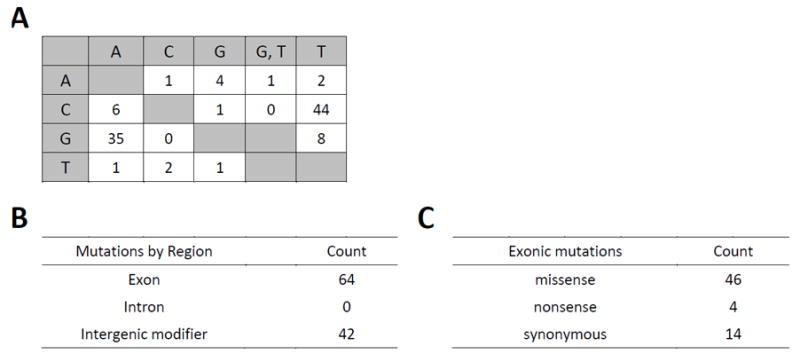
(A) Matrix showing the base changes for all new SNPs (N=106) in the benomyl pool.
(B) Mutations by region for all new SNPs in the rapamycin pool.
(C) Distribution of exonic mutation types (N=64)
Figure 6. Distribution of gene level allele counts for rapamycin pool, including those with GL-AC = 0.
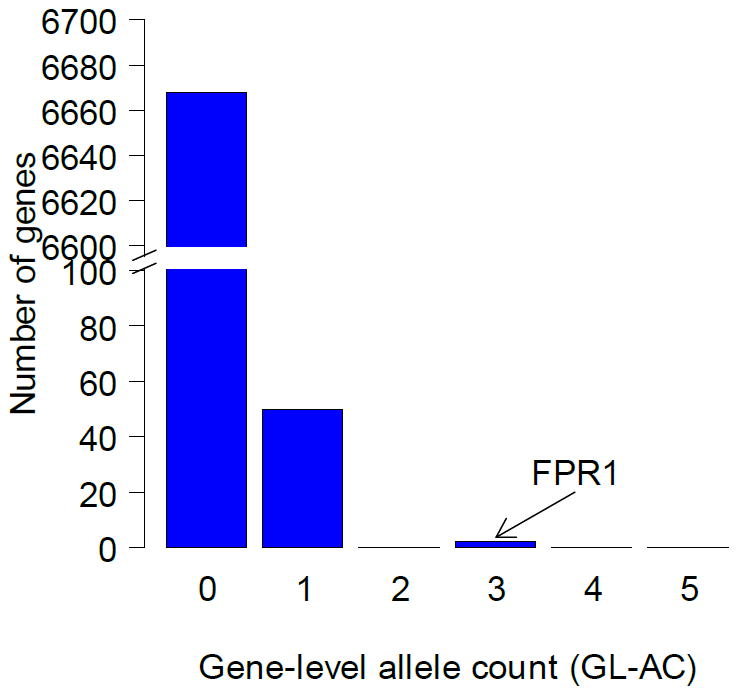
Table 2. Sequencing results for rapamycin pool.
Highest ranking genes based on (gene level allele count) GL-AC.
| gene | position | alternate reads/ total reads | AC | p-value (AC) | GL-AC | p-value (GL-AC) | mutation | |
|---|---|---|---|---|---|---|---|---|
| 1 | FPR1 | chrXIV:372100 | 38/63 | 3 | 1.40E-09 | 3 | 0.002 | F43I, F43L |
| 2 | MZM | chrIV:1436553 | 38/177 | 1 | 1 | 1 | 1 | A113S |
| 3 | DCR2 | chrXII:847561 | 33/156 | 1 | 1 | 1 | 1 | G522R |
| 4 | ITC1 | chrVII:258208 | 44/212 | 1 | 1 | 1 | 1 | R168C |
| 5 | ARO2 | chrVII:226630 | 26/136 | 1 | 1 | 1 | 1 | P78S |
| : | : | |||||||
| : | : | |||||||
| : | : | |||||||
| 16 | TOR1 | chrX:565331 | 33/204 | 1 | 1 | 1 | 1 | S1972R |
| : | : | |||||||
| : | : | |||||||
| : | : | |||||||
| 50 | NTE1 | chrXIII:157425 | 12/221 | 1 | 1 | 1 | 1 | D278E |
Discussion
This proof-of-concept study provides a demonstration of the use of MUTseq for identifying drug targets in yeast. Analysis of the sequencing data from pools of benomyl- and rapamycin-resistant mutants resulted in a ranked list of genes for each drug, at the top of which were their known targets, TUB2 and FPR1. Using MUTseq to confirm the target(s) of benomyl revealed three genes with GL-AC counts of five or more, with TUB2, the gene that encodes benomyl’s known target, β-tubulin, at the top of the list. Interestingly, the most frequent alternate allele in TUB2 that we identified corresponds to an Arg-to-Cys mutation at position 241 in β-tubulin, which is at the same site as a mutation (Arg to His) previously found in a screen for benomyl resistance[37].
Applying MUTseq to the antifungal, rapamycin revealed the gene FPR1 (GL-AC=2), which encodes the homolog of the rapamycin- and FK506 binding protein FKBP12. Since four of the five strains in the rapamycin-resistant pool were selected from a set of spontaneous mutants, there were considerably fewer SNPs than in the benomyl pool. While the mutations that we identified in FPR1 (F43I/L) has not been reported previously, in the crystal structure of the complex with FKBP12 (PDB 1FKB)[38], Phe43 projects directly into the FK506/rapamycin binding pocket. The SNP that we found in TOR1 corresponds to the same mutation at Ser1972 that had been shown previously to confer resistance to rapamycin in yeast[16].
In the absence of selection, the likelihood of finding any SNP with an AC of greater than 2 in non-exonic bases in a pool of 9 strains is very low. It is unlikely that two or more of the benomyl mutants are clones since the EMS protocol used to introduce mutations does not allow for a recovery time after EMS treatment, precluding the cells from replicating and producing clones. A more likely explanation for unexpectedly large number of alternate alleles with AC = 2 and AC = 3 is that during pooling and library preparation prior to sequencing, one of the mutants had become disproportionately represented in the pool (e.g., from differential PCR amplification). Such differences in the relative contributions of strains within the pool, however, would have less of an impact on the interpretation of SNPs with higher AC values, especially ones that are rare. The only SNP with an AC = 4 in the benomyl pool was the R241C mutation in TUB2. If any such high-AC SNPs had arisen from an overrepresented strain, we would have expected other SNPs from this strain with similarly high AC values. GL-AC metric helps to mitigate against this source of error by providing an independent, gene-level analysis of SNPs based on the fact that multiple amino acid changes in the same protein can confer resistance to a drug.
In our selection of resistant mutants we found that the optimal selection conditions, as well as the mutation rates under each condition, were different for the two drugs tested. We obtained no spontaneous benomyl-resistant mutants, but isolated many EMS-derived mutants. In contrast, the majority of the rapamycin-resistant mutants that we identified were spontaneous, and the mutation rate for rapamycin was about 10-fold less than that of benomyl. This suggests that a variety of mutagenesis methods should be employed for each new drug to increase the likelihood of finding a constellation of resistance alleles for each drug. For example, UV-irradiation and proofreading-deficient polδ mutants show different mutation specificities that are both somewhat orthogonal to that of EMS. Pools of resistant mutants derived from a variety of mutagens would increase the effective genome size available to absorb neutral mutations, while increasing the significance of any genes identified with high GL-AC values.
Of course the use of MUTseq requires that the compound of interest is lethal toward S. cerevisiae. While a given drug of interest may not be lethal toward wt yeast, or even toward classic MDR mutants like pdr1Δ, it may be possible to identify a yeast deletion mutant that is sensitive to the drug. An initial genome-wide search for sensitive haploid deletion mutants could be performed for a given compound using available techniques; such deletion mutants would provide the necessary genetic background for mutant selection, and in addition, the genome-wide sensitivity data could be useful in downstream MOA studies. In order to mitigate against identifying MDR resistance mutations in such cases, it would be advisable to knock out the gene that confers specific resistance in a pdr1Δ background.
Our results show that the sequencing of resistant strains of S. cerevisiae using NGS shows promise as a general method for identifying small molecule targets in this organism. The unbiased approach of prioritizing mutations, and the known targets that were uncovered in doing so, shows that this MUTseq can be applied to the discovery of new targets of novel compounds. In addition we show that by using the pdr1Δ background strain and screening for multi-drug resistance, we can minimize the occurrence of confounding MDR mutations.
Supplementary Material
Footnotes
In a previously published paper[41] the term “Mut-seq was used to describe a method to probe essential amino acids in the T7 bacteriophage genome. In contrast, our method, “MUTseq”, is an acronym to describe our method, “Mutagenesis to Uncover Targets by deep Sequencing”.
References
- 1.Swinney DC, Anthony J. How were new medicines discovered? Nature reviews Drug discovery. 2011;10(7):507–19. doi: 10.1038/nrd3480. [DOI] [PubMed] [Google Scholar]
- 2.Wacker SA, Houghtaling BR, Elemento O, Kapoor TM. Using transcriptome sequencing to identify mechanisms of drug action and resistance. Nat Chem Biol. 2012;8(3):235–7. doi: 10.1038/nchembio.779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lomenick B, Hao R, Jonai N, Chin RM, Aghajan M, Warburton S, Wang J, Wu RP, Gomez F, Loo JA, Wohlschlegel JA, Vondriska TM, Pelletier J, Herschman HR, Clardy J, Clarke CF, Huang J. Target identification using drug affinity responsive target stability (DARTS) Proc Natl Acad Sci U S A. 2009;106(51):21984–9. doi: 10.1073/pnas.0910040106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Menacho-Marquez M, Murguia JR. Yeast on drugs: Saccharomyces cerevisiae as a tool for anticancer drug research. Clinical &translational oncology : official publication of the Federation of Spanish Oncology Societies and of the National Cancer Institute of Mexico. 2007;9(4):221–8. doi: 10.1007/s12094-007-0043-2. [DOI] [PubMed] [Google Scholar]
- 5.Lopez A, Parsons AB, Nislow C, Giaever G, Boone C. Chemical-genetic approaches for exploring the mode of action of natural products. Progress in drug research Fortschritte der Arzneimittelforschung Progres des recherches pharmaceutiques. 2008;66237:239–71. doi: 10.1007/978-3-7643-8595-8_5. [DOI] [PubMed] [Google Scholar]
- 6.Giaever G, Shoemaker DD, Jones TW, Liang H, Winzeler EA, Astromoff A, Davis RW. Genomic profiling of drug sensitivities via induced haploinsufficiency. Nat Genet. 1999;21(3):278–83. doi: 10.1038/6791. [DOI] [PubMed] [Google Scholar]
- 7.Giaever G, Chu AM, Ni L, Connelly C, Riles L, Veronneau S, Dow S, Lucau-Danila A, Anderson K, Andre B, Arkin AP, Astromoff A, El-Bakkoury M, Bangham R, Benito R, Brachat S, Campanaro S, Curtiss M, Davis K, Deutschbauer A, Entian KD, Flaherty P, Foury F, Garfinkel DJ, Gerstein M, Gotte D, Guldener U, Hegemann JH, Hempel S, Herman Z, Jaramillo DF, Kelly DE, Kelly SL, Kotter P, LaBonte D, Lamb DC, Lan N, Liang H, Liao H, Liu L, Luo C, Lussier M, Mao R, Menard P, Ooi SL, Revuelta JL, Roberts CJ, Rose M, Ross-Macdonald P, Scherens B, Schimmack G, Shafer B, Shoemaker DD, Sookhai-Mahadeo S, Storms RK, Strathern JN, Valle G, Voet M, Volckaert G, Wang CY, Ward TR, Wilhelmy J, Winzeler EA, Yang Y, Yen G, Youngman E, Yu K, Bussey H, Boeke JD, Snyder M, Philippsen P, Davis RW, Johnston M. Functional profiling of the Saccharomyces cerevisiae genome. Nature. 2002;418(6896):387–91. doi: 10.1038/nature00935. [DOI] [PubMed] [Google Scholar]
- 8.Parsons AB, Lopez A, Givoni IE, Williams DE, Gray CA, Porter J, Chua G, Sopko R, Brost RL, Ho CH, Wang J, Ketela T, Brenner C, Brill JA, Fernandez GE, Lorenz TC, Payne GS, Ishihara S, Ohya Y, Andrews B, Hughes TR, Frey BJ, Graham TR, Andersen RJ, Boone C. Exploring the mode-of-action of bioactive compounds by chemical-genetic profiling in yeast. Cell. 2006;126(3):611–25. doi: 10.1016/j.cell.2006.06.040. [DOI] [PubMed] [Google Scholar]
- 9.Hoon S, Smith AM, Wallace IM, Suresh S, Miranda M, Fung E, Proctor M, Shokat KM, Zhang C, Davis RW, Giaever G, St Onge RP, Nislow C. An integrated platform of genomic assays reveals small-molecule bioactivities. Nat Chem Biol. 2008;4(8):498–506. doi: 10.1038/nchembio.100. [DOI] [PubMed] [Google Scholar]
- 10.Luesch H, Wu TY, Ren P, Gray NS, Schultz PG, Supek F. A genome-wide overexpression screen in yeast for small-molecule target identification. Chem Biol. 2005;12(1):55–63. doi: 10.1016/j.chembiol.2004.10.015. [DOI] [PubMed] [Google Scholar]
- 11.Butcher RA, Schreiber SL. A small molecule suppressor of FK506 that targets the mitochondria and modulates ionic balance in Saccharomyces cerevisiae. Chem Biol. 2003;10(6):521–31. doi: 10.1016/s1074-5521(03)00108-x. [DOI] [PubMed] [Google Scholar]
- 12.Ho CH, Magtanong L, Barker SL, Gresham D, Nishimura S, Natarajan P, Koh JL, Porter J, Gray CA, Andersen RJ, Giaever G, Nislow C, Andrews B, Botstein D, Graham TR, Yoshida M, Boone C. A molecular barcoded yeast ORF library enables mode-of-action analysis of bioactive compounds. Nat Biotechnol. 2009;27(4):369–77. doi: 10.1038/nbt.1534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Huang Z, Chen K, Zhang J, Li Y, Wang H, Cui D, Tang J, Liu Y, Shi X, Li W, Liu D, Chen R, Sucgang RS, Pan X. A functional variomics tool for discovering drug-resistance genes and drug targets. Cell reports. 2013;3(2):577–85. doi: 10.1016/j.celrep.2013.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lum PY, Armour CD, Stepaniants SB, Cavet G, Wolf MK, Butler JS, Hinshaw JC, Garnier P, Prestwich GD, Leonardson A, Garrett-Engele P, Rush CM, Bard M, Schimmack G, Phillips JW, Roberts CJ, Shoemaker DD. Discovering modes of action for therapeutic compounds using a genome-wide screen of yeast heterozygotes. Cell. 2004;116(1):121–37. doi: 10.1016/s0092-8674(03)01035-3. [DOI] [PubMed] [Google Scholar]
- 15.Heitman J, Movva NR, Hall MN. Targets for cell cycle arrest by the immunosuppressant rapamycin in yeast. Science. 1991;253(5022):905–9. doi: 10.1126/science.1715094. [DOI] [PubMed] [Google Scholar]
- 16.Cafferkey R, Young PR, McLaughlin MM, Bergsma DJ, Koltin Y, Sathe GM, Faucette L, Eng WK, Johnson RK, Livi GP. Dominant missense mutations in a novel yeast protein related to mammalian phosphatidylinositol 3-kinase and VPS34 abrogate rapamycin cytotoxicity. Mol Cell Biol. 1993;13(10):6012–23. doi: 10.1128/mcb.13.10.6012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kunz J, Henriquez R, Schneider U, Deuter-Reinhard M, Movva NR, Hall MN. Target of rapamycin in yeast, TOR2, is an essential phosphatidylinositol kinase homolog required for G1 progression. Cell. 1993;73(3):585–96. doi: 10.1016/0092-8674(93)90144-f. [DOI] [PubMed] [Google Scholar]
- 18.Brown EJ, Albers MW, Shin TB, Ichikawa K, Keith CT, Lane WS, Schreiber SL. A mammalian protein targeted by G1-arresting rapamycin-receptor complex. Nature. 1994;369(6483):756–8. doi: 10.1038/369756a0. [DOI] [PubMed] [Google Scholar]
- 19.Inokoshi J, Tomoda H, Hashimoto H, Watanabe A, Takeshima H, Omura S. Cerulenin-resistant mutants of Saccharomyces cerevisiae with an altered fatty acid synthase gene. Molecular &general genetics : MGG. 1994;244(1):90–6. doi: 10.1007/BF00280191. [DOI] [PubMed] [Google Scholar]
- 20.Wu L, Pan J, Thoroddsen V, Wysong DR, Blackman RK, Bulawa CE, Gould AE, Ocain TD, Dick LR, Errada P, Dorr PK, Parkinson T, Wood T, Kornitzer D, Weissman Z, Willis IM, McGovern K. Novel small-molecule inhibitors of RNA polymerase III. Eukaryot Cell. 2003;2(2):256–64. doi: 10.1128/EC.2.2.256-264.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gustafson G, Davis G, Waldron C, Smith A, Henry M. Identification of a new antifungal target site through a dual biochemical and molecular-genetics approach. Curr Genet. 1996;30(2):159–65. doi: 10.1007/s002940050115. [DOI] [PubMed] [Google Scholar]
- 22.Andries K, Verhasselt P, Guillemont J, Gohlmann HW, Neefs JM, Winkler H, Van Gestel J, Timmerman P, Zhu M, Lee E, Williams P, de Chaffoy D, Huitric E, Hoffner S, Cambau E, Truffot-Pernot C, Lounis N, Jarlier V. A diarylquinoline drug active on the ATP synthase of Mycobacterium tuberculosis. Science. 2005;307(5707):223–7. doi: 10.1126/science.1106753. [DOI] [PubMed] [Google Scholar]
- 23.Singh-Babak SD, Babak T, Diezmann S, Hill JA, Xie JL, Chen YL, Poutanen SM, Rennie RP, Heitman J, Cowen LE. Global analysis of the evolution and mechanism of echinocandin resistance in Candida glabrata. PLoS Path. 2012;8(5):e1002718. doi: 10.1371/journal.ppat.1002718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Timmermann B, Jarolim S, Russmayer H, Kerick M, Michel S, Kruger A, Bluemlein K, Laun P, Grillari J, Lehrach H, Breitenbach M, Ralser M. A new dominant peroxiredoxin allele identified by whole-genome re-sequencing of random mutagenized yeast causes oxidant-resistance and premature aging. Aging (Milano) 2010;2(8):475–86. doi: 10.18632/aging.100187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Brachmann CB, Davies A, Cost GJ, Caputo E, Li J, Hieter P, Boeke JD. Designer deletion strains derived from Saccharomyces cerevisiae S288C: a useful set of strains and plasmids for PCR-mediated gene disruption and other applications. Yeast. 1998;14(2):115–32. doi: 10.1002/(SICI)1097-0061(19980130)14:2<115::AID-YEA204>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- 26.Woehrmann MH, Gassner NC, Bray WM, Stuart JM, Lokey S. HALO384: a halo-based potency prediction algorithm for high-throughput detection of antimicrobial agents. J Biomol Screen. 2010;15(2):196–205. doi: 10.1177/1087057109355060. [DOI] [PubMed] [Google Scholar]
- 27.Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods. 2012;9(4):357–9. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, DePristo MA. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20(9):1297–303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.DePristo MA, Banks E, Poplin R, Garimella KV, Maguire JR, Hartl C, Philippakis AA, del Angel G, Rivas MA, Hanna M, McKenna A, Fennell TJ, Kernytsky AM, Sivachenko AY, Cibulskis K, Gabriel SB, Altshuler D, Daly MJ. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet. 2011;43(5):491–8. doi: 10.1038/ng.806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gassner NC, Tamble CM, Bock JE, Cotton N, White KN, Tenney K, St Onge RP, Proctor MJ, Giaever G, Nislow C, Davis RW, Crews P, Holman TR, Lokey RS. Accelerating the discovery of biologically active small molecules using a high-throughput yeast halo assay. J Nat Prod. 2007;70(3):383–90. doi: 10.1021/np060555t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Carvajal E, van den Hazel HB, Cybularz-Kolaczkowska A, Balzi E, Goffeau A. Molecular and phenotypic characterization of yeast PDR1 mutants that show hyperactive transcription of various ABC multidrug transporter genes. Molecular &general genetics : MGG. 1997;256(4):406–15. doi: 10.1007/s004380050584. [DOI] [PubMed] [Google Scholar]
- 32.Balzi E, Goffeau A. Yeast multidrug resistance: the PDR network. J Bioenerg Biomembr. 1995;27(1):71–6. doi: 10.1007/BF02110333. [DOI] [PubMed] [Google Scholar]
- 33.Woehrmann MH, Gassner NC, Bray WM, Stuart JM, Lokey S. HALO384: a halo-based potency prediction algorithm for high-throughput detection of antimicrobial agents. J Biomol Screen. 15(2):196–205. doi: 10.1177/1087057109355060. [DOI] [PubMed] [Google Scholar]
- 34.Lynch M, Sung W, Morris K, Coffey N, Landry CR, Dopman EB, Dickinson WJ, Okamoto K, Kulkarni S, Hartl DL, Thomas WK. A genome-wide view of the spectrum of spontaneous mutations in yeast. Proc Natl Acad Sci U S A. 2008;105(27):9272–7. doi: 10.1073/pnas.0803466105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gerstein AC, Chun HJ, Grant A, Otto SP. Genomic convergence toward diploidy in Saccharomyces cerevisiae. PLoS Genet. 2006;2(9):e145. doi: 10.1371/journal.pgen.0020145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shiwa Y, Fukushima-Tanaka S, Kasahara K, Horiuchi T, Yoshikawa H. Whole-Genome Profiling of a Novel Mutagenesis Technique Using Proofreading-Deficient DNA Polymerase delta. International journal of evolutionary biology. 2012;2012:860797. doi: 10.1155/2012/860797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Thomas JH, Neff NF, Botstein D. Isolation and characterization of mutations in the beta-tubulin gene of Saccharomyces cerevisiae. Genetics. 1985;111(4):715–34. doi: 10.1093/genetics/111.4.715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Van Duyne GD, Standaert RF, Schreiber SL, Clardy J. Atomic Structure of the Rapamycin Human Immunophilin FKBP-12 Complex. J Am Chem Soc. 1991;113:7433–7434. [Google Scholar]
- 39.Helliwell SB, Wagner P, Kunz J, Deuter-Reinhard M, Henriquez R, Hall MN. TOR1 and TOR2 are structurally and functionally similar but not identical phosphatidylinositol kinase homologues in yeast. Mol Biol Cell. 1994;5(1):105–18. doi: 10.1091/mbc.5.1.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lorenz MC, Heitman J. TOR mutations confer rapamycin resistance by preventing interaction with FKBP12-rapamycin. J Biol Chem. 1995;270(46):27531–7. doi: 10.1074/jbc.270.46.27531. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.