

Abstract
Hyperglycemia leads to vascular smooth muscle cell (VSMC) dedifferentiation and enhances responses to IGF-I. Prior studies showed that hyperglycemia stimulated NADPH oxidase 4 (Nox4) synthesis, and IGF-I facilitated its recruitment to a signaling complex where it oxidized src, leading to AKT and MAPK activation. To determine the mechanism that led to these changes, we analyzed the roles of p62 (sequestrosome1) and PKCζ. Hyperglycemia induced a 4.9 ± 1.0-fold increase in p62/PKCζ association, and disruption of PKCζ/p62 using a peptide inhibitor or p62 knockdown reduced PKCζ activation (78 ± 6%). 3-Phosphoinoside–dependent protein kinase 1 was also recruited to the p62 complex and directly phosphorylated PKCζ, leading to its activation (3.1 ± 0.4-fold). Subsequently, activated PKCζ phosphorylated p65 rel, which led to increased Nox4 synthesis. Studies in diabetic mice confirmed these findings (6.0 ± 0.4-fold increase in p62/PKCζ) and their disruption of attenuated Nox4 synthesis (76 ± 9% reduction). PKCζ/p62 activation stimulated inflammatory cytokine production and enhanced IGF-I–stimulated VSMC proliferation. These results define the molecular mechanism by which PKCζ is activated in response to hyperglycemia and suggest that this could be a mechanism by which other stimuli such as cytokines or metabolic stress function to stimulate NF-κB activation, thereby altering VSMC sensitivity to IGF-I.—Xi, G., Shen, X., Wai, C., Vilas, C. K., Clemmons, D. R. Hyperglycemia stimulates p62/PKCζ interaction, which mediates NF-κB activation, increased Nox4 expression, and inflammatory cytokine activation in vascular smooth muscle.
Keywords: PDK1, p65 rel, diabetes, TNF-α, IGF-I
Exposure of vascular smooth muscle cells (VSMCs) to hyperglycemia results in altered sensitivity to peptide growth factors (1, 2). The response to IGF-I is significantly enhanced by cellular exposure to high glucose in diabetic animals (3). This results in the enhancement of multiple signaling events that lead to stimulation of cell proliferation and migration. In response to hyperglycemia, the protein that normally acts as an intermediary between activating the IGF-I receptor and downstream signaling (insulin receptor substrate-1) is down-regulated, and the IGF-I receptor utilizes an alternative molecular scaffold, Src homology 2 (SH2) domain-containing protein tyrosine phosphatase substrate 1 (SHPS-1), to assemble the signaling components that activate the PI3K and MAPK pathways (4). The IGF-I receptor phosphorylates tyrosine residues located in the SHPS-1 cytoplasmic domain (5). These phosphotyrosines form a binding site for the tyrosine phosphatase, SHP-2, which then recruits c-Src whose activation is required for stimulation of the PI3K and MAPK pathways (6). Although Src is recruited to SHPS-1, full Src activation requires its oxidation (7). Src colocalizes on SHPS-1 with the NADPH oxidase 4 (Nox4), and Nox4 oxidizes Src, leading to its activation (8). Our studies have shown that in order for Nox4 to be translocated to the SHPS-1 scaffold, cells must be exposed to hyperglycemia for 6 h. During this time, Nox4 synthesis is stimulated, and this is mediated in part through activation of NF-κB (8). In addition to increased Nox4 synthesis, activation of PKCζ is also required for Src oxidation, and exposure to a PKCζ inhibitor inhibits the ability of hyperglycemia to stimulate Nox4 synthesis (9). Prior studies have shown that cytokine exposure, hyperglycemia, and induction of oxidative stress activate the NF-κB pathway, but detailed analysis of the exact molecular components that are necessary for NF-κB activation in response to hyperglycemia or increased reactive oxygen species (ROS) generation has not been determined (10–13).
Cytokine induction of NF-κB activation in vascular endothelial cells has been shown to require p62 (also termed sequestrosome1) (14). Hyperglycemia stimulates PKCζ activation and increases p62, but the molecular mechanism by which these changes occur has not been characterized (15, 16). These results raise the question whether a direct interaction between p62 and PKCζ might be involved in hyperglycemia-mediated activation of NF-κB and induction of Nox4 synthesis. These studies were undertaken to determine if hyperglycemia stimulated a change in the p62/PKCζ interaction in VSMCs and if disrupting this interaction attenuated stimulation of Nox4 synthesis. In addition, we identified 3-phosphoinoside–dependent protein kinase 1 (PDK1) as the kinase responsible for PKCζ activation and showed that PKCζ phosphorylated P65 rel, leading to subsequent NF-κB activation, enhanced cytokine production, and increased cell proliferation.
MATERIALS AND METHODS
Human IGF-I was a gift from Genentech (South San Francisco, CA, USA). Immobilon-P membranes, a PDK1 inhibitor (GSK227434), and an anti-Src antibody were purchased from EMD Millipore (Billerica, MA, USA). DMEM containing 1.0 or 4.5 g glucose per liter, streptomycin, and penicillin were purchased from Life Technologies (Grand Island, NY, USA). Antibodies against phospho-AKT, AKT, phospho-Erk1/2, Erk1/2, phospho-Src (Tyr419), phospho-PKCζ (Thr410), phospho-PDK1 (Ser241), PDK1, and TNF-α were from Cell Signaling Technology, Inc. (Beverly, MA, USA). The anti–phospho-p65 (Ser311), p65, PKCζ, and p62 antibodies were purchased from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA, USA). The anti-Nox4, IL-6, and Ki67 antibodies were purchased from Abcam Inc. (Cambridge, MA, USA). All other reagents were purchased from Sigma-Aldrich (St. Louis, MO, USA) unless otherwise stated. A synthetic peptide containing the cell-permeability sequence of protein transduction domain and the underlined (PB-1 domain) sequence from p62 (YARAAARQARASLTVKAYLLGKE), hereafter referred to as a disrupting peptide, and a control peptide (YARAAARQARAKEVYLSLAGTLK) were synthesized by the Protein Chemistry Core Facility at the University of North Carolina, Chapel Hill. The sequences and purity of the peptides were confirmed by mass spectrometry.
Cell culture
VSMCs were isolated from the aortic explants obtained from 3-wk-old pigs and were maintained as described previously (17). Cells were maintained in DMEM high-glucose (4.5 g/L) or normal-glucose (1 g/L) growth medium with 10% fetal bovine serum (FBS) (HyClone Laboratories, Logan, UT, USA), 100 μg/ml streptomycin, and 100 U/ml penicillin, and they were used between passages 5 and 14. Prior to the initiation of the experiments conducted in vitro, the cultures were washed with serum-free DMEM [serum-free medium (SFM)] containing 5 mM glucose, then fresh SFM containing 5 or 25 mM glucose was added for 16 h. At that time, the treatments listed were added to SFM for the times indicated. In some experiments, the cultures were incubated in 5 mM glucose SFM for 16 h, then SFM containing 25 or 5 mM glucose was added for another 6 h. Subsequently, the treatments were added to this medium unless otherwise stated.
Construction of a plasmid containing short hairpin RNA for silencing p62 and establishment of smooth muscle cells expressing p62 short hairpin RNA and control short hairpin RNA
In order to silence p62, a plasmid containing short hairpin RNA (shRNA) and the target sequence GCTCCTACAGACCAAGAATTA was constructed using the Block-iT U6 RNAi Kit (Life Technologies) described previously (18). The shRNA template for LacZ was used as a control. 293FT cells (Life Technologies) were prepared for generation of virus stocks, and VSMCs expressing p62 shRNA or control shRNA were established using procedures described previously (18).
Immunoprecipitation and immunoblotting
The cell monolayers were lysed in a modified RIPA buffer (18). Immunoprecipitation was performed by incubating 0.5 mg cell lysate protein with 1 μg of each of the following antibodies: anti-p62 or anti–phospho-p65 (Ser311) or anti-Src at 4°C overnight. Immunoblotting was performed as described previously (18) using a dilution of 1:500 for anti-p62, Nox4, PDK1, and phospho-p65 (Ser311) antibodies and 1:1000 for anti-PKCζ, phospho-PKCζ (Thr410), phospho-AKT (Ser473), phospho-Erk1/2, AKT, Erk1/2, p65, and β-actin antibodies. The proteins were visualized using ECL (Thermo Fisher Scientific, Rockford, IL, USA).
PKCζ kinase assay
PKCζ kinase activity was measured following a procedure described previously (15). A peptide substrate (ERMRPRKRQGSVRRRV) was synthesized by the Protein Chemistry Core Facility at the University of North Carolina (Chapel Hill, NC, USA).
Measurement of intracellular ROS level and Src oxidation level
Intracellular ROS was measured using a DFC-DA (Life Technologies) assay as described previously (9). Src oxidation was measured using a modified OxyBlot Protein Detection Kit (EMD Millipore). Following the immunoprecipitation of Src, the protein was released from the protein G beads (EMD Millipore, Billerica, MA, USA) using 12% sodium dodecyl sulfate. A total of 5 μg protein was derivatized with 20 μl 1× dinitrophenylhydrazine at room temperature for 15 min. The reaction was stopped by adding 15 μl neutralization solution, and then the proteins were separated using an 8% polyacrylamide gel.
Induction of hyperglycemia in mice and preparation of aortas for analysis
All mouse experiments were approved by the Institutional Animal Care and Use Committee of the University of North Carolina, Chapel Hill. Hyperglycemia was induced in C57/B6 mice (Taconic Biosciences, Inc., Hudson, NY, USA) using the low-dose streptozotocin (19). Normal mice (n = 12) had serum glucose concentrations at 98 ± 18 mg/dl and body weight at 27.2 ± 1.1 g. Diabetic mice had serum glucose concentrations at 416 ± 12 mg/dl and body weight at 26.3 ± 0.5 g. The disrupting peptide (n = 12) (4 mg/kg) or control peptide (n = 12) (4 mg/kg) was administered i.p. for 2 d (24 h and 60 min before death). IGF-I (1 mg/kg) was administered i.p. 24 h (n = 12) before death when measuring Ki67 staining. The mouse aorta samples were prepared following the procedure described previously (8). The lysate protein concentrations were measured using a BCA Protein Assay (Thermo Fisher Scientific). Equal amounts of protein were used in each analysis.
Immunohistochemistry
The aortas from mice were fixed with 4% paraformaldehyde overnight, and paraffin-embedded sections were prepared by the University of North Carolina histology core facility. An immunohistochemistry-paraffin protocol described previously (9) was followed to stain the Ki67-positive nuclei. A DAPI-containing mounting medium (Vector Laboratories, Burlingame, CA, USA) was used to stain the total nuclei. The Ki67-positive nuclei and total nuclei were counted in the aortic rings and expressed as the percentage of positive nuclei.
Statistical analysis
The results that are shown in all experiments are the representatives of 3 separate experiments and expressed as the means ± sd. The Student’s t test was used to compare differences between control and 1 treatment or control cells and 1 mutant for some in vitro experiments. One- or 2-way ANOVA was applied for all data obtained from in vivo studies or when multiple treatments or multiple cell types were compared using data from in vitro studies. P ≤ 0.05 was considered statistically significant.
RESULTS
Hyperglycemia stimulates p62 expression and PKCζ activation
To determine if hyperglycemic stress stimulated an increase in p62/PKCζ association, initially, we determined the effect of hyperglycemia regulating each of these proteins. Exposure of VSMCs maintained in 5–25 mM glucose resulted in a time-dependent increase in p62 (Fig. 1A). Following a 3 h incubation, there was a 2.2 ± 0.4-fold increase in p62 that changed only slightly at later time points. Phosphorylation of Thr410 is required for PKCζ activation; therefore, we determined the time course of PKCζ Thr410 phosphorylation. There was a 2.0 ± 0.5-fold increase in PKCζ Thr410 phosphorylation at 3 h, which increased slightly thereafter and was maintained at 24 h (Fig. 1B). To determine if this resulted in enhanced enzymatic activity, we utilized a PKCε-derived peptide substrate that has been shown to be specifically phosphorylated in vitro by PKCζ (15). Immunoprecipitation of PKCζ following cellular exposure to hyperglycemia showed that the enzymatic activity peaked at 6 h, similar to the time course of Thr410 phosphorylation (Fig. 1C).
Figure 1.

Hyperglycemia increases p62 and PKCζ activation in VSMCs. VSMCs were cultured in DMEM containing normal glucose (5 mM) plus 10% FBS then serum deprived for 16 h before exposure to 25 mM glucose for the indicated times. A) Cell lysates were immunoblotted (IB) with an anti-p62 antibody. To control for loading, the blot was stripped and reprobed with an anti–β-actin antibody. B) Cell lysates were immunoblotted with an anti-pThr410 PKCζ antibody. The blot was stripped and reprobed with an anti-PKCζ antibody. The value of each bar is the ratio of the scan values of the p62 or pThr410 PKCζ bands divided by the values of β-actin or the PKCζ bands. C) PKCζ was immunoprecipitated with an anti-PKCζ antibody, and the immune complexes were used to measure in vitro PKCζ kinase activity following the procedure described in Materials and Methods. To control for PKCζ input, after immunoprecipitation, the amount of immune complex from each treatment was adjusted for PKCζ as determined by immunoblotting with anti-PKCζ. High glucose (HG) was 25 mM, and normal glucose (NG) was 5 mM. *P < 0.05 and **P < 0.01 indicate significant differences between 2 treatments.
Hyperglycemia stimulates PKCζ and PDK1 recruitment to p62, and PDK1 activates PKCζ
Based on these results, we determined whether hyperglycemia induced a direct p62/PKCζ interaction. Cells maintained in 5.0 mM glucose had minimal PKCζ/pp62 association; however, following exposure to hyperglycemia for 6 h, there was a 4.9 ± 1.0-fold increase (P < 0.01) (Fig. 2A). In order to determine the specificity of this interaction, we utilized a cell-permeable peptide whose sequence was derived from the PB-1 domain of p62 that interacts directly with a distinct motif contained in PKCζ (20). Exposure to this peptide during hyperglycemia resulted in disruption of p62/PKCζ association (Fig. 2A); however, it did not affect cell viability (Supplemental Fig. 1). To determine the physiologic significance of this disruption, cells were exposed to the peptide then immunoblotted for PKCζ pThr410. Although p62 was induced during hyperglycemia, in cells exposed to this peptide PKCζ activation was inhibited (78 ± 6% reduction; P < 0.01) (Fig. 2B). That disruption of PKCζ/p62 resulted in the direct loss of enzymatic activity was shown using a PKCζ in vitro kinase assay (Fig. 2C).
Figure 2.
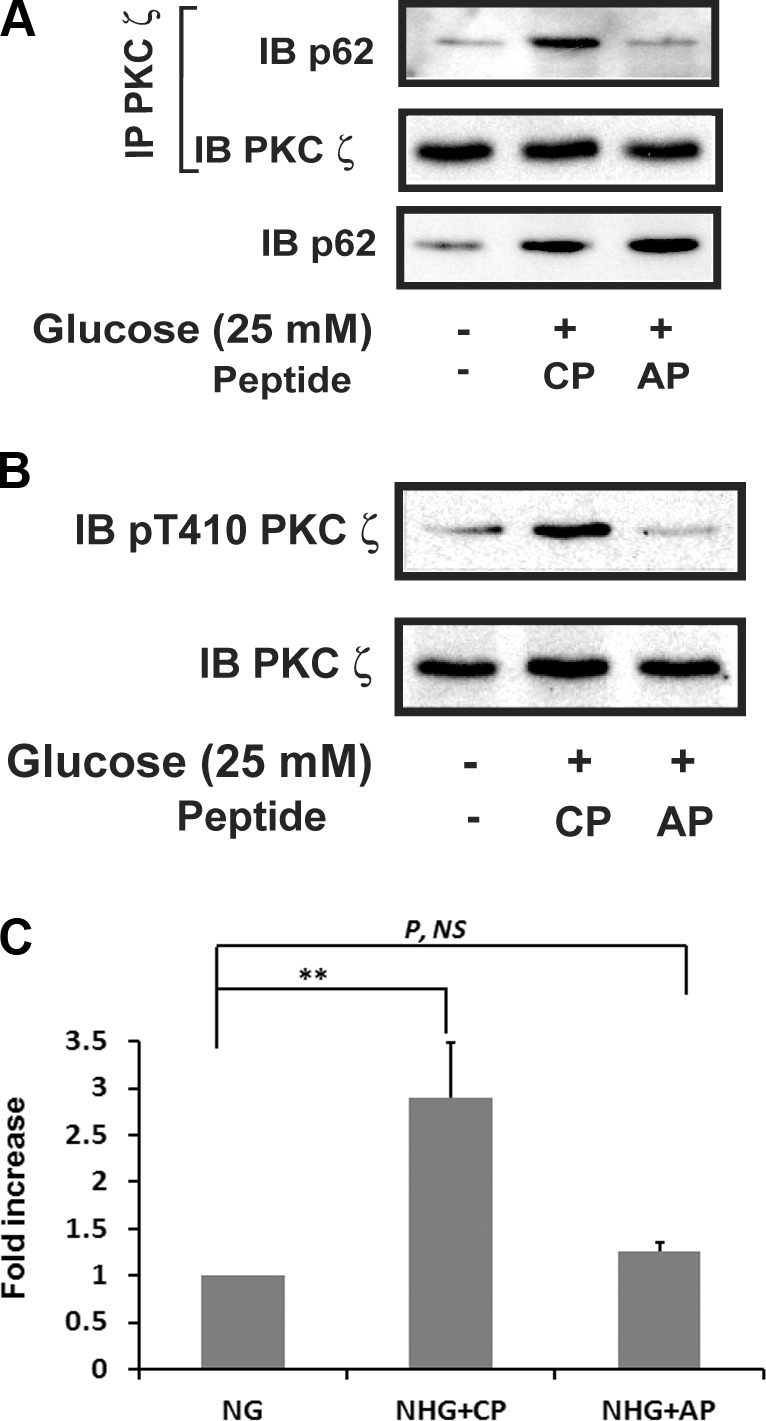
Disruption of p62 and PKCζ association impairs hyperglycemia-stimulated PKCζ activation. VSMCs were cultured in DMEM containing normal glucose (5 mM) plus 10% FBS then serum deprived for 16 h before exposure to SFM with 25 mM glucose for 6 h in the presence of a control peptide (CP) or a disrupting peptide (AP) (10 μg/ml). A) Cell lysates were immunoprecipitated (IP) with an anti-PKCζ antibody and immunoblotted with an anti-p62 antibody. The blot was reprobed with an anti-PKCζ antibody. The same amount of lysate was immunoblotted with an anti-p62 antibody. B) Cell lysates were immunoblotted (IB) with an anti-pThr410 PKCζ antibody. To control for loading, the blots were stripped and reprobed with an anti-PKCζ antibody. C) PKCζ in vitro kinase activity was measured following the procedure described in Fig. 1C. P, NS, no significant difference in P value. **P < 0.01 indicates significant differences between 2 treatments.
To confirm the importance of the p62/PKCζ interaction, we used an shRNA to knock down p62 and determined its effect on PKCζ activation. As shown in Fig. 3A, the shRNA successfully decreased p62 by >90%. When cells that had undergone p62 knockdown were exposed to hyperglycemia, there was no significant increase in p62 protein (Fig. 3B) and PKCζ Thr410 phosphorylation (Fig. 3C). Therefore, the results of these knockdown experiments confirmed the results obtained using peptide disruption and show that PKCζ/p62 interaction is required for PKCζ activation in response to hyperglycemia. To further demonstrate the importance of p62/PKCζ complex formation for hyperglycemia-induced PKCζ activation, we determined the level of pThr410 PKCζ that was associated with p62 immunocomplex and the level in the supernatant after p62 immunoprecipitation. The results showed abundant activated PKCζ in the p62 immunoprecipitate but no detectable PKCζ pThr410 in the supernatant, even though a large amount of inactive PKCζ was present in the supernatant after immunoprecipitation of p62 (Fig. 3D).
Figure 3.

Knockdown or deletion of p62 prevents hyperglycemia-stimulated PKCζ activation. A) VSMCs expressing the shRNA targeting LacZ (Ctrl Si) and p62 (p62 Si) were cultured in DMEM containing 25 mM glucose. Cell lysates were immunoblotted (IB) with an anti-p62 antibody. To control for loading, the blots were stripped and reprobed with anti–β-actin. B and C) VSMCs expressing Ctrl Si or p62 Si were cultured in DMEM containing normal glucose (5 mM, NG) plus 10% FBS then serum deprived for 16 h before exposure to 25 mM glucose (NHG) for 6 h or maintained in NG. Cell lysates were immunoblotted with an anti-p62 antibody and reprobed with an anti–β-actin antibody (B) or immunoblotted with anti-pThr410 PKCζ, anti-PKCζ, or anti-PDK1 antibodies (C). D) VSMCs were cultured as in (B) and (C). Cell lysates were immunoprecipitated (IP) with an anti-p62 antibody. The immune complexes (Pellet) and supernatant (Super.) were immunoblotted with anti-pThr410 PKCζ and anti-PKCζ antibodies.
To determine the mechanism by which p62 binding to PKCζ led to PKCζ activation, we determined if a serine/threonine kinase was recruited to the p62/PKCζ complex in response to high glucose. Prior studies have shown that PDK1 directly phosphorylates PKCζ in vitro (21); therefore, we investigated whether hyperglycemia could stimulate PDK1 recruitment to p62. Hyperglycemia led to increased p62/PDK1 association (2.4 ± 0.3; P < 0.05) (Fig. 4A), and the PDK1 that was recruited was activated (e.g., phospho-Ser241) (Fig. 4B). To determine if PDK1 that was recruited to p62 could phosphorylate PKCζ, we repeated the experiment in the presence of a PDK1 inhibitor. Addition of GSK227434 (1 μM) inhibited PDK1 activation, and more importantly, it completely inhibited hyperglycemia-induced PKCζ Thr410 phosphorylation (71 ± 5% reduction compared to no inhibitor; P < 0.001), and the band intensity was not different compared with cells exposed to normal glucose (an 18 ± 16% difference; P value was nonsignificant) (Fig. 4C). To confirm the importance of PDK1 recruitment to p62 for PKCζ activation, we analyzed PKCζ/PDK1 association in normal and p62 knockdown cells. In control cells exposed to hyperglycemia, PKCζ/PDK1 association was increased (3.5 ± 0.6-fold; P < 0.01) compared with cells exposed to normal glucose. In contrast, in the p62 knockdown cells, hyperglycemia did not induce PDK1/PKCζ association (Fig. 4D). To determine if PKCζ/PDK1 association required their recruitment to the p62 scaffold, we disrupted p62/PKCζ using the PB-1 domain peptide. This resulted in a 72 ± 12% decrease in PDK1/PKCζ association (Fig. 4E).
Figure 4.

PDK1 phosphorylates PKCζ contained in the p62 complex during hyperglycemia. VSMCs were cultured in DMEM containing NG plus 10% FBS then serum deprived for 16 h before exposure to 25 mM glucose (NHG) for 6 h or maintained in NG. A and B) Cell lysates were immunoprecipitated (IP) with an anti-p62 antibody and immunoblotted (IB) with an anti-PDK1 or PDK1 (Ser241) antibody. To control for loading, the same amount of cell lysate was immunoblotted with an anti-PDK1 or anti-p62 antibody. C) Different concentrations of GSK227434 were added 30 min before a 6 h exposure to SFM containing 25 mM glucose. Cell lysates were immunoblotted with anti-pThr410 PKCζ and PDK1 (Ser241) antibodies. The blots were stripped and reprobed with an anti-PKCζ or anti-PDK1 antibody. D) VSMCs expressing the shRNA targeting LacZ (Ctrl Si) and p62 (p62 Si) were cultured in DMEM containing normal glucose (5 mM, NG) plus 10% FBS then serum deprived for 16 h before exposure to 25 mM glucose (NHG) for 6 h or maintained in NG. Cell lysates were immunoprecipitated with an anti-PKCζ antibody and immunoblotted with an anti-PDK1 antibody. To control for loading, the blots were stripped and reprobed with an anti-PKCζ antibody. E) Cell lysates were immunoprecipitated with an anti-PKCζ antibody and immunoblotted with an anti-PDK1 antibody. To control for loading, the blots were reprobed with an anti-PKCζ antibody.
PKCζ phosphorylates p65 rel/Ser311, leading to NF-κB activation and Nox4 synthesis
To determine how the p62/PKCζ interaction was linked to enhanced Nox4 synthesis, we determined if disrupting p62/PKCζ would alter phosphorylation of p65 rel Ser311 because this results in NF-κB activation. There was a major increase (2.7 ± 0.5-fold; P < 0.01) in p65 rel Ser311 phosphorylation after a 3 h exposure to hyperglycemia (Fig. 5A). Addition of a PKCζ inhibitor blocked this increase in p65 rel Ser311 phosphorylation (75 ± 10% reduction; P < 0.01) (Fig. 5B). To determine if PKCζ acted directly to phosphorylate p65 rel, we immunoprecipitated p65 rel then immunoblotted for PKCζ. As shown in Fig. 5C, p65 rel/PKCζ association was stimulated in the presence of hyperglycemia (2.5 ± 0.7-fold; P < 0.05), and blocking PKCζ recruitment to p62 inhibited p65 rel/PKCζ association. To confirm that PDK1-mediated activation of PKCζ was required, we added GSK227434 and measured p65 rel phosphorylation. As shown in Supplemental Fig. 2, high-glucose–induced p65 rel phosphorylation was inhibited. To determine the functional significance of this interaction, we disrupted p62/PKCζ using the PB-1 domain and measured p65 rel Ser311 phosphorylation in the presence of hyperglycemia. This resulted in marked attenuation of p65 rel phosphorylation (72 ± 5% reduction; P < 0.01) (Fig. 5D). Furthermore, immunoprecipitation of PKCζ followed by immunoblotting for phospho-p65 rel showed that the p65 that coprecipitated after high-glucose exposure was phosphorylated, which was impaired when p62/PKCζ interaction was disrupted (Fig. 5E). To confirm this result, we utilized cells with p62 knockdown and determined that this also attenuated p65 rel/PKCζ association and p65 rel phosphorylation in response to hyperglycemia (Fig. 5F, G).
Figure 5.

PKCζ activation mediates hyperglycemia-stimulated p65 Ser311 phosphorylation. A and B) VSMCs were cultured in DMEM containing normal glucose (5 mM) plus 10% FBS then serum deprived for 16 h before exposure to 25 mM glucose for the indicated time points (A) or 6 h in the presence or absence of a PKCζ inhibitor (B). Cell lysates were immunoblotted (IB) with an anti-pSer311 p65 antibody. To control for loading, the blots were stripped and reprobed with an anti-p65 antibody. C–E) VSMCs were cultured in DMEM containing normal glucose (5 mM) plus 10% FBS then serum deprived for 16 h before exposure to 25 mM glucose (NHG) for 6 h in the presence of a control peptide (CP) or a disrupting peptide (AP) (10 μg/ml). F and G) VSMCs expressing the shRNA targeting LacZ (Ctrl Si) and p62 (p62 Si) were cultured in DMEM containing normal glucose (5 mM, NG) plus 10% FBS then serum deprived for 16 h before treatment with 25 mM glucose (NHG) for 6 h or maintained in NG. Cell lysates were immunoprecipitated (IP) with anti-p65 (C and F) or anti-pSer311 p65 antibody (E) and immunoblotted with an anti-PKCζ antibody. The same amount of each lysate was immunoblotted with an anti-PKCζ antibody as a protein input control. D and G) Cell lysates were immunoblotted with an anti-pSer311 p65 antibody. To control for loading, the blots were stripped and reprobed with an anti-p65 antibody. Each bar is the ratio of the scan value of the pSer311 p65 band divided by the p65 band. p, NS, no significant difference in P value. *P < 0.05, **P < 0.01, and ***P < 0.001 indicate significant differences between 2 treatments.
To determine the biologic significance of p65 rel phosphorylation, we analyzed Nox4 expression because we had shown previously that this required NF-κB pathway activation. Exposure to hyperglycemia resulted in significant enhancement of Nox4 synthesis, and this was completely inhibited either by exposure to the PB-1 peptide or knockdown of p62 (Fig. 6A, B). Additionally, p62/PKCζ association was required for hyperglycemia-induced increases in IL-6 and TNF-α, proteins whose expression is known to be regulated by NF-κB. However, knockdown of p62 did not affect high-glucose–induced general ROS generation (Fig. 6C). To determine the consequences of inhibiting p62/PKCζ association for IGF-I–mediated signaling, cells that were exposed to the disrupting peptide or in which p62 had been knocked down were stimulated with IGF-I, and Src Tyr419 phosphorylation (an index of Src activation) was analyzed. Both of these manipulations inhibited IGF-I–stimulated Src activation (Fig. 6D, E). Importantly, disrupting p62/PKCζ association impaired IGF-I–stimulated Src oxidation, which is dependent upon an increase in Nox4 expression and its colocalization with Src on the SHPS-1 scaffold that catalyzes Src oxidation and activation (8, 9). Because we had shown previously that inhibition of NF-κB inhibited Nox4 synthesis, we determined whether this altered Src activation. Addition of Bay 11-7082, an inhibitor of NF-κB, inhibited IGF-I–stimulated Src activation in high glucose (Fig. 6F). To determine the consequences for IGF-I signaling, we examined AKT and Erk activation. As shown in Fig. 6G, H, the activation of both was significantly attenuated in response to disruption of p62/PKCζ association or p62 knockdown.
Figure 6.

p62 and PKCζ association is required for hyperglycemia to stimulate NF-κB pathway-dependent proteins and for IGF-I–stimulated downstream signaling. A, D, and G) VSMCs were cultured in DMEM containing normal glucose (5 mM) plus 10% FBS then serum deprived for 16 h before treatment with 25 mM glucose (NHG) for 6 h in the presence of a control peptide (CP) or a disrupting peptide (AP) (10 μg/ml). B, C, E, and H) VSMCs expressing shRNA targeting LacZ (Ctrl Si) or p62 (p62 Si) were cultured in DMEM containing normal glucose (5 mM, NG) plus 10% FBS then serum deprived for 16 h before treatment with 25 mM glucose (NHG) for 6 h or maintained in NG. IGF-I (100 ng/ml) was added [1 min treatment for (D and (E), or 10 min treatment for (G) and (H)] before cells were harvested. A and B) Cell lysates were immunoblotted (IB) using an anti-Nox4, anti–IL-6, or anti–TNF-α antibody. To control for loading, the blots were stripped and reprobed with an anti-β-actin antibody. C) ROS was measured as described in Materials and Methods. D and E) Cell lysates were immunoblotted with an anti-p419 Src antibody. Cell lysates were also immunoprecipitated (IP) with an anti-Src antibody and immunoblotted with an anti-DNP antibody (D). To control for loading, the blots were stripped and reprobed with an anti-Src antibody. F) VSMCs were cultured in DMEM containing normal glucose (5 mM) plus 10% FBS then serum deprived for 16 h before treatment with 25 mM glucose (NHG) for 6 h. Prior to 25 mM glucose exposure, cells were preincubated with or without Bay 11-7082 (10 μM), an NF-κB inhibitor (NF-κB in), for 30 min. After 6 h high-glucose exposure, IGF-I (100 ng/ml) was added for 1 min treatment. Cell lysates were immunoblotted with an anti-pTyr419 Src antibody. To control for loading, the blots were reprobed with an anti-Src antibody. G and H) Cell lysates were immunoblotted with an anti-pAKT (Ser473) or anti-pErk1/2 antibody. To control for loading, the blots were stripped and reprobed with an anti-AKT or anti-Erk1/2 antibody. p, NS, no significant difference in P value. *P < 0.05, **P < 0.01, and ***P < 0.001 indicate significant differences between 2 treatments.
p62 recruitment of PKCζ mediates increased Nox4 during hyperglycemia in vivo
To determine the significance of the p62/PKCζ/PDK1 interaction for signaling in vivo, mice were made diabetic then exposed to the p62/PKCζ disrupting peptide. The aortas were isolated, and the effect of inhibiting the p62/PKCζ interaction on signaling in aortic smooth muscle cells (SMCs) was determined. Aortas from diabetic mice showed a major increase in p62 and p62/PKCζ association compared to aortas from nondiabetic animals [5.1 ± 0.6-fold (P < 0.001) and 6.0 ± 0.4-fold (P < 0.001), respectively] (Fig. 7A, B). Injection of the cell-permeable PB-1 domain peptide resulted in significant disruption of PKCζ/p62 interaction (Fig. 7B). Importantly, this peptide also inhibited PDK1/PKCζ association (Fig. 7C). As predicted, this resulted in inhibition of PKCζ Thr410 phosphorylation (86 ± 8% reduction; P < 0.001) (Fig. 7D). Injection of disrupting peptide also resulted in attenuation of p65 rel Ser311 phosphorylation in diabetic animals (Fig. 7E). Assessment of Nox4 expression showed that it was significantly increased in the diabetic animals, and following exposure to the dissociating peptide, its expression was reduced to a level that was no greater than nondiabetic animals (Fig. 7F). Increased expression of IL-6 and TNF-α was present in diabetic mice, and this was inhibited by the disrupting peptide. To determine the physiologic consequences of p62/PKCζ inhibition, IGF-I–stimulated Ki67 labeling was quantified. The results showed that IGF-I stimulated a 36 ± 6% increase in diabetic mice, and the disrupting peptide significantly reduced this response by 80 ± 7% (P < 0.001) (Fig. 7G).
Figure 7.

p62 recruitment of PDK1 and PKCζ activation mediate increased Nox4 and cytokine expression in diabetic mice. The 12-wk-old male C57/B6 mice were injected with streptozotocin (DM) or citrate buffer (NM) and were maintained as in Materials and Methods. Aortic extracts were prepared using the procedure described in Materials and Methods. A) Aortic extracts were immunoblotted (IB) using an anti-p62 antibody. The blot was stripped and reprobed with anti-β-actin antibody as a loading control. B) Aortic extracts were immunoprecipitated (IP) using anti-p62 antibody and immunoblotted using an anti-PKCζ antibody. The same amount of each lysate was immunoblotted with an anti-PKCζ antibody to control for a protein input. C) Aortic extracts were immunoprecipitated using an anti-PKCζ antibody and immunoblotted using an anti-PDK1 antibody. The blots were reprobed with an anti-PKCζ antibody as a loading control. D–F) Aortic extracts were immunoblotted using an anti-pThr410 PKCζ (D) or anti-pSer311 p65 antibody (E) or an anti-Nox4, anti–IL-6, or anti–TNF-α (F) antibody. The blots were stripped and reprobed with an anti-PKCζ or anti-p65 or anti–β-actin antibody as a loading control, respectively. G) The aortas were fixed with 4% paraformaldehyde overnight for preparation of paraffin-embedded sections. After staining with an anti-Ki67 antibody and DAPI, the number of proliferating cells and total nuclei in each section (total 16 sections) were counted and expressed as the percentage of Ki67-positive nuclei. The means ± sd from 4 mice per treatment group (4 sections measured per mouse) are shown graphically. CP, control peptide; AP, disrupting peptide; p, NS, no significant difference in P value. ***P < 0.001 indicates significant differences when the 2 treatments were compared.
DISCUSSION
Exposure of VSMCs to hyperglycemia leads to multiple changes secondary to increased oxidative stress. Hyperglycemia stimulates increased synthesis of the Nox4, which leads to enhanced downstream signaling and VSMC proliferation or migration in response to IGF-I. Nox4 is recruited to the cell membrane-associated scaffold, SHPS-1, in association with Src. This results in localized oxidation of Src directly by Nox4 on the SHPS-1 scaffold, which enhances its kinase activity leading to stimulation of both the PI3K and MAPK pathways (8, 9). Our prior studies showed that the hyperglycemia-induced change in Nox4 could be blocked by inhibiting NF-κB activation (8). Because of the importance of increased Nox4 synthesis for activating Src and thereby enhancing the cellular response to IGF-I, we determined the mechanism by which hyperglycemia leads to NF-κB pathway activation. The results clearly demonstrate that hyperglycemia increases the molecular chaperone p62, sequestrosome1 (Fig. 8). p62 subsequently recruits the serine/threonine kinases PDK1 and PKCζ. PDK1 phosphorylates PKCζ, resulting in its activation. This conclusion is supported by our results showing that the addition of a PDK1 inhibitor blocked PKCζ activation. Similarly, inhibition of the recruitment of PKCζ or constitutively active PDK1 to p62 inhibited PKCζ activation. Activated PKCζ phosphorylated p65 rel Ser311, an event that is required for activation of NF-κB (22). Inhibition of PKCζ/p62 association or knockdown of p62 inhibited the hyperglycemia-induced increase in p65 rel Ser311 phosphorylation, Nox4 synthesis, as well as IGF-I–stimulated Src activation and VSMC replication. Importantly, PKCζ/p62 association was required to stimulate p65 rel Ser311 phosphorylation in aortas of diabetic animals, and disruption of PKCζ/p62 inhibited the increases in IL-6 and TNF-α, proteins whose synthesis is stimulated in response to NF-κB. Taken together, the results definitively demonstrate that hyperglycemia leads to PDK1 and PKCζ recruitment to p62 and PDK1 phosphorylation of PKCζ, thereby allowing PKCζ to activate the NF-κB pathway leading to increased Nox4 synthesis. Nox4 is then translocated to the SHPS-1 signaling complex resulting in Src activation in response to IGF-I and enhanced downstream signaling (9).
Figure 8.
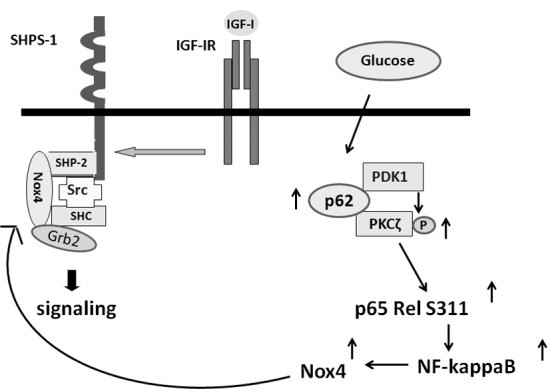
Mechanism of hyperglycemia-mediated activation of increased NF-κB and enhanced IGF-I signaling. During hyperglycemia, p62 is increased, which results in PDK1 and PKCζ recruitment followed by PDK1 activation of PKCζ. Activated PKCζ phosphorylates p65 rel Ser311, leading to NF-κB activation, resulting in increased Nox4 expression. In response to IGF-I, Nox4 is recruited to the SHPS-1 signaling complex, where it oxidizes Src. Activated Src stimulates downstream signaling through both the MAPK and PI3K/AKT pathways.
Prior reports had shown that high glucose induced increases in p62, PKCζ Thr410 phosphorylation, and PKCζ enzymatic activity (23) over a similar time course, but whether the increase in p62 played a role in PKCζ activation had not been analyzed. Prior studies showed that high-glucose activation of Pyk2 and phospholipase D in adipocytes and skeletal muscle resulted in PKCζ activation (24), and glucose infusion into rats resulted in PKCζ translocation from the cytosol to the plasma membrane where it was subsequently activated (25). Glucose-induced increases in ROS were also implicated in PKCζ activation (26). Zhang et al. (27) showed that superoxide ions stimulated PKCζ activation in endothelial cells, and this led to induction of NADPH oxidase. Taken together, these studies showed that hyperglycemia induces PKCζ activation, but the signaling events that mediated PKCζ activation and its access to substrates were not determined. Similarly, Sugimoto et al. (28) reported that rat SMCs exposed to 30 mM glucose had increased p62 and other stimuli that induce oxidative stress such as high-fat feeding inducing p62 expression in skeletal (11) or cardiac muscle (29). These findings were consistent with our results and suggested that hyperglycemia induced an increase in p62 in response to ROS-generated stress (30). Oxidative stress inhibits autophagosome/lysosome fusion, and this inhibits p62 degradation; therefore, it is possible that hyperglycemia is regulating p62 in VSMCs through this mechanism (16). Based on these reports, we determined if high glucose induced p62/PKCζ association and if that altered PKCζ activation. Our results showed that high glucose induced p62/PKCζ association, but more importantly, they showed that specific disruption of their association in VSMCs in culture or in diabetic mice led to the loss of PKCζ activation.
Although one study had demonstrated that p62 association with PKCζ activated PKCζ, it did not define the mechanism of activation (13). Direct binding of p62 to PKCζ in vitro does not alter its kinase activity (31). Because p62 can bind multiple proteins simultaneously, we determined whether a kinase was recruited to p62 that could directly phosphorylate PKCζ. PDK1 is constitutively active in VSMCs, and its activity does not increase in response to hyperglycemia (32); however, PDK1 had been shown to be recruited to the cell membrane fraction in response to oxidative stress. Furthermore, Le Good et al. (21) demonstrated that PDK1 could directly phosphorylate Thr410 in PKCζ in vitro and that knockdown of PDK1 resulted in a 53% reduction in PKCζ phosphorylation. Therefore, we conducted coimmunoprecipitation studies and showed that constitutively activated PDK1 is also recruited to p62 in response to hyperglycemia. More importantly, in the p62 knockdown cells, there was no association between PDK1 and PKCζ as well as inhibition of PKCζ phosphorylation. Similarly, a PDK1 inhibitor reduced PKCζ activation. We conclude that activated PDK1 and inactive PKCζ are recruited to p62 in response to hyperglycemia, thereby facilitating PKCζ and subsequently p65 rel activation. The results establish that PDK1 is the kinase that activates PKCζ and demonstrate that p62 mediates their interaction.
The molecular basis of p62/PKCζ interaction has been well characterized. Structural analysis shows that the type II PB-1 domain of p62 binds directly to the type I OPCA domain of PKCζ (20). Mutagenesis has been utilized to determine the importance of interactions through these domains and to show that inhibition of PKCζ/p62 interaction suppresses the inflammatory response to cytokine stimulation of NF-κB (33). Decreased binding of PKCζ to p62 through the OPCA motif mutations also resulted in attenuated PKCζ kinase activity (34). Our results using a cell-permeable peptide that contains the p62 PB-1 domain clearly show that the increase in PKCζ/p62 association that occurs in response to hyperglycemia is mediated through this specific molecular interaction.
A variety of other stimuli induce the association of these 2 proteins leading to functional consequences that are dependent on the particular cell type and the composition of the complex of proteins that is localized on the p62/PKCζ scaffold. Stimulation of endothelial cells with TNF-α induced p62/PKCζ (13). However, in contrast to our findings, this did not lead to NF-κB activation but rather activation of JNK and caspase-3, which resulted in enhanced apoptosis. Notably, that study showed increased PKCζ activation, but it also showed inhibition of p65 rel Ser311 phosphorylation (13). In contrast, Zhang et al. (34) demonstrated that p62 and PKCζ formed a complex with JNK, which resulted in enhanced NF-κB activation in response to TNF receptor-associated factor 6 activation (35), and disruption of PKCζ/p62 association using ursolic acid was shown to inhibit TNF-α–stimulated activation of NF-κB (36).
To assess the consequences of high-glucose–induced PKCζ activation, we analyzed its role in mediating p65 rel Ser311 phosphorylation because PKCζ can directly phosphorylate p65 rel in vitro (37), and diacylglycerol-induced PKCζ activation led to p65 rel Ser311 phosphorylation and NF-κB activation. Our studies confirmed those findings and further showed that deletion of p62 resulted in impaired p65 rel phosphorylation. An important difference in our study and the study that analyzed TNF-α induction of p62/PKCζ in endothelium (13) is the presence of high glucose. A prior study showed clearly that high glucose led not only to a constitutive increase in NF-κB activation in VSMCs but also to enhanced responsiveness to peptide growth factors that was NF-κB dependent (38). Similarly, high glucose also induced p22phox and Nox4 expression in pulmonary artery SMCs, which required enhanced NF-κB activity (39).Taken together with our findings, these results suggest that p62 induction of PKCζ activation may result in different outcomes depending upon the type of stimulus that is present.
Importantly, we also demonstrated that NF-κB pathway activation in response to PKCζ activation led to increased Nox4 synthesis and Src oxidation. These are both required for VSMC proliferation in response to IGF-I, and disruption of p62/PKCζ led to decreased PKCζ activation, Nox4 synthesis, and IGF-I–stimulated VSMC proliferation in vivo. p62/PKCζ also stimulated an increase in production of the inflammatory cytokines IL-6 and TNF-α. Both of these cytokines have been implicated in the development of atherosclerosis, and their expression is increased in VSMCs exposed to hyperglycemia. These cytokines have been shown to enhance the proliferative response of coronary artery SMCs to IGF-I (40). Additionally, because these cytokines also activate NF-κB, their induction by p62 could potentially lead to a positive feedback loop, further enhancing atherosclerotic lesion development (41).
In summary, our findings support a role for hyperglycemia-induced formation of a critical complex of signaling proteins on the p62 scaffold. Assembly of this complex results in the recruitment of PKCζ and PDK1, leading directly to PKCζ activation by PDK1. Activated PKCζ phosphorylates p65 rel Ser311, thereby activating NF-κB, which leads to increased Nox4 synthesis that is necessary to oxidize Src kinase and thereby enhance PI3K and MAPK activation leading to increased VSMC proliferation. Because multiple other stimuli such as high-fat feeding, cytokine secretion, or other activators of oxidative stress may alter the sensitivity of SMCs to stimulation by growth factors such as IGF-I, future studies need to determine if any of these other extracellular stimuli result in activation of these signaling elements leading to enhanced Nox4 synthesis and selective protein oxidation.
Acknowledgments
The authors thank Ms. Laura Lindsey (University of North Carolina, Chapel Hill) for her help in preparing the manuscript. This work was supported by a grant (AG-02331) from the U.S. National Institutes of Health. D.R.C. is a guarantor of this manuscript. X.S. was supported by a grant from the National Natural Science Foundation of China (No. 31271983). G.X. contributed to research design, performed the experiments, and wrote/edited the manuscript. X.S., C.W., and C.K.V. performed the experiments. D.R.C. wrote and edited the manuscript, and contributed to research design and discussion of the results. The authors declare no conflicts of interest.
Glossary
- FBS
fetal bovine serum
- Nox4
NADPH oxidase 4
- PDK1
3-phosphoinoside–dependent protein kinase 1
- ROS
reactive oxygen species
- SFM
serum-free medium
- SH2
Src homology 2
- SHPS-1
Src homology 2 domain-containing protein tyrosine phosphatase substrate 1
- shRNA
short hairpin RNA
- SMC
smooth muscle cell
- VSMC
vascular smooth muscle cell
Footnotes
This article includes supplemental data. Please visit http://www.fasebj.org to obtain this information.
REFERENCES
- 1.Lan T. H., Huang X. Q., Tan H. M. (2013) Vascular fibrosis in atherosclerosis. Cardiovasc. Pathol. 22, 401–407 [DOI] [PubMed] [Google Scholar]
- 2.Fiorentino T. V., Prioletta A., Zuo P., Folli F. (2013) Hyperglycemia-induced oxidative stress and its role in diabetes mellitus related cardiovascular diseases. Curr. Pharm. Des. 19, 5695–5703 [DOI] [PubMed] [Google Scholar]
- 3.Maile L. A., Busby W. H., Nichols T. C., Bellinger D. A., Merricks E. P., Rowland M., Veluvolu U., Clemmons D. R. (2010) A monoclonal antibody against alphaVbeta3 integrin inhibits development of atherosclerotic lesions in diabetic pigs. Sci. Transl. Med. 2, 18ra11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Radhakrishnan Y., Busby W. H. Jr, Shen X., Maile L. A., Clemmons D. R. (2010) Insulin-like growth factor-I-stimulated insulin receptor substrate-1 negatively regulates Src homology 2 domain-containing protein-tyrosine phosphatase substrate-1 function in vascular smooth muscle cells. J. Biol. Chem. 285, 15682–15695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Radhakrishnan Y., Shen X., Maile L. A., Xi G., Clemmons D. R. (2011) IGF-I stimulates cooperative interaction between the IGF-I receptor and CSK homologous kinase that regulates SHPS-1 phosphorylation in vascular smooth muscle cells. Mol. Endocrinol. 25, 1636–1649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lieskovska J., Ling Y., Badley-Clarke J., Clemmons D. R. (2006) The role of Src kinase in insulin-like growth factor-dependent mitogenic signaling in vascular smooth muscle cells. J. Biol. Chem. 281, 25041–25053 [DOI] [PubMed] [Google Scholar]
- 7.Block K., Eid A., Griendling K. K., Lee D. Y., Wittrant Y., Gorin Y. (2008) Nox4 NAD(P)H oxidase mediates Src-dependent tyrosine phosphorylation of PDK-1 in response to angiotensin II: role in mesangial cell hypertrophy and fibronectin expression. J. Biol. Chem. 283, 24061–24076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Xi G., Shen X. C., Wai C., Clemmons D. R. (2013) Recruitment of Nox4 to a plasma membrane scaffold is required for localized reactive oxygen species generation and sustained Src activation in response to insulin-like growth factor-I. J. Biol. Chem. 288, 15641–15653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Xi G., Shen X., Maile L. A., Wai C., Gollahon K., Clemmons D. R. (2012) Hyperglycemia enhances IGF-I-stimulated Src activation via increasing Nox4-derived reactive oxygen species in a PKCζ-dependent manner in vascular smooth muscle cells. Diabetes 61, 104–113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Li H., Peng W., Zhuang J., Lu Y., Jian W., Wei Y., Li W., Xu Y. (2013) Vaspin attenuates high glucose-induced vascular smooth muscle cells proliferation and chemokinesis by inhibiting the MAPK, PI3K/Akt, and NF-κB signaling pathways. Atherosclerosis 228, 61–68 [DOI] [PubMed] [Google Scholar]
- 11.Panzhinskiy E., Ren J., Nair S. (2013) Protein tyrosine phosphatase 1B and insulin resistance: role of endoplasmic reticulum stress/reactive oxygen species/nuclear factor kappa B axis. PLoS One 8, e77228 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 12.Sun X., Belkin N., Feinberg M. W. (2013) Endothelial microRNAs and atherosclerosis. Curr. Atheroscler. Rep. 15, 372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kim G. Y., Nigro P., Fujiwara K., Abe J., Berk B. C. (2012) p62 binding to protein kinase C ζ regulates tumor necrosis factor α-induced apoptotic pathway in endothelial cells. Arterioscler. Thromb. Vasc. Biol. 32, 2974–2980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.McManus S., Roux S. (2012) The adaptor protein p62/SQSTM1 in osteoclast signaling pathways. J. Mol. Signal. 7, 1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kwan J., Wang H., Munk S., Xia L., Goldberg H. J., Whiteside C. I. (2005) In high glucose protein kinase C-zeta activation is required for mesangial cell generation of reactive oxygen species. Kidney Int. 68, 2526–2541 [DOI] [PubMed] [Google Scholar]
- 16.Huang C., Lin M. Z., Cheng D., Braet F., Pollock C. A., Chen X. M. (2014) Thioredoxin-interacting protein mediates dysfunction of tubular autophagy in diabetic kidneys through inhibiting autophagic flux. Lab. Invest. 94, 309–320 [DOI] [PubMed] [Google Scholar]
- 17.Gockerman A., Clemmons D. R. (1995) Porcine aortic smooth muscle cells secrete a serine protease for insulin-like growth factor binding protein-2. Circ. Res. 76, 514–521 [DOI] [PubMed] [Google Scholar]
- 18.Xi G., Shen X., Clemmons D. R. (2008) p66shc negatively regulates insulin-like growth factor I signal transduction via inhibition of p52shc binding to Src homology 2 domain-containing protein tyrosine phosphatase substrate-1 leading to impaired growth factor receptor-bound protein-2 membrane recruitment. Mol. Endocrinol. 22, 2162–2175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Maile L. A., Capps B. E., Miller E. C., Aday A. W., Clemmons D. R. (2008) Integrin-associated protein association with SRC homology 2 domain containing tyrosine phosphatase substrate 1 regulates igf-I signaling in vivo. Diabetes 57, 2637–2643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wilson M. I., Gill D. J., Perisic O., Quinn M. T., Williams R. L. (2003) PB1 domain-mediated heterodimerization in NADPH oxidase and signaling complexes of atypical protein kinase C with Par6 and p62. Mol. Cell 12, 39–50 [DOI] [PubMed] [Google Scholar]
- 21.Le Good J. A., Ziegler W. H., Parekh D. B., Alessi D. R., Cohen P., Parker P. J. (1998) Protein kinase C isotypes controlled by phosphoinositide 3-kinase through the protein kinase PDK1. Science 281, 2042–2045 [DOI] [PubMed] [Google Scholar]
- 22.Duran A., Diaz-Meco M. T., Moscat J. (2003) Essential role of RelA Ser311 phosphorylation by zetaPKC in NF-kappaB transcriptional activation. EMBO J. 22, 3910–3918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hirai T., Chida K. (2003) Protein kinase Czeta (PKCzeta): activation mechanisms and cellular functions. J. Biochem. 133, 1–7 [DOI] [PubMed] [Google Scholar]
- 24.Bandyopadhyay G., Sajan M. P., Kanoh Y., Standaert M. L., Quon M. J., Reed B. C., Dikic I., Farese R. V. (2001) Glucose activates protein kinase C-zeta/lambda through proline-rich tyrosine kinase-2, extracellular signal-regulated kinase, and phospholipase D: a novel mechanism for activating glucose transporter translocation. J. Biol. Chem. 276, 35537–35545 [DOI] [PubMed] [Google Scholar]
- 25.Zhang L., Pang S., Deng B., Qian L., Chen J., Zou J., Zheng J., Yang L., Zhang C., Chen X., Liu Z., Le Y. (2012) High glucose induces renal mesangial cell proliferation and fibronectin expression through JNK/NF-κB/NADPH oxidase/ROS pathway, which is inhibited by resveratrol. Int. J. Biochem. Cell Biol. 44, 629–638 [DOI] [PubMed] [Google Scholar]
- 26.Xia L., Wang H., Goldberg H. J., Munk S., Fantus I. G., Whiteside C. I. (2006) Mesangial cell NADPH oxidase upregulation in high glucose is protein kinase C dependent and required for collagen IV expression. Am. J. Physiol. Renal Physiol. 290, F345–F356 [DOI] [PubMed] [Google Scholar]
- 27.Zhang M., Song P., Xu J., Zou M. H. (2011) Activation of NAD(P)H oxidases by thromboxane A2 receptor uncouples endothelial nitric oxide synthase. Arterioscler. Thromb. Vasc. Biol. 31, 125–132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sugimoto R., Warabi E., Katayanagi S., Sakai S., Uwayama J., Yanagawa T., Watanabe A., Harada H., Kitamura K., Noguchi N., Yoshida H., Siow R. C., Mann G. E., Ishii T. (2010) Enhanced neointimal hyperplasia and carotid artery remodelling in sequestosome 1 deficient mice. J. Cell. Mol. Med. 14(6B), 1546–1554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Guo R., Zhang Y., Turdi S., Ren J. (2013) Adiponectin knockout accentuates high fat diet-induced obesity and cardiac dysfunction: role of autophagy. Biochim. Biophys. Acta 1832, 1136–1148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chen Y., Azad M. B., Gibson S. B. (2009) Superoxide is the major reactive oxygen species regulating autophagy. Cell Death Differ. 16, 1040–1052 [DOI] [PubMed] [Google Scholar]
- 31.Diaz-Meco M. T., Moscat J. (2012) The atypical PKCs in inflammation: NF-κB and beyond. Immunol. Rev. 246, 154–167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shen X., Xi G., Radhakrishnan Y., Clemmons D. R. (2010) PDK1 recruitment to the SHPS-1 signaling complex enhances insulin-like growth factor-i-stimulated AKT activation and vascular smooth muscle cell survival. J. Biol. Chem. 285, 29416–29424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ren J., Wang J., Wang Z., Wu J. (2014) Structural and biochemical insights into the homotypic PB1-PB1 complex between PKCζ and p62. Sci. China Life Sci. 57, 69–80 [DOI] [PubMed] [Google Scholar]
- 34.Zhang P., Chan J., Dragoi A. M., Gong X., Ivanov S., Li Z. W., Chuang T. H., Tuthill C., Wan Y., Karin M., Chu W. M. (2005) Activation of IKK by thymosin alpha1 requires the TRAF6 signalling pathway. EMBO Rep. 6, 531–537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Huang H. C., Huang C. Y., Lin-Shiau S. Y., Lin J. K. (2009) Ursolic acid inhibits IL-1beta or TNF-alpha-induced C6 glioma invasion through suppressing the association ZIP/p62 with PKC-zeta and downregulating the MMP-9 expression. Mol. Carcinog. 48, 517–531 [DOI] [PubMed] [Google Scholar]
- 36.Leitges M., Sanz L., Martin P., Duran A., Braun U., García J. F., Camacho F., Diaz-Meco M. T., Rennert P. D., Moscat J. (2001) Targeted disruption of the zetaPKC gene results in the impairment of the NF-kappaB pathway. Mol. Cell 8, 771–780 [DOI] [PubMed] [Google Scholar]
- 37.Kai M., Yasuda S., Imai S., Toyota M., Kanoh H., Sakane F. (2009) Diacylglycerol kinase alpha enhances protein kinase Czeta-dependent phosphorylation at Ser311 of p65/RelA subunit of nuclear factor-kappaB. FEBS Lett. 583, 3265–3268 [DOI] [PubMed] [Google Scholar]
- 38.Yerneni K. K., Bai W., Khan B. V., Medford R. M., Natarajan R. (1999) Hyperglycemia-induced activation of nuclear transcription factor kappaB in vascular smooth muscle cells. Diabetes 48, 855–864 [DOI] [PubMed] [Google Scholar]
- 39.Wedgwood S., Lakshminrusimha S., Czech L., Schumacker P. T., Steinhorn R. H. (2013) Increased p22(phox)/Nox4 expression is involved in remodeling through hydrogen peroxide signaling in experimental persistent pulmonary hypertension of the newborn. Antioxid. Redox Signal. 18, 1765–1776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gupta G. K., Dhar K., Del Core M. G., Hunter W. J. III, Hatzoudis G. I., Agrawal D. K. (2011) Suppressor of cytokine signaling-3 and intimal hyperplasia in porcine coronary arteries following coronary intervention. Exp. Mol. Pathol. 91, 346–352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sprague A. H., Khalil R. A. (2009) Inflammatory cytokines in vascular dysfunction and vascular disease. Biochem. Pharmacol. 78, 539–552 [DOI] [PMC free article] [PubMed] [Google Scholar]